
Clinical Presentation, Diagnosis and Treatment of TTR Amyloidosis
Abstract
Systemic amyloidosis can be hereditary or acquired with autosomal dominant mutations in the transthyretin gene (TTR) being the most common cause of hereditary amyloidosis. ATTRm amyloidosis is a multi-system disorder with cardiovascular, peripheral and autonomic nerve involvement that can be difficult to diagnose due to phenotypic heterogeneity. This review will focus on the neuropathic manifestations of ATTRm, the genotype-phenotype variability, the diagnostic approach and the recent therapeutic advances in this disabling condition.
INTRODUCTION
There are more than 30 proteins that can cause localised or systemic amyloidosis; 12 of which acquire amyloidogenicity from a germline mutation (Table 1 outlines the characteristics of the more common amyloidogenic proteins). Of the hereditary amyloidosis, transthyretin (TTR) is the most prevalent subtype with more than 100 pathogenic TTR mutations reported to date. TTR is amyloidogenic in both wild-type and hereditary forms (ATTRwt and ATTRm are the approved nomenclature for wild type and hereditary ATTR amyloidosis, respectively) [8]. TTR is primarily synthesized in the liver and is a 127-residue homotetrameric protein that carries thyroxine and retinol-binding protein [1]. Dissociation of TTR followed by aggregation and misfolding of the oligomers and monomers causes formation of insoluble amyloid fibrils which deposit systemically resulting in peripheral and/or autonomic neuropathy, and other systemic manifestations, particularly cardiomyopathy [9]. Significant progress in the treatment of ATTRm has been made with exciting developments in gene silencing therapies. This review will discuss the clinical features of ATTRm neuropathy and highlight therapeutic developments in the field.
Table 1
A summary of the common types of amyloid and their clinical features [5, 10–12]
| Form of amyloidosis | Acquired or hereditary | Underlying diagnosis | Precursor protein | Organ Involvement | Treatment | |||||||
| Peripheral Nervous System | Autonomic Nervous System | Heart | Kidney | Liver | GIT | Eyes | Tongue | |||||
| AL | Acquired | Plasma cell dyscrasia | Monoclonal immunoglobulin light chain | ++ | ++ | +++ | +++ | ++ | ++ | – | +++ | Chemotherapy/ASCT |
| ATTRm | Hereditary | Mutations in TTR gene | Mutant TTR | +++ | +++ | ++ | +/– | – | – | ++ | – | Liver transplant for younger patients with V30MATTR, TTR stabilisers or genetic therapies |
| ATTRwt | Acquired | Wild-type TTR | + | – | +++ | – | – | – | – | – | Supportive | |
| AA | Acquired | Inflammatory disorders | SAA | – | ++ | +/– (late) | +++ | +(Late) | + | – | +/– | Suppression of inflammation |
| AFib | Hereditary | Mutations in fibrinogen α-chain gene | Mutant fibrinogen | – | – | +/– | +++ | +/– | – | – | – | Supportive, organ transplant |
| AAPoA1 | Hereditary | Mutations in apolipoprotein A1 gene | MutantApoA1 | + | – | + | ++ | ++ | – | – | – | Supportive, organ transplant |
| ALys | Hereditary | Mutations in lysozyme gene | Mutant lysozyme | – | – | +/– | +++ | ++ | ++ | – | – | Supportive, organ transplant |
| AGel | Hereditary | Mutations in gelsolin gene | Mutant gelsolin | ++(Cranial) | – | + | + | – | – | – | – | Supportive |
| Aβ2M | Acquired or hereditary | Long-term dialysis | β2M | – (CTS) | – | +/– | – | +/– | – | – | – | Supportive, renal transplant |
Abbreviations: AA = amyloid A, AApoA1 = apolipoprotein A1 amyloid, Aβ2M =β2-microglobulin amyloid, AFib = fibrinogen A α-chain amyloid, AGel = gelsolin amyloid, AL = amyloid light chain, ALys = lysozyme amyloid. ASCT = autologous stem cell transplant, ATTR = amyloid transthyretin, CTS- Carpal tunnel syndrome, GIT = Gastrointestinal, SAA = serum amyloid A. +++ very common, ++ common, + less common, +/– rare, – does not occur, or not applicable.
CLINICAL PRESENTATION OF ATTR
ATTRm amyloidosis is a rare disease with diverse clinical manifestations that is in part determined by the genotype. Given this complexity, there can be a delay in diagnosis of up to 4 years from symptom onset for patients with ATTRm presenting with a peripheral neuropathy and up to 8 years for patients presenting with a cardiomyopathy [13]. Carpal tunnel syndrome (CTS) can be the initial symptom in up to 33% of patients with a mean period of 4–6 years before other organs are clinically involved [14]. Patients then usually develop a peripheral and autonomic neuropathy, and often cardiac involvement. As TTR is also produced within the choroid plexus and the epithelium of the retina, central nervous system (CNS) manifestations can also rarely occur, and are more common for some disease-causing mutations that have a predilection for the CNS, such as ATTRL12P [15, 16].
Peripheral neuropathy
V30M is the most common ATTR mutation worldwide and is endemic in Portugal, Japan and Sweden [17]. The early-onset phenotype, with onset in the 20s to 40s, was first described in Portugal and was previously known as familial amyloid polyneuropathy (FAP). Patients initially present with a length-dependent, painful, small-fibre neuropathy progressing over time to a generalised sensorimotor neuropathy. In contrast, cases from non-endemic areas, and late-onset presentations of many mutations may present with involvement of all sensory modalities. Some mutations have a faster disease progression than ATTRV30M, such as the ATTRL55P where patients may present with a peripheral neuropathy as early as 19 years of age and die within 3 to 8 years [18]. Amyloidosis can also present atypically as an upper-limb predominant neuropathy, a radiculopathy or a myopathy [5–7].
