
Disability and Contextual Factors in Patients with Amyotrophic Lateral Sclerosis - A Three-Year Observational Study
Abstract
Background:
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by muscle weakness and wasting. Observational natural history studies can give information on body function/structure impairments, activity limitations and participation restrictions, i.e. disability. Information needed to plan and develop care and support.
Objective:
To describe and explore disease severity and impairments, activity limitations, participation restrictions and contextual factors over time. In specific, to explore concurrent presence of cognitive impairment, fatigue, anxiety, depression and pain, and whether these impairments were related to disease severity.
Methods:
In this three-year observational study, 60 patients with ALS were included at baseline. Follow-ups were performed every 6 months unless participants had deceased or declined participation. Data was collected from medical records, and by study-specific and standardized questionnaires administrated during home visits.
Results:
Regardless of disease severity; fatigue, anxiety, depression and pain were present in patients with ALS at each data collection. Approximately one-third experienced two or more of these impairments concurrently, i.e. at the same time point. Cognitive impairment could not be assessed in many patients due to their physical impairments. Disease severity was not associated with fatigue, anxiety, depression or pain.
Conclusions:
Patients with ALS need, throughout the course of the disease, to be regularly screened for commonly present impairments, activity limitations and participation restrictions so that person-centered interventions can be applied at the right time.
INTRODUCTION
The impact of a disease can be described from the perspective of body, individual and society by the International Classification of Functioning, Disability and Health (ICF), that was launched by the World Health Organization in 2001 [1]. Neutral aspects are expressed as functioning whereas disability indicates problems and summarises body function/structure impairments, activity limitations, and participation restrictions. Disability is always an interaction between contextual factors, i.e. personal and environmental factors, and the disease [1]. Descriptions of disease associated disability give a broader and more meaningful picture of individuals’ and populations’ experience of health.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of upper and lower motor neurons. The pooled worldwide standardized incidence is 1.7 per 100 000 person-years, 2.0 for men and 1.4 for women [2]. There are, however, subcontinent differences with higher rates of ALS reported from Europe, e.g. from Sweden with an age-standardized incidence of 3.0 per 100 000 person-years [3]. Incidence rates increase by age and reach a peak at 70–74 years [4]. The disease can be classified depending on site of onset. Most common is spinal onset where symptoms start in limb muscles. Approximately one-third of patients with ALS have bulbar onset, i.e. symptoms start in muscles that control speech, chewing and swallowing [4]. Most patients with ALS live for approximately 2–4 years from symptom onset, though some may survive for decades [5]. Predictors of longer survival are younger age at diagnosis and spinal onset [5].
The revised ALS functional rating scale (ALSFRS-R) [6] is commonly used in research and clinical practice as a marker of ALS disease severity. The scale captures impairments in speech, salivation, swallowing and respiratory functions; and activity limitations in mobility and self-care. The ALSFRS-R measures disease severity from the patient’s perspective and gives a broad picture of the impact of the motor neuron degeneration. One way to facilitate the clinical interpretation of the scale is to categorize the scores into stages/levels of severity [7, 8].
Besides muscle weakness and atrophy, other impairments may occur such as cognitive impairments [9, 10], fatigue [11, 12], anxiety [13, 14], depression [11, 13, 14] and pain [15, 16]. The reported frequencies of these impairments in ALS vary greatly, from around 20%up to approximately 50–60%[9, 11–13, 15, 16]. The large variability is likely due to differences in study designs, settings, sample sizes and the used measures. Thus, there are uncertainties about the occurrence of these impairments during the disease course, and especially how cognitive impairment, fatigue, anxiety, depression and pain concurrently occur in patients with ALS. Further, less is known regarding the relation to disease severity and whether there is a change over time. Longitudinal studies are scarce but have indicated that cognitive impairment and fatigue might be associated with disease severity [10–12] in contrast to anxiety and depression [14]. As for pain, findings from cross-sectional studies are contradictive regard to the relationship with disease severity [15, 16].
Although it is generally agreed that ALS impacts patients’ everyday life, studies describing activity and participation in ALS are surprisingly few. However, both activity limitations and participation restrictions have been reported and associations with physical functioning and disease severity have been identified [8, 17, 18].
We initiated a three-year observational study in patients with ALS in 2012 with the aim to describe and explore disease severity and impairments, activity limitations, participation restrictions and contextual factors over time. In specific we wanted to explore concurrent presence of cognitive impairment, fatigue, anxiety, depression and pain, and whether these impairments were related to disease severity.
MATERIALS AND METHODS
Participants
Participants were recruited from the ALS clinic at the Karolinska University Hospital, Stockholm, Sweden. Inclusion criteria were an ALS diagnosis according to the revised El Escorial criteria [19] and an age over 18 years. Twenty of 102 identified patients were excluded due to being in a terminal stage (n = 11), having dementia (n = 4) or inability to understand Swedish (n = 5). Eligible patients received an invitation letter whereof 19 declined participation and three did not answer, thus, 60 patients were included at baseline. These patients were thereafter contacted every six months up to three years after baseline unless they declined participation or deceased. The study was approved by the ethical review board in Stockholm, registration number: 2012/842-31/2, and procedures were conducted in accordance with the Declaration of Helsinki.
Procedures
Data collections were performed at baseline (T1) and every six months (T2-T7) up to three years after baseline, and informed signed consent was retrieved at each time point. Data was collected from medical records, and by study-specific and standardized questionnaires administrated during home visits. Each home visit lasted approximately 2.5 hours and was performed by either one out of three physiotherapists or by an occupational therapist. All therapists had clinical experience of ALS and were trained to perform data collections. Some baseline data in the present study has been published elsewhere [20].
Measures
Medical records were used to retrieve information on site of onset, time since diagnosis and medications. Study-specific questionnaires were used for data collection on contextual factors, i.e. age, sex, education level, living conditions, work status, ventilation, gastrostomy, aids (for mobility, activities of daily living (ADL) and communication), home adaptations, home care or personal assistance, and transport services.
Disease severity was assessed with the ALSFRS-R [6]. This 12-item scale ranges from 0 to 48, where higher scores indicate less severity. The ALSFRS-R score was categorized into three stages/levels of severity, i.e. mild (score 37–48), moderate (score 25–36), and severe (score 0–24), as previously suggested [7].
