
Evaluation of Children with SMA Type 1 Under Treatment with Nusinersen within the Expanded Access Program in Germany
Abstract
Background:
Spinal muscular atrophy (SMA) is a neuromuscular disorder characterized by muscle weakness and muscle atrophy. Nusinersen acts as a splicing modifier and has recently been approved for intrathecal treatment of SMA.
Objective:
Prior to approval, nusinersen was provided to patients with SMA type 1 in Germany within an Expanded Access Program (EAP). In contrast to previous clinical trials, children of different age groups and different stages of the disease were treated with nusinersen.
Methods:
We conducted a prospective, longitudinal data collection of patients treated with nusinersen within the EAP in seven neuromuscular centers in Germany. Standardized assessments including CHOP-INTEND and HINE-2 motor milestones were performed at baseline and 60 and 180 days after start of treatment.
Results:
Data from 61 SMA type 1 patients (mean age 21.08 months, range 1–93) were available for analysis. After six months of treatment, 47 children (77.0%) improved by ≥4 points in CHOP INTEND score. Mean change in CHOP INTEND score was 9.0±8.0 points. Nineteen patients (31.1%) improved by ≥2 points in HINE-2 motor milestones. Regression analysis revealed age at onset of treatment as major determinant of change in CHOP INTEND from baseline.
Conclusion:
When analyzing a broad spectrum of SMA type 1 patients, many children showed an improvement of motor function after six months of treatment with nusinersen, which is generally not expected within the natural course of the disease. Long-term observation and follow-up of patients with later onset types of SMA are crucial to understand the clinical impact of treatment with nusinersen.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal-recessive disorder caused by a homozygous deletion in the survival motor neuron (SMN) 1 gene on chromosome 5q13 and has an incidence of about 1:10,000. It is characterized by degeneration of the anterior horn cells of the spinal cord resulting in muscle atrophy and proximal muscle weakness. Depending on severity of symptoms and age of onset, SMA was classified into five different subtypes. SMA type 1 is the most common (50–60%) and also the most severe subtype of SMA. Per definition, onset of symptoms is before 6 months of age and mortality of these children is mainly caused by respiratory involvement. From the time point of diagnosis, infants with SMA type 1 rarely achieve improvements of motor function or acquire motor developmental milestones [1].
Within the last years, there has been a promising approach for the development of novel drugs intervening the pathophysiology of SMA. Among these, nusinersen is the first drug approved to treat SMA [2]. This antisense oligonucleotide (ASO) acts as splicing modifier targeting the intronic splicing silencer N1 in SMN2 [3, 4]. SMN2 is a centromeric copy of SMN1 that produces transcripts of SMN protein lacking exon 7 because of a C-to-T transition in SMN2 [5], resulting in an alternatively spliced truncated and non-functional SMN protein (SMNΔ7) [5, 6]. Thus, increasing SMN2 exon 7 inclusion using an ASO as splicing modifier leads to expression of more full-length and functional SMN protein [7]. The results of a phase III randomized, double-blind and sham-controlled clinical trial showed an improvement in motor milestones and event-free survival (time to death or use of permanent assisted ventilation) in infants with SMA type 1 that were treated with nusinersen compared to the control group [8]. Amongst others, inclusion criteria for this phase III clinical trial were onset of symptoms ≤6 months of age, age of ≤7 months at screening and a normal oxygen saturation without ventilator support [8].
Nusinersen is administered intrathecally. It has been approved in the United States since 12/2016 and in Europe since 06/2017. Prior to approval in Europe, nusinersen was provided to patients with SMA type 1 within an Expanded Access Program (EAP). In contrast to the previous clinical trials, children of different age groups and different stages of the disease were treated with nusinersen within the EAP.
PATIENTS AND METHODS
In Germany, treatment with nusinersen was provided to children with SMA type 1 within the EAP from 11/2016 until 06/2017. Inclusion criteria to participate in the EAP were presence of a genetically proven 5q SMA, documented onset of clinical signs and symptoms before 6 months of age and that patients’ care met the guidelines published in 2007 as consensus statement for standard of care in SMA [9]. The following exclusion criteria were defined: participation in an ongoing clinical trial with nusinersen or in a prior nusinersen study, previous exposure to nusinersen, history of brain- or spinal cord disease that would interfere with lumbar puncture procedures or cerebrospinal fluid (CSF) circulation, presence of an implanted shunt for CSF drainage or implanted central nervous system (CNS) catheter, previous or current participation in a clinical trial with an investigational gene therapy for SMA, or participation in a study with an investigational therapy for SMA within the past six months.