Autonomic neuropathy
ATTRm amyloidosis often involves early autonomic involvement and overall, up to 75% of patients with ATTRm develop symptoms of an autonomic neuropathy, affecting the cardiac, gastrointestinal, and genitourinary systems [19]. Autonomic symptoms can be very disabling with a high morbidity and the most worrying autonomic manifestation is arrhythmias; and sudden death has been reported. The severity of cardiovascular autonomic impairment is unrelated to the severity of the peripheral neuropathy. Orthostatic hypotension is a troublesome symptom that can present with non-specific symptoms of fatigue, reduced exercise tolerance and vague dizziness. Gastrointestinal symptoms caused by amyloid infiltration of the mesenteric plexus include gastroparesis, dysmotility, constipation and diarrhoea (often nocturnal initially). Genitourinary dysfunction can include urinary retention, nocturia, incomplete emptying and frequency; erectile dysfunction is very common in male patients. Pupillomotor and sudomotor functions can also be impaired [20].
Other systemic manifestations
Certain TTR mutations primarily cause cardiac amyloidosis, the most common worldwide being ATTRV122I, which is present in approximately 4% of African Americans and causes a restrictive cardiomyopathy. ATTRT60A can present with cardiac involvement alone but with time both cardiac and peripheral nerve involvement is common. Cardiomyopathy is more common with late-onset ATTRV30M cases than the classic early-onset presentation [21]. Oculoleptomeningeal amyloidosis, associated with, but not limited to, ATTRL12P can present with a range of neurological signs and symptoms including headaches, seizures, subarachnoid haemorrhage, and hearing or visual loss [15]. Ocular abnormalities including vitreous deposits are reported in approximately 10% of patients with ATTRV30M. Significant, unexplained weight loss of more than 10% of bodyweight is a common manifestation of systemic amyloidosis [5].
Other hereditary amyloidosis that can cause neuropathies
Mutations in ApoA1 gene have been associated with systemic amyloidosis mainly causing renal failure. However, one mutation, AApoAIG26R, can cause a length-dependent, sensorimotor neuropathy similar to the neuropathy associated with ATTRm [22]. Gelsolin related amyloidosis (also referred to as Finnish type amyloidosis) initially presents as a lattice corneal dystrophy followed by a progressive cranial neuropathy causing bilateral facial weakness [23].
PHENOTYPIC VARIATIONS WITHIN GENOTYPES
Penetrance
The development of disease-modifying treatments raises questions about screening and timely access to treatments. Hence, knowing the penetrance of different mutations and the reasons for varying clinical manifestations is relevant in clinical practice. ATTRV30M amyloidosis has variable disease penetrance depending on geographic location and age of onset. In Sweden, where the age of onset is later than in Portugal, penetrance of ATTRV30M is reported to be less than 50%, compared with 80% penetrance in the early-onset Portuguese population. Penetrance of ATTRT60A is difficult to define as age of onset of disease is in the 60s, and patients may die from other causes before manifesting the disease [24, 25].
INVESTIGATIONS
Diagnosing neuropathy
Depending on the presenting symptoms, patients may see a range of physicians prior to a diagnosis of ATTRm amyloidosis. Neurologists are generally referred patients with symptoms suggestive of a peripheral neuropathy or those with a known diagnosis of amyloidosis to be investigated for a clinical or subclinical neuropathy. Initially, patients may only have clinical features of a small fibre neuropathy (SFN), and conventional nerve conduction studies (NCS) may be normal. There are quantitative and qualitative methods to measure small fibre dysfunction including quantitative sensory testing (QST), which is a psychophysical test of small fibre function. Quantification of intraepidermal nerve fibres in a skin biopsy is the gold standard for diagnosing SFN and has a sensitivity and specificity of around 90% [26]. As the peripheral neuropathy progresses, large fibres are affected. Classically, NCS demonstrate an axonal, sensorimotor, length-dependent neuropathy with frequent median nerve entrapment at the wrists. However, in both ATTRm and AL amyloidosis, slow conduction with prolonged distal motor latencies can be seen which may lead to a neurophysiological diagnosis of a demyelinating neuropathy and subsequently a clinical diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP) [27].
Autonomic neuropathy
Symptoms of autonomic dysfunction are a common feature of ATTRm related neuropathy. Autonomic function tests can help diagnose dysautonomia and investigate sudomotor, cadiovagal and adrenergic function using tests that assess physiologic or neurochemical function in response to a change in the environment [28]. Cardiovagal function is commonly assessed by quantifying the heart rate response to deep breathing and to the Valsalva manoeuvre, with a loss of heart rate variability suggesting cardiovagal dysfunction. This is complemented by the head-up tilt test and measuring plasma catecholamines in response to the tilt to test for orthostatic hypotension. An attenuated plasma catecholamines increase suggests sympathetic dysfunction. Orthostatic hypotension is defined as a sustained reduction of systolic blood pressure of at least 20 mmHg or diastolic blood pressure of 10 mmHg within 3 min of standing or head-up tilt to at least 60 degrees on a tilt table [29]. Pupillometry, urodynamics and gastrointestinal motility studies or manometry can be performed to assess autonomic dysfunction in these organs [30].
Investigate cause of neuropathy
If the clinical suspicion for ATTRm is high, for example, if a patient has a painful, axonal neuropathy, dominant family history, cardiac and/or autonomic symptoms or an ethnicity with a high prevalence of ATTRm, investigations for the cause of the neuropathy may be very limited. ATTRm amyloidosis can be excluded if genetic testing confirms wild-type TTR, as sequence analysis of the gene detects more than 99% of pathogenic variants [31]. An online database (http://www.amyloidosismutations.com/main_menu.html) provides an updated list of amyloidogenic mutations and their phenotypes [24]. In the absence of diabetes, the development of a neuropathy with autonomic involvement should always raise the possibility of amyloidosis. In more ambiguous cases, a broad, routine approach to investigating neuropathies may be taken, especially to exclude acquired causes.