Cognition was assessed with the Montreal Cognitive Assessment (MoCA), a brief screening tool evaluating short-term and working memory, visuospatial and executive functions, attention, concentration, language and orientation [21]. Fatigue was assessed with the Checklist Individual Strength (CIS) fatigue subscale [22]. This eight-item subscale ranges from 8 to 56. Higher scores indicate more impairment and a score ≥35 was used for categorization of fatigue [12]. Anxiety and depression were assessed with the anxiety and depression subscales of the Hospital Anxiety and Depression Scale (HADS) [23]. Each seven-item subscale ranges from 0 to 21, with higher scores indicating more impairment. A score ≥8 was used for categorization of a possible or probable case of anxiety and depression, respectively [23]. Pain was screened for by the question “Throughout our lives, most of us have had pain from time to time (such as minor headaches, sprains, and toothaches). Have you had pain other than these everyday kinds of pain the last week?”. Participants answering “Yes” were asked to rate their pain on average using a Numeric Rating Scale (NRS) ranging from 0 (no pain) to 10 (worst imaginable pain). A score ≥4 was, in accordance with our hospital guidelines, considered to indicate clinically relevant pain and used for categorization of impairment.
Personal ADL (P-ADL) was assessed with Katz P-ADL index and instrumental ADL (I-ADL) with the Katz I-ADL index [24]. The Katz P-ADL index consists of six items; feeding, continence, transfer, toileting, dressing and bathing, and the Katz I-ADL index of four items; cooking, cleaning, shopping and transportation. Each item is assessed based on level of dependency and scored 0 if the patient is dependent and 1 if independent. Total scores can range from 0 to 6 and from 0 to 4 for the Katz P-ADL and I-ADL index, respectively. Patients scoring less than full sum scores were categorized as dependent and having activity limitations. Frequency of social and lifestyle activities was assessed with the Frenchay Activities Index (FAI) [25]. This 15-item scale can range from 0 to 45. Higher scores indicate a higher frequency of social and lifestyle activities. Patients with scores ≤25th percentile of age and sex predicted normative values were categorized as below norm [26] and having participation restrictions.
Data analysis
Statistical analyses of baseline (T1) data between patients in the study at T7 and those who had declined participation or died after T1 were performed with Chi-square test for sex and Mann-Whitney U-test for age, time since diagnosis and disease severity (ALSFRS-R score).
Descriptive statistics were used to present data, i.e. frequency, percentage, median and interquartile range (IQR). Results on cognitive impairment were excluded as 60–80%of patients were unable to complete the MoCA due to physical disability (unable to perform pen-and-paper items). The described cut-off values were used for categorization of presence of an impairment, i.e. fatigue, anxiety, depression and clinically relevant pain. To describe concurrent presence of impairments, each patient’s total number of impairments was calculated. Descriptive statistics were used to explore relations between impairments with the three levels of disease severity. The non-parametric Spearman rank correlation test was used to explore associations between impairments (CIS fatigue subscale, HADS, pain NRS) and disease severity (ALSFRS-R). The scores from CIS fatigue subscale, HADS and pain NRS were inverted in these analyses to facilitate interpretation. Thus, higher scores indicated a better functioning in all measures. Correlation coefficients (rs) were considered to be low if having absolute values ≤0.49, high when ≥0.70, and moderate when in between low and high [27]. The level of significance was set to p < 0.05 and analyses were performed with IBM SPSS Statistics 24 (for Windows).
RESULTS
Twelve of the 60 patients included at baseline (T1) were still in the study three years later (T7). There was a significant difference (p = 0.048) in time since diagnosis at baseline between these 12 patients (median time 41 months) and the other 48 (median time 16 months), but no significant differences in sex, age or disease severity. Twenty-five patients died between data collections during study period and 23 patients declined participation, the most common given reasons for declined participation were disease-related such as being too fatigued or being in a terminal stage. There were no significant differences in baseline data concerning time since diagnosis, sex, age or disease severity between the 12 patients who were still in the study at T7 and those 23 who had declined participation at some time-point. Seven of these 23 patients were alive, but not participating, at T7. Figure 1 presents a flow chart of patient inclusion and retention during the three-year study period.
