
Myo-Glyco disease Biology: Genetic Myopathies Caused by Abnormal Glycan Synthesis and Degradation
Abstract
Glycosylation is a major form of post-translational modification and plays various important roles in organisms by modifying proteins or lipids, which generates functional variability and can increase their stability. Because of the physiological importance of glycosylation, defects in genes encoding proteins involved in glycosylation or glycan degradation are sometimes associated with human diseases. A number of genetic neuromuscular diseases are caused by abnormal glycan modification or degeneration. Heterogeneous and complex modification machinery, and difficulties in structural and functional analysis of glycans have impeded the understanding of how glycosylation contributes to pathology. However, recent rapid advances in glycan and genetic analyses, as well as accumulating genetic and clinical information have greatly contributed to identifying glycan structures and modification enzymes, which has led to breakthroughs in the understanding of the molecular pathogenesis of various diseases and the possible development of therapeutic strategies. For example, studies on the relationship between glycosylation and muscular dystrophy in the last two decades have significantly impacted the fields of glycobiology and neuromyology. In this review, the basis of glycan structure and biosynthesis will be briefly explained, and then molecular pathogenesis and therapeutic concepts related to neuromuscular diseases will be introduced from the point of view of the life cycle of a glycan molecule.
STRUCTURE AND CELL BIOLOGY OF GLYCANS – AN OVERVIEW
Glycosylation is a post-translational modification where sugars (monosaccharides) or sugar chains (oligosaccharides) are covalently bound to proteins or lipids. In principle, the term glycan refers to compounds consisting of several monosaccharides linked through glycosidic bonds; however, the term glycan is generally used to refer the carbohydrate portion of a glycoconjugate, such as a glycoprotein and glycolipid. Currently, hundreds of monosaccharides are known in nature, but nine are generally found in mammalian glycans as common monosaccharides (Supplementary Figure 1). Monosaccharides are the minimum unit of glycans, like amino acids and nucleotides in protein or nucleic acids, respectively. Monosaccharides are often abbreviated, for example, Glc for glucose and GlcNAc for N-acetylglucosamine. Simple symbols are also used: blue circle for Glc and blue square for GlcNAc. These abbreviations and symbols are summarized in Supplementary Figure 1A. Monosaccharides can bind together via a glycosidic bond. Amino acids and nucleotides are linearly linked in a unidirectional format (N-terminus ⟶ C-terminus via a peptide bond; 5′⟶ 3′via a phosphodiester bond), but oligosaccharides can produce more diverse and complex structures. This complexity has been a big hurdle for a deeper understanding of glycan structures and biosynthetic mechanisms. A determinant of the divergent structure of glycans arises from a chemical property of the glycosidic bond. The asymmetric center of monosaccharides is called the anomeric carbon (Supplementary Figure 1B). The glycosidic bond is formed between this anomeric carbon and a hydroxyl group (-OH) of another monosaccharide (acceptor monosaccharide) and yields two stereoisomers, with α- and β-linkage forms. In addition, because monosaccharides contain several hydroxyl groups, anomeric carbon in theory can form glycosidic bonds with any of the hydroxyl groups in the acceptor monosaccharides, resulting in several regioisomers. For example, in the case of disaccharides consisting of two Glc residues, Glcβ1-6Glc refers to the first Glc residue linked to 6-position of the second Glc residue via a β-linkage, whereas Glcα1–4Glc refers to the first Glc residue linked to 4-position of Glc residue via an α-linkage (Supplementary Figure 1B). Thus, even with the same monosaccharide composition, different linkage formats produce diverse structures. As described above, the description of glycans includes the order of monosaccharides and the linkage types, but sometimes the type of linkage is omitted when just referring the order and composition of an oligosaccharide. An acceptor monosaccharide can allow more than two glycosidic linkage, resulting in branched structures. Sometimes, monosaccharides are further modified with phosphates or sulfates, conferring further diversity to glycan structures and functions. Oligosaccharides have reducing and non-reducing termini, analogous to the N-and C-terminal ends of proteins. The reducing terminus of an oligosaccharide bears a free anomeric center. The term reducing terminus is often used to refer to the side engaged with proteins.
Glycosidic bonds can be formed with amino acids through a hydroxyl group in threonine (Thr) and serine (Ser), or an amide nitrogen in asparagine (Asn). Two major types of glycan modifications on proteins are the N-and O-linked types, which vary in terms of how the glycans are covalently attached to a protein. N-linked glycans refer to sugar chains linked to an Asn via a GlcNAc residue at the reducing end. O-linked glycans refer to sugar chains linked to a Ser or Thr residue. An N-acetylgalactosamine (GalNAc) residue is frequently seen at the reducing end of O-glycan, but other monosaccharides are also seen at the reducing end of O-glycans, such as Man, Glc, and fucose (Fuc) residues. Proteoglycans are proteins that are modified with glycosaminoglycan (GAG) chains on Ser residues. GAGs are linear polymers of repeating disaccharide units, which define a form of GAGs such as chondroitin and heparin. A glycosylphosphatidylinositol (GPI) anchor is a glycan bridge between phosphatidylinositol in the plasma membrane and a phosphoethanolamine-modified protein, which serves as a linker to anchor proteins to the cell surface. The clear majority of glycolipids in vertebrates are glycosphingolipids (GSLs), which consist of a glycan attached via a Glc or Gal unit to the lipid moiety, ceramide. GSLs are further classified as neutral, sialylated, or sulfated. Gangliosides, which are GSLs containing Sia residues and abundantly seen in the nervous system, are well known to be associated with neurological diseases [1]. For example, accumulation of gangliosides, caused by defects in degradation, are associated with several neurological diseases such as GM1 gangliosidosis and Tay–Sachs disease. In addition, some forms of Guillain–Barre syndrome involves auto-antibodies against gangliosides [2].
Table 1
Events in a glycan’s life cycle are associated with human diseases. Some examples of the diseases, responsible genes, and functions are illustrated
| Life events of a glycan | Disease | Functions of responsible genes |
| Glycan precursor synthesis (Sugar nucleotide metabolism) | Congenital myasthenic syndrome GNE myopathy | Sugar nucleotide synthesis, N-Glycan precursor synthesis Sugar nucleotide synthesis |
| Glycosylation | Congenital disorder of glycosylation | Sugar nucleotide synthesis, Sugar nucleotide transporter, N-Glycan precursor synthesis, N-glycosylation, GPI-anchor synthesis, GAG synthesis, etc. |
| Muscular dystrophy | O-Mannosylation, O-Glucosylation | |
| Glycan degradation | Lysosomal storage disease | Lysosomal enzymes for glycan degradation, Lysosomal structural proteins |
Glycan biosynthesis occurs through multistep processes, in which glycosyltransferases play primary roles, for example, in assembling a monosaccharide into linear or branched sugar chains or linking a monosaccharide or oligosaccharide to proteins or lipids. Basically, most glycosyltransferases show strict donor, acceptor, and linkage specificities. Thus, many enzymes, generally acting sequentially, are required to complete the synthesis of glycans. Protein glycosylation usually takes place in the endoplasmic reticulum (ER) or the Golgi apparatus. Most glycosyltransferases use activated forms of monosaccharides, often nucleotide sugars and in some cases lipid-phosphate-linked sugars, as donor substrates for glycan elongation (thus, they are referred to as glycosylation precursors). Glycans also undergo trimming or modification such as sulfation and are then transported to where they function properly. Degradation of glycans typically occurs in lysosomes and is mediated by glycosidases. As explained so far, diversity in glycan structure and function can build upon the balance of players involved in all processes during the life of glycans (e.g., sugar precursor, glycosyltransferase, trafficking, or degradation) and genetic defects in such processes are associated with human diseases (Table 1). In addition, cellular circumstance including metabolism, aging, stress, and tumor environment can affect sugar chain structures. For readers who want to understand glycobiology in greater detail, see the on-line textbook at https://www.ncbi.nlm.nih.gov/books/NBK1908/.