Amyloidosis is a histologic diagnosis; however, a nerve biopsy is not always needed if the neurological phenotype is concordant with the diagnosis, no other cause for a neuropathy is found and the patient has a pathogenic mutation in the TTR gene. In these cases, to diagnose systemic amyloidosis, a less invasive method can be used such as subcutaneous abdominal fat aspirate or labial salivary gland biopsy, which have a sensitivity of 50–80% and 90%, respectively [2–4]. Sural nerve biopsies have a sensitivity cited as high as 86%, but it can be difficult to confirm the diagnosis in any tissue due to patchy deposition and repeated biopsies, or biopsies from different sites may be required [32]. Congo-red positive areas of the biopsy undergo immunohistochemistry for typing of the amyloid protein (TTR vs. other), and if this is equivocal, the biopsy undergoes laser microdissection followed by mass spectrometry [33]. These tests to identify protein type can be performed on any affected tissue including salivary gland, nerve, rectal mucosa, endomyocardial biopsy specimens and tenosynovial tissues obtained at carpal tunnel release surgery. Sequencing of the TTR gene is required to differentiate between ATTRwt and ATTRm.
Systemic involvement of ATTRm
Identifying the extent of amyloidosis is important for management and prognosis. There are specialised imaging modalities that are sensitive in identifying organ deposition and the pattern of uptake can provide clues to the type of amyloid. Serum amyloid P (SAP) is a glycoprotein found in all types of amyloid deposits. SAP scintigraphy uses radiolabelled SAP as a tracer to quantify and identify amyloid deposition, but has poor visualisation of heart, peripheral nerve and the CNS. SAP scintigraphy has high sensitivity, 90%, in AA and AL amyloidosis [34], but only 48% in ATTR amyloidosis [35]. A radionuclide tracer with greater sensitivity in ATTR amyloidosis is 99 m-technetium-3,3, -diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD). It has a sensitivity and specificity of identifying cardiac ATTR amyloid deposits of 91% and 82%, respectively [36].
DIFFERENTIAL DIAGNOSES
The most common misdiagnosis of amyloidosis affecting the peripheral nerves is CIDP as patients with ATTRm can have slowed conduction on neurophysiology and raised CSF protein. In a study of 150 patients with ATTRm, 42 (32%) had been misdiagnosed, 30 (61%) of which were initially diagnosed as CIDP, and 2% as vasculitic neuropathy [37]. Out of the patients misdiagnosed as CIDP, seven patients fulfilled EFNS/PNS criteria for definite CIDP with conduction velocities as low as 33 m/s in the upper limbs. Also, as amyloid can have patchy deposition, it can also be misdiagnosed as a radiculopathy or plexopathy. As such there should be a high suspicion for ATTRm in patients diagnosed with CIDP that do not respond to immunomodulatory treatment [38]. AL-amyloidosis can also be difficult to distinguish from ATTRm as both may initially present with isolated CTS and up to 65% of patients with AL-amyloidosis develop symptoms of an autonomic neuropathy [19]. The coexistence of monoclonal gammopathy of undetermined significance (MGUS) with ATTRm or ATTRwt is recognised, especially in older people, but not widely appreciated. In a study of 57 patients with ATTRV122I amyloidosis, aged between 50–90, median age 71 years, 49% had abnormal serum free light chain ratios and/or paraprotein on immunofixation, suggesting neurologists may see abnormal haematological investigations commonly in these patients [39]. Therefore, even in the presence of ATTRm, AL-amyloidosis is possible and vice versa. Hence, biopsies from different sites and organs may be necessary and typing the amyloid fibrils is essential.
PROGNOSIS
Natural history studies of untreated ATTRm show that late-onset ATTRV30M cases require a single point stick to mobilise within 4 years of diagnosis and a wheelchair within 7 years compared with 10 and 17 years, respectively, for patients with the classic ATTRV30M presentation [40]. Cardiac biomarkers, specifically N-terminal pro b-type natriuretic peptide (NT-proBNP) is an independent predictor of mortality in AL, ATTRm and ATTRwt amyloidosis [41]. In one study of 116 patients with untreated ATTRm, the four year survival for ATTRV30M, ATTRT60A and ATTRV122I, a predominantly cardiac phenotype, was 79%, 40% and 16%, respectively [42]. There is a significant difference between time to death in the early compared to late-onset ATTRV30M, median number of years is 16.9 vs. 6.8, respectively [40].
SYMPTOMATIC TREATMENT
Patients with ATTRm require a multidisciplinary approach to their disease and symptomatic treatment remains very important. Troublesome symptoms include neuropathic pain, increasing weakness, autonomic dysfunction, especially orthostatic hypotension and altered bowel habit, and cardiac symptoms of heart failure, arrhythmias or heart block frequently needing a pacemaker or other devices. When considering neuropathic pain agents, care must be sought before using drugs that can affect cardiac conduction or cause anticholinergic side effects.