We conducted a prospective, longitudinal data collection of SMA patients who started treatment with nusinersen within the EAP and fulfilled the criteria of SMA type 1 with an onset of symptoms ≤6 months of age and no ability to sit independently. In cooperation with the German patient organization for neuromuscular disorders (Deutsche Gesellschaft für Muskelkranke, DGM), patients with SMA type 1 were identified and allocated to the different neuromuscular centers that participated in the EAP.
Treatment with nusinersen
Intrathecal injections with nusinersen were performed on treatment days 1, 15, 30, 60 and 180. Prior to drug approval, dosage of nusinersen was age-dependent analogous to the preceding clinical trials: 9.6 mg (0–90 days), 10.3 mg (91–182 days), 10.8 mg (183–365 days), 11.3 mg (366–730 days) and 12 mg (>731 days). After approval, dosage was 12 mg for all children independent of age.
Data collection
Data were collected in seven different German neuromuscular centers during routine patient visits. As characteristics prior to treatment, we documented age at onset of symptoms, age at start of treatment, sex, use of ventilator support, nutritional status, additional therapies, concomitant medication, and SMN2 copy number, if available. Data regarding respiratory and nutritional status, adverse events, and the subjective impression of parents were evaluated during every patient visit. In addition, a clinical examination including vital and growth parameters was performed. As functional outcome measures, we used the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) [10] and the Hammersmith Infant Neurological Examination (HINE) [11]. Two-day central training workshops were organized for participating physiotherapists.
Primary endpoint
The primary endpoint of our study was change in CHOP INTEND score from baseline. The CHOP INTEND was developed and validated to evaluate the severely limited repertoire of motor skills of infants with SMA type 1 [10]. The score ranges from 0 to 64 points. Natural history studies reveal a mean CHOP INTEND score of 21.4 points in infants with SMA type 1 [12]. CHOP INTEND was performed at baseline and 60 and 180 days after start of treatment.
Secondary endpoints
As secondary endpoint, we used section 2 of the HINE (HINE-2) to investigate the achievement of motor milestones with a score ranging from 0 to 26 [11, 13]. We evaluated seven categories of motor milestones excluding the category of voluntary grasp. According to the previous clinical trials, a motor milestone response was defined as improvement in at least one category (increase ≥1 point in categories head control, rolling, sitting, crawling, standing or walking, or increase ≥2 points in category kicking) and more categories with improvement than categories with worsening. Children who died were considered to have no motor response. The HINE-2 was performed during every patient visit. Furthermore, we evaluated changes in respiratory and nutritional status as well as parents’ impression regarding improvements or worsening in motor and respiratory function as secondary endpoints. To evaluate parents’ impression, we used a Likert scale with five categories (marked improvement, slight improvement, no changes, slight worsening, and marked worsening).
Statistical analysis
Descriptive data analyses were performed by calculation of absolute frequencies and percentages. Continuous data were analyzed as mean±standard deviation. Statistical analyses were performed with SAS 9.2 and R [14]. Univariate and multivariate regression analyses were applied to evaluate the effect of age at onset of symptoms, age at onset of treatment, SMN2 copy number, use of ventilator support, nutritional status prior to treatment and CHOP INTEND and HINE-2 score prior to treatment and change in CHOP INTEND score from baseline. Multivariate regression analysis was performed with a backward selection procedure with p = 0.1. Additionally, a Spearman correlation analysis between parents’ impression about changes in motor function and the change in CHOP INTEND score from baseline was performed. A p-value of <0.01 was considered statistically significant. In case of missing data on treatment day 180, we used the last observation carried forward approach.
RESULTS
Characteristics prior to treatment
Data of children with SMA type 1 treated with nusinersen were collected in seven different neuropediatric departments in Germany. In total, 61 patients fulfilled the criteria of an onset of symptoms ≤6 months of age and no ability to sit independently. Baseline characteristics are shown in Table 1. Mean age at onset of symptoms was 2.78±1.72 months and mean age at diagnosis was 5.84±3.56 months. Age at start of treatment was mean 21.08±20.23 months ranging between 1 and 93 months. Prior to treatment, 26 children (42.6%) had no ventilator support, 17 children (27.9%) required non-invasive ventilation (NIV) less than 16 hours per day, 6 children (9.8%) used NIV more than 16 hours per day, and 12 patients (19.7%) had already undergone tracheostomy. In 34 patients (55.7%), a feeding tube or gastrostomy was used and in 12 patients (19.7%) body weight was below the third percentile. As concomitant medication with a potential influence on motor function, the following drugs were named: salbutamole in 6 children (9.8%), and valproate and biocarn in 2 patients (3.3%). Physiotherapy was performed regularly in 46 children (75.4%), speech therapy was performed in 13 children (21.3%) and occupational therapy in 5 children (8.2%).
