
Phase 1 Study of Edasalonexent (CAT-1004), an Oral NF-κB Inhibitor, in Pediatric Patients with Duchenne Muscular Dystrophy
Abstract
Background:
Edasalonexent is an orally administered small molecule designed to inhibit NF-κB, which is activated from infancy in Duchenne muscular dystrophy and is central to causing muscle damage and preventing muscle regeneration.
Objective:
Evaluate the safety, tolerability, pharmacokinetics and exploratory pharmacodynamics of three doses of edasalonexent in ambulatory males ≥4 to <8 years of age with genetically confirmed Duchenne muscular dystrophy.
Methods:
This was a 1-week, open-label, multiple-dose study with 3 sequential ascending doses (33, 67 and 100 mg/kg/day) of edasalonexent administered under different dietary conditions to 17 males with a mean age of 5.5 years.
Results:
All doses of edasalonexent were well tolerated, with no serious adverse events, no drug discontinuations and no dose reductions. The majority of adverse events were mild, and the most common adverse events were gastrointestinal (primarily diarrhea). Edasalonexent was rapidly absorbed with peak levels observed 2–6 hours after dosing and exposures appeared to increase nearly proportionally to dose for the 2 lower and all 3 doses under low-fat and high-fat meal conditions, respectively. Only minor plasma accumulation of edasalonexent was observed with 7 days of dosing. After treatment with edasalonexent for 7 days, levels of NF-κB-regulated genes and serum proteins were decreased.
Conclusions:
This first report of edasalonexent oral administration for one week in male pediatric patients with Duchenne muscular dystrophy showed that treatment was well tolerated and inhibited NF-kB pathways.
INTRODUCTION
Duchenne muscular dystrophy (DMD) results from mutations in the X chromosome DMD gene coding for the cytoskeletal protein dystrophin and is the most common genetic neuromuscular disease of childhood, affecting 1 in 3,500–6000 male births [1, 2]. DMD manifests clinically as progressive symmetric muscle weakness and degeneration with loss of contractile function and is uniformly fatal with death most often resulting from cardio-pulmonary complications in early- to mid-adulthood [3].
Treatment options for DMD management are limited. Eteplirsen is a DMD-specific therapy approved in the US for patients with confirmed dystrophin mutations amenable to exon 51 skipping, which affect approximately 13% of the DMD population [4]. The glucocorticoid deflazacort is approved for patients with DMD older than 5 years of age. Glucocorticoid treatment with prednisone or deflazacort improves cardiopulmonary function and delays loss of motor milestones [5–9], but is associated with multiple side effects that can limit treatment, including Cushingoid facies and habitus, growth inhibition/pubertal delay, behavioral changes and osteoporosis, particularly with daily dosing [6, 10]. Glucocorticoids also can disrupt expression of genes involved in muscle degradation and regeneration [11] leading to chronic myopathies that contribute to proximal muscle weakness [12]. Among 1564 US DMD registry patients surveyed through 2011, 36% had never used glucocorticoids and 10% discontinued their use [13].
An unmet need remains for DMD disease-modifying drugs that are well tolerated and effective regardless of dystrophin mutation, and that can be used in combination with drugs directly targeting dystrophin expression. Edasalonexent (CAT-1004, [N-(2-((4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenamido) ethyl)-2-hydroxybenzamide]), is a bifunctional, orally administered novel small molecule that covalently links two bioactives: salicylic acid and docosahexaenoic acid (DHA) [14]. Edasalonexent synergistically leverages the ability of the two compounds to inhibit the transcription factor NF-κB [14]. In a Phase 1 study in adults, NF-κB activity in peripheral mononuclear cells was inhibited following a single dose of edasalonexent but not by equimolar doses of salicylic acid and DHA [15]. Chronic activation of NF-κB is a key driver of muscle degeneration and suppression of muscle regeneration in DMD [16–20], which occurs early in the disease process and precedes loss of muscle function [21]. Salicylic acid prevents NF-κB mediated muscle atrophy and decreases protein catabolism in muscle [22–24]. DHA has been shown to upregulate anti-inflammatory pathways and suppress pro-inflammatory pathways via modulation of NF-κB activity [25, 26]. In addition, DHA is metabolized intracellularly to anti-inflammatory eicosanoids [27–30]. Edasalonexent is endocytosed and hydrolyzed by intracellular fatty acid amide hydrolase (FAAH) to release salicylic acid and DHA in the intracellular compartment [14], thus having a potential advantage of selectively delivering higher doses in target muscle cells where FAAH is abundant. In human skeletal myoblast cultures using radiolabeled edasalonexent, the three major metabolites were identified as linker-salicylic acid, salicylic acid, and salicyluric acid (i.e., glycine conjugate of salicylic acid) [31]. Multiple metabolites of the DHA moiety (via oxidation, or oxidation and conjugation pathways), were identified in primary human hepatocytes [31].
Edasalonexent inhibits NF-κB p65-dependent inflammatory responses as well as downstream pro-inflammatory genes modulated by p65 in the golden retriever DMD model [32]. Preclinical studies of a closely related compound inhibiting NF-κB showed reduction in muscle degeneration and promotion of muscle regeneration and function in both mice and dogs [32]. In Phase 1 studies in adults, edasalonexent was well tolerated and showed rapid and saturable oral absorption with minimal accumulation after multiple doses, and inhibited activated NF-κB pathways [15]. Food increased plasma exposures of both edasalonexent and salicyluric acid, an intracellularly formed major metabolite of salicylic acid. The primary objective of this Phase 1 portion of a Phase 1/2 study (NCT02439216) was to assess the safety and tolerability of edasalonexent with a secondary objective to assess the PK of edasalonexent in pediatric patients with DMD under different dietary conditions in order to provide guidance for dosing in the Phase 2 portion of the study. The Phase 2 portion of the study was planned to examine the effect of edasalonexent on muscle magnetic resonance imaging (MRI) and age-appropriate functional measures.
METHODS
Study participants
All subjects gave written informed consent prior to study participation. The study was approved by independent Ethics Committees at all 3 sites and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines as set forth by the International Conference on Harmonization (ICH) and the U.S. Code of Federal Regulations.
Males ≥4 to <8 years of age with a diagnosis of DMD based on clinical phenotype and DMD mutation were enrolled if they were able to ambulate independently with or without assistive devices. Only patients adequately vaccinated or with acquired immunity for Varicella virus and vaccinated for influenza were included. Exclusion criteria included corticosteroid use within 6 months of the study since glucocorticoids may modulate NF-κB activity and could thus interfere with attribution of edasalonexent effects. Patients with prior or ongoing medical conditions that could affect patient safety or impair assessment of study results were excluded.
Baseline disease status assessments included those used in DMD clinical trials and natural history studies [33–38] such as age-appropriate timed function tests, [(including the 10 m walk/run, time to climb 4 stairs, and time to stand), the North Star Ambulatory Assessment (NSAA), which is a multi-component functional assessment developed for DMD, and a caregiver-reported quality of life and physical function measure [Pediatric Outcomes Data Collection Instrument (PODCI)]. Timed functional assessments, the NSAA, and quantitative muscle testing were administered by trained physical therapists. These assessments serve as baseline measurements to determine disease progression during the period up to the baseline of the Phase 2 portion of the study.