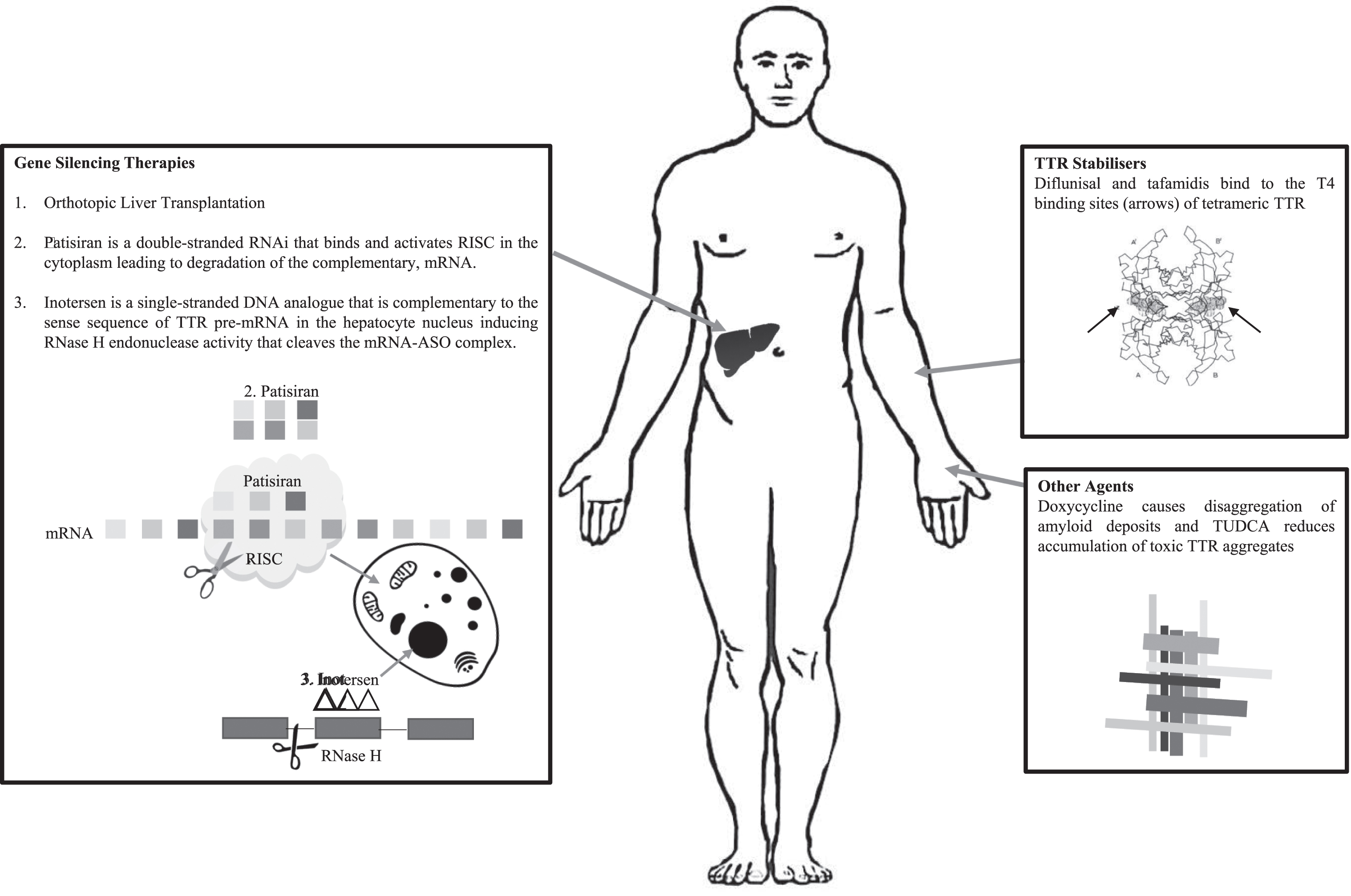
DISEASE MODIFYING THERAPIES (See Fig. 1 FOR Summary)
TTR exists as a stable tetramer, the dissociation of which into monomers is required for amyloid deposition. In addition, there is emerging evidence that mutant TTR undergoes selective proteolytic cleavage that predisposes the TTR tetramer to dissociate, particularly when exposed to shear forces such as those in the heart and the carpal tunnel [43]. Current disease modifying therapies for ATTRm aim to either stabilise the TTR tetramer (Diflunisal and Tafamidis) or to reduce mutant TTR production (liver transplantation or gene-silencing therapies).
TREATMENTS AVAILABLE PRIOR TO 2018
Liver transplantation
The liver produces the majority of circulating TTR and therefore orthotopic liver transplantation (OLT) is an attractive treatment strategy for ATTRm as it significantly reduces the production of mutant protein. In carefully selected patients and experienced centres, the 10-year survival following OLT is 75–78% in early-onset ATTRV30M [44]. Non-V30M patients, however, have a worse 5 and 10-year survival following OLT of 59% and 44%, respectively, and late-onset ATTRV30M survival is significantly reduced compared to early-onset patients [44]. Other major prognostic factors for the success of OLT include age and duration of disease before transplantation. OLT rarely leads to regression of autonomic or peripheral neuropathy but does slow rate of progression, and in some cases, leads to stability. The limitations of OLT include the requirement for surgery and the associated mortality; the incidence of the main causes of death of ATTRm patients receiving OLT are comparable to patients being transplanted for other reasons, except for cardiac-related deaths which are much greater in the ATTRm population [44]. Also, there is ongoing synthesis of ATTRm from retinal pigment epithelium and choroid plexus, and the progression of ATTRwt deposition, especially in the myocardium, with up to 18.6% of patients developing a cardiomyopathy post OLT [45]. Nevertheless, the experience with OLT has encouraged the development of other disease-modifying therapies aimed at reducing TTR production from the liver.
Diflunisal
Diflunisal is one of two TTR stabilisers used in clinical practice, the other being tafamidis. It is a nonsteroidal anti-inflammatory drug that binds to the thyroxine (T4) binding sites of tetrameric TTR thereby reducing dissociation and misfolding, and subsequent formation of amyloid fibrils [46]. The efficacy of difunisal, 500 mg a day, in patients with ATTRm and peripheral or autonomic neuropathy was demonstrated in a randomised, double-blind, placebo-controlled study conducted of 130 patients over 2 years. Diflunisal reduced the rate of progression of neurological impairment and preserved quality of life (QOL) compared to placebo [47]. Diflunisal is not licenced for ATTRm in Australia, EU, UK or USA but off-label use is managed by specialist centres.