Table 1
Baseline demographics prior to treatment of all children including subgroups according to SMN2 copy number*
| All Children N = 61 | Children with ≤2 SMN2 copies N = 38 | Children with ≥3 SMN2 copies N = 20 | Unknown SMN2 copy number N = 3 | |
| Sex | ♂ 31/♀ 30 | ♂ 17/♀ 21 | ♂ 11/♀ 9 | ♂ 3 |
| Age at symptom onset (months) | 2.78 (range 0–6) | 2.53 (range 0–6) | 3.5 (0–6) | 1.0 (0–3) |
| Age at start of treatment (months) | 21.08 (range 1–93) | 22.08 (range 2–93) | 21.85 (range 2–50) | 3.33 (range 1–6) |
| Ventilator support | ||||
| – No ventilator support | 26 (42.6%) | 14 (36.8%) | 10 (50%) | 2 (66.7%) |
| – NIV <16 h/day | 17 (27.9%) | 9 (23.7%) | 7 (35%) | 1 (33.3%) |
| – NIV >16 h/day | 6 (9.8%) | 5 (13.2%) | 1 (5%) | 0 (0%) |
| – Tracheostomy | 12 (19.7%) | 10 (26.3%) | 2 (10%) | 0 (0%) |
| Feeding tube or gastrostomy | 34 (55.7%) | 23 (60.5%) | 8 (40%) | 2 (66.7%) |
| Body weight | ||||
| – <3rd percentile | 12 (19.7%) | 7 (18.4%) | 5 (25%) | 3 (100%) |
| – >3rd percentile | 49 (80.3%) | 31 (81.6%) | 15 (75%) | 0 (0%) |
| CHOP INTEND score | 22.3 (range 1–50) | 19.5 (range 1–46) | 27.7 (range 1–50) | 22.3 (range 10–30) |
| HINE-2 score | 0.8 (range 0–8) | 0.5 (range 0–8) | 1.4 (range 0–6) | 0.3 (range 0-1) |
*Data are listed as n (%) or mean±standard deviation.
All children had genetically confirmed 5q-SMA. In our cohort, 38 children (62.3%) had ≤2 SMN2 copies and 20 children (32.8%) had ≥3 SMN2 copies. In 3 patients (4.9%), SMN2 copy number was unknown.
Regarding motor function in our cohort prior to treatment, CHOP INTEND score was 22.3±13.9 ranging from 0 to 50 and the HINE-2 score was 0.8±1.7 ranging from 0 to 8. Characteristics prior to treatment are summarized in Table 1, with an additional division into subgroups according to SMN2 copy number.
After six months of treatment, all but one child (98.3%) were alive. This one child died after treatment day 60 due to respiratory failure at the age of 5 months. In two children, treatment was delayed about 2 or 3 months between treatment day 1 and 30 due to respiratory failure in one child and a post-lumbar puncture syndrome in the other child. Five children continued treatment at another institution after treatment day 60, so that data for treatment day 180 were not available.
Primary endpoint
Mean CHOP INTEND score after six months of treatment was 31.2±16.2 with an improvement of 9.0±8.0 points. Eight children (13.1%) showed an improvement of 1 to 4 points, 17 children (27.9%) an improvement of 5 to 9 points, 16 children (26.2%) an improvement of 10 to 14 points and 11 children (18.0%) an improvement of ≥15 points in the CHOP INTEND score. Children with ≤2 SMN2 copies had a lower CHOP INTEND score prior to treatment (≤2 SMN2 copies: 19.5±12.8 vs. ≥3 SMN2 copies: 27.7±15.1), but the change in CHOP INTEND score from baseline was comparable to children with ≥3 SMN2 copies (≤2 SMN2 copies: 8.1±7.0 vs. ≥3 SMN2 copies: 8.2±5.3). Regarding age at onset of treatment, we observed a greater change in CHOP INTEND score from baseline in children aged ≤7 months compared to older children (children ≤7 months: change in CHOP INTEND score: 14.4±9.2; children >7 months of age: change in CHOP INTEND score 7.0±6.6). In children requiring permanent ventilator support or tracheostomy prior to treatment, CHOP INTEND score at baseline was 9.4±9.1 and change in CHOP INTEND score was 5.6±7.5. In 11 children, the last observation endpoint was on treatment day 60. In one child, CHOP INTEND was only performed prior to treatment. Results of CHOP INTEND score according to age at onset of treatment are shown in Table 2 and Fig. 1.