Information on patient characteristics and contextual factors is presented in Table 1. Patients’ median age was around 60 years, approximately half were men, and spinal onset was most common. A few (four patients at baseline of whom three were still in the study at T7) patients’ time since diagnosis exceeded 120 months and, thus, they had been living with the disease for more than a decade. Most patients were cohabiting with a family member in their own home, and only three patients stayed in residential care facilities. Disease severity progressed during the study period and none were classified as having mild disease after two years. Most patients had disease modifying drugs (riluzole), and anti-depressive medication was prescribed to approximately one-third. Although non-invasive ventilation was more frequently used, three to five patients had invasive ventilation. Over time, almost all patients reported having aids, home adaptations, home care/personal assistance and transportation services.
Fig.1
Flow chart of patient inclusion and retention during the three-year observational study, i.e. at baseline (T1) and each six-month follow-up (T2-T7). Patients who are listed as lost to follow-up have later rejoined and participated in the next data collection.
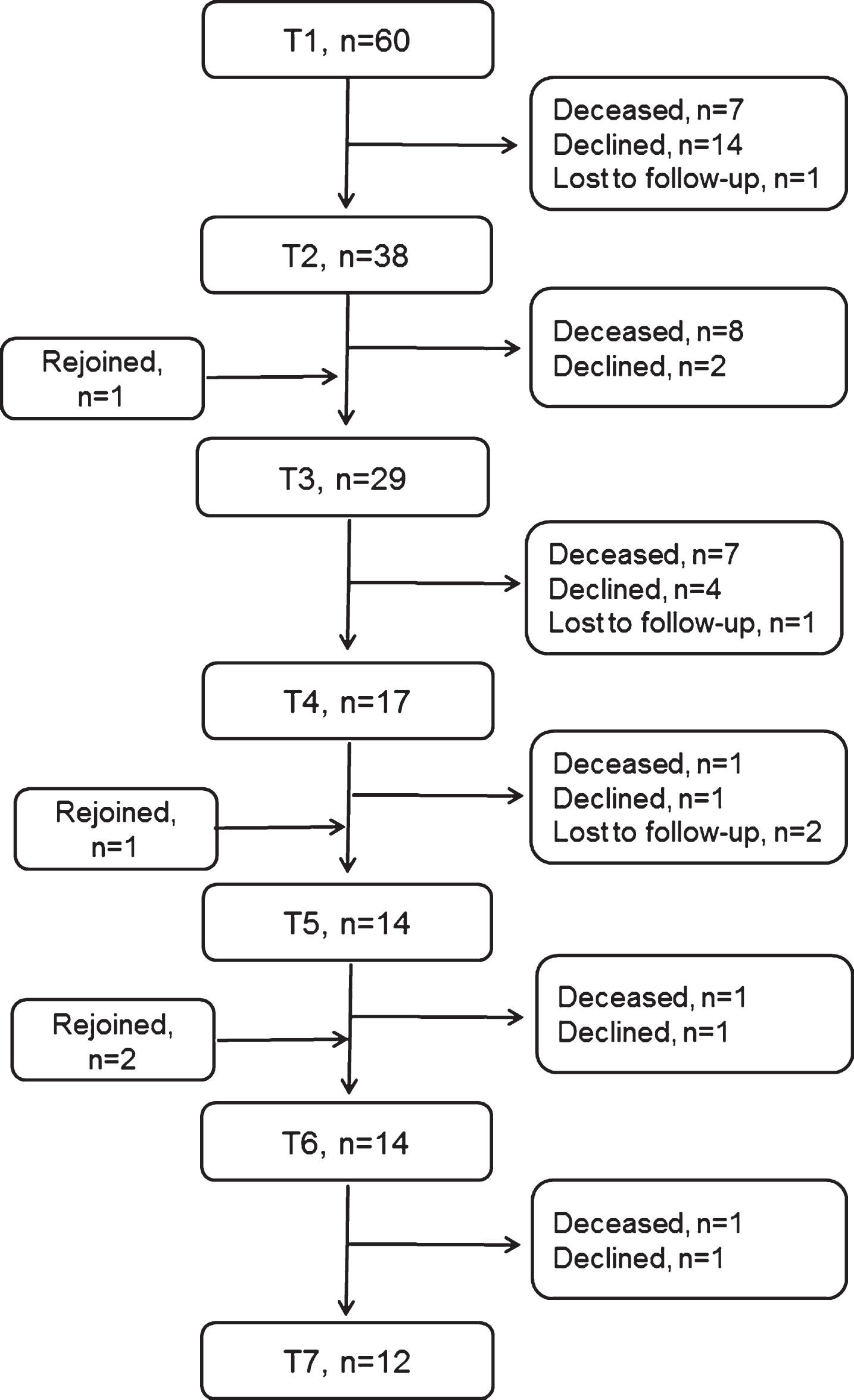
Table 1
Information on patient characteristics and contextual factors at baseline (T1) and each six-month follow-up (T2-T7). N represents the number of patients participating at each time point
| T1 (N = 60) | T2 (N = 38) | T3 (N = 29) | T4 (N = 17) | T5 (N = 14) | T6 (N = 14) | T7 (N = 12) | |
| Age, years, median (IQR) | 61 (55–69) | 60 (54–67) | 61 (53–66) | 61 (48–68) | 63 (58–68) | 62 (50–67) | 63 (48–68) |
| Men, n (%) | 32 (53) | 21 (55) | 17 (59) | 11 (65) | 10 (71) | 8 (57) | 6 (50) |
| University education, n (%) | 22 (37) | 11 (29) | 9 (31) | 6 (35) | 5 (36) | 5 (36) | 5 (42) |
| Living with family member, n (%) | 45 (75) | 31 (82) | 23 (79) | 14 (82) | 12 (86) | 12 (86) | 10 (83) |
| Working, n | 8 | 4 | 2 | 2 | − | − | − |
| Time since diagnosis, months, median (IQR) | 17 (8–35) | 24 (14–57) | 35 (20–82) | 52 (32–107) | 60 (36–123) | 65 (45–129) | 77 (50–152) |
| Spinal onset, n (%) | 45 (75) | 30 (79) | 25 (86) | 16 (94) | 13 (93) | 13 (93) | 11 (92) |
| ALSFRS-R, median (IQR) score 0–48 | 29 (20–37) | 29 (18–34) | 25 (12–33) | 27 (16–33) | 23 (11–32) | 19 (3–26) | 15 (5–27) |
| −mild, n (%) score 37–48 | 20 (33) | 6 (16) | 5 (17) | 1 (6) | 1 (7) | − | − |
| −moderate, n (%) score 25–36 | 17 (28) | 19 (50) | 10 (35) | 9 (53) | 4 (29) | 6 (43) | 4 (33) |
| −severe, n (%) score 0–24 | 23 (39) | 13 (34) | 14 (48) | 7 (41) | 9 (64) | 8 (57) | 8 (67) |
| Medications | |||||||
| −disease modifying, n (%) | 46 (77) | 29 (76) | 23 (79) | 13 (77) | 12 (86) | 12 (86) | 10 (83) |
| −symptomatic, n (%) | 27 (45) | 22 (58) | 15 (52) | 10 (59) | 7 (50) | 8 (57) | 10 (83) |
| −anti-depressive, n (%) | 16 (27) | 11 (29) | 14 (48) | 3 (18) | 5 (36) | 3 (21) | 4 (33) |
| Ventilation | |||||||
| −non-invasive, n (%) | 18 (30) | 11 (29) | 5 (17) | 3 (18) | 3 (21) | 3 (21) | 3 (25) |
| −invasive, n (%) | 4 (7) | 3 (8) | 3 (10) | 3 (18) | 3 (21) | 5 (36) | 4 (33) |
| −Gastrostomy, n (%) | 18 (30) | 11 (29) | 11 (38) | 5 (29) | 6 (43) | 7 (50) | 7 (58) |
| Aids | |||||||
| −mobility, n (%) | 49 (82) | 35 (92) | 28 (97) | 16 (94) | 13 (93) | 14 (100) | 12 (100) |
| −activities of daily living, n (%) | 43 (72) | 31 (82) | 24 (83) | 15 (88) | 12 (86) | 13 (93) | 12 (100) |
| −communication, n (%) | 36 (60) | 23 (61) | 17 (59) | 11 (65) | 11 (79) | 13 (93) | 11 (92) |
| Home adaptations, n (%) | 34 (57) | 27 (71) | 23 (79) | 13 (77) | 12 (86) | 13 (93) | 12 (100) |
| Home care, personal assistance, n (%) | 31 (52) | 24 (63) | 23 (79) | 12 (71) | 10 (71) | 11 (79) | 10 (83) |
| Transport service, n (%) | 40 (67) | 32 (84) | 27 (93) | 17 (100) | 14 (100) | 14 (100) | 12 (100) |
IQR, interquartile range; ALSFRS-R, ALS Functioning Rating Scale-Revised.