Tafamidis
Tafamidis is a newer drug specifically developed to stabilise the TTR tetramer by also binding to the T4 binding sites. Tafamidis has been approved in over 40 countries for treatment of ATTRm neuropathy. In the UK, tafamidis is licenced for the treatment of ATTRV30M with polyneuropathy, and it has orphan drug status in the USA, EU and Australia [48]. A multicentre, randomised, double-blind, placebo-controlled study of tafamidis in 128 ATTRV30M patients completed in May 2009 over 18 months failed to meet its co-primary endpoints [49]. These results, however, were confounded by a greater than expected dropout in the treatment group due to the availability of OLT. All participants who underwent OLT were deemed to be non-responders in the intention to treat (ITT) analysis thereby under-powering the trial. However, when the patients who completed the study were analysed for the co-primary endpoints, 60% of the patients in the tafamidis group were Neuropathy Impairment Score-Lower Limb (NIS-LL) responders compared with 38.1% in the placebo group, and the deterioration from baseline in Norfolk Quality of Life Questionnaire-Diabetic Neuropathy (Norfolk QOL-DN) questionnaire scores was 0.1 point in the tafamidis group compared with 8.9 points in the placebo group [49]. A prospectively planned, interim analysis was then conducted on patients included in a 12-month, open-label extension of the above study and a single-arm, open label study in 18 non-ATTRV30M patients [50]. Mean, cumulative tafamidis exposure was 5.1 years in the ATTRV30M patients who had 30 months of tafamidis treatment, 3.5 years in the placebo arm who received tafamidis in the open-label extension only, and 3.6 years in the non-ATTRV30M patients. All tafamidis groups had slower rate of deterioration as measured by NIS-LL than natural history data [47]. A 30-month, phase 3, placebo-controlled, randomised study published in 2018 investigated the efficacy and safety of two doses of tafamidis, 20 mg and 80 mg daily, in 441 patients with either ATTRwt or ATTRm amyloidosis related cardiomyopathy [51]. The safety profile was similar between the tafamidis and placebo arms. All-cause mortality, rate of cardiovascular-related hospitalisations and decline in functional capacity and QOL were lower in the tafamidis groups compared with the placebo group [51]. No serious adverse events were reported. This is promising for the treatment of the cardiac involvement in TTR amyloidosis.
Doxycycline
The combination of doxycycline and tauroursodeoxycholic acid (TUDCA), a biliary acid, reduced TTR tissue deposition and SAP in ATTRV30M amyloidosis mouse models [52]. The two agents are complementary in their targets, doxycycline causes disaggregation of amyloid deposits and TUDCA reduces accumulation of toxic TTR aggregates, but only works on non-fibrillar TTR deposits. A phase II open-label study completed in October 2015, evaluated the efficacy and safety of this combination over 12 months in 20 patients, 17 patients with varying genotypes of ATTRm, and 3 with ATTRwt. 7 patients completed the 12-month treatment trial, 5 patients with a polyneuropathy had less than 2-point increase in NIS-LL, no patients had a worsening of modified Body Mass Index (mBMI) and no patients had a worsening of echocardiographic findings or heart failure symptoms. Doxycycline has orphan drug status in the EU through the European Medicines Authority (EMA). There is an ongoing phase 3 randomised study (ClinicalTrials.gov Identifier: NCT03481972) investigating doxycycline and TUDCA compared with standard therapy alone in ATTRm or ATTRwt cardiac amyloidosis.
NEW TREATMENTS FROM TRIALS PUBLISHED IN 2018
In 2018, two novel treatments for ATTRm amyloidosis related neuropathy, patisiran and inotersen (formerly IONIS-TTRRx/ISIS 420915) attained EMA and United States Food and Drug Administration (FDA) approval following two successful phase-3 trials both published in New England Journal of Medicine in July, 2018 [53–55]. Both drugs are genetic therapies that suppress ATTRwt and ATTRm synthesis in the liver through different but similar mechanisms and represent a paradigm shift in the management of this devastating disease.
Patisiran
Patisiran is a double-stranded synthetic ribonucleic acid molecule (RNAi) that targets hepatocytes in the liver and binds and activates the RNA-induced silencing complex (RISC) leading to degradation of the complementary, TTR messenger RNA (mRNA), thereby reducing protein translation for both mutant and wild-type TTR [53, 56] (See Fig. 1). Patisiran is administered as an intravenous infusion every 21 days.
Fig.1
Treatments for ATTRm and Their Targets.
The APOLLO trial was a 18-month, randomised, double-blind, multicentre, placebo-controlled trial in 225 patients (approximately 50% had ATTRV30M) with ATTRm amyloidosis related peripheral neuropathy [53]. This study included patients with prior treatment with tetramer stabiliser and patients with varying levels of disability, ranging from sensory symptoms to the requirement of 1 or 2 sticks to mobilise. All primary and secondary endpoints were met; the primary endpoint was a change from baseline in modified NIS + 7 (mNIS + 7) between groups. At the end of 18 months, the least-squares mean change in mNIS + 7 from baseline was – 6.0 in the patisiran group compared with 28.0 in the placebo arm. This was significant for all subgroups, including age, genotype, clinical severity, previous tetramer stabiliser usage, and presence of cardiomyopathy. Patisiran resulted in a median of 81% decrease in serum TTR from baseline which was consistent across age, gender and genotype [53]. There were more infusion related reactions in patients receiving patisiran but the incidence of serious adverse events and deaths were similar in the two groups [53]. 186 of the eligible 187 patients who completed the study were enrolled in to the open-label extension study expected to be completed in 2022.
Inotersen
Inotersen is a single-stranded deoxynucleotide analogue (DNA) complementary to the sequence of TTR pre-mRNA. The hybridisation of Inotersen to the pre-mRNA induces RNase H endonuclease activity that cleaves the mRNA-ASO complex thereby inhibiting production of mutant and wild-type TTR protein [54–57] (See Fig. 1). Inotersen is administered as a weekly subcutaneous injection.