Table 2
Contingency table showing CHOP INTEND baseline scores and changes after six months of treatment according to age at onset of treatment and SMN2 copy number*
| ≤2 SMN2 copies | ≥3 SMN2 copies | all SMN2 copies | |
| ≤7 months of age** | n = 11 | n = 3 | n = 17 |
| Score at baseline | 29.9±8.3 | 30.3±5.7 | 28.6±8.4 |
| Change in score | 11.8±4.4 | 12.5±6.4 | 14.4±9.2 |
| >7 months of age | n = 27 | n = 17 | n = 44 |
| Score at baseline | 15.3±12.0 | 27.2±16.3 | 19.9±14.8 |
| Change in score | 6.6±7.4 | 7.7±5.2 | 7.0±6.6 |
| Both age groups | n = 38 | n = 20 | n = 61 |
| Score at baseline | 19.5±12.8 | 27.7±15.1 | 22.3±13.9 |
| Change in score | 8.1±7.0 | 8.2±5.3 | 9.0±8.0 |
*Data are listed as mean±standard deviation. In one child CHOP INTEND was only performed prior to treatment and in 11 children the last observation endpoint was on treatment day 60. **In 3 children with age ≤7 months SMN2 copy number was unknown.
Fig.1
Longitudinal CHOP INTEND score according to age at onset of treatment. Colors are for visualization only.
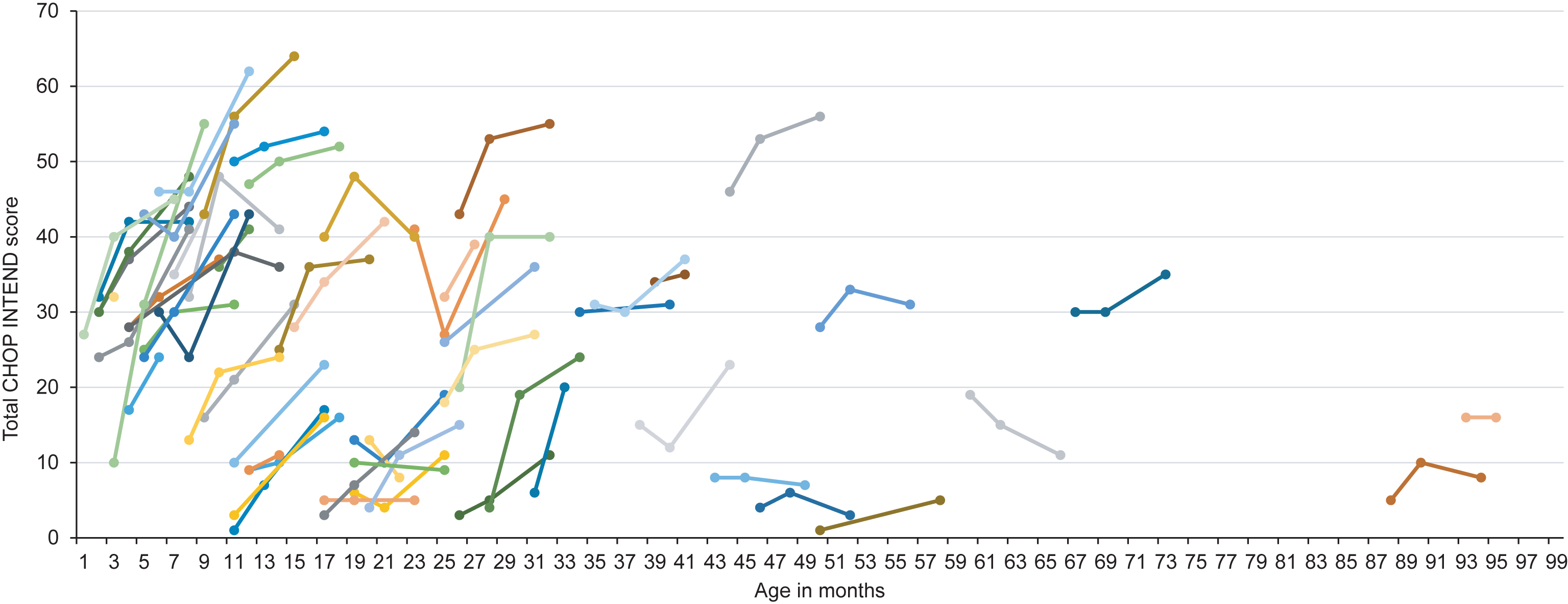
Univariate regression analysis revealed age at onset of treatment as major determinant of change in CHOP INTEND score from baseline (parameter estimate –0.146, 95% CI –0.227; –0.006, p = 0.0006). Backward selection of multivariate regression analysis showed age at onset of treatment as only significant variable.