GLYCOSYLATION PRECURSORS AND NEUROMUSCULAR DISEASES
Most glycosyltransferases use nucleotide sugars (e.g., UDP-Gal, GDP-Fuc, or CMP-Sia) or lipid-linked sugars (e.g., dolichol-phosphate-Man [Dol-P-Man] or Dol-P-Glc) to transfer monosaccharides to acceptor substrates such as proteins, lipids, and sugar chains. Activation processes that yield nucleotide sugars include coupling reactions of glycosyl-1-phosphate and nucleoside triphosphates (e.g., Glc-1-P + UTP⟶UDP-Glc, or Man-1-P + GTP⟶GDP-Man). A nucleotide-exchange reaction or the direct conversion of sugar moiety by an epimerase also produces forms of nucleotide sugars (e.g., UDP-GalNAc↔UDP-GlcNAc). Nucleotide sugars are synthesized in the cytoplasm or nucleus, and are then delivered by nucleotide sugar transporters into the ER or the Golgi lumen, where most glycosylation takes place. Details of the biosynthesis and interconversion of sugar nucleotides are reviewed elsewhere [3], and here, I will introduce two cases of neuromuscular diseases that are associated with sugar nucleotide metabolism.
Congenital myasthenic syndromes (CMSs) are a group of genetic disorders in which signal transmission at neuromuscular junctions (NMJs) is impaired [4, 5]. A common clinical feature of CMSs is fatigable weakness, but there are clinical variations in this condition, such as the age of onset, affected muscles, or decremental electromyographic response. Currently, mutations in at least 30 genes are known to cause CMSs and several of them are involved in glycosylation. In 2011, mutations in the GFPT1 gene were identified in families with an autosomal recessive CMS [6]. GFPT1 encodes glutamine-fructose-6-phosphate transamidase 1, an enzyme in the hexosamine biosynthesis pathway that catalyzes the conversion of fructose-6-phosphate and glutamine into glucosamine-6-phosphate and glutamate, respectively (Supplementary Figure 2A). Subsequent to glucosamine-6-phosphate production is the formation of UDP-GlcNAc, which can be further used to produce UDP-GalNAc and CMP-Sia. Thus, defects in GFPT1 are expected to affect several glycosylation pathways, leading to abnormal glycosylation of proteins or lipids in NMJs, and consequently, compromised NMJ development, stability, and function [6–8]. Other CMS-causing genes, such as ALG2, ALG14, DPAGT1, and GMPPB, are also known to participate in glycosylation (Supplementary Figure 3A). ALG2, ALG14, and DPAGT1 are involved in producing dolichol-linked precursors that are used for protein N-glycosylation [4, 9]. GMPPB is involved in the synthesis of GDP-Man. GDP-Man is used as a Man donor (for N-glycan precursor molecules) and for producing Dol-P-Man, which serves as a Man donor for both N-glycan precursors and O-Man glycosylation. Mutations in GMPPB are also associated with muscular dystrophy (see below).
GNE myopathy is an autosomal recessive myopathy caused by mutations in the GNE gene that encodes a single protein with UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities, which function in the Sia-biosynthesis pathway [10, 11]. GNE myopathy has also been referred to as distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. GNE myopathy is characterized clinically by weakness of the distal muscle of the leg and pathologically by the presence of rimmed vacuoles and intracellular protein aggregations [12]. The GNE enzyme catalyzes the conversions of UDP-GlcNAc to N-acetylmannosamine (ManNAc) and ManNAc to ManNAc-6-phosphate (Supplementary Figure 2B). GNE mutations reduce either or both enzyme activities, resulting in reduced Sia levels in the serum and skeletal muscle [13]. Sia, one of the acidic sugars carrying carboxylates, is abundant in vertebrate glycoproteins and plays diverse roles in cell biology such as cell–cell contacts and cell–pathogen recognition. The molecular mechanism whereby abnormal sialylation causes GNE myopathy is still not fully understood, but it has been hypothesized that hypo-sialylation causes the misfolding of some glycoproteins, which could be targets of autophagic degradation and serve as cores for the formation of protein aggregates [13]. The Gne gene knock-out (KO) in mice resulted in embryonic lethality [14], but transgenic expression of the GNE D167V mutant, one of the most prevalent mutations among Japanese patients, in Gne-KO mice recapitulated the biochemical, clinical, and histopathological features of GNE myopathy [15]. A Japanese group successfully showed that the administration of a Sia-related compound, N-acetylneuramic acid (NeuAc), its glycosyl conjugate sialyllactose, or its precursor ManNAc increased Sia levels and ameliorated muscle pathology in Gne-KO/D167V-transgenic mice [16, 17], indicating the therapeutic potential of Sia-related compounds. Indeed, several clinical trials with metabolic supplementation of Sia-related compounds are currently ongoing [12].
GLYCOSYLATION AND DISEASES – (1) CONGENITAL DISORDER OF GLYCOSYLATION
Protein N-glycosylation refers to the covalent attachment of sugar chain to proteins via a GlcNAcβ1-Asn linkage. Many secreted and membrane-bound proteins carry N-glycans. Protein N-glycosylation participates in various physiological processes, such as protein folding, stability, trafficking, localization, and protein–protein interactions. N-glycosylation processing is roughly divided into the following three steps. N-glycan synthesis begins in the ER where monosaccharides assemble to form a specific structure on a lipid-like compound (Dol-P), based on the activities of a series of glycosyltransferases encoded by the Asn-linked glycosylation (ALG)-pathway genes (Supplementary Figure 3A). Then, the N-glycan precursor is transferred en bloc to proteins by the multisubunit oligosaccharyltransferase (OST) complex. Finally, the N-glycan undergoes processing such as trimming by glycosidases and decoration with sugars by glycosyltransferases to the matured forms. N-glycan synthesis and processing occur in the ER and Golgi apparatus and involve dozens of proteins. In addition to glycosyltransferases and glycosidases, these processes require enzymes that produce sugar precursors (nucleotide sugars and Dol-P-sugars), flippases that translocate Dol-P-oligosaccharide or Dol-P-sugar across the ER membrane, or transporters that deliver sugar nucleotides into the Golgi lumen (Supplementary Figure 3B).
The term congenital disorder of glycosylation (CDG) has been used in a narrow sense to refer to genetic diseases caused by defects in the N-glycosylation pathway, but currently includes genetic disorders caused by abnormal O-glycosylation of proteins, GAG modification, and GPI and glycolipids syntheses. CDGs have been described as CDG-type I or type II depending on molecular phenotypes, but are currently described using the responsible gene name with a CDG suffix, such as PMM2-CDG. Over 100 distinct CDGs are known and most of the cases have neurological abnormalities. Although the clinical phenotypes range from mild to severe, patients typically have developmental delay, intellectual disabilities, hypotonia, seizures, or metabolic abnormalities. As mentioned above, several enzymes mediate N-glycosylation and, thus, mutations in any gene involved in common N-glycosylation processing may result in a CDG. Here, I will briefly introduce how just one gene deficiency affects glycosylation and how a metabolic glycosylation pathway can serve as a therapeutic target by illustrating some CDGs as examples. For a detailed overview of N-glycosylation pathways, please refer to excellent reviews [18–20] or the online textbook at https://www.ncbi.nlm.nih.gov/books/NBK1908/. In addition, glycan structural analysis using mass spectrometry has been applied for diagnosing CDGs associated with defects in the N-glycosylation pathway [19, 21, 22].