The NEURO-TTR trial was a 15-month, randomised, double-blind, multicentre, placebo-controlled, trial in 172 patients (approximately 50% had ATTRV30M) with ATTRm amyloidosis related peripheral neuropathy [54]. This study included patients with prior treatment with tetramer stabiliser and patients with varying levels of disability. Inotersen met both of its primary endpoints, there was significantly less decline in the neuropathy and QOL measure in the inotersen group compared with the placebo group [54]. At the end of the 15 months, patients who received inotersen had an average increase of 5.8 points from baseline in mNIS + 7, compared with 25.5 points in the placebo group, and 1.0 point increase in the Norfolk QOL-DN score compared with 12.7 points in the placebo arm. This was significant across all of the subgroups, including genotype, age, race, previous exposure to TTR stabilisers and presence of cardiomyopathy. Patients receiving inotersen had a median reduction in serum TTR levels of 79% compared to baseline. Thrombocytopenia (platelet count less than 140,000 per cubic millimetre) occurred in 54% of patients receiving inotersen resulting in a fatal intracranial haemorrhage in one patient. As a result, regular platelet monitoring was introduced, and no further episodes of severe thrombocytopenia were encountered. Glomerulonephritis occurred in three patients in the inotersen group, two of which improved with corticosteroid treatment [54]. 139 patients completed the study period of which 135 were enrolled in to the open-label extension study expected to be completed in 2022.
COMPLICATIONS AND LIMITATIONS OF DISEASE-MODIFYING TREATMENTS
The name transthyretin reflects the numerous roles of the protein, TRANSport of THYroid hormones and RETINol-binding protein [58]. Patients receiving these treatments therefore require vitamin A supplementation and monitoring of thyroid function. In addition, gene silencing therapies have off-target effects and toxicities related to their chemical structure, rather than the nucleotide sequence. These include activation of the complement and coagulation cascades when administered systemically which may explain the infusion-site reactions [59]. Inotersen and patisiran do not cross the blood-brain barrier, and therefore, synthesis of TTR by the choroid plexus is not affected by these treatments. This may prove to be relevant as the complications of ongoing CNS ATTRm deposition after OLT are well-documented. In one case study of 87 patients with ATTRV30M who had OLT, after an average of 14.6 years, 31% of patients had focal neurological episodes classified clinically as focal seizures, aura-like episodes, or transient ischaemic attacks (MRI was contraindicated in all patients) and 5 patients had strokes [16]. There are also reports of the development or progression of vitreous opacities (reflecting ongoing retinal TTR production) and amyloid deposits in the pupillary margin that could lead to glaucoma after OLT [60].
IMPLICATIONS FOR CLINICAL PRACTICE
Early diagnosis of ATTRm is essential for timely access to treatment. The availability of gene silencing treatments raises issues regarding genetic screening and management of asymptomatic individuals. For example, patients with ATTRm may develop carpal tunnel syndrome with deposition of amyloid in the flexor retinaculum several decades before the development of a more generalised neuropathy or cardiomyopathy. The optimum time to commence treatment will need careful consideration and may have some genotype/ethnic specificities.
CONCLUSION
The development and success of gene silencing therapies in ATTRm amyloidosis is a breakthrough for adult-onset, neurodegenerative diseases. Slowing disease progression of this disabling, hereditary illness provides great hope for patients and treating teams. There are uncertainties about the long-term clinical benefit, when to initiate treatment and how to incorporate these treatments in to the current algorithms, however, they are a very welcome addition to presently available therapies. Accurate and timely diagnosis of ATTRm amyloidosis has become increasingly important as these therapies become imminently available around the world. Efficiently diagnosing patients requires first and foremost having a high clinical suspicion, an awareness of the systemic nature of the disease and an understanding of the available diagnostic techniques.
COMPETING INTERESTS
AMR has received support from Alnylam UK Limited to attend scientific meetings and an honorarium for speaking at a sponsored symposium. MMR has or has had consultancies with Alnylam Pharmaceuticals and Ionis Pharmaceuticals and MMR and ML were involved in the NEURO-TTR Trial (ClinicalTrials.gov Identifier: NCT01737398).
MMR designed the study, MK collected data and wrote a draft of the paper. AMR and ML provided detailed written edits and multiple further drafts of the review for publication. All authors work fulfilled the following: substantial contributions to the conception or design of the work or the acquisition, analysis or interpretation of data; drafting the work or revising it critically for important intellectual content; gave final approval of the version published; agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
ACKNOWLEDGMENTS
AMR is funded by a Wellcome Trust Postdoctoral Fellowship for Clinicians (110043/Z/15/Z).
MMR is grateful to the Medical Research Council (MRC), MRC Centre grant (G0601943), and the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) for their support. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
REFERENCES