Secondary endpoints
Regarding motor milestones (HINE-2), 21 patients (34.4%) showed a motor response as defined in the methods section. Fifteen patients (24.6%) showed an improvement of 2 to 4 points, whereas 4 children (6.6%) showed an improvement of ≥5 points. Mean HINE-2 score after six months of treatment was 2.5±3.3 with a mean improvement of 1.4±2.1 points. Four children (6.6%) achieved full head control, and 2 children (3.3%) were able to sit independently. After six months of treatment, none of the children were able to stand or walk independently. Children with ≥3 SMN2 copies had higher HINE-2 scores prior to treatment (≤2 SMN2 copies: 0.5±1.7 vs. ≥3 SMN2 copies: 1.4±1.9). In 7 patients, treatment day 60 was the last observation endpoint and in 6 children HINE-2 was not performed. Results of HINE-2 according to age at onset of treatment are shown in Fig. 2.
Fig.2
Longitudinal HINE-2 score according to age at onset of treatment. Colors are for visualization only.
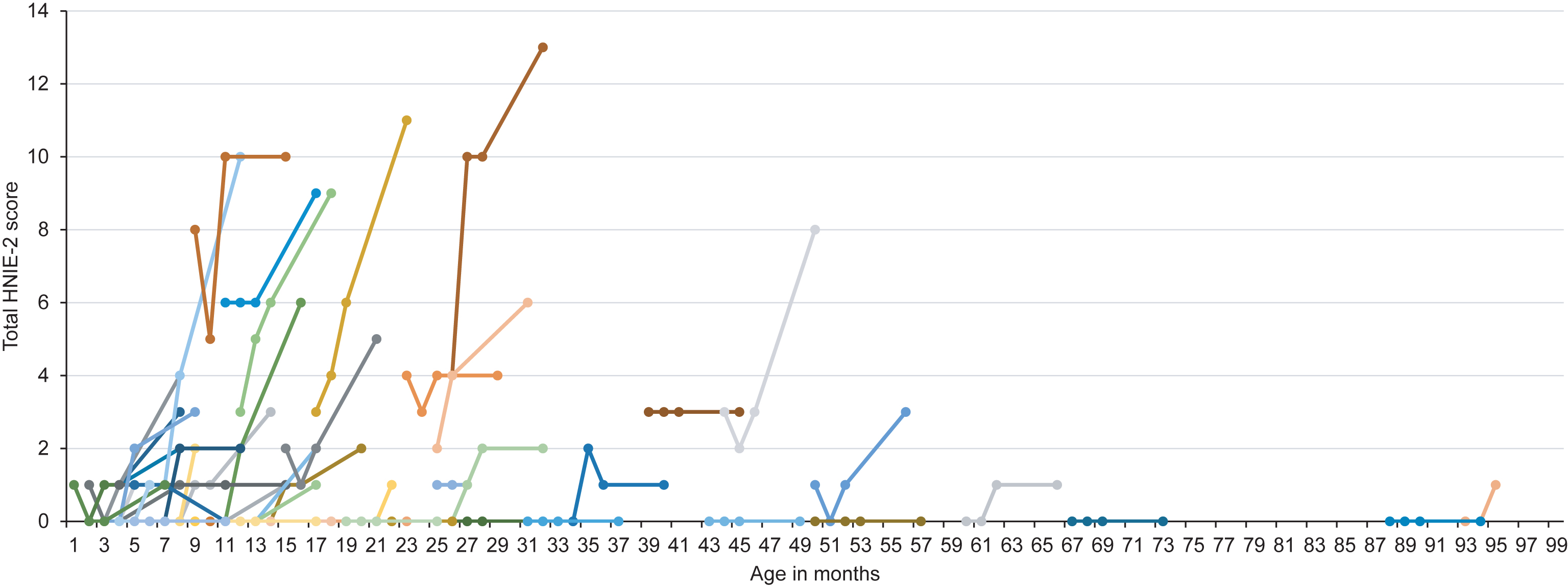
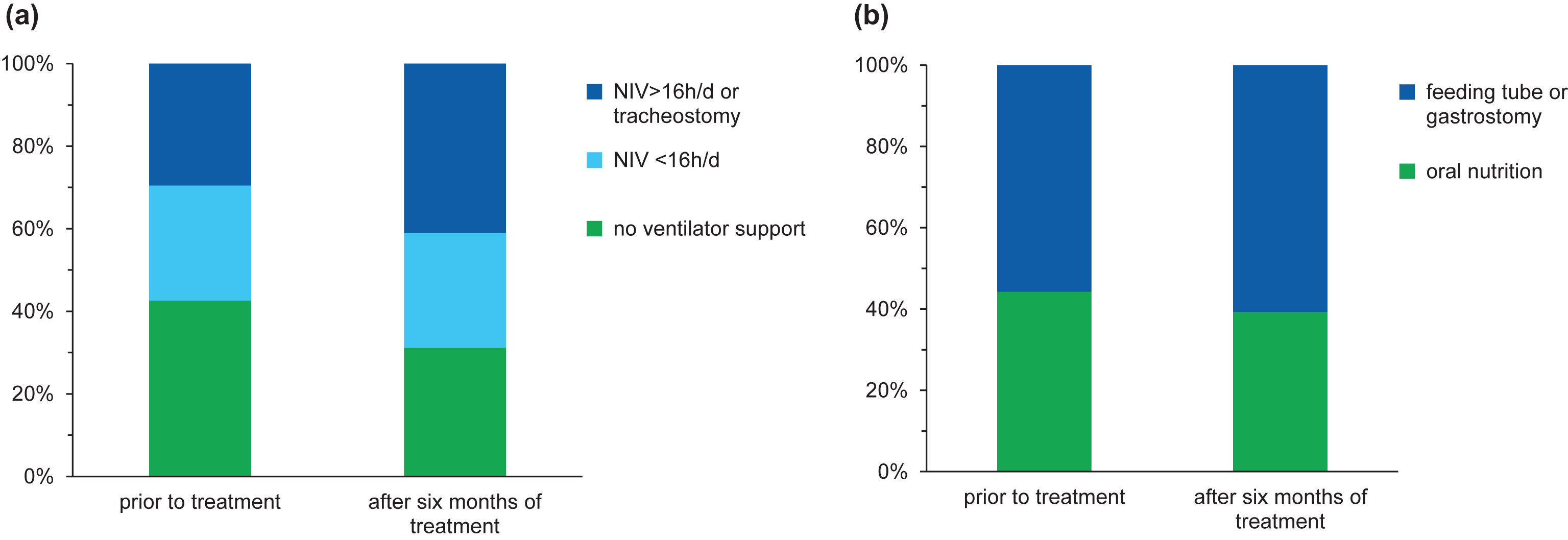
After six months of treatment, 19 children (31.1%) did not require any ventilator support, whereas 6 children (9.8%) started with NIV (<16 hours per day) during the EAP. Three children (4.9%) underwent tracheostomy and four children (6.6%) additionally required NIV >16 hours per day on treatment day 180 (see Fig. 3). An improvement was reported in four children (6.6%): in three of them the time of ventilator use was reduced up to eight hours per day and in one child with a milder phenotype prior to treatment, NIV could be terminated after treatment day 30. Regarding nutrition, a gastrostomy was performed in five children (8.2%) during EAP, whereas 24 patients (39.3%) still did not require any nutritional support after six months of treatment (see Fig. 3).
Fig.3
Data at baseline and after six months of treatment regarding ventilator support (a) and nutrition (b).
We asked parents about their impression regarding improvements or worsening in motor and respiratory function. After six months of treatment, parents of 28 children (45.9%) reported a marked improvement and parents of 29 children (47.5%) a slight improvement in motor function, whereas three parents (4.9%) did not observe any changes and only one parent (1.6%) observed a slight worsening. Regarding respiratory function an improvement was reported in 16 children (26.2%). Among these, only 4 parents (6.6%) indicated a marked improvement. Forty parents (65.6%) did not observe a change and five parents (8.2%) reported a worsening in respiratory function. Correlation analysis of the parents’ impression regarding motor function and the change in CHOP INTEND score from baseline revealed no significant correlation (correlation coefficient 0.313, p = 0.015).
Safety
Lumbar punctures were performed without any severe complications and nusinersen was administered intrathecally in all children on all treatment days. Sedation was used during 31.8% of lumbar punctures.