Table 2
Results from measures regarding impairments (CIS fatigue, HADS anxiety, HADS depression, pain NRS), activity limitations (Katz P-ADL, Katz I-ADL) and participation restrictions (FAI) at baseline (T1) and each six-month follow-up (T2-T7). N represents the number of patients participating at each time point and n indicates the number of patients having answered the questionnaires
| T1 (N = 60) | T2 (N = 38) | T3 (N = 29) | T4 (N = 17) | T5 (N = 14) | T6 (N = 14) | T7 (N = 12) | |
| CIS fatigue, score 8–56 | |||||||
| −median (IQR) | 38 (26–43) | 38 (30–44) | 35 (25–43) | 33 (24–44) | 30 (21–40) | 32 (25–42) | 34 (23–46) |
| −fatigue ≥35, yes/no, n | 31/20 | 21/15 | 13/13 | 7/10 | 6/8 | 4/9 | 6/6 |
| HADS anxiety, score 0–21 | |||||||
| −median (IQR) | 5 (2–8) | 4 (1–8) | 4 (1–6) | 3 (1–7) | 2 (1–5) | 3 (1–6) | 3 (0–7) |
| −anxiety ≥8, yes/no, n | 14/42 | 9/27 | 3/24 | 2/15 | 2/12 | 0/14 | 2/10 |
| HADS depression, score 0–21 | |||||||
| −median (IQR) | 4 (2–7) | 5 (2–9) | 4 (2–7) | 4 (1–8) | 4 (2–8) | 5 (2–7) | 3 (2–7) |
| −depression ≥8, yes/no, n | 12/44 | 10/26 | 3/24 | 4/13 | 5/9 | 3/11 | 2/10 |
| −Pain NRS, score 0–10 | |||||||
| −median (IQR) | 5 (3–7) | 5 (3–6) | 5 (4–7) | 6 (4–7) | 4 (4-5) | 5 (4-5) | 4 (3–5) |
| −pain ≥4, yes/no, n | 14/7 | 12/4 | 11/3 | 6/2 | 3/0 | 5/1 | 3/3 |
| Katz P-ADL, score 0-6 | |||||||
| −median (IQR) | 4 (1–6) | 3 (2–5) | 2 (1–5) | 2 (1–3) | 2 (1–4) | 1 (0–3) | 1 (0-1) |
| −dependent ≤5, yes/no, n | 41/19 | 29/9 | 25/4 | 16/1 | 12/2 | 14/0 | 12/0 |
| −Katz-I-ADL, score 0–4 | |||||||
| −median (IQR) | 0 (0–2) | 0 (0-1) | 0 (0-0) | 0 (0-0) | 0 (0-0) | 0 (0-0) | 0 (0-0) |
| −dependent ≤3, yes/no, n | 52/8 | 37/1 | 28/1 | 16/1 | 14/0 | 14/0 | 12/0 |
| FAI, score 0–45 | |||||||
| −median (IQR) | 12 (5–26) | 7 (4–18) | 8 (4–14) | 7 (4–11) | 6 (3–9) | 4 (2–8) | 3 (2–5) |
| −below norm*, yes/no, n | 47/13 | 35/3 | 27/2 | 16/1 | 14/0 | 14/0 | 12/0 |
IQR, interquartile range; CIS fatigue, Checklist Individual Strength fatigue subscale; HADS, Hospital Anxiety and Depression Scale; NRS Numeric Rating Scale; Katz-P, Katz personal ADL index; Katz-I, Katz instrumental ADL index; FAI, Frenchay Activities Index. *≤25th percentile of age and sex predicted normative values.
Results from measures regarding impairments (CIS fatigue, HADS anxiety, HADS depression, pain NRS), activity limitations (Katz P-ADL, Katz I-ADL) and participation restrictions (FAI) are presented in Table 2. Results on cognitive impairment were excluded as 60–80% of patients were unable to complete the MoCA due to physical disability (unable to perform pen-and-paper items). Most patients answered the CIS fatigue subscale. Around half of them were categorized as having fatigue, and there was a trend of higher presence of fatigue during the first year compared to later time points. Almost all patients completed the HADS, and approximately one-fourth were categorized as having anxiety and depression at T1 and T2. At later time points, the presence of anxiety seemed to decline while the proportions of patients categorized as having depression was about the same. The use of anti-depressive medical treatment was found in both those over and under the cut-off for a possible or probable case of depression. However, over time between one (8%at T7) and eight (14%at T1) patients were categorized as a case, but did not have anti-depressive medication. Perceived pain was reported by 21%to 50%of patients depending on time point. Most of these patients were categorized as having clinically relevant pain, i.e. scored ≥4 on the NRS. Almost all had some form of medical pain treatment, but many also reported non-pharmacological interventions like repositioning, massage and active or passive movements. All patients answered the Katz P-ADL and I-ADL indexes and the FAI. Activity limitations were common, and almost all patients were categorized as dependent in P-ADL and I-ADL after one year. Likewise, participation restrictions were present in most patients already at baseline, and all were categorized as below norm after 1.5 years.
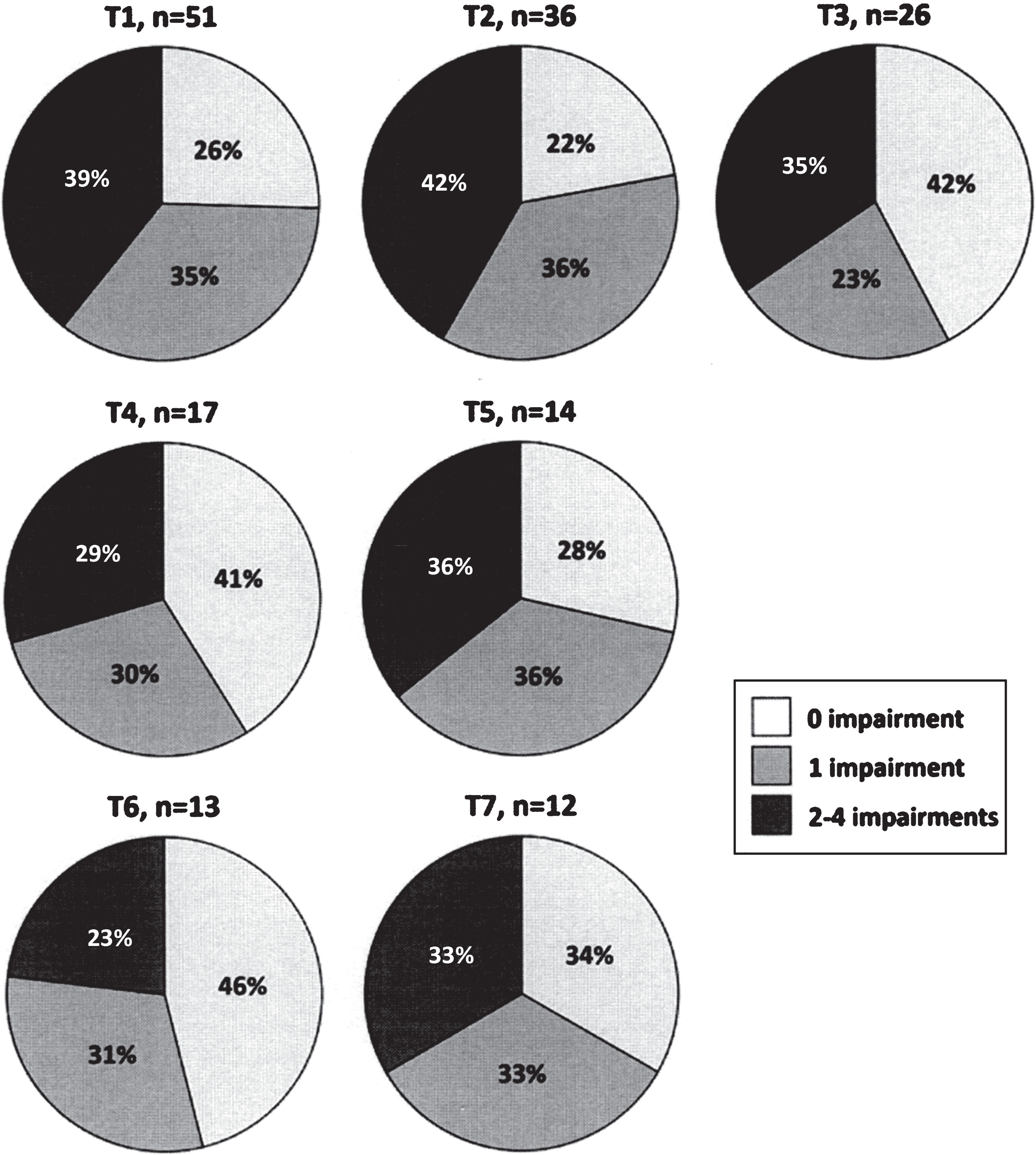
Presence of impairments in relation to the three levels of disease severity is presented in Fig. 2. Patients having fatigue, anxiety, depression and clinically relevant pain were found in all disease severity levels, i.e. in those categorized as having mild, moderate or severe disease. The concurrent presence of impairments during the study period is presented in Fig. 3. Approximately one-third of the patients had between two and four concurrent impairments at each time point.