Study design
This was a 1-week, open-label, multiple-dose study with 3 sequential ascending doses of edasalonexent under different dietary conditions (i.e., administration with low-fat and high-fat meals) designed to assess tolerability and determine dosing regimen that would be used in the Phase 2 portion of the study. Edasalonexent was supplied as 100 mg and 250 mg softgel capsules. To maximize exposure for safety assessments in this first study in pediatric patients, edasalonexent was administered under high fat meal conditions for most of the first week (Days 2–6). A factorial design was used to assess PK under a randomized sequence of high-and low-fat meal conditions on Days 1 and 7. This design permitted assessment of both dosing duration and dietary conditions in a way that minimized time and sample volumes for young patients.
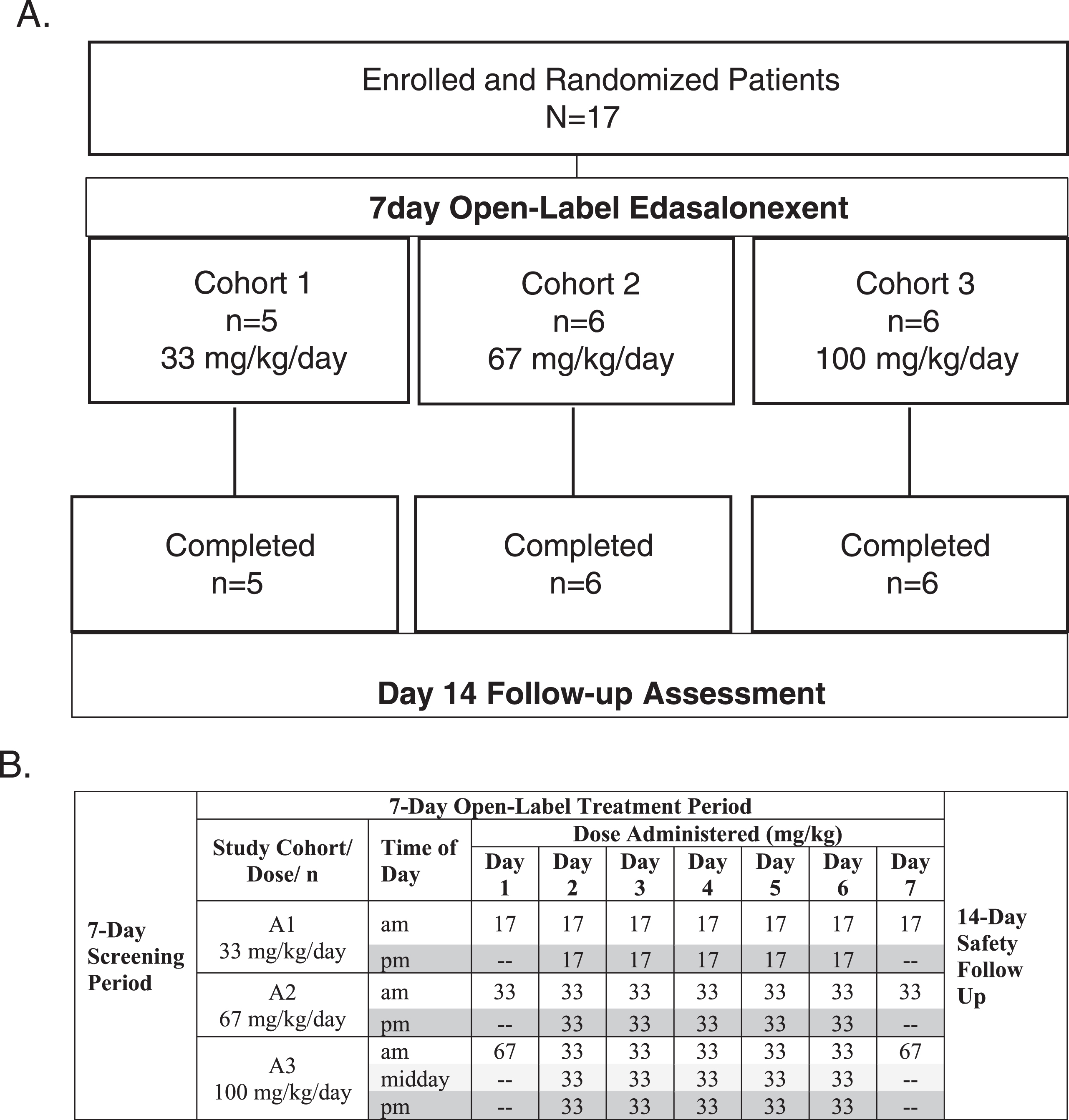
Following 1-week screening to assess eligibility and a baseline assessment to characterize DMD disease status, single oral doses of 17, 33, or 67 mg/kg/day were administered to patients in Cohorts 1, 2 and 3, respectively, on the mornings of Days 1 and 7. Patients within each cohort were randomized 1:1 to receive edasalonexent with a low-fat meal (approximately 0.4 g/kg) on Day 1 and a high-fat meal (approximately 1 g/kg) on Day 7, or a high-fat meal on Day 1 and a low-fat meal on Day 7. This factorial design was based on previous data in adults showing minimal accumulation over 14 days. Pooling Day 1 and 7 data for high- vs. low-fat meals allowed assessment of PK to plan dietary guidance in future clinical trials. Plasma samples were collected up to 24 hours post-dose on Days 1 and 7 for evaluation of PK. In addition, a 24-hour and a 9-hour interval urine collection on Days 1 and 7 post-dose, respectively, were performed for evaluation of metabolite urine PK parameters. On Days 2 through 6, patients received edasalonexent with high-fat meals at total daily doses of 33, 67 and 100 mg/kg/day. Study design and dosing schedule are shown in (Fig. 1). A safety follow-up visit occurred 14 days after the last dose.
Fig. 1.
A. Study Design and Disposition of Patients, B. Dosing Schedule for Day 1–7.
Endpoints
Safety and tolerability of edasalonexent, and the effects of a low- and high-fat meal on exposure following edasalonexent dosing were the primary endpoints. Exploratory endpoints included pharmacodynamic evaluation of levels of serum proteins, and expression of NF-κB-regulated genes in whole blood.
Assessments
Blood and urine samples
Blood samples for determination of plasma concentrations of edasalonexent, and its metabolites salicyluric acid, linker-salicylic acid, linker-DHA and salicylic acid were drawn on Days 1 and 7 for up to 24 hours post-dose. Urine collections were performed on Day 1 (24 hours) and on Day 7 (from pre-dose until 9 hours post-dose) to determine metabolites. Blood samples for evaluation of biomarkers of inflammation and muscle damage and gene expression were collected on Day 1 and Day 7 (pre-dose and 2 hours post-dose).
Pharmacokinetics
Edasalonexent, salicyluric acid, linker-salicylic acid, linker-DHA and salicylic acid in blood and salicylic acid, linker-salicylic acid, and salicyluric acid were determined using validated high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) methods at Celerion (Lincoln, NE). The lower limits of quantitation (LLOQ) values were 1.00, 2.50, 2.00, 2.00 and 100 ng/mL for edasalonexent, salicyluric acid, linker-salicylic acid, linker-DHA, and salicylic acid in blood, respectively. LLOQ values in urine were 200, 20.0, and 20.0 ng/mL for salicylic acid, linker-salicylic acid, and salicyluric acid, respectively.
Noncompartmental PK parameters were calculated including AUCinf, AUClast, Cmax, tmax. and terminal elimination half-life (t1/2). Oral clearance (CL/F), and apparent volume of distribution (Vz/F) were determined for Day 1 dosing only.
Urine PK parameters were calculated including amount of metabolite excreted in the urine during the collection interval (Ae) and percentage of the dose excreted in the urine (% Fe) on a molar basis.