PMM2-CDG is the most prevalent form of CDG and clinical features include intellectual disabilities, seizures, hypotonia, microcephaly, cerebellar atrophy/hypoplasia, strabismus, and stroke-like episodes [23]. PMM2 encodes phosphomannomutase 2 catalyzing the conversion of Man-6-phosphate to Man-1-phosphate, which is then used to generate the Man donors GDP-Man and Dol-P-Man (Supplementary Figure 3A). GDP-Man and Dol-P-Man serve as Man donors for synthesizing the N-glycan precursor on Dol-p; thus, PMM2 mutations primarily affect N-glycosylation.
Proper glycosylation is established based on the flux of several enzyme reactions and metabolites. Thus, manipulation of the flux is a possible therapeutic option. PGM1-CDG is associated with abnormal glycosylation, glycogenesis, and glycolysis due to defects in phosphoglucomutase-1 (PGM1). Affected adults predominantly show symptoms of primary muscle disease, whereas pediatric cases are associated with variable clinical symptoms including midline facial defects, growth retardation, myopathy, and progressive cardiac failure [24, 25]. The PGM1 enzyme interconverts Glc-1-phosphate and Glc-6-phosphate (Supplementary Figure 2C). Glc-1-phosphate serves as a substrate in the production of UDP-Glc. UDP-Glc is used for glycogen synthesis, protein glycosylation, and UDP-Gal production. UDP-Gal is made from UDP-Glc by the exchange of Gal-1-phospate for Glc-1-phosphate. Thus, PGM1 deficiency changes the intracellular metabolic balance of glycosylation precursors [24]. In the cell of patients with PGM1-CDG, the level of Glc-1-phosphate is remarkably increased, which leads to a decreased UDP-Gal/UDP-Glc ratio and abnormal glycosylation. Supplementation with Gal, which is converted to Gal-1-phosphate, increased the level of UDP-Gal and improved glycosylation. Most importantly, dietary supplementation with Gal in patients resulted in changes that were suggestive of clinical improvement [25].
Another example is SLC35C1-CDG, which is also referred to as leukocyte adhesion deficiency (LAD II) [26]. Patients with SLC35C1-CDG presents with recurrent episodes of infections, persistent leukocytosis, and mental and growth retardation. SLC35C1 encodes a GDP-Fuc transporter in the Golgi membrane and, thus, SLC35C1 mutations reduce the level of GDP-Fuc in the Golgi lumen, thereby affecting fucosylation (Supplementary Figure 3B). Interestingly, oral supplementation with Fuc improved clinical manifestations in a few patients with SLC35C1-CDG [27, 28]. Intracellular Fuc can be converted into Fuc-1-phosphate and then GDP-Fuc. Thus, elevated GDP-Fuc levels induced by Fuc supplementation possibly compensated for a reduced affinity of disease-causing SLC35C1 mutant protein for GDP-Fuc. The observation of patients that do not respond to Fuc treatment suggests that therapeutic benefits might depend on the mutation background.
It is noted that mutations in the genes responsible for CDG sometimes cause other distinct diseases such as CMSs (as mentioned above) and muscular dystrophies (see next section). Determinants of how mutations in a particular gene lead to CMS or muscular dystrophy rather than CDG are unclear, but certain mutations may selectively or preferentially affect N-glycosylation or O-mannosylation pathways or target proteins.
GLYCOSYLATION AND DISEASES – (2) MUSCULAR DYSTROPHY
O-glycosylation refers to mono-or oligosaccharide modifications on Ser or Thr residues of proteins. Perhaps O-GalNAc glycans (mucin-type O-glycan) represent the best-characterized O-glycans, although other monosaccharides (such as Fuc, Glu, GlcNAc, and Man) can be attached to proteins. Defects in O-glycosylation are also sometimes associated with human diseases. In this section, I will introduce muscular dystrophies caused by defects in O-Man or O-Glc modifications.
Muscular dystrophy is a genetic disease that is characterized by progressive muscle weakness and degeneration. Currently, over 50 distinct disease forms and responsible genes are known. Dystroglycanopathy is a collective term for referring to a group of congenital and limb-girdle types of muscular dystrophy that are caused by abnormal glycosylation of dystroglycan. Dystroglycan is a cellular receptor for basement membrane proteins or synaptic proteins such as laminins, agrin, and neurexin [29]. O-Mannosyl glycosylation is essential for the ligand-binding activity of dystroglycan, and the most genes responsible for dystroglycanopathy encode enzymes involved in the synthesis of O-Man glycan [30]. Figure 1 summarizes the structure of O-Man glycan on dystroglycan and the functions of dystroglycanopathy-related gene products. In this section, I will introduce the history of dystroglycanopathy research and then describe recent progress in identifying a novel glycan unit, ribitol-phosphate (RboP), and several therapeutic strategies.
Fig.1
Schematic representation of the structures of O-Man glycans on dystroglycan and modification enzymes. Dystroglycan is modified with O-Man glycans, namely CoreM1 and CoreM3. A repeating unit of GlcA-Xyl at the terminal end of CoreM3 serves as a ligand-binding moiety. This repeating unit and CoreM3 are linked by tandem RboP groups. Dol-P-Man and CDP-Rbo serve as donor substrates for O-mannosylation and RboP transfer, respectively. Enzymes responsible for these modifications and muscular dystrophy are illustrated with blue rectangle.
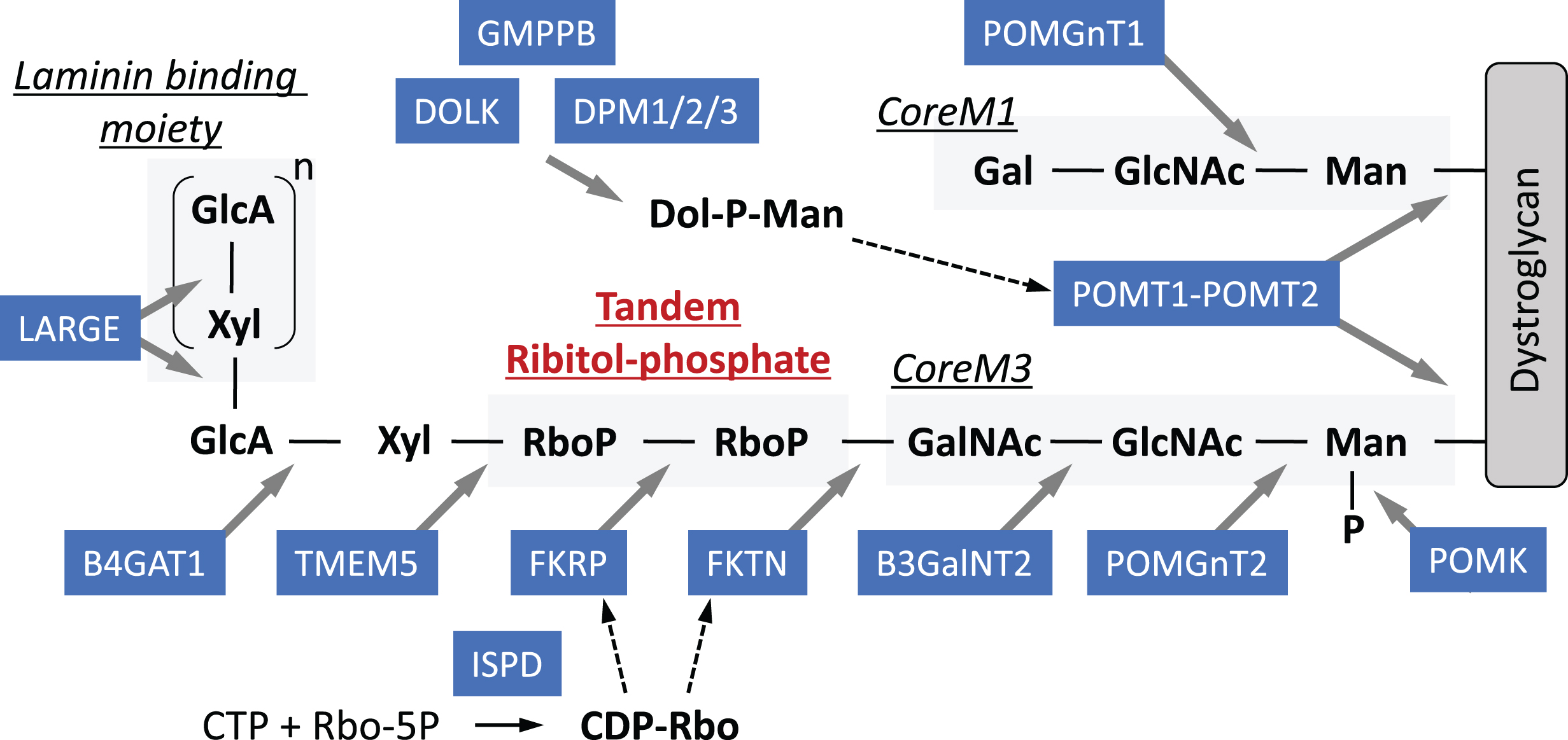
In the early 2000s, it was shown that abnormal glycosylation of dystroglycan with reduced ligand-binding activity was associated with muscle–eye–brain (MEB) disease, Fukuyama muscular dystrophy (FCMD), and Walker–Warburg syndrome (WWS) [31]. These three diseases are characterized by severe congenital forms of muscular dystrophy with brain malformation (micropolygyria of the cerebrum and cerebellum, and type II lissencephaly), mental retardation, and eye involvement (myopia, cataracts, abnormal eye movement, pale optic discs, and retinal detachment). Subsequently, less severe forms of muscular dystrophy with abnormal glycosylation of dystroglycan were reported such as limb-girdle type 2I. At that moment, a unique O-Man-type glycan (Sia-Gal-GlcNAc-Man-O), later referred to as CoreM1 glycan, was known to be present in dystroglycan [32]. A protein complex of protein O-mannosyl-transferase (POMT) 1 and POMT2, causative gene products for WWS, catalyzes the initial transfer of Man to dystroglycan as an O-Man transferase, using Dol-P-Man as a donor substrate [33]. Protein O-linked-Manβ-1,2-N-acetylglucosaminyltransferase 1 (POMGNT1), the causative gene product for MEB, catalyzes the second modification, which is the formation of a GlcNAc-Man linkage [34].