Fig.2
Presence of impairments (i.e. fatigue, anxiety, depression and clinically relevant pain) in relation to the three levels of disease severity (mild, moderate and severe) at baseline (T1) and each six-month follow-up (T2-T7). The y-axis shows the number of patients categorized as having an impairment.
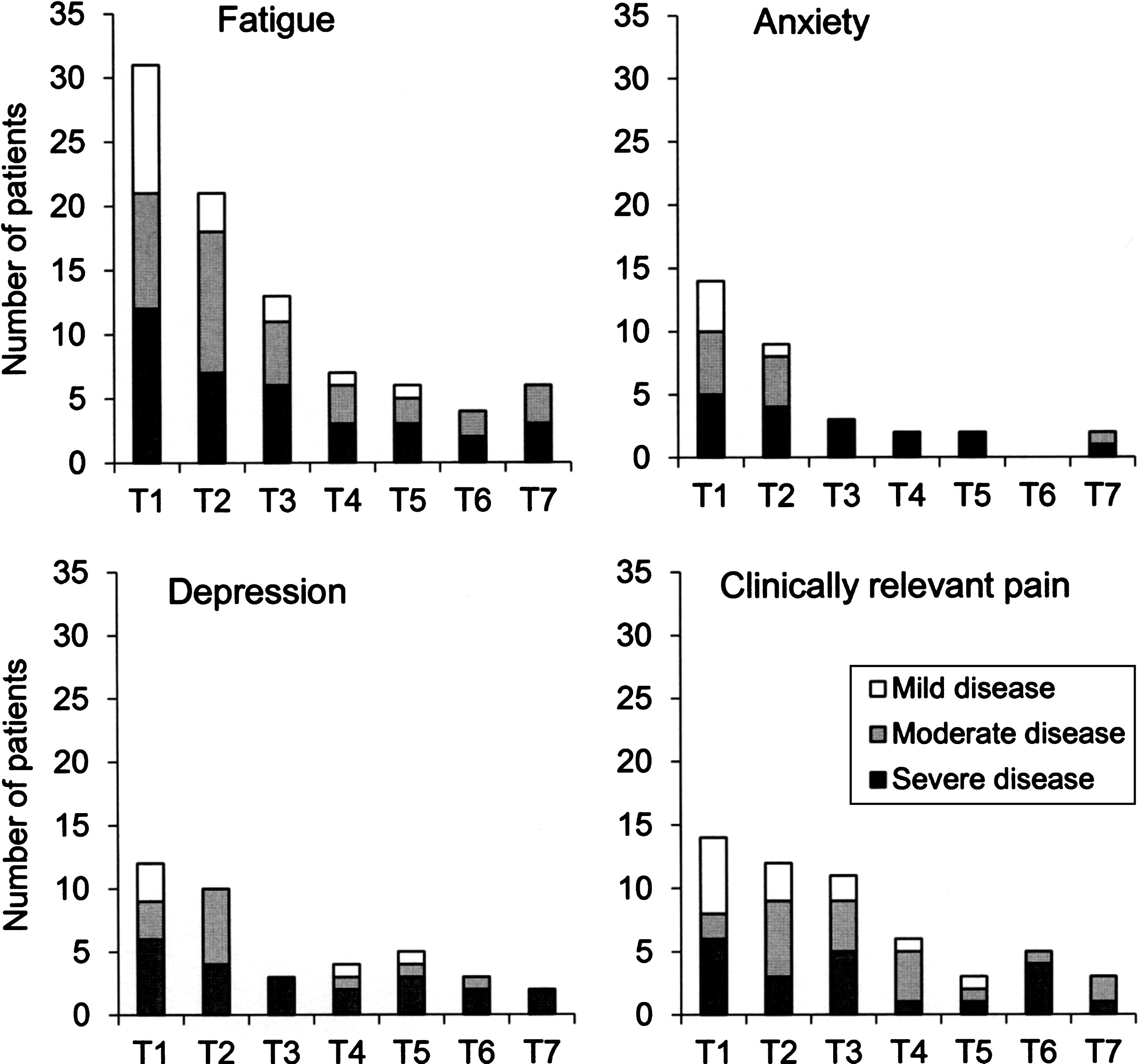
Fig.3
Concurrent presence of impairments (fatigue, anxiety, depression and clinically relevant pain) at baseline (T1) and each six-month follow-up (T2-T7).
Results of associations between impairments (CIS fatigue subscale, HADS anxiety, HADS depression, pain NRS) and disease severity (ALSFRS-R) are presented in Table 3. In general, correlation coefficients were non-significant and considered to be low.