[1] | Lim KH , Dasari AKR , Hung I , Gan Z , Kelly JW , Wemmer DE . Structural Changes Associated with Transthyretin Misfolding and Amyloid Formation Revealed by Solution and Solid-State NMR. Biochemistry. (2016) ;55: (13):1941–4. |
[2] | Do Amaral B , Coelho T , Sousa A , Guimarães A . Usefulness of labial salivary gland biopsy in familial amyloid polyneuropathy Portuguese type. Amyloid. (2009) ;16: (4):232–8. 10.3109/13506120903421850 |
[3] | Lechapt-Zalcman E , Authier FJ , Creance A , Voisin MC , Gherardi RK . Labial salivary gland biopsy for diagnosis of amyloid polyneuropathy. Muscle and Nerve. (1999) ;22: (1):105–7. |
[4] | Adams D , Suhr OB , Hund E , Obici L , Tournev I , Campistol JM , et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. (2016) ;29: : S14–26. 10.1097/WCO.0000000000000289 |
[5] | Théaudin M , Lozeron P , Algalarrondo V , Lacroix C , Cauquil C , Labeyrie C , et al. Upper limb onset of hereditary transthyretin amyloidosis is common in non-endemic areas. Eur J Neurol. (2018) ;ene: . 13845. 10.1111/ene.13845 |
[6] | Carr AS , Pelayo-Negro AL , Jaunmuktane Z , Scalco RS , Hutt D , Evans MRB , et al.. Transthyretin V122I amyloidosis with clinical and histological evidence of amyloid neuropathy and myopathy. Neuromuscul Disord. (2015) ;25: (6):511–5. 10.1016/j.nmd.2015.02.001 |
[7] | Planté-Bordeneuve V , Said G . Familial amyloid polyneuropathy. Lancet Neurol. (2011) ;10: (12):1086–97. 10.1016/S1474-4422(11)70246-0 |
[8] | Sipe JD , Benson MD , Buxbaum JN , Ikeda SI , Merlini G , Saraiva MJM , et al. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis Nomenclature Guidelines. Amyloid. (2016) ;23: (4):209–13. 10.1080/13506129.2016.1257986 |
[9] | Planté-Bordeneuve V , Ferreira A , Lalu T , Zaros C , Lacroix C , Adams D , et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology. (2007) ;69: (7):693–8. |
[10] | Parman Y , Adams D , Obici L , Galán L , Guergueltcheva V , Suhr OB , et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe. Curr Opin Neurol. (2016) ;29: : S3–13. 10.1097/WCO.0000000000000288 |
[11] | Gertz MA . Immunoglobulin light chain amyloidosis: Update on diagnosis, prognosis, and treatment. Am J Hematol. (2018) ;93: (9):1169–80. 10.1002/ajh.25149 . |
[12] | Liewluck T , Milone M . Characterization of isolated amyloid myopathy. Eur J Neurol. (2017) ;24: (12):1437–45. |
[13] | Hawkins PN , Ando Y , Dispenzeri A , Gonzalez-Duarte A , Adams D , Suhr OB . Evolving landscape in the management of transthyretin amyloidosis. Ann Med. (2015) ;47: (8):625–38. |
[14] | Sekijima Y , Ueda M , Koike H , Misawa S , Ishii T , Ando Y . Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: Red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. (2018) ;13: (1):6. 10.1186/s13023-017-0726-x |
[15] | McColgan P , Viegas S , Gandhi S , Bull K , Tudor R , Sheikh F , et al. Oculoleptomeningeal Amyloidosis associated with transthyretin Leu12Pro in an African patient. J Neurol. (2015) ;262: (1):228–34. |
[16] | Maia LF , Magalhães R , Freitas J , Taipa R , Pires MM , Osório H , et al. CNS involvement in V30M transthyretin amyloidosis: Clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry. (2015) ;86: (2):159–67. |
[17] | Rapezzi C , Quarta CC , Obici L , Perfetto F , Longhi S , Salvi F , et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: An Italian perspective. Eur Heart. (2013) ;34: (7):520–8. 10.1093/eurheartj/ehs123 |
[18] | Jacobson D , McFarlin D , Kane I , Buxbaum J . Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. (1992) ;89: (3):353–6. 10.1007/BF00220559 |
[19] | Pearson KT , Vota S . Amyloidosis and its management: Amyloid neuropathies. Curr Probl Cancer. (2016) ;40: (5-6):198–208. |
[20] | Gonzalez-Duarte A . Autonomic involvement in hereditary transthyretin amyloidosis (hATTR amyloidosis). Clin Auton Res. (2018) ; (0123456789). 10.1007/s10286-018-0514-2 |
[21] | Alavi A , French LE , Davis MD , Brassard A , Kirsner RS . Pyoderma Gangrenosum: An Update on Pathophysiology, Diagnosis and Treatment. Am J Clin Dermatol. (2017) ;18: (3):355–72. |
[22] | Nichols WC , Gregg RE , Brewer HB , Benson MD . A mutation in apolipoprotein A-I in the Iowa type of familial amyloidotic polyneuropathy. Genomics. (1990) ;8: (2):318–23. 10.1016/0888-7543(90)90288-6 |
[23] | Perfetto F , Moggi-Pignone A , Livi R , Tempestini A , Bergesio F , Matucci-Cerinic M . Systemic amyloidosis: A challenge for the rheumatologist. Nat Rev Rheumatol. (2010) ;6: (7):417–29. 10.1038/nrrheum.2010.84 |
[24] | Wechalekar AD , Gillmore JD , Hawkins PN . Systemic amyloidosis. Lancet. (2016) ;387: (10038):2641–54. 10.1016/S0140-6736(15)01274-X |
[25] | Reilly MM , Staunton H , Harding AE . Familial amyloid polyneuropathy (TTR ala 60) in north west Ireland: A clinical, genetic, and epidemiological study. J Neurol Neurosurg Psychiatry. (1995) ;59: (1):45–9. |
[26] | Terkelsen AJ , Karlsson P , Lauria G , Freeman R , Finnerup NB , Jensen TS . The diagnostic challenge of small fibre neuropathy: Clinical presentations, evaluations, and causes. Lancet Neurol. (2017) ;16: (11):934–44. 10.1016/S1474-4422(17)30329-0 |
[27] | Lozeron P , Mariani L-L , Dodet P , Beaudonnet G , Théaudin M , Adam C , et al. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology. (2018) ;91: (2):e143–52. 10.1212/WNL.0000000000005777 |
[28] | Low PA , Tomalia VA , Park K-J . Autonomic Function Tests: Some Clinical Applications. J Clin Neurol. (2013) ;9: (1):1. 10.3988/jcn.2013.9.1.1 |
[29] | Freeman R , Wieling W , Axelrod FB , Benditt DG , Benarroch E , Biaggioni I , et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. (2011) ;21: (2):69–72. |
[30] | Weimer LH . Autonomic Testing. Neurologist. (2010) ;16: (4):215–22. 10.1097/NRL.0b013e3181cf86ab |
[31] | Sekijima Y . Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. (2015) ;86: (9):1036–43. 10.1136/jnnp-2014-308724 |