In the 2010s, a novel glycan structure GalNAc-GlcNAc-Man-O (namely CoreM3 glycan) was identified and this finding led to the discovery that several dystroglycanopathy-related gene products function as glycosyltransferases [35, 36]. A GAG-like repeating structure consisting of a disaccharide unit (glucuronic acid [GlcA]-Xyl) was also identified and, importantly, this repeat was shown to serve as a ligand-binding domain. This repeating unit is synthesized by the bifunctional enzyme LARGE, which has two glycosyltransferase activities (e.g., α3-xylosyltransferase and β3-glucuronyltransferase activities) [37]. In 2016, our group identified a tandemly connected RboP between CoreM3 and GlcA-Xyl repeats [38]. Ribitol (Rbo) is a sugar alcohol that was previously not known to be used in mammals, but was known to be present in bacterial cell walls as an RboP polymer. Tandem RboP and RboP-GalNAcunits are linked through a phosphodiester bond, not a glycosyl bond. The tandem RboP structure is synthesized by the sequential action of two different RboP transferases, fukutin (FKTN) and fukutin-related protein (FKRP), which are encoded by the FCMD and LGMD2I genes, respectively, both of which use CDP-ribitol (CDP-Rbo) as the donor substrate. For more details regarding sugar chain structures and modification enzymes, please refer to our previous reviews [29, 30].
Strictly speaking, Rbo is not a sugar, but a precursor of RboP modification is the nucleotide-linked compound CDP-Rbo, as is the case with other sugar precursors used for glycosylation. CDP-Rbo is synthesized from cytosolic free Rbo-5-phosphate and CTP by theisoprenoid domain-containing protein (ISPD) – mutations in which were also identified in patients with dystroglycanopathy [38–40]. Defects in the synthesis pathway for the O-mannosylation precursor Dol-P-Man have also been implicated in dystroglycanopathy. Genes identified as causative for dystroglycanopathy include DPMs, DOLK, and GMPPB and defects in these genes likely reduce the cellular level of Dol-P-Man, affecting O-mannosylation. However, Dol-P-Man is also used as a Man donor during N-glycosylation (Supplementary Figure 3A) and in GPI anchor biosynthesis. In fact, defects in the Dol-P-Man pathway have been associated with CDG with N-glycosylation defects. As discussed in the section related to CMS and CDG, the mechanism by which mutations in the Dol-P-Man-synthesis pathway selectively or preferentially affect O-mannosylation or N-glycosylation may reflect the impact of mutations on enzyme activities and/or patients’ genetic backgrounds, which should be clarified in the future.
Dystroglycan plays crucial roles in maintaining the physical strength of the muscle plasma membrane in a glycosylation-dependent manner [41]. Thus, abnormal glycosylation renders muscles prone to contraction-induced injuries (disease-triggering membrane fragility). In addition, dystroglycan also plays important roles in the maintenance of viability of satellite cells (stem cells required for muscle regeneration) and the proliferation and differentiation activities of myoblasts [42]. Conditional deletion of mouse Fktn in muscle precursor cells impaired muscle regeneration, which underlies severe muscle pathology in dystroglycanopathy. However, viral-mediated selective gene expression in mature myofibers ameliorated the pathology, suggesting that protecting disease-triggering membrane fragility is a potential target for therapeutic intervention [42]. Dystroglycan glycosylation also plays critical roles in brain development by maintaining the physical stability of the glia limitans/basement-membrane complex in a glycosylation-dependent manner. Several types of dystroglycanopathy model mice exhibited pathological abnormalities such as neuron over-migration, basement membrane breakdown, and cerebral hemisphere fusion [43, 44], indicating that these abnormalities underlie the cause of brain malformation. We recently reported that gene rescue in the developing embryo brain suppressed the disease-associated phenotype, which opens new avenues of intervention for therapeutic strategies related to central nervous system abnormalities [44]. FCMD is the most prevalent dystroglycanopathy in Japan and most patients carry a retrotransposal insertion in the FKTN 3′-untranslated region [45]. This insertion contains a strong splice-acceptor site, which induces an alternative donor site in the last exon, and thus causes abnormal mRNA splicing (exon trapping) [46]. Introduction of antisense oligonucleotide targeting to prevent pathogenic exon trapping restores normal FKTN protein production and dystroglycan glycosylation in FCMD patient cells and model mice. Thus, this exon trapping strategy may serve as a radical approach for clinically treating FCMD.
In ISPD-deficient dystroglycanopathy, reduction of cellular CDP-Rbo is a cause of abnormal glycosylation, and thus CDP-Rbo supplementation could be used as a therapeutic strategy. In fact, supplementing ISPD-deficient cells with CDP-Rbo rescued dystroglycan glycosylation [38]. Although the biosynthetic pathway of CDP-Rbo has not been revealed yet, ribitol inclusion in drinking water increases muscle CDP-Rbo levels in wild-type mice and FKRP L448P mutant mice [40, 47]. In the latter case, surprisingly, abnormal glycosylation of dystroglycan and dystrophic pathology were improved [47]. Together, these findings suggest that the metabolic pathway for CDP-Rbo production could also be a therapeutic target, but more careful analyses are required. For example, it is necessary to clarify players in the biosynthesis pathway for Rbo-5-phosphate (a CDP-Rboprecursor). Enzymological characterization of mutant FKTN or FKRP enzymes would help determine whether their activities could be restored by increased CDP-Rbo.