Table 3
Results of associations between impairments (CIS fatigue, HADS anxiety, HADS depression, Pain NRS) and disease severity (ALSFRS-R) at baseline (T1) and each six-month follow-up (T2-T7). The scores from CIS fatigue subscale, HADS and pain NRS were inverted in these analyses to facilitate interpretation. Thus, higher scores indicate a better functioning in all measures
| ALSFRS-R | T1 | T2 | T3 | T4 | T5 | T6 | T7 | |
| CIS fatigue | rs | 0.17 | 0.05 | 0.19 | −0.03 | −0.20 | −0.29 | −0.42 |
| p-value | 0.24 | 0.79 | 0.35 | 0.91 | 0.50 | 0.34 | 0.17 | |
| n | 51 | 36 | 26 | 17 | 14 | 13 | 12 | |
| HADS anxiety | rs | 0.00 | 0.16 | 0.40 | 0.19 | 0.10 | 0.08 | −0.13 |
| p-value | 0.99 | 0.35 | 0.04 | 0.47 | 0.73 | 0.79 | 0.69 | |
| n | 56 | 36 | 27 | 17 | 14 | 14 | 12 | |
| HADS depression | rs | 0.22 | 0.07 | 0.26 | 0.11 | 0.02 | 0.18 | 0.46 |
| p-value | 0.10 | 0.68 | 0.20 | 0.66 | 0.95 | 0.54 | 0.14 | |
| n | 56 | 36 | 27 | 17 | 14 | 14 | 12 | |
| Pain NRS | rs | 0.04 | −0.07 | −0.27 | −0.37 | −0.87 | 0.88 | −0.74 |
| p-value | 0.87 | 0.80 | 0.35 | 0.37 | 0.33 | 0.02 | 0.10 | |
| n | 21 | 16 | 14 | 8 | 3 | 6 | 6 |
rs, Spearman’s correlation coefficient; n, sample size; ALSFRS-R, ALS Functioning Rating Scale-Revised; CIS fatigue, Checklist Individual Strength; HADS, Hospital Anxiety and Depression Scale; NRS Numeric Rating Scale.
DISCUSSION
The main findings of this study were that fatigue, anxiety, depression and pain were present over time in patients with ALS, and that these impairments were found regardless of disease severity. Further, that approximately one-third experienced two or more of these impairments concurrently, i.e. at the same data collection time point.
Patient characteristics (e.g. age, sex, site of onset) in our study cohort were in agreement with reports from other European population-based studies [3–5] and thus, strengthens the external validity. As shown before [5], some patients had a slowly progressive ALS and a 10-year survival already at baseline. We found that the proportion of patients with bulbar onset decreased over time, which could be explained by the longer survival in patients with spinal onset [5]. That most patients, despite increased disease severity, lived in their own home during the three-year study period could be due to the Swedish disability policy and health care system, which ensure that support and services are provided for people with disability. Accordingly, the provision of aids, home adaptations, home care, personal assistance, and transportation services increased over time. These findings are probably general for those living in countries with similar health care systems but might not reflect the situation for patients with ALS in countries where the healthcare system is organized, managed, and financed in a very different way, e.g. the United States of America. In agreement with current European clinical management guidelines [28], patients had the disease modifying drug riluzole, non-invasive or invasive ventilation, and gastrostomy for enteral nutrition.
Between 60–80%of our patients were unable to complete the MoCA and data on cognitive impairment was therefore not reported. The MoCA was chosen as it was available in Swedish and used in the clinic at the time when the study was planned. However, even though MoCA might detect cognitive impairment and has been used in ALS [29–31], it had obvious limitations. As many other cognitive tests, MoCA relies on certain physical capabilities which makes it less useful in ALS. A disease-specific screening test like the Edinburgh Cognitive and Behavioural ALS Screen [32] might overcome these shortcomings and would be interesting to use in future studies.
Fatigue was a common impairment at all time points, though we did not find an increase over time as other report [11, 12]. This might be due to differences in follow-up time and sample characteristics, our sample had for example a longer disease duration. Fatigue is a multidimensional concept including physiological (central and peripheral fatigue) and psychological dimensions (depression, concentration and motivational influences) [33]. It can be speculated that physiological and psychological dimensions of fatigue might have a greater impact in early stages of the disease due to compensatory mechanisms of the nervous system and the obvious psychological strain related to be diagnosed with ALS. However, it is also possible that patients become less physically active over time due to muscle weakness, and thus less fatigued. An explanation in line with findings that patients with ALS experience fatigue as use-dependent reversible muscle weakness and whole-body tiredness [34]. A recent survey-study in patients with ALS report that fatigue is the most prevalent impairment and at the same time the least treated [35]. There is, unfortunately, not enough evidence to make conclusions about effectiveness of pharmacological or non-pharmacological treatments [36]. Thus, high-quality studies on intervention to improve fatigue in ALS are much needed.
We used the HADS to screen for anxiety and depression and found like others that scores seemed to decrease over time [14, 37]. However, even if the proportion of patients categorized as having anxiety was larger at the two first data collection time points, there were cases even at later time-points. In addition, some patients over the cut-off score for depression lacked medical treatment. Thus, systematic and regular screening for anxiety and depression is important to identify those in need of interventions. Although no formal controlled studies have been performed, clinical guidelines recommend pharmacological treatment [28, 38]. Non-pharmacological interventions, such as meditation training and cognitive behavioural therapy, might also be considered as these treatments seem to ameliorate psychological distress in patients with ALS [39, 40]. Further, knowledge and awareness of coping strategies is important when care and support is planned as coping has been found to be associated with both anxiety and depression [41, 42].
As highlighted in recent years [15, 16], pain is often present in patients with ALS. Information on pain frequency and intensity has previously been based on cross-sectional studies. Thus, our findings, that pain occurred in as many as 50%over time and that a considerable number of patients scored over the cut-off for clinically relevant pain, provide new important knowledge. Many factors might contribute to pain in ALS, e.g. muscle cramps, spasticity, contractures, imposed stress on the musculoskeletal system due to muscle weakness and immobilisation, etc. It is therefore important to thoroughly assess patients on a regular basis so that proper interventions can be applied.
Not only have we shown that fatigue, anxiety, depression and pain were frequently present in patients with ALS, about one third had two or more of these impairments at the same time, i.e. concurrent impairments. Furthermore, the impairments were present regardless of disease severity. This clinically important result indicates the necessity for screening patients repeatedly over time. Many patients had both symptomatic and anti-depressive medications, but there were still cases of fatigue, anxiety, depression and clinically relevant pain. Given the multidimensional origin and perception of these impairments [13, 16, 33, 34] other non-pharmacological approaches are also recommended [28, 43].
According to the ICF, activity is defined as “the execution of a task or action by an individual” and participation as “a person’s involvement in a life situation” [1]. Studies on activity and participation in patients with ALS are surprisingly scare and to our knowledge mainly based on cross-sectional data [8, 18]. We found that activity limitations in P- and I-ADL, and participation restrictions were present already at baseline and, as expected, increased over time. The FAI was used to map frequency of social and lifestyle activities, a questionnaire much based on physical performance and developed before the use of internet for e.g. shopping and social interactions. For this reason, our results must be interpreted with some caution as patients might have been able to keep their social roles and participate in a fulfilling life using new technology and provided aids, services and support.