Safety
Safety assessments included clinical laboratory evaluations (hematology, chemistry, coagulation, adrenal function, and urinalysis), physical examinations, adverse events, 12 lead ECGs, and vital signs (blood pressure, pulse, temperature, and respiration). Vital signs, physical exams, clinical labs, and ECGs were assessed/conducted at baseline (predose), throughout the treatment period, and at the end of dosing. Adverse events were monitored throughout treatment and at the follow-up assessment.
Pharmacodynamics
Gene expression analysis of a predefined group of 200 NF-κB-regulated genes (Broad Institute curated HALLMARK NF-κB gene-set [39]) was evaluated via mRNA Sequencing (Illumina) on RNA isolated from whole blood (PAXgene Blood mRNA Kit, PreAnalytix).
Statistics
Plasma concentrations of edasalonexent and metabolites were summarized by treatment group and day using descriptive statistics, as were urine metabolite parameters. The effect of low- versus high-fat meal on edasalonexent PK parameters was evaluated by comparing ln-transformed PK parameters Cmax, AUClast, and AUCinf using an analysis of variance (ANOVA) model using PROC MIXED of SAS ® (Version 9.1.3), with sequence, study day (Day 1 or Day 7), meal type as fixed factors, and patient within sequence as a random factor.
Descriptive statistics were reported for all safety assessments and no inferential statistics were performed. ECG results were classified as normal, abnormal not clinically significant, or abnormal clinically significant. Descriptive statistics were calculated for pharmacodynamic data as well as for the difference from baseline and differences between groups. Two-tailed Wilcoxon matched-pairs signed rank test was used to compare changes from baseline in gene expression.
RESULTS
Study participants
Eighteen patients were screened, and 17 were enrolled from June 2015 to December 2015, with one patient failing screening due to difficulty with trial procedures. None of the 17 participants had received glucocorticoid therapy prior to enrollment. Figure 1A shows the progression of enrolled patients in the study. All enrolled patients were able to swallow capsules and all completed 7 days of treatment without treatment interruption, dose reduction, or early withdrawal. Six patients received 100 mg capsules and 11 received 250 mg capsules. Patient compliance as assessed by diary and observation was >99%.
Demographics and baseline disease characteristics are shown in Table 1. The majority of patients were white and mean age (SD) was 5.5 (0.94) years. At baseline, functional assessments showed reduction in ambulation, endurance and quality of life consistent with previous reports for this patient population and in comparison to healthy age-matched controls [40–42]. (Normative data for timed assessments: 10 m walk/run: 3.4 seconds; 4-stair climb: 1.4 seconds; and TTS: 2.1 seconds)
Table 1
Patient Demographics and Baseline Disease Assessments
| Edasalonexent Cohort | All Patients | |||
| 33 mg/kg/day | 67 mg/kg/day | 100 mg/kg/day | ||
| N = 5 | N = 6 | N = 6 | N = 17 | |
| Age, mean years (SD) | 5.2 (1.10) | 5.5 (0.55) | 5.7 (1.21) | 5.5 (0.94) |
| Race n (%) | ||||
| White | 5 (100) | 6 (100) | 6 (100) | 17 (100) |
| Ethnicity n (%) | ||||
| Hispanic or Latino | 0 | 0 | 1 (16.7) | 1 (5.9) |
| Not Hispanic or Latino | 4 (80) | 6 (100) | 5 (83.3) | 15 (88.2) |
| Unknown | 1 (20) | 0 | 0 | 1 (5.9) |
| Weight, mean kg (SD) | 17.9 (1.87) | 19.6 (3.41) | 22.9 (7.86) | 20.3 (5.49) |
| Baseline Functional Assessments, mean (SD) | ||||
| 10 m walk/run speed (m/sec) | 0.17 (0.05) | 0.19 (0.04) | 0.16 (0.02) | 0.17 (0.04) |
| 10 m walk/run time (sec) | 6.6 (1.9) | 5.4 (0.9) | 6.3 (0.8) | 6.1 (1.3) |
| 4 SC speed (stairs/sec) | 0.2 (0.1) | 0.3 (0.1) | 0.2 (0.1) | 0.3 (0.1) |
| 4 SC time (sec) | 5.4 (2.7) | 3.7 (1.9) | 5.0 (1.6) | 4.6 (2.1) |
| TTS speed (lifts/sec) | 0.2 (0.06) | 0.2 (0.07) | 0.2 (1.1) | 0.2 (0.08) |
| TTS time (sec) | 6.3 (2.4) | 5.3 (2.9) | 6.1 (3.0) | 5.9 (2.7) |
| NSAA total score | 18 (5) | 21 (3) | 19 (6) | 20 (5) |
| PODCI basic mobility and transfer score | 77.6 (19.8) | 77.5 (3.6) | 74.6 (23.0) | 76.5 (16.4) |
| PODCI global score | 63.8 (17.4) | 69.0 (7.5) | 68.6 (13.6) | 67.3 (12.5) |
Pharmacokinetics
Plasma concentrations of edasalonexent and metabolites were determined predose and from 1 to 24 hours after dosing on Days 1 and 7. Plasma pharmacokinetic parameters on Days 1 and 7 are summarized in Table 2. Mean plasma edasalonexent and salicyluric acid concentrations over time on Day 1 and Day 7 for the three dose cohorts are shown in (Fig. 2A and B). Edasalonexent was rapidly absorbed with median tmax ranging from 1.5 to 2.1 hours on Days 1 and 7 for all 3 doses with the exception of 17 mg/kg dose on Day 1 where median tmax was 6.1 hours. Only minor accumulation of edasalonexent was observed following 7 days of repeated oral dosing. Elimination was variable, and dose-independent. Mean t1/2 ranged from 5.4 to 10.4 hours across 3 doses. Apparent volume of distribution and clearance estimates at both 33 and 67 mg/kg doses on Day 1 were high.
Table 2
Edasalonexent and Plasma Pharmacokinetic Parameters on Days 1 and 7 Following Single and Multiple Edasalonexent Doses (Pooled Low-Fat and High-Fat Results)
| Edasalonexent Dose (mg/kg) | ||||||
| Day 1 | Day 7 | |||||
| 17 | 33 | 67 | 17 | 33 | 67 | |
| N = 5 | N = 6 | N = 4 | N = 5 | N = 6 | N = 6 | |
| Edasalonexent | ||||||
| Cmax (ng/mL) | 48±26 | 365±250 | 450±114 | 75±60 | 340±169 | 529±248 |
| AUClast (ng.h/mL) | 340±262 | 1130±592 | 1500±520 | 311±146 | 1340±430 | 2100±824 |
| t1/2 (h) | – | 6.3±0.8 | 5.4±1.1 | 10.4±1.5 | 10.3±5.2 | 8.7±2.3 |
| CL/F (L/h) | – | 615±215 | 1040±705 | – | – | – |
| Vz/F (L) | – | 5620±2450 | 8690±6570 | – | – | – |
| tmax (h) | 6 (4, 9) | 2 (2, 2) | 2 (1, 3) | 2 (2, 4) | 1.5 (1, 2) | 2 (1, 4) |
| Salicyluric acid | ||||||
| Cmax (ng/mL) | 47±26 | 180±86 | 228±67 | 85±64 | 180±64 | 214±82 |
| AUClast (ng.h/mL) | 412±211 | 726±256 | 1200±723 | 556±383 | 961±307 | 1170±349 |
| t1/2 (h) | 15.8±5.6 (n = 2) | 9.8±2.3 (n = 5) | 5.8±2.8 (n = 2) | 23.1 (n = 1) | 12.5±5.6 (n = 4) | 9.8±4.4 (n = 5) |
| tmax (h) | 6 (4, 9) | 2 (2, 2) | 2 (2, 2) | 2 (2, 4) | 2 (2, 4) | 2 (1, 4) |
| Linker-salicylic acid | ||||||
| Cmax (ng/mL) | – | 19±14 | 23±12 | 15 (n = 1) | 20±14 | 29±25 |
| AUClast (ng.h/mL) | – | 38±38 | 50±35 | 7 (n = 1) | 53±44 | 67±42 |
| tmax (h) | – | 2 (2, 2) | 2 (2, 2) | 2 (n = 1) | 2 (2, 2) | 2 (2, 4) |
SD, standard deviation; AUClast, area under the plasma concentration-time curve from time 0 to time of last quantifiable concentration; CL/F, apparent oral clearance; Cmax, maximum observed plasma concentration; tmax, time to Cmax; t1/2, terminal half-life; Vz/F, apparent volume of distribution. All data expressed as the mean±SD except for tmax expressed as the median (min, max).