In 2016, another form of muscular dystrophy associated with abnormal glycosylation was reported. A mutation in POGLUT1 was identified in a family with autosomal recessive limb-girdle muscular dystrophy [48]. POGLUT1 encodes the protein O-glucosyltransferase 1, which catalyzes the O-glucosylation of Notch, a famous signaling molecule that is widely involved in animal development. Notch is post-translationally modified with O-Glc and O-Fuc, and alteration of glycosylation affected Notch activities. The Notch pathway played roles in satellite cells, and disruption of Notch signaling in satellite cells recapitulated the muscular dystrophic phenotype in mice [49]. The disease-causing mutation reduced POGLUT1 activity, which impaired Notch signaling and consequently decreased myoblast proliferation and differentiation, as well as the number of satellite cells [48].
Very recently, mutations in the membrane-trafficking proteins TRAPPC11 and GOSR2 were reported in muscular dystrophy patients that were associated with abnormal dystroglycan glycosylation [50]. TRAPPC11 is a component of the transport protein particle (TRAPP) III complex, which is involved in cargo transport from the ER to the ER–Golgi intermediate compartment (ERGIC). Mutations in TRAPPC11 have been also reported in patients presenting with myopathy and intellectual disability or CDG [51, 52]. TRAPPC11 mutant proteins disrupted the Golgi apparatus architecture and delayed exit from the Golgi to the cell surface [51]. Mutations in TRAPPC11 found in CDG patients affected both N-and O-glycosylation [52]. Since the Golgi is the major site for protein glycosylation, defects in the Golgi morphology and trafficking may result in abnormal protein glycosylation. However, in the latest case, glycosylation analysis was normal, suggesting other functional roles for TRAPPC11 [50]. GOSR2 encodes a Golgi Qb-SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) protein, which localizes in the cis-Golgi and mediates docking and fusion of vesicles from the ER. Human mutations in GOSR2 were originally reported in patients with progressive myoclonus epilepsy (PME) with early ataxia [53]. A mutation associated with PME altered Golgi localization of GOSR2, although the cellular glycosylation status was not examined. In the recent report, the GOSR2 mutation was shown not to alter membrane trafficking. It is widely thought that defects in Golgi homeostasis via abnormal architecture or trafficking are associated with glycosylation diseases [54], but recent examination of the mutations in TRAPPC11 and GOSR2 indicate that they may selectively affect dystroglycan glycosylation. The determinant mechanism explaining how mutations in these membrane-trafficking proteins in some cases selectively affect glycosylation of dystroglycan or in other cases affect global N-and O-glycosylation is unknown. A possible explanation is that trafficking proteins may also play specific roles in glycosylation, for example, by assuring proper trafficking of enzymes or by forming a zone where they function appropriately for the O-Man-glycosylation pathway (e.g., the O-Man-glycosylation zone). Thus, some mutations could selectively affect dystroglycan, and other mutations could deteriorate trafficking functions more severely, thus affecting global glycosylation.
GLYCAN DEGRADATION AND NEUROMUSCULAR DISEASES
Glycans are turned over, like other components in living cells. Most glycan turnover occurs by endocytosis, and degradation is generally performed stepwise by multiple glycosidases in the lysosome. Exoglycosidases, which cleave the glycosidic bond from the non-reducing end, basically recognize one monosaccharide in a specific anomeric linkage. Modifications on the non-reducing terminal sugar (e.g. acetate, sulfate, or phosphate) have to be removed by other enzymes (e.g. esterases, sulfatases, or phosphatases) before reactions can proceed with exoglycosidases. In contrast to exoglycosidases, endoglycosidases cleave internal glycosidic linkages and thus release long oligosaccharides from conjugated molecules, including proteins or lipids. These variations in the properties of degradation enzymes drive complexity in glycan degradation and synthesis. Monosaccharide units arising from degradation are typically exported from the lysosome to the cytosol and then reutilized for producing glycan precursors. Lysosomal enzymes are N-glycosylated, bearing Man-6-phosphate on the sugar chain. Man-6-phosphate serves as a signal for proteins being transported into the lysosome, after recognition by the transmembrane Man-6-phosphate receptor (Supplementary Figure 3B). This machinery constitutes the Man-6-phosphate pathway. There is a group of genetic diseases associated with decreased activities of lysosomal enzymes, consequently leading to accumulation of their substrates as undigested fragments in the lysosome; thus, this group of diseases is referred to collectively as lysosomal storage diseases (LSDs) [1, 55]. Currently, approximately 50 types of LSDs are known. Not only lysosomal enzymes, but also defects in lysosomal structural proteins can cause LSDs, such as Danon disease [56]. The accumulation of undigested macromolecules causes enlargement of the lysosomes and reduces the supply of products necessary for biosynthesis and energy metabolism. In addition, defects in lysosome functions can alter many cellular processes, including lysosomal pH regulation, synaptic release, endocytosis, exocytosis, and vesicle maturation [1, 55].
Clinical manifestations vary among LSD types. Many LSDs present with a range of severity, but hepatomegaly and splenomegaly are the most common symptoms. Over half of all LSDs are accompanied by central nervous system involvement, such as progressive cognitive and motor decline, and neuronal degeneration. The variation in pathology is thought to depend on the cell type and the cellular balance of synthesis and turnover of the macromolecule or its precursors. For example, sialylated glycolipid gangliosides are abundant in neurons, and thus LSDs caused by accumulated gangliosides are preliminary brain disorders (GM1 gangliosidosis and GM2 gangliosidosis). Pompe disease is caused by an accumulation of glycogen in the lysosome, and its clinical phenotypes (particularly in cardiac and skeletal muscles) are supported by the importance of glycogen for muscle tissues.
Pompe disease (also known as glycogenosis), the only LSD among glycogen storage diseases, is caused by mutations in α-glucosidase (GAA), a lysosomal enzyme that hydrolyzes α1,4-and α1,6-linkages of glycogen to glucose. It is proposed that lysosomal glycogen clearance probably has no genuine metabolic function, but serves to dispose of glycogen that is accidentally taken up into lysosomes through autophagy [57]. Glycogen-filled lysosomes due to the deficiency of GAA might impair the general functions of lysosomes [57] and/or causes muscle damage [58] possibly via lysosomal rupture and the release of toxic substances into the cytosol [59]. The clinical spectrum of Pompe disease can be categorized into classic infantile, childhood, and adult forms. The infantile form presents with generalized hypotonia, muscle weakness, hypertrophic cardiomyopathy, and respiratory failure. The adult form is characterized by a relatively slow progressive proximal and axial muscle weakness. Histopathologically, most infantile and childhood forms exhibit fibers occupied by huge vacuoles that contain basophilic amorphous periodic acid Schiff-positive materials, but in late-onset form, such vacuoles may be present only in a few fibers [58, 59]. Other glycogen storage diseases with myopathy include McArdle disease (glycogenosis type V) and Cori disease (glycogenosis type III), among others. The responsible gene products for McArdle disease and Cori disease function as a glycogen phosphorylase and a glycogen-debranching enzyme, respectively, both of which are coordinately involved in glycogenolysis in the cytoplasm [60].