Impairments like fatigue, anxiety, depression and pain were not significantly associated with disease severity as captured by the ALSFRS-R scale. This is a clinically important finding highlighting the need to screen for these impairments regardless of disease severity. Thus, even though the ALSFRS-R is commonly used, and regarded as reliable and clinically meaningful, health care professionals must be aware of its shortcomings to give an overall picture of disability in ALS.
Studying patients with ALS over time is accompanied with difficulties due to the progressive nature of the disease. Although we recruited patients from probably the largest cohort in Sweden, the study sample was considerably reduced already after one year, which is a limitation of the study. Furthermore, this hampered the possibilities of performing statistical analyses, and presentation of data is therefore mainly descriptive. Patient compliance in data collections were in general high. This could be due to the use of home-visits as these were highly appreciated by patients. Although the used outcome measures were all standardized and previously used in ALS and other neurological diseases, some had definite flaws. Thus, in future studies we propose that cognition, activity and participation would be better to explore with measures that are not based on physical capability.
Our results that fatigue, anxiety, depression and pain are persistent impairments in ALS, that one-third of patients have two or more concurrent impairments, and that these findings are regardless of disease severity indicate the importance and necessity for systematic and regular screening for these impairments so that proper intervention can be applied. The time frame for screening should be more frequent than the six-month interval used in the present study and preferably adapted to the individual variation in disability and disease progression rate. Further, health care professionals need to monitor whether patients perceive activity limitations and participation restrictions to timely provide the aids, support, and services needed. In a Swedish context, the newly started internet-based Swedish Motor Neuron Disease Quality Registry [44] address several of these issues and has the aim to assure high-quality care for all patients with ALS. In conclusion, regardless of disease severity, patients with ALS need to be regularly screened for commonly present impairments, and activity limitations and participation restrictions, so that person-centered interventions are applied at the right time.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors thank the patients with ALS who participated in the study. We acknowledge help from Anne Zachau, Rayomand Press and Lars-Olof Ronnevi with the recruitment of ALS patients, Gunilla Cröde Widsell in data collections, and from professor Lotta Widén Holmqvist, statistician Elisabeth Berg and Lisbet Broman. This work was supported by the Swedish Research Council; under grant 521-2014-3196; Neuro Sweden; KID funding, Karolinska Institutet under grant 3-1233/2013; and the Strategic Research Programme in Care Sciences, Karolinska Institutet.
REFERENCES
[1] | International classification of functioning disability and health (ICF). Geneva: World Health Organization; (2001) . |
[2] | Marin B , Boumediene F , Logroscino G , Couratier P , Babron MC , Leutenegger AL , et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int J Epidemiol. (2017) ;46: (1):57–74 10.1093/ije/dyw061 |
[3] | Fang F , Valdimarsdottir U , Bellocco R , Ronnevi LO , Sparen P , Fall K , et al. Amyotrophic lateral sclerosis in Sweden, 1991-2005. Arch Neurol. (2009) ;66: (4):515–519 10.1001/archneurol.2009.13 |
[4] | Logroscino G , Traynor BJ , Hardiman O , Chio A , Mitchell D , Swingler RJ , et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. (2010) ;81: (4):385–90 10.1136/jnn2009.183525 |
[5] | Pupillo E , Messina P , Logroscino G , Beghi E . Long-term survival in amyotrophic lateral sclerosis: A population-based study. Ann Neurol. (2014) ;75: (2):287–297. doi: 10.1002/ana.24096 |
[6] | Cedarbaum JM , Stambler N , Malta E , Fuller C , Hilt D , Thurmond B , et al. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. (1999) ;169: (1-2):13–21. |
[7] | Geng D , Ou R , Miao X , Zhao L , Wei Q , Chen X , et al. Patients’ self-perceived burden, caregivers’ burden, and quality of life for ALS patients: A cross-sectional study. J Clin Nurs. (2016) . doi: 10.1111/jocn.13667 |
[8] | Mioshi E , Lillo P , Kiernan M , Hodges J . Activities of daily living in motor neuron disease: Role of behavioural and motor changes. J Clin Neurosci. (2012) ;19: (4):552–556. doi: 10.1016/j.jocn.2011.07.042 |
[9] | Montuschi A , Iazzolino B , Calvo A , Moglia C , Lopiano L , Restagno G , et al. Cognitive correlates in amyotrophic lateral sclerosis: A population-based study in Italy. J Neurol Neurosurg Psychiatry. (2015) ;86: (2):168–173. doi: 10.1136/jnnp-2013-307223 |
[10] | Elamin M , Bede P , Byrne S , Jordan N , Gallagher L , Wynne B , et al. Cognitive changes predict functional decline in ALS: A population-based longitudinal study. Neurology. (1590) ;80: (17):7. doi: 10.1212/WNL.0b013e31828f18ac |
[11] | McElhiney MC , Rabkin JG , Gordon PH , Goetz R , Mitsumoto H . Prevalence of fatigue and depression in ALS patients and change over time. J Neurol Neurosurg Psychiatry. (1146) ;80: (10):9. doi: 10.1136/jnn2008.163246 |
[12] | Panitz S , Kornhuber M , Hanisch F . The checklist individual strength (CIS20-R) in patients with amyotrophic lateral sclerosis - A longitudinal study. Acta Neurol Scand. (2015) . doi: 10.1111/ane.12349 |
[13] | Carvalho TL , de Almeida LM , Lorega CM , Barata MF , Ferreira ML , de Brito-Marques PR , et al. Depression and anxiety in individuals with amyotrophic lateral sclerosis: A systematic review. Trends Psychiatry Psychother. (2016) ;38: (1):1–5. doi: 10.1590/2237-6089-2015-0030 |
[14] | Olsson AG , Markhede I , Strang S , Persson LI . Differences in quality of life modalities give rise to needs of individual support in patients with ALS and their next of kin. Palliat Support Care. (2010) ;8: (1):75–82. doi: 10.1017/s1478951509990733 |
[15] | Chio A , Canosa A , Gallo S , Moglia C , Ilardi A , Cammarosano S , et al. Pain in amyotrophic lateral sclerosis: A population-based controlled study. Eur J Neurol. (2012) ;19: (4):551–555. doi: 10.1111/j.1468-1331.2011.03540.x |