Fig. 2.
Mean Plasma Concentrations versus Time and Plasma Edasalonexent AUC. A. Mean edasalonexent plasma concentrations Day 1 and Day 7 following single edasalonexent doses of 17 (squares), 33 (triangles) or 67 (circles) mg/kg. B. Mean salicyluric acid plasma concentrations Day 1 and Day 7 following single edasalonexent doses of 17 (squares), 33 (triangles) or 67 (circles) mg/kg. C. Pooled Day 1 and Day 7 AUC and Cmax after single daily doses of edasalonexent at 17, 33 and 67 mg/kg administered with low-fat and high-fat meals.
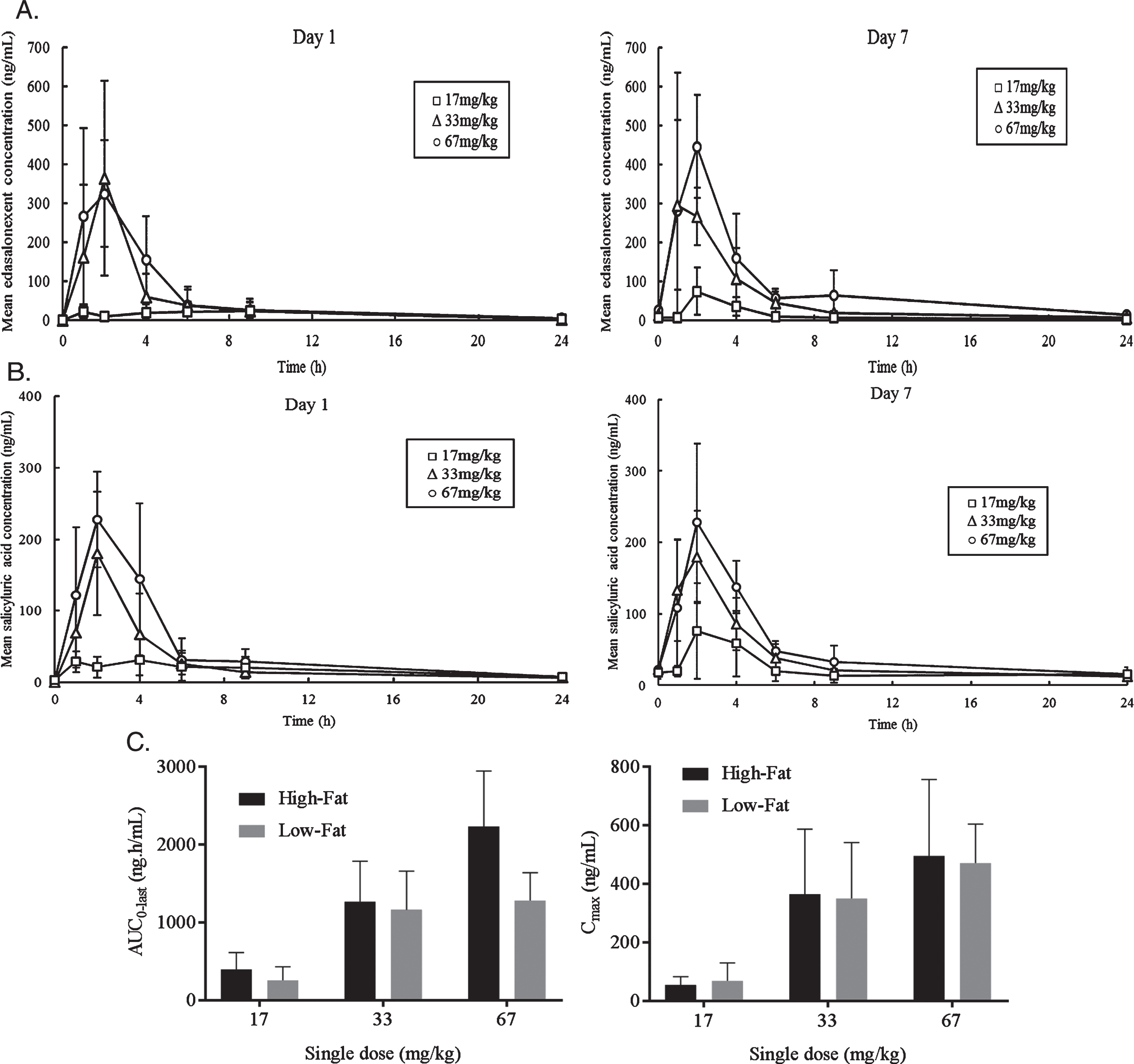
Plasma pharmacokinetic parameters under high-fat and low-fat meal conditions are summarized in (Table 3). Edasalonexent was rapidly absorbed, with mean plasma tmax values ranging from 1.9 to 4.2 hours and from 1.7 to 5.1 hours across dose levels under low-fat and high-fat meal conditions, respectively. Mean terminal half-life values were similar between low-fat (6.6 to 11.5 hours) and high-fat (6.7 to 9.8 hours) meal conditions. AUC and Cmax under low-fat and high-fat meal conditions for Day 1 and Day 7 pooled data are shown in (Fig. 2C). When given with a high-fat meal, edasalonexent exposures appeared to increase nearly proportionally to dose for all 3 doses (single doses of 17, 33, and 67 mg/kg). When edasalonexent was administered with a low-fat meal, AUC and Cmax increased from 17 to 33 mg/kg dose and appeared to plateau at 33 and 67 mg/kg doses.