Several strategies have been developed for treating LSDs. Enzyme-replacement therapy (ERT) is based on replacement of the mutant protein with its normal version to restore function. ERT relies on the Man-6-phosphate pathway for efficient delivery of recombinant enzymes, which are modified with Man-6-phosphate on the N-glycans, from outside of cells to the lysosome via cell surface Man-6-phosphate receptors. ERT is currently available for several LSDs such as Gaucher disease, Fabry disease, some types of mucopolysaccharidosis, and Pompe disease [55]. A limitation of ERT is that the enzymes cannot penetrate the blood-brain barrier so that ERTs do not affect symptoms of the central nervous system. The effectiveness of ERTs also depends on the severity of the disease when treatment is initiated [1, 59].
Substrate reduction therapy (SRT) using small molecules aims at the inhibition of enzymes located upstream of the mutated disease-causing protein to reduce production of substrates of the mutated proteins. Other strategies using small molecules include enzyme-enhancement therapy and pharmacological chaperone therapy aiming at enhancing the residual enzyme activity of the mutated proteins [1, 61]. Independent of the type of accumulated substances or mutated genes, enhancing lysosomal function can be a therapeutic option with broader applicability across LSDs. For example, it has been reported that overexpression of transcription factor EB (TFEB), which regulates the coordinated transcriptional behavior of most lysosomal genes, induced lysosomal biogenesis and increased the degradation of complex molecules [62]. δ-Tocopherol, a natural form of vitamin E, has been reported to reduce pathological phenotypes in patient fibroblasts from several distinct LSDs by enhancing lysosomal exocytosis [63].Very recently, fetal intracranial adeno-associated viral vector-mediated gene delivery for glucocerebrosidase was reported to ameliorate neurodegeneration seen in a mouse model for neuronopathic Gaucher disease [64]. This group also demonstrated the feasibility of ultrasound-guided global gene transfer to fetal macaque brains. Together, not only ERT, but also other strategies with gene replacement or small molecules hopefully will be developed and approved to overcome the hurdles of neurological problems of LSDs in the future.
CONCLUSION
The relationship between neuromuscular diseases and glycan’s abnormalities presents the need for establishing the new field of “myo-glyco disease biology.” This review provides a comprehensive summary of the nature of glycans and introduces several neuromuscular diseases that are caused by abnormalities in glycan synthesis and degradation (Table 1 and Supplementary Table 1). Understanding the molecular basis of the life cycle of glycans can help in clarifying disease pathogenesis and developing therapeutic strategies. Many diseases directly caused by abnormal glycosylation are single-gene disorders, which can feasibly treated with gene therapy. However, many hurdles impede clinical applications in gene therapy, although progress has certainly been made. A systematic and integrated understanding of glycosylation pathways related to disease-causing gene products or biological characteristics of mutated enzymes may yield unexpected therapeutic approaches. Sometimes therapeutic target molecules may be safe and low-cost natural or metabolic products. There are some established treatments for neuromuscular diseases such as LSDs, and the contributions of basic research in glycobiology were no doubt immeasurable. Although the significance of glycans is widely recognized, it is unfortunately true that difficulties in structural and functional analyses have limited advances. However, considering recent rapid technological advances in glycan and glycome analysis and accumulation of large information such as genetic and clinical database, the challenges of glycoscience are no longer high obstacles. Rather, I believe, there is big potential for new breakthroughs in understanding the molecular mechanisms of pathogenesis and developing therapeutic interventions.
ACKNOWLEDGEMENTS, INCLUDING SOURCES OF SUPPORT
The author thanks Dr. Tamao Endo for fruitful comments and discussion. This work was supported by the Japan Agency for Medical Research and Development (AMED; grant numbers JP18ek0109197 and JP18gm0810010) and the Japan Society for the Promotion of Science (JSPS; grant numbers 18H02616 and 17H06421).
CONFLICT OF INTEREST
The author has no conflict of interest to report.
SUPPLEMENTARY MATERIALS
[1] The Supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-180369.
REFERENCES
[1] | Xu M , Motabar O , Ferrer M , Marugan JJ , Zheng W , Ottinger EA . Disease models for the development of therapies for lysosomal storage diseases. Ann N Y Acad Sci. (2016) ;1371: (1):15–29. |
[2] | Kuwabara S , Yuki N . Axonal Guillain-Barré syndrome: Concepts and controversies. Lancet Neurol. (2013) ;12: (12):1180–8. |
[3] | Blondeel EJM , Aucoin MG . Supplementing glycosylation: A review of applying nucleotide-sugar precursors to growth medium to affect therapeutic recombinant protein glyco-form distributions. Biotechnol Adv. (2018) ;36: (5):1505–23. |
[4] | Engel AG , Shen XM , Selcen D , Sine SM . Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. (2015) ;14: (4):420–34. |
[5] | Nicole S , Azuma Y , Bauché S , Eymard B , Lochmüller H , Slater C . Congenital myasthenicsyndromes or inherited disorders of neuromuscular transmission: Recent discoveries and open questions. J Neuromuscul Dis. (2017) ;4: (4):269–84. |
[6] | Senderek J , Müller JS , Dusl M , Strom TM , Guergueltcheva V , Diepolder I , Laval SH , Maxwell S , Cossins J , Krause S , Muelas N , Vilchez JJ , Colomer J , Mallebrera CJ , Nascimento A , Nafissi S , Kariminejad A , Nilipour Y , Bozorgmehr B , Najmabadi H , Rodolico C , Sieb JP , Steinlein OK , Schlotter B , Schoser B , Kirschner J , Herrmann R , Voit T , Oldfors A , Lindbergh C , Urtizberea A , von der Hagen M , Hü bner A , Palace J , Bushby K , Straub V , Beeson D , Abicht A , Lochmüller H . Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. (2011) ;88: (2):162–72. |
[7] | Zoltowska K , Webster R , Finlayson S , Maxwell S , Cossins J , Müller J , Lochmüller H , Beeson D . Mutations in GFPT1 that underlie limb-girdle congenital myasthenic syndrome result in reduced cell-surface expression of muscle AChR. Hum Mol Genet. (2013) ;22: (14):2905–13. |
[8] | Issop Y , Hathazi D , Khan MM , Rudolf R , Weis J , Spendiff S , Slater CR , Roos A , Lochmüller H . GFPT1 deficiency in muscle leads to myasthenia and myopathy in mice. Hum Mol Genet. (2018) ;27: (18):3218–32. |
[9] | Belaya K , Rodríguez Cruz PM , Liu WW , Maxwell S , McGowan S , Farrugia ME , Petty R , Walls TJ , Sedghi M , Basiri K , Yue WW , Sarkozy A , Bertoli M , Pitt M , Kennett R , Schaefer A , Bushby K , Parton M , Lochmüller H , Palace J , Muntoni F , Beeson D . Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain. (2015) ;138: (Pt9):2493–504. |