During the EAP, 53 adverse events were reported. Among these, 29 (54.7%) were classified as serious adverse events leading to death, hospitalization or prolonged hospitalization. Respiratory tract infections were the most frequently observed adverse events (58.5%). Furthermore, acute respiratory failure (15.1%), other infections (7.5%), skin reactions (5.7%), epileptic seizures (1.9%), acute hypoglycemia (1.9%), neutropenia (1.9%), or edema and hair loss (1.9%) were reported. As procedural related adverse events, post-lumbar puncture syndrome occurred in three children (4.9%). One child that already required invasive ventilator support due to disease progression suffered from acute desaturation during sedation for lumbar puncture. Adverse events are summarized in Table 3.
Table 3
Serious and severe adverse events*
| Adverse events | n (%) |
| Serious adverse events** | 29 (54.7%) |
| Severe adverse events | 24 (45.3%) |
| Respiratory tract infections | 31 (58.5%) |
| Acute respiratory failure | 8 (15.1%) |
| Other infections | 4 (7.5%) |
| Post-lumbar puncture syndrome | 3 (5.7%) |
| Skin reaction | 3 (5.7%) |
| Epileptic seizures | 1 (1.9%) |
| Acute hypoglycemia | 1 (1.9%) |
| Neutropenia | 1 (1.9%) |
| Edema and hair loss | 1 (1.9%) |
*Data are listed as n (%). **Serious adverse events were defined as symptoms leading to death, hospitalization or prolonged hospitalization.
DISCUSSION
In our prospective open-label study evaluating six months of treatment with nusinersen in a broad spectrum of patients with SMA type 1, we observed an improvement in motor function in many patients.
Comparing our results with the previous clinical trials for infantile onset SMA, we could detect similar results in CHOP INTEND score with a responder rate (improvement ≥4 points) of 77% versus 71% in the previous phase III clinical trial [8]. Improvements in CHOP-INTEND scores were very rarely observed in the sham-control group of the phase III trial and in previous natural history studies [1, 15]. Regarding motor milestones, 34.4% of children in our cohort showed a motor response, compared to 51% in the phase III clinical trial [8]. Motor milestones such as full head control or independent sitting were achieved only by a small percentage of children in our cohort. While the CHOP INTEND was developed to evaluate very limited motor skills [10], a much larger improvement in motor function is necessary to reach milestones on the HINE-2 scale, thus explaining the higher amount of patients improving in the CHOP INTEND.
The phase II clinical trial showed a mean change in CHOP INTEND score of 15.2 points in children that received 12 mg of nusinersen equivalent dose [16]. This finding is comparable to our results regarding the subgroup of children aged less than 7 months at start of treatment. In older children of our cohort, the change in CHOP INTEND score was lower indicating that response to treatment strongly correlates with age at onset of treatment. Preclinical data from animal models [17–19] and first results from a clinical trial with pre-symptomatic treatment of infants with SMA [20] also provide strong evidence for a critical therapeutic time window for delivery of SMN-targeted therapies. Different methods for a newborn screening have been developed to diagnose SMA from DNA extracted from newborn blood spots [21, 22] and there is an effort in different countries over the world to include screening for SMA into newborn screening programs. The implementation of a newborn screening would allow pre-symptomatic diagnosis in many cases.
In addition, treatment was offered to patients with SMA type 1 in various stages of the disease. Children that already required permanent ventilator support or tracheostomy prior to treatment showed lower changes in CHOP INTEND score compared to the overall cohort. Furthermore, the final follow-up was already performed after six months of treatment as compared to 13 months in the previous phase III clinical trial. These factors might also explain the observed differences in motor response.
The overall parents’ impression about motor function was an improvement in 93.4% of children after six months of treatment, but the correlation between parents’ impression and change in CHOP INTEND score from baseline was not significant. In several cases, parents described improvements in motor function in children with changes less than 4 points or even a worsening in CHOP INTEND score. Here, parents reported small changes in spontaneous movements that are not captured performing the CHOP INTEND or even a more stable course of the disease as improvement in motor function. Parents’ high expectations to the new drug treatment might have influenced the positive assessment of their child. Thus, further research is needed to evaluate the impact of changes in CHOP INTEND score on daily life and on quality of life in children with SMA type 1, which are not as obvious as changes in motor milestones.