Table 3
Edasalonexent Plasma Pharmacokinetic Parameters under Low-Fat and High-Fat Meal Conditions (Pooled Day 1 and Day 7 Results)
| Dose | Meal Fat Content | Cmax (ng/mL) | AUC0–last (h.ng/mL) | AUC0 - ∞ (h.ng/mL) | tmax (h) | t1/2 (h) | |
| Mean (SD)/N | Mean (SD)/N | Mean (SD)/N | Mean (SD)/N | Mean (SD)/N | |||
| Edasalonexent | 17 mg/kg | High Fat | 54 (28)/5 | 395 (218)/5 | 306 (44.4)/2 | 5.1 (3)/5 | 8.8 (0.1)/2 |
| Low Fat | 69 (61)/5 | 256 (176)/5 | 390 (222)/3 | 4.2 (2.9)/5 | 11.5 (0.3)/3 | ||
| 33 mg/kg | High Fat | 355 (236)/6 | 1230 (538)/6 | 1550 (763)/3 | 1.7 (0.5)/6 | 9.8 (5.6)/3 | |
| Low Fat | 350 (190)/6 | 1240 (520)/6 | 1200 (555)/5 | 1.9 (0.4)/6 | 6.6 (1.0)/5 | ||
| 67 mg/kg | High Fat | 503 (258)/6 | 2260 (710)/6 | 2400 (812)/6 | 2.0 (1.1)/6 | 6.7 (1.9)/6 | |
| Low Fat | 490 (97.0)/4 | 1260 (228)/4 | 1340 (296)/4 | 2.0 (0.04)/4 | 8.4 (3.2)/4 | ||
| Salicyluric acid | 17 mg/kg | High Fat | 43.2 (16.4)/5 | 442 (192)/5 | 549 (70.3)/2 | 4.7 (2.9)/5 | 21.4 (2.4)/2 |
| Low Fat | 87.9 (64.5)/5 | 526 (404)/5 | 416 (NA)/1 | 4.2 (2.9)/5 | 11.8 (NA)/1 | ||
| 33 mg/kg | High Fat | 159 (83.0)/6 | 779 (285)/6 | 1090 (419)/3 | 2.3 (0.8)/6 | 11.8 (6.9)/3 | |
| Low Fat | 201 (59.0)/6 | 908 (318)/6 | 1040 (352)/6 | 2.0 (0.05)/6 | 10.6 (2.6)/6 | ||
| 67 mg/kg | High Fat | 227 (89.4)/6 | 1340 (503)/6 | 1450 (532)/5 | 2.4 (0.8)/6 | 7.4 (4.3)/5 | |
| Low Fat | 208 (47.1)/4 | 935 (413)/4 | 1490 (209)/2 | 1.8 (0.4)/4 | 11.7 (2.51)/2 | ||
| Linker-salicylic acid | 17 mg/kg | High Fat | – | – | – | – | – |
| Low Fat | 14.6 (NA)/1 | 7.30 (NA)/1 | – | 2.03 (NA)/1 | – | ||
| 33 mg/kg | High Fat | 14.7 (11.3)/6 | 35.3 (36.0)/6 | – | 2.00 (0.00)/6 | – | |
| Low Fat | 24.0 (14.5)/6 | 55.1 (43.9)/6 | 137/1 | 2.02 (0.0456)/6 | 1.84 (NA)/1 | ||
| 67 mg/kg | High Fat | 23.8 (13.3)/6 | 61.2 (26.8)/6 | – | 2.37(0.806)/6 | – | |
| Low Fat | 30.9 (28.8)/4 | 58.6 (56.1)/4 | – | 2.03 (0.0397)/4 | – |
SD, standard deviation; AUClast, area under the serum concentration-time curve from time 0 to time of last quantifiable concentration; AUC0-∞, area under the plasma concentration-time curve from time 0 to infinity; Cmax, maximum observed plasma concentration; tmax, time to Cmax; t½, terminal half-life; NA not applicable.
Metabolites of edasalonexent, including linker-salicylic acid, linker-DHA, free salicylic acid and its metabolite salicyluric acid were also measured. As shown in (Table 2), only minor accumulation of salicyluric acid and linker-salicylic acid was observed following 7 days of repeated oral dosing of edasalonexent. Elimination was variable, and dose-independent. Mean t1/2 of salicyluric acid ranged from 5.8 to 23.1 hours across 3 doses. Similar results for metabolite PK parameters were observed when edasalonexent was administered under low-fat and high-fat meal conditions (Table 3). Salicyluric acid levels mirrored the edasalonexent mean concentration profile and exposure was comparable in molar concentrations. Linker-DHA was not detectable and linker-salicylic acid levels were much lower than either edasalonexent or salicyluric acid levels. Salicylic acid was only sporadically detectable above the lower limit of quantitation.
In urine, a maximum of 4% of the dose was excreted as salicyluric acid, with very low (<0.1%) amounts as linker-salicyluric acid or salicylic acid. Amounts of salicyluric acid generally increased in a dose proportional manner.
Safety and tolerability
During the 7 days of dosing, 30 treatment-emergent adverse events were reported by 12/17 (70.5%) patients. All events were mild in severity except for 3 events of moderate severity (fall, upper abdominal pain and diarrhea in 3 individual patients). Gastrointestinal complaints were the most commonly reported events (17 events in 10 patients). Diarrhea was the most common event (7 events in 4 patients) followed by soft stool (3 events in 3 patients), constipation (3 events in 1 patient), vomiting (2 events in 2 patients), and upper abdominal pain (2 events in 2 patients). The list of all adverse events are shown in (Table 4). Diarrhea (typically described as soft stool) was common at the total daily dose of 100 mg/kg/day, but none of the patients required or received concomitant medications as treatment.
Table 4
All Treatment-Emergent Adverse Events
| Edasalonexent Cohort | All Patients | |||
| 33 mg/kg/day | 67 mg/kg/day | 100 mg/kg/day | ||
| N = 5 | N = 6 | N = 6 | N = 17 | |
| Adverse Event | # Patients (%) | |||
| Any adverse event | 2 (40) | 5 (83.3) | 5 (83.3) | 12 (70.6) |
| Diarrhea | 0 | 0 | 4 (66.7) | 4 (23.5) |
| Soft feces | 1 (20.0) | 1 (16.7) | 1 (16.7) | 3 (17.6) |
| Upper abdominal pain | 1 (20.0) | 0 | 1 (16.7) | 2 (11.8) |
| Vomiting | 0 | 1 (16.7) | 1 (16.7) | 2 (11.8) |
| Nasopharyngitis | 0 | 1 (16.7) | 1 (16.7) | 2 (11.8) |
| Anxiety | 0 | 1 (16.7) | 0 | 1 (5.9) |
| Coagulation time prolonged | 0 | 1 (16.7) | 0 | 1 (5.9) |
| Constipation | 0 | 1 (16.7) | 0 | 1 (5.9) |
| Contact dermatitis | 1 (20.0) | 0 | 0 | 1 (5.9) |
| Enuresis | 0 | 1 (16.7) | 0 | 1 (5.9) |
| Fall | 0 | 1 (16.7) | 0 | 1 (5.9) |
| Headache | 0 | 0 | 1 (16.7) | 1 (5.9) |
| Hyperhidrosis | 0 | 0 | 1 (16.7) | 1 (5.9) |
| Increased appetite | 0 | 1 (16.7) | 0 | 1 (5.9) |
| Musculoskeletal pain | 0 | 0 | 1 (16.7) | 1 (5.9) |
| Swelling face | 0 | 0 | 1 (16.7) | 1 (5.9) |
There were no serious adverse events, dosing interruptions, dose reductions, or discontinuations due to adverse events. Vital signs, blood chemistry, hematology, coagulation and ECG parameters remained similar to baseline values during the 7 days of edasalonexent administration and at the Day 21 follow-up assessment. As expected in patients with DMD, ALT, AST, CK, and LDH levels were elevated from baseline through Day 7 in all patients regardless of cohort/dose level, with no apparent trends related to treatment or dose. Most (10/17, 58.8%) patients also presented with ECG abnormalities at baseline, including 2 patients (1 each in Cohorts 1 and 2) with biventricular hypertrophy and ECG abnormalities assessed as clinically significant at screening. No patient with a normal, or non-clinically significant abnormal ECG at baseline developed a clinically significant ECG abnormality during the study.