[10] | Eisenberg I , Avidan N , Potikha T , Hochner H , Chen M , Olender T , Barash M , Shemesh M , Sadeh M , Grabov-Nardini G , Shmilevich I , Friedmann A , Karpati G , Bradley WG , Baumbach L , Lancet D , Asher EB , Beckmann JS , Argov Z , Mitrani-Rosenbaum S . The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. (2001) ;29: (1):83–7. |
[11] | Hinderlich S , Stäsche R , Zeitler R , Reutter W . A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. (1997) ;272: (39):24313–8. |
[12] | Nishino I , Carrillo-Carrasco N , Argov Z . GNE myopathy: Current update and future therapy. J NeurolNeurosurg Psychiatry. (2015) ;86: (4):385–92. |
[13] | Noguchi S , Keira Y , Murayama K , Ogawa M , Fujita M , Kawahara G , Oya Y , Imazawa M , Goto Y , Hayashi YK , Non-aka I , Nishino I . Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sia-lylation in distal myopathy with rimmed vacuoles. J Biol Chem. (2004) ;279: (12):11402–7. |
[14] | Schwarzkopf M , Knobeloch KP , Rohde E , Hinderlich S , Wiechens N , Lucka L , Horak I , Reutter W , Horstkorte R . Sialylation is essential for early development in mice. Proc Natl AcadSci USA. (2002) ;99: (8):5267–70. |
[15] | Malicdan MC , Noguchi S , Nonaka I , Hayashi YK , Nishino I . A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. (2007) ;16: (22):2669–82. |
[16] | Malicdan MC , Noguchi S , Hayashi YK , Nonaka I , Nishino I . Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. (2009) ;15: (6):690–5. |
[17] | Yonekawa T , Malicdan MC , Cho A , Hayashi YK , Nonaka I , Mine T , Yamamoto T , Nishino I , Noguchi S . Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain. (2014) ;137: (Pt10):2670–9. |
[18] | Freeze HH , Eklund EA , Ng BG , Patterson MC . Neurological aspects of human glycosylation disorders. Annu Rev Neurosci. (2015) ;38: :105–25. |
[19] | Van Scherpenzeel M , Willems E , Lefeber DJ . Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconj J. (2016) ;33: (3):345–58. |
[20] | Ng BG , Freeze HH . Perspectives on glycosylation and its congenital disorders. Trends Genet. (2018) ;34: (6):466–76. |
[21] | Wada Y . Mass spectrometry for congenital disorders of gly-cosylation, CDG. J Chromatogr B AnalytTechnol Biomed Life Sci. (2006) ;838: (1):3–8. |
[22] | Sturiale L , Barone R , Garozzo D . The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis. (2011) ;34: (4):891–9. |
[23] | Matthijs G , Schollen E , Pardon E , Veiga-Da-Cunha M , Jaeken J , Cassiman JJ , Van Schaftingen E . Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet. (1997) ;16: (1):88–92. |
[24] | Morava E . Galactose supplementation in phosphoglu-comutase-1 deficiency; review and outlookfor a novel treatable CDG. Mol Genet Metab. (2014) ;112: (4):275–9. |
[25] | Tegtmeyer LC , Rust S , van Scherpenzeel M , Ng BG , Losfeld ME , Timal S , Raymond K , He P , Ichikawa M , Veltman J , Huijben K , Shin YS , Sharma V , Adamowicz M , Lammens M , Reunert J , Witten A , Schrapers E , Matthijs G , Jaeken J , Rymen D , Stojkovic T , Laforêt P , Petit F , Aumaître O , Czarnowska E , Piraud M , Podskarbi T , Stanley CA , Matalon R , Burda P , Seyyedi S , Debus V , Socha P , Sykut-Cegielska J , van Spronsen F , de Meirleir L , Vajro P , DeClue T , Ficicioglu C , Wada Y , Wevers RA , Vanderschaeghe D , Callewaert N , Fingerhut R , van Schaftingen E , Freeze HH , Morava E , Lefeber DJ , Marquardt T . Multiple pheno-types in phosphoglucomutase 1 deficiency. N Engl J Med. (2014) ;370: (6):533–42. |
[26] | Liibke T , Marquardt T , Etzioni A , Hartmann E , von Figura K , Körner C . Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet. (2001) ;28: (1):73–6. |
[27] | Lühn K , Marquardt T , Harms E , Vestweber D . Discontinuation of fucose therapy in LADII causes rapid loss of selectin ligands and rise of leukocyte counts. Blood. (2001) ;97: (1):330–2. |
[28] | Hidalgo A , Ma S , Peired AJ , Weiss LA , Cunningham-Rundles C , Frenette PS . Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood. (2003) ;101: (5):1705–12. |
[29] | Kanagawa M , Toda T . Ribitol-phosphate-a newly identified posttranslational glycosylation unit in mammals: Structure, modification enzymes and relationship to human diseases. J Biochem. (2018) ;163: (5):359–69. |
[30] | Kanagawa M , Toda T . Muscular dystrophy with ribitol-phosphate deficiency: A novel post-translational mechanism in dystroglycanopathy. J Neuromuscul Dis. (2017) ;4: (4):259–67. |
[31] | Michele DE , Barresi R , Kanagawa M , Saito F , Cohn RD , Satz JS , Dollar J , Nishino I , Kelley RI , Somer H , Straub V , Mathews KD , Moore SA , Campbell KP . Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. (2002) ;418: (6896):417–22. |
[32] | Chiba A , Matsumura K , Yamada H , Inazu T , Shimizu T , Kusunoki S , Kanazawa I , Kobata A , Endo T . Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha-dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of alpha-dystroglycan with laminin. J Biol Chem. (1997) ;272: (4):2156–62. |
[33] | Manya H , Chiba A , Yoshida A , Wang X , Chiba Y , Jigami Y , Margolis RU , Endo T . Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl AcadSci USA. (2004) ;101: (2):500–5. |
[34] | Yoshida A , Kobayashi K , Manya H , Taniguchi K , Kano H , Mizuno M , Inazu T , Mitsuhashi H , Takahashi S , Takeuchi M , Herrmann R , Straub V , Talim B , Voit T , Topaloglu H , Toda T , Endo T . Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. (2001) ;1: (5):717–24. |
[35] | Yoshida-Moriguchi T , Yu L , Stalnaker SH , Davis S , Kunz S , Madson M , Oldstone MB , Schachter H , Wells L , Campbell KP . O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. (2010) ;327: (5961):88–92. |
[36] | Yoshida-Moriguchi T , Willer T , Anderson ME , Ven-zke D , Whyte T , Muntoni F , Lee H , Nelson SF , Yu L , Campbell KP . SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science. (2013) ;341: (6148):896–9. |
[37] | Inamori K , Yoshida-Moriguchi T , Hara Y , Anderson ME , Yu L , Campbell KP . Dystroglycan function requires xylosyl-and glucuronyltransferase activities of LARGE. Science. (2012) ;335: (6064):93–6. |
[38] | Kanagawa M , Kobayashi K , Tajiri M , Manya H , Kuga A , Yamaguchi Y , Akasaka-Manya K , Furukawa JI , Mizuno M , Kawakami H , Shinohara Y , Wada Y , Endo T , Toda T . Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. (2016) ;14: (9):2209–23. |
[39] | Riemersma M , Froese DS , van Tol W , Engelke UF , Kopec J , van Scherpenzeel M , Ashikov A , Krojer T , von Delft F , Tessari M , Buczkowska A , Swiezewska E , Jae LT , Brummelkamp TR , Manya H , Endo T , van Bokhoven H , Yue WW , Lefeber DJ . Human ISPD is a cytidyltransferase required for dystroglycan O-mannosylation. Chem Biol. (2015) ;22: (12):1643–52. |