[16] | Stephens HE , Lehman E , Raheja D , Yang C , Walsh S , McArthur DB , et al. Pain in amyotrophic lateral sclerosis: Patient and physician perspectives and practices. Amyotroph Lateral Scler Frontotemporal Degener. (2015) ;17: (1–2):21–29. doi: 10.3109/21678421.2015.1074701 |
[17] | De Groot IJ , Post MW , Van Heuveln T , Van Den Berg LH , Lindeman E . Measurement of decline of functioning in persons with amyotrophic lateral sclerosis: Responsiveness and possible applications of the Functional Independence Measure, Barthel Index, Rehabilitation Activities Profile and Frenchay Activities Index. Amyotroph Lateral Scler. (2006) ;7: (3):167–672. doi: 10.1080/14660820600640620 |
[18] | Van Groenestijn AC , Schroder CD , Kruitwagen-Van Reenen ET , Van Den Berg LH , Visser-Meily JMA . Participation restrictions in ambulatory amyotrophic lateral sclerosis patients: Physical and psychological factors. Muscle Nerve. (2017) ;56: (5):912–918. doi: 10.1002/mus.25574 |
[19] | Brooks BR , Miller RG , Swash M , Munsat TL World Federation of Neurology Research Group on Motor Neuron D El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) ;1: (5):293–9. |
[20] | Sandstedt P , Johansson S , Ytterberg C , Ingre C , Holmqvist LW , Kierkegaard M . Predictors of health-related quality of life in people with amyotrophic lateral sclerosis. J Neurol Sci. (2016) ;370: :269–273. doi: 10.1016/j.jns.2016.09.034 |
[21] | Nasreddine ZS , Phillips NA , Bedirian V , Charbonneau S , Whitehead V , Collin I , et al. The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J Am Geriatr Soc. (2005) ;53: (4):695–699. doi: 10.1111/j.1532-5415.2005.53221.x |
[22] | Beurskens AJ , Bultmann U , Kant I , Vercoulen JH , Bleijenberg G , Swaen GM . Fatigue among working people: Validity of a questionnaire measure. Occup Environ Med. (2000) ;57: (5):353–7. |
[23] | Zigmond AS , Snaith RP . The hospital anxiety and depression scale. Acta Psychiatr Scand. (1983) ;67: (6):361–70. |
[24] | Asberg KH , Sonn U . The cumulative structure of personal and instrumental ADL. A study of elderly people in a health service district. Scand J Rehabil Med. (1989) ;21: (4):171–7. |
[25] | Wade DT , Legh-Smith J , Langton Hewer R . Social activities after stroke: Measurement and natural history using the Frenchay Activities Index. Int Rehabil Med. (1985) ;7: (4):176–81. |
[26] | Turnbull JC , Kersten P , Habib M , McLellan L , Mullee MA , George S . Validation of the Frenchay Activities Index in a general population aged 16 years and older. Arch Phys Med Rehabil. (2000) ;81: (8):1034–8. |
[27] | Domholdt E . Physical therapy research: Principles and applications, 2. ed. Philadelphia: Saunders; (2000) . |
[28] | Andersen PM , Abrahams S , Borasio GD , de Carvalho M , Chio A , Van Damme P , et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol. (2012) ;19: (3):360–375. doi: 10.1111/j.1468-1331.2011.03501.x |
[29] | Osborne RA , Sekhon R , Johnston W , Kalra S . Screening for frontal lobe and general cognitive impairment in patients with amyotrophic lateral sclerosis. J Neurol Sci. (2014) ;336: (1–2):191–196. doi: 10.1016/j.jns.2013.10.038 |
[30] | Ohta Y , Sato K , Takemoto M , Takahashi Y , Morihara R , Nakano Y , et al. Behavioral and affective features of amyotrophic lateral sclerosis patients. J Neurol Sci. (2017) ;381: :119–125. 10.1016/j.jns.2017.08.024 |
[31] | Liu S , Huang Y , Tai H , Zhang K , Wang Z , Shen D , et al. Excessive daytime sleepiness in Chinese patients with sporadic amyotrophic lateral sclerosis and its association with cognitive and behavioural impairments. J Neurol Neurosurg Psychiatry. (2018) . doi: 10.1136/jnnp-2018-318810 |
[32] | Abrahams S , Newton J , Niven E , Foley J , Bak TH . Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2014) ;15: (1–2):9–14. doi: 10.3109/21678421.2013.805784 |
[33] | Zwarts MJ , Bleijenberg G , van Engelen BG . Clinical neurophysiology of fatigue. Clin Neurophysiol. (2008) ;119: (1):2–10. doi: 10.1016/j.clinph.2007.09.126 |
[34] | Gibbons CJ , Thornton EW , Young CA . The patient experience of fatigue in motor neurone disease. Front Psychol. (2013) ;4: :788.10.3389/fpsyg.2013.00788 |
[35] | Nicholson K , Murphy A , McDonnell E , Shapiro J , Simpson E , Glass J , et al. Improving symptom management for people with amyotrophic lateral sclerosis. Muscle Nerve. (2018) ;57: (1):20–24. 10.1002/mus.25712 |
[36] | Gibbons C , Pagnini F , Friede T , Young CA . Treatment of fatigue in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev (2018) ;1: :Cd011005. 10.1002/14651858.CD011005.pub2 |
[37] | Jakobsson Larsson B , Ozanne AG , Nordin K , Nygren I . A prospective study of quality of life in amyotrophic lateral sclerosis patients. Acta Neurol Scand. (2017) . doi: 10.1111/ane.12774 |
[38] | Miller RG , Jackson CE , Kasarskis EJ , England JD , Forshew D , Johnston W , et al. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. (2009) ;73: (15):1227–33. doi: 10.1212/WNL.0b013e3181bc01a4 |
[39] | Pagnini F , Marconi A , Tagliaferri A , Manzoni GM , Gatto R , Fabiani V , et al. Meditation training for people with amyotrophic lateral sclerosis: A randomized clinical trial. Eur J Neurol. (2017) ;24: (4):578–586. doi: 10.1111/ene.13246 |
[40] | van Groenestijn AC , Schroder CD , Visser-Meily JM , Reenen ET , Veldink JH , van den Berg LH . Cognitive behavioural therapy and quality of life in psychologically distressed patients with amyotrophic lateral sclerosis and their caregivers: Results of a prematurely stopped randomized controlled trial. Amyotroph Lateral Scler Frontotemporal Degener. (2015) ;16: (5–6):309–15. 10.3109/21678421.2015.1038276 |
[41] | Jakobsson Larsson B , Nordin K , Nygren I . Coping with amyotrophic lateral sclerosis; from diagnosis and during disease progression. J Neurol Sci. (2016) ;361: :235–242. doi: 10.1016/j.jns.2015.12.042 |
[42] | Jakobsson Larsson B , Nordin K , Askmark H , Nygren I . Coping strategies among patients with newly diagnosed amyotrophic lateral sclerosis. J Clin Nurs (2014) ;23: (21–22):3148–55. doi: 10.1111/jocn.12557 |
[43] | Radunovic A , Mitsumoto H , Leigh PN . Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol. (2007) ;6: (10):913–925. doi: 10.1016/s1474-4422(07)70244-2 |
[44] | Registries SN . Motor Neuron Disease [homepage on the Internet]. Stockholm: 2018. [cited 2018 Apr 12]. http://www.neuroreg.se/en.html/motor-neuron-disease. |