Pharmacodynamics
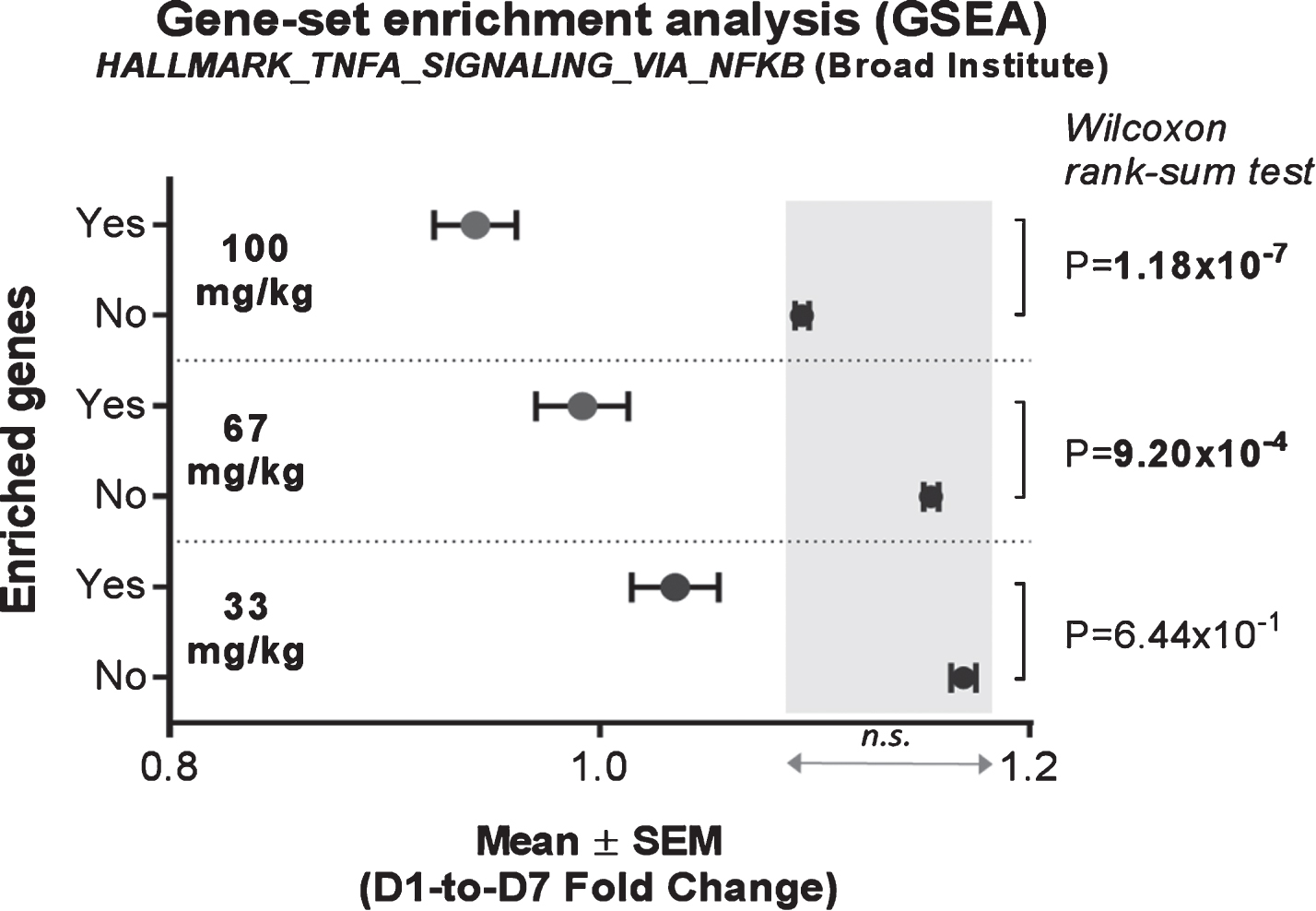
In order to monitor direct short-term activity of edasalonexent on NF-κB in circulating blood cells, expression of NF-κB target genes were assessed in mRNA from whole blood of patients at baseline and after administration of edasalonexent for 1 week. Compared with baseline, the NF-κB-target enriched gene-set was significantly inhibited at the 67 and 100 mg/kg/day doses with mean fold-change difference of –0.1622, and –0.1518, respectively, when compared to the unenriched genes (Fig. 3A). There was no significant difference in unenriched genes among the different dose groups.
Fig. 3.
Effect of Edasalonexent on NF-κB: Change in Expression of NF-κB-Target Genes Following Edasalonexent Treatment. Whole blood mRNA was isolated at baseline and after 1 week of edasalonexent administration. Gene expression was measured in gene sets with (unshaded area) and without (shaded area) enrichment for NF-κB-regulated genes.
DISCUSSION
Contraction-induced damage in muscles of patients with DMD results in chronic activation of inflammatory responses [16]. Chronic activation of NF-κB is initiated early in the disease process, preceding loss of muscle function and [21], and drives muscle degeneration and suppresses muscle regeneration [16–20]. NF-κB signaling pathways are most prominently elevated in skeletal muscles undergoing mechanical stress, (e.g., posterior leg muscles) [43], which show disproportionate inflammation, muscle degeneration, and fibrosis in animal models [44, 45] and in patients [38, 46]. In contrast, muscles of DMD patients exposed to minimal mechanical stress (e.g., thigh gracilis and sartorius muscles and extraocular muscles), while dystrophin-deficient, have less NF-κB activation and also significantly less pathology [44–46]. Inhibition of NF-κB pathways in mdx mice [18, 47, 48] significantly improves histology, function, and muscle regeneration, even with a partial reduction in NF-κB activity achieved by removal of only 1 allele of the p65 subunit of NF-κB [18]. Therefore, targeting NF-κB in DMD may provide a disease-modifying therapeutic approach that is independent of the specific mutation in the dystrophin gene.
Edasalonexent inhibits activated NF-κB pathways and was well tolerated during Phase 1 studies in adults [15]. In this initial one-week study in boys with DMD, edasalonexent was generally well-tolerated, with all patients completing the 1-week study without serious adverse events or dose reductions. Baseline laboratory and ECG abnormalities consistent with clinical observations in DMD patients [49, 50] were not changed following edasalonexent treatment. Studies of longer duration are needed to fully evaluate the safety of edasalonexent in boys with DMD.
Pharmacokinetic assessments were consistent with intact absorption and intracellular cleavage of edasalonexent to salicylic acid, which is subsequently converted to salicyluric acid, and DHA. Edasalonexent exposure was dose dependent and exposure was greater at the higher dose (i.e., 67 mg/kg) when administered with a high-fat meal compared to a low-fat meal, consistent with data reported in healthy adults. Exposures of edasalonexent were generally consistent with those observed in healthy adults on a mg/kg basis [15]. Preliminary population pharmacokinetic modeling showed PK of edasalonexent was not impacted by age (data not shown). Salicyluric acid profiles mirrored the edasalonexent mean concentration profile. Similar to those observed in healthy adults, mean exposures of salicyluric acid were approximately 1.4- to 4.5-fold higher on molar basis than those of edasalonexent [15]. Linker-DHA and linker-salicylic acid, which would be expected to remain intracellular, were not detectable, or only sporadically detectable in plasma. Consistent with plasma observations, very low amounts of linker-salicylic acid and salicylic acid was excreted in urine. Urinary salicyluric acid profiles in pediatric patients with DMD were consistent with previous observations in adults on a mg/kg basis [15].