[40] | Gerin I , Ury B , Breloy I , Bouchet-Seraphin C , Bolsee J , Halbout M , Graff J , Vertommen D , Muccioli GG , Seta N , Cuisset JM , Dabaj I , Quijano-Roy S , Grahn A , van Schaftingen E , Bommer GT . ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto a-dystroglycan. Nat Commun. (2016) ;7: :11534. |
[41] | Han R , Kanagawa M , Yoshida-Moriguchi T , Rader EP , Ng RA , Michele DE , Muirhead DE , Kunz S , Moore SA , Ian-naccone ST , Miyake K , McNeil PL , Mayer U , Oldstone MB , Faulkner JA , Campbell KP . Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of alpha-dystroglycan. Proc Natl AcadSci USA. (2009) ;106: (31):12573–9. |
[42] | Kanagawa M , Yu CC , Ito C , Fukada S , Hozoji-Inada M , Chiyo T , Kuga A , Matsuo M , Sato K , Yamaguchi M , Ito T , Ohtsuka Y , Katanosaka Y , Miyagoe-Suzuki Y , Naruse K , Kobayashi K , Okada T , Takeda S , Toda T . Impaired viability of muscle precursor cells in muscular dystrophy with glycosylation defects and amelioration of its severe phenotype by limited gene expression. Hum Mol Genet. (2013) ;22: (15):3003–15. |
[43] | Moore SA , Saito F , Chen J , Michele DE , Henry MD , Messing A , Cohn RD , Ross-Barta SE , Westra S , Williamson RA , Hoshi T , Campbell KP . Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. (2002) ;418: (6896):422–5. |
[44] | Sudo A , Kanagawa M , Kondo M , Ito C , Kobayashi K , Endo M , Minami Y , Aiba A , Toda T . Temporal requirement of dystroglycan glycosylation during brain development and rescue of severe cortical dysplasia via gene delivery in the fetal stage. Hum Mol Genet. (2018) ;27: (7):1174–85. |
[45] | Kobayashi K , Nakahori Y , Miyake M , Matsumura K , Kondo-Iida E , Nomura Y , Segawa M , Yoshioka M , Saito K , Osawa M , Hamano K , Sakakihara Y , Nonaka I , Nakagome Y , Kanazawa I , Nakamura Y , Tokunaga K , Toda T . An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. (1998) ;394: (6691):388–92. |
[46] | Taniguchi-Ikeda M , Kobayashi K , Kanagawa M , Yu CC , Mori K , Oda T , Kuga A , Kurahashi H , Akman HO , DiMauro S , Kaji R , Yokota T , Takeda S , Toda T . Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. (2011) ;478: (7367):127–31. |
[47] | Cataldi MP , Lu P , Blaeser A , Lu QL . Ribitol restores functionally glycosylated a-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat Commun. (2018) ;9: (1):3448. |
[48] | Servián-Morilla E , Takeuchi H , Lee TV , Clarimon J , Mavil-lard F , Area-Gómez E , Rivas E , Nieto-González JL , Rivero MC , Cabrera-Serrano M , Gómez-Sánchez L , Martínez-López JA , Estrada B , Marquez C , Morgado Y , Suárez-Calvet X , Pita G , Bigot A , Gallardo E , Fernández-Chacón R , Hirano M , Haltiwanger RS , Jafar-Nejad H , Paradas C . A mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med. (2016) ;8: (11):1289–309. |
[49] | Lin S , Shen H , Jin B , Gu Y , Chen Z , Cao C , Hu C , Keller C , Pear WS , Wu L . Brief report: Blockade of Notch signaling in muscle stem cells causes muscular dystrophic phenotype and impaired muscle regeneration. Stem Cells. (2013) ;31: (4):823–8. |
[50] | Larson AA , Baker PR 2nd , Milev MP , Press CA , Sokol RJ , Cox MO , Lekostaj JK , Stence AA , Bossler AD , Mueller JM , Prematilake K , Tadjo TF , Williams CA , Sacher M , Moore SA . TRAPPC11 and GOSR2 mutations associate with hypoglycosylation of a-dystroglycan and muscular dystrophy. Skelet Muscle. (2018) ;8: (1):17. |
[51] | Bögershausen N , Shahrzad N , Chong JX , von Kleist-Retzow JC , Stanga D , Li Y , Bernier FP , Loucks CM , Wirth R , Puffenberger EG , Hegele RA , Schreml J , Lapointe G , Keupp K , Brett CL , Anderson R , Hahn A , Innes AM , Suchowersky O , Mets MB , Nürnberg G , McLeod DR , Thiele H , Waggoner D , Altmüller J , Boycott KM , Schoser B , Nürnberg P , Ober C , Heller R , Parboosingh JS , Wollnik B , Sacher M , Lamont RE . Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet. (2013) ;93: (1):181–90. |
[52] | Matalonga L , Bravo M , Serra-Peinado C , García-Pelegrí E , Ugarteburu O , Vidal S , Llambrich M , Quintana E , Fuster-Jorge P , Gonzalez-Bravo MN , Beltran S , Dopazo J , Garcia-Garcia F , Foulquier F , Matthijs G , Mills P , Ribes A , Egea G , Briones P , Tort F , Giros M . Mutations in TRAPPC11 are associated with a congenital disorder of glycosylation. Hum Mutat. (2017) ;38: (2):148–51. |
[53] | Corbett MA , Schwake M , Bahlo M , Dibbens LM , Lin M , Gandolfo LC , Vears DF , O’Sullivan JD , Robertson T , Bayly MA , Gardner AE , Vlaar AM , Korenke GC , Bloem BR , de Coo IF , Verhagen JM , Lehesjoki AE , Gecz J , Berkovic SF . A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet. (2011) ;88: (5):657–63. |
[54] | Freeze HH , Chong JX , Bamshad MJ , Ng BG . Solving glycosylation disorders: Fundamental approaches reveal complicated pathways. Am J Hum Genet. (2014) ;94: (2):161–75. |
[55] | Schultz ML , Tecedor L , Chang M , Davidson BL . Clarifying lysosomal storage diseases. Trends Neurosci. (2011) ;34: (8):401–10. |
[56] | Nishino I , Fu J , Tanji K , Yamada T , Shimojo S , Koori T , Mora M , Riggs JE , Oh SJ , Koga Y , Sue CM , Yamamoto A , Murakami N , Shanske S , Byrne E , Bonilla E , Nonaka I , DiMauro S , Hirano M . Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. (2000) ;406: (6798):906–10. |
[57] | Kilimann MW , Oldfors A . Glycogen pathways in disease: New developments in a classical field of medical genetics. J Inherit Metab Dis. (2015) ;38: (3):483–7. |
[58] | Angelini C . Spectrum of metabolic myopathies. Biochim-BiophysActa. (2015) ;1852: (4):615–21. |
[59] | Lim JA , Li L , Raben N . Pompe disease: From pathophys-iology to therapy and back again. Front Aging Neurosci. (2014) ;6: :177. |
[60] | Adeva-Andany MM , González-Lucán M , Donapetry-García C , Fernández-Fernández C , Ameneiros-Rodríguez E . Glycogen metabolism in humans. BBA Clin. (2016) ;5: :85–100. |
[61] | Suzuki Y . Chaperone therapy update: Fabry disease, GM1-gangliosidosis and Gaucher disease. Brain Dev. (2013) ;35: (6):515–23. |
[62] | Sardiello M , Palmieri M , di Ronza A , Medina DL , Valenza M , Gennarino VA , Di Malta C , Donaudy F , Embrione V , Pol-ishchuk RS , Banfi S , Parenti G , Cattaneo E , Ballabio A . A gene network regulating lysosomal biogenesis and function. Science. (2009) ;325: (5939):473–7. |
[63] | Xu M , Liu K , Swaroop M , Porter FD , Sidhu R , Firnkes S , Ory DS , Marugan JJ , Xiao J , Southall N , Pavan WJ , Davidson C , Walkley SU , Remaley AT , Baxa U , Sun W , McKew JC , Austin CP , Zheng W . δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J Biol Chem. (2012) ;287: (47):39349–60. |
[64] | Massaro G , Mattar CNZ , Wong AMS , Sirka E , Buckley SMK , Herbert BR , Karlsson S , Perocheau DP , Burke D , Heales S , Richard-Londt A , Brandner S , Huebecker M , Priestman DA , Platt FM , Mills K , Biswas A , Cooper JD , Chan JKY , Cheng SH , Waddington SN , Rahim AA . Fetal gene therapy for neurodegenerative disease of infants. Nat Med. (2018) ;24: (9):1317–23. |