[32] | Simmons Z , Specht CS . The neuromuscular manifestations of amyloidosis. J Clin Neuromuscul Dis. (2010) ;11: (3):145–57. |
[33] | Leung N , Nasr SH , Sethi S . How I Treat amyloidosis: The importance of accurate diagnosis and amyloid typing. Blood. (2012) ;120: (16):3206–13. 10.1182/blood-2012-03-413682 |
[34] | Sachchithanantham S , Wechalekar AD . Imaging in systemic amyloidosis. Br Med Bull. (2013) ;107: (1):41–56. |
[35] | Hazenberg BPC , Van Rijswijk MH , Piers DA , Lub-De Hooge MN , Vellenga E , Haagsma EB , et al. Diagnostic performance of123I-labeled serum amyloid P component scintigraphy in patients with amyloidosis. Am J Med. (2006) ;119: (4). |
[36] | Rapezzi C , Quarta CC , Guidalotti PL , Longhi S , Pettinato C , Leone O , et al. Usefulness and limitations of99mTc-3, 3-diphosphono-1, 2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging. (2011) ;38: (3):470–8. |
[37] | Cortese A , Vegezzi E , Lozza A , Alfonsi E , Montini A , Moglia A , et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: Avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. (2017) ;88: (5):457–8. |
[38] | Conceição I , González-Duarte A , Obici L , Schmidt HHJ , Simoneau D , Ong ML , et al. Red-flag symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. (2016) ;21: (1):5–9. |
[39] | Phull P , Sanchorawala V , Connors LH , Doros G , Ruberg FL , Berk JL , et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. (2018) ;25: (1):62–7. 10.1080/13506129.2018.1436048 |
[40] | Mariani LL , Lozeron P , Théaudin M , Mincheva Z , Signate A , Ducot B , et al. Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. (2015) ;78: (6):901–16. 10.1002/ana.24519 |
[41] | Damy T , Jaccard A , Guellich A , Lavergne D , Galat A , Deux JF , et al. Identification of prognostic markers in transthyretin and AL cardiac amyloidosis*. Amyloid. (2016) ;23: (3):194–202. |
[42] | Arruda-Olson AM , Zeldenrust SR , Dispenzieri A , Gertz MA , Miller FA , Bielinski SJ , et al. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid. (2013) ;20: (4):263–8. 10.3109/13506129.2013.845745 |
[43] | Marcoux J , Mangione PP , Porcari R , Degiacomi MT , Verona G , Taylor GW , et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. (2015) ;7: (10):1337–49. 10.15252/emmm.201505357 |
[44] | Ericzon BG , Wilczek HE , Larsson M , Wijayatunga P , Stangou A , Pena JR , et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation. (2015) ;99: (9):1847–54. |
[45] | Yamamoto S , Wilczek HE , Nowak G , Larsson M , Oksanen A , Iwata T , et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): A single-center experience over 16 years. Am J Transplant. (2007) ;7: (11):2597–604. 10.1111/j.1600-6143.2007.01969.x |
[46] | Adams D , Cauquil C , Labeyrie C , Beaudonnet G , Algalarrondo V , Théaudin M . TTR kinetic stabilizers and TTR gene silencing: A new era in therapy for familial amyloidotic polyneuropathies. Expert Opin Pharmacother. (2016) ;17: (6):791–802. 10.1517/14656566.2016.1145664 |
[47] | Berk JL , Suhr OB , Obici L , Sekijima Y , Zeldenrust SR , Yamashita T , et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA - J Am Med Assoc. (2013) ;310: (24):2658–67. 10.1001/jama.2013.283815 |
[48] | Joint Formulary Committee. Tafamidis [Internet]. British National Formulary (BNF). |
[49] | Coelho T , Maia LF , Da Silva AM , Cruz MW , Planté-Bordeneuve V , Lozeron P , et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology. (2012) ;79: (8):785–92. 10.1212/WNL.0b013e3182661eb1 |
[50] | Barroso FA , Judge DP , Ebede B , Li H , Stewart M , Amass L , et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: Results up to 6 years. Amyloid. (2017) ;24: (3):194–204. 10.1080/13506129.2017.1357545 |
[51] | Maurer MS , Schwartz JH , Gundapaneni B , Elliott PM , Merlini G , Waddington-Cruz M , et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. (2018) ;379: (11): NEJMoa1805689. 10.1056/NEJMoa1805689 |
[52] | Cardoso I , Martins D , Ribeiro T , Merlini G , Saraiva MJ . Synergy of combined Doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: Studies in FAP mouse models. J Transl Med. (2010) ;8: (1):74. 10.1186/1479-5876-8-74 |
[53] | Adams D , Gonzalez-Duarte A , O’Riordan WD , Yang C-C , Ueda M , Kristen AV , et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. (2018) ;379: (1):11–21. 10.1056/NEJMoa1716153 |
[54] | Benson MD , Waddington-Cruz M , Berk JL , Polydefkis M , Dyck PJ , Wang AK , et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. (2018) ;379: (1):22–31. 10.1056/NEJMoa1716793 |
[55] | Grosenbach DW , Honeychurch K , Rose EA , Chinsangaram J , Frimm A , Maiti B , et al. Oligonucleotide Drugs for Transthyretin Amyloidosis. N Engl J Med. (2018) ;379: (21):2085–6. 10.1056/NEJMc1810994 |
[56] | Rossor AM , Reilly MM , Sleigh JN . Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol. (2018) ;18: (2):126–31. 10.1136/practneurol-2017-001764 |
[57] | Ghosh R , Tabrizi SJ . Gene suppression approaches to neurodegeneration. Alzheimer’s Res Ther. (2017) ;9: (1):1–13. 10.1186/s13195-017-0307-1 |
[58] | Alshehri B , D’Souza DG , Lee JY , Petratos S , Richardson SJ . The diversity of mechanisms influenced by transthyretin in neurobiology: Development, disease and endocrine disruption. J Neuroendocrinol. (2015) ;27: (5):303–23. 10.1111/jne.12271 |
[59] | Jason TLH , Koropatnick J , Berg RW . Toxicology of antisense therapeutics. Toxicol Appl Pharmacol. (2004) ;201: (1):63–83. 10.1016/j.taap.2004.04.017 |
[60] | Ando Y , Terazaki H , Nakamura M , Ando E , Haraoka K , Yamashita T , et al. A different amyloid formation mechanism: De novo oculoleptomeningeal amyloid deposits after liver transplantation. Transplantation. (2004) ;77: (3):345–9. |