Edasalonexent plasma levels were consistent with those previously measured in adults at which inhibition of NF-κB was seen [15], and exceeded plasma levels measured in the mdx mouse associated with reduced muscle inflammation and degenerating fibers, and increased regenerating muscle fibers [32]. Contraction-induced muscle damage and associated muscle inflammation can lead to a small increase in measures of systemic inflammation in DMD [51]. The increased systemic inflammation is likely to be the result of NF-κB activation in circulating peripheral blood mononuclear cells induced by intracellular muscle components released during damage [52].
As an exploratory biomarker to assess effects on NF-κB signaling, whole blood gene expression was assessed by mRNA sequencing. Compared with baseline, a prespecified NF-κB signaling pathway gene set was significantly downregulated following treatment with edasalonexent for 7 days at the 2 higher doses of 67 and 100 mg/kg with a statistically significant dose response using regression analysis. This shows that with short-term dosing, edasalonexent can directly reduce the levels of elevated NF-κB in circulating DMD mononuclear cells prior to any changes observable in muscle. In contrast to whole blood mRNA, which is derived from circulating immune cells in the blood, levels of protein in circulation can originate from damaged tissues, such as the detection of elevated levels of muscle-specific proteins in DMD serum [53]. Longer term treatment with edasalonexent would be expected to lower circulating levels of these inflammation and muscle-damage associated proteins, concurrent with expected improvement in muscle pathology. Since removal of just 1 allele of the p65 subunit of NF-κB was sufficient to improve pathology in mdx mice, complete inhibition of NF-κB may not be necessary for improvement in muscle pathology and function [18]. The ability of edasalonexent to block NF-κB and maintain or allow replenishing of the progenitor population even partially, could have disease-modifying impact in patients with DMD. Since inhibition of NF-κB by edasalonexent is independent of the underlying dystrophin mutation, edasalonexent may potentially be effective in all patients with DMD. This could lead to use either as monotherapy or in combination with dystrophin-targeting or myostatin-targeting-therapies to augment reduction in muscle inflammation and degeneration, and enhanced muscle regeneration.
The patient population had decreased ambulation and endurance similar to similarly-aged DMD patients [40, 42], which were reduced compared to historical data for healthy subjects of similar age. PODCI scores were similar to previously reported data for young DMD patients and were 20–30% lower than scores in healthy boys of similar ages [41]. There is a great need for interventions in this patient population that could impact the natural decline in ambulation and strength expected over time [54].
Conclusions
This first study in pediatric patients with DMD showed that edasalonexent was well-tolerated and inhibited NF-κB pathways after one week of dosing. These results support further clinical development of edasalonexent and the dose regimen selection in Part B (12-week double-blind, placebo-controlled) of the study in pediatric patients with DMD.
DECLARATIONS OF INTEREST
EF and RSF are principal investigators in clinical trials for Catabasis and have received financial support for conduct of the study, and also served as advisory board members for Catabasis. KV, SY, and GT are investigators on research studies for Catabasis and have received financial support for conduct of the study. HLS is a consultant for Catabasis and has also served as an advisory board member for Catabasis. MM, HL, AN, PB, AF and JD are employees of Catabasis Pharmaceuticals, Inc.
ACKNOWLEDGMENTS
The authors thank the subjects for their participation and the staff of the clinical research organizations (site) for conduct of the studies. The authors gratefully acknowledge colleagues at Catabasis Pharmaceuticals, Inc. for their contributions to the development of edasalonexent. All authors participated in the design, conduct, and analysis of the study and approved the final manuscript. Catabasis Pharmaceuticals, Inc. provided financial support for the conduct of the research and data analyses. Patrice C. Ferriola, PhD, of KZE PharmAssociates, LLC provided assistance in preparation of the manuscript and was funded by Catabasis Pharmaceuticals, Inc.
REFERENCES
[1] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: (3):304–13. |
[2] | Moat SJ , Bradley DM , Salmon R , Clarke A , Hartley L . Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. (2013) ;21: (10):1049–53. |
[3] | Kieny P , Chollet S , Delalande P , Le Fort M , Magot A , Pereon Y , et al. Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011. Ann Phys Rehabil Med. (2013) ;56: (6):443–54. |
[4] | Nelson SF , Miceli MC . FDA approval of eteplirsen for muscular dystrophy. Jama. (2017) ;317: (14):1480. |
[5] | Goto M , Komaki H , Takeshita E , Abe Y , Ishiyama A , Sugai K , et al. Long-term outcomes of steroid therapy for Duchenne muscular dystrophy in Japan. Brain Dev. (2016) ;38: (9):785–91. |
[6] | Wong BL , Rybalsky I , Shellenbarger KC , Tian C , McMahon MA , Rutter MM , et al. Long-term outcome of interdisciplinary management of patients with duchenne muscular dystrophy receiving daily glucocorticoid treatment. J Pediatr. (2017) ;182: :296–303.e1. |
[7] | Biggar WD , Harris VA , Eliasoph L , Alman B . Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. (2006) ;16: (4):249–55. |
[8] | Kim S , Zhu Y , Romitti PA , Fox DJ , Sheehan DW , Valdez R , et al. Associations between timing of corticosteroid treatment initiation and clinical outcomes in Duchenne muscular dystrophy. Neuromuscul Disord. (2017) . |
[9] | Matthews E , Brassington R , Kuntzer T , Jichi F , Manzur AY . Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. (2016) ;(5):Cd003725. |
[10] | Ricotti V , Ridout DA , Scott E , Quinlivan R , Robb SA , Manzur AY , et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. (2013) ;84: (6):698–705. |
[11] | Hanaoka BY , Peterson CA , Horbinski C , Crofford LJ . Implications of glucocorticoid therapy in idiopathic inflammatory myopathies. Nat Rev Rheumatol. (2012) ;8: (8):448–57. |
[12] | Schakman O , Gilson H , Kalista S , Thissen J . Mechanisms of muscle atrophy induced by glucocorticoids. Hormone Res. 72: (Suppl 1):36–41. |
[13] | Rangel V , Martin AS , Peay HL . Duchenne connect registry report. PLoS Curr. (2012) ;4: :RRN1309. |
[14] | Vu CB , Bemis JE , Benson E , Bista P , Carney D , Fahrner R , et al. Synthesis and characterization of fatty acid conjugates of niacin and salicylic acid. J Med Chem. (2016) ;59: (3):1217–31. |
[15] | Donovan JM , Zimmer M , Offman E , Grant T , Jirousek M . A novel NF-kappaB inhibitor, edasalonexent (CAT-1004), in development as a disease-modifying treatment for patients with duchenne muscular dystrophy: Phase 1 safety, pharma-cokinetics, and pharmacodynamics in adult subjects. J Clin Pharmacol. (2017) ;57: (5):627–39. |
[16] | Rosenberg AS , Puig M , Nagaraju K , Hoffman EP , Vil-lalta SA , Rao VA , et al. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. (2015) ; 7: (299):299rv4. |
[17] | Shin J , Tajrishi MM , Ogura Y , Kumar A . Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol. (2013) ;45: (10):2266–79. |
[18] | Acharyya S , Villalta SA , Bakkar N , Bupha-Intr T , Janssen PM , Carathers M , et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. (2007) ;117: (4):889–901. |
[19] | Li H , Malhotra S , Kumar A . Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl). (2008) ;86: (10):1113–26. |
[20] | Peterson JM , Bakkar N , Guttridge DC . NF-kappaB signaling in skeletal muscle health and disease. Curr Top Dev Biol. (2011) ;96: :85–119. |
[21] | Chen YW , Nagaraju K , Bakay M , McIntyre O , Rawat R , Shi R , et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. (2005) ;65: (6):826–34. |
[22] | Yin MJ , Yamamoto Y , Gaynor RB . The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. (1998) ;396: (6706):77–80. |
[23] | Kopp E , Ghosh S . Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. (1994) ;265: (5174):956–9. |
[24] | Cai D , Frantz JD , Tawa NE Jr , Melendez PA , Oh BC , Lidov HG , et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. (2004) ;119: (2):285–98. |
[25] | Williams-Bey Y , Boularan C , Vural A , Huang NN , Hwang IY , Shan-Shi C , et al. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-kappaB activation and enhancing autophagy. PLoS One. (2014) ;9: (6):e97957. |
[26] | Zwart SR , Pierson D , Mehta S , Gonda S , Smith SM . Capacity of omega-3 fatty acids or eicosapentaenoic acid to counteract weightlessness-induced bone loss by inhibiting NF-kappaB activation: From cells to bed rest to astronauts. J Bone Miner Res. (2010) ;25: (5):1049–57. |
[27] | Vedin I , Cederholm T , Freund-Levi Y , Basun H , Garlind A , Irving GF , et al. Effects of DHA-rich n-3 fatty acid supplementation on gene expression in blood mononuclear leukocytes: The OmegAD study. PLoS One. (2012) ;7: (4):e35425. |
[28] | Simopoulos AP . Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. (2002) ;21: (6): 495–505. |
[29] | Bouwens M , van de Rest O , Dellschaft N , Bromhaar MG , de Groot LC , Geleijnse JM , et al. Fish-oil supplementation induces antiinflammatory gene expression profiles in human blood mononuclear cells. Am J Clin Nutr. (2009) ;90: (2): 415–24. |
[30] | Oh DY , Talukdar S , Bae EJ , Imamura T , Morinaga H , Fan W , et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. (2010) ;142: (5):687–98. |
[31] | Liu H , Ting A , Lee D , Zimmer M , D W , R D , et al. In vitro and in vivo metabolite profiling and identification of edasalonexent (CAT-1004), a bioconjugate of salicylic acid (SA) and docosahexaenoic acid (DHA) using SMART linker technology, in New England Drug Metabolism and Discussion Summer Symposium. Waltham, MA, (2017) . |
[32] | Hammers DW , Sleeper MM , Forbes SC , Coker CC , Jirousek MR , Zimmer M , et al. Disease-modifying effects of orally bioavailable NF-kappaB inhibitors in dystrophin-deficient muscle. JCI Insight. (2016) ;1: (21):e90341. |
[33] | Henricson EK , Abresch RT , Cnaan A , Hu F , Duong T , Arrieta A , et al. The cooperative international neuro-muscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. (2013) ;48: (1):55–67. |
[34] | Hollingsworth KG , Garrood P , Eagle M , Bushby K , Straub V . Magnetic resonance imaging in Duchenne muscular dystrophy: Longitudinal assessment of natural history over 18 months. Muscle Nerve. (2013) ;48: (4):586–8. |
[35] | McDonald CM , Henricson EK , Abresch RT , Florence J , Eagle M , Gappmaier E , et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: Reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. (2013) ;48: (3):357–68. |
[36] | McDonald CM , Henricson EK , Abresch RT , Han JJ , Escolar DM , Florence JM , et al. The cooperative international neuromuscular research group Duchenne natural history study-a longitudinal investigation in the era of glucocor-ticoid therapy: Design of protocol and the methods used. Muscle Nerve. (2013) ;48: (1):32–54. |
[37] | Scott E , Eagle M , Mayhew A , Freeman J , Main M , Shee-han J , et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int. (2012) ;17: (2):101–9. |
[38] | Willcocks RJ , Arpan IA , Forbes SC , Lott DJ , Senesac CR , Senesac E , et al. Longitudinal measurements of MRI-T2 in boys with Duchenne muscular dystrophy: Effects of age and disease progression. Neuromuscul Disord. (2014) ;24: (5):393–401. |
[39] | Liberzon A , Birger C , Thorvaldsdottir H , Ghandi M , Mesirov Jill P , Tamayo P . The molecular signatures database hallmark gene set collection. Cell Systems. (2015) ;1: (6):417–25. |
[40] | Bushby K , Connor E . Clinical outcome measures for trials in Duchenne muscular dystrophy: Report from International Working Group meetings. Clinical investigation. (2011) ;1: (9):1217–35. |
[41] | Henricson E , Abresch R , Han JJ , Nicorici A , Goude Keller E , de Bie E , et al. The 6-minute walk test and person-reported outcomes in boys with duchenne muscular dystrophy and typically developing controls: Longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. (2013) ;5. |
[42] | Mazzone E , Martinelli D , Berardinelli A , Messina S , D'Amico A , Vasco G , et al. North star ambulatory assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord. (2010) ;20: (11):712–6. |
[43] | Kumar A , Boriek AM . Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. (2003) ;17: (3):386–96. |
[44] | Hu X , Blemker S . Musculoskeletal simulation can help explain selective muscle degeneration in Duchenne muscular dystrophy. Muscle Nerve. (2015) ;52: :174–82. |
[45] | Porter JD , Merriam AP , Leahy P , Gong B , Khanna S . Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum Mol Genet. (2003) ;12: (15):1813–21. |
[46] | Forbes SC , Willcocks RJ , Triplett WT , Rooney WD , Lott DJ , Wang DJ , et al. Magnetic resonance imaging and spec-troscopy assessment of lower extremity skeletal muscles in boys with Duchenne muscular dystrophy: A multicenter cross sectional study. PLoS One. (2014) ;9: (9):e106435. |
[47] | Lu A , Proto JD , Guo L , Tang Y , Lavasani M , Tilstra JS , et al. NF-kappaB negatively impacts the myogenic potential of muscle-derived stem cells. Mol Ther. (2012) ;20: (3):661–8. |
[48] | Mourkioti F , Kratsios P , Luedde T , Song YH , Delafontaine P , Adami R , et al. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest. (2006) ;116: (11):2945–54. |
[49] | Korones DN , Brown MR , Palis J . "Liver function tests" are not always tests of liver function. Am J Hematol. (2001) ;66: (1):46–8. |
[50] | Takami Y , Takeshima Y , Awano H , Okizuka Y , Yagi M , Matsuo M . High incidence of electrocardiogram abnormalities in young patients with duchenne muscular dystrophy. Pediatr Neurol. (2008) ;39: (6):399–403. |
[51] | Cruz-Guzman Odel R , Rodriguez-Cruz M , Escobar Cedillo RE . Systemic inflammation in duchenne muscular dystrophy: Association with muscle function and nutritional status. Biomed Res Int. (2015) ;2015: :891972. |
[52] | Rayavarapu S , Coley W , Nagaraju K . An update on pathogenic mechanisms of inflammatory myopathies. Curr Opin Rheumatol. (2011) ;23: (6):579–84. |
[53] | Hathout Y , Brody E , Clemens PR , Cripe L , DeLisle RK , Furlong P , et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. (2015) ;112: (23):7153–8. |
[54] | Mazzone ES , Pane M , Sormani MP , Scalise R , Berardinelli A , Messina S , et al. 24 month longitudinal data in ambulant boys with Duchenne muscular dystrophy. PLoS One. (2013) ;8: (1):e52512. |