SMN2 copy number correlates inversely with the severity of the disease [23, 24]. In a Spanish cohort of 625 SMA patients, all children with one SMN2 copy and 88% of those with two SMN2 copies showed a phenotype of SMA type 1 [25]. Children with three SMN2 copies are likely to develop type 1 (6%), type 2 (57%) or type 3 (37%), so that prediction of phenotype in these cases is still challenging [25]. In our cohort, children with ≥3 SMN2 copies showed a milder phenotype with higher CHOP INTEND and HINE-2 scores prior to treatment compared with children having ≤2 SMN2 copies. In addition, the need for ventilator support or a feeding tube was less in children with ≥3 SMN2 copies. However, we documented a similar change in CHOP INTEND score from baseline in both groups, indicating that the change in CHOP INTEND score from baseline did not depend on SMN2 copy number.
In our cohort, we did not observe major improvements in respiratory function. After six months of treatment, more patients received NIV or permanent ventilation as compared to start of treatment, and three patients underwent tracheostomy during the observation period. Most of the parents did not report any changes in respiratory function, although in some cases they perceived a more stable respiratory situation, especially during episodes of respiratory infections. Concerning nutrition, there was also a slight increase of patients requiring tube feeding or gastrostomy during the six months treatment period. The information gathered during this study does not allow a clear judgment whether changes in ventilator support and tube feeding are due to progression of the disease or whether this reflects a more proactive approach to provide NIV or a feeding tube to patients with SMA type 1 after start of treatment with nusinersen. However, it can be concluded that for most patients of our cohort, six months of treatment did not allow to reduce ventilator support or the need for tube feeding.
Although this study does not provide evidence comparable to a randomized controlled trial, the results indicate that even in advanced stages of the disease, nusinersen can lead to improvement of motor function as measured by CHOP INTEND. Within the SMArtCARE project we aim to collect further data to evaluate if the effect size increases with prolonged treatment periods and to what extent these changes affect patients’ and caregivers’ quality of life. Our findings also add to the increasing body of evidence that early diagnosis and initiation of treatment is fundamental for patients with infantile onset spinal muscular atrophy.
CONFLICT OF INTEREST
JK received research funding and/or compensation for presentations and consultancy from Avexis, Biogen, Ionis Pharmaceuticals, Novartis, and Roche. AH, AP, CV, SS, and SV received compensations for presentations and training activities from Biogen. OS received compensations for presentation from Biogen and Santhera. JJ received compensation for presentation from PTC Therapeutics and consultancy fees from Sarepta Therapeutics, Biogen GmbH, PTC Therapeutics.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
This research was funded by Initiative SMA (DGM e.V.). Training workshops for participating physiotherapists were supported by Biogen. The authors acknowledge Gabriele Ihorst for statistical support and Brunhilde Wirth for genetic testing of SMN2 copy numbers.
REFERENCES
[1] | Sanctis R de , Coratti G , Pasternak A , Montes J , Pane M , Mazzone ES , et al. Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord. (2016) ;26: (11):754–9. |
[2] | Pechmann A , Kirschner J . Diagnosis and new treatment avenues in spinal muscular atrophy. Neuropediatrics. (2017) ;48: (4):273–81. |
[3] | Passini MA , Bu J , Richards AM , Kinnecom C , Sardi SP , Stanek LM , et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. (2011) ;3: (72):72ra18. |
[4] | Singh NK , Singh NN , Androphy EJ , Singh RN . Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. (2006) ;26: (4):1333–46. |
[5] | Lorson CL , Androphy EJ . An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. (2000) ;9: (2):259–65. |
[6] | Lefebvre S , Bürglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: (1):155–65. |
[7] | Hua Y , Sahashi K , Hung G , Rigo F , Passini MA , Bennett CF , et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. (2010) ;24: (15):1634–44. |
[8] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. (2017) ;377: (18):1723–32. |
[9] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. (2007) ;22: (8):1027–49. |
[10] | Glanzman AM , Mazzone E , Main M , Pelliccioni M , Wood J , Swoboda KJ , et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscul Disord. (2010) ;20: (3):155–61. |
[11] | Haataja L , Mercuri E , Regev R , Cowan F , Rutherford M , Dubowitz V , et al. Optimality score for the neurologic examination of the infant at 12 and 18 months of age. J Pediatr. (1999) ;135: (2 Pt 1):153–61. |
[12] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol. (2016) ;3: (2):132–45. |
[13] | Bishop KM , Montes J , Finkel RS . Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological Exam-Part Experience from a nusinersen clinical study. Muscle Nerve. (2018) ;57: (1):142–6. |
[14] | R Development Core Team, editor. R: A language and environment for statistical computing. Vienna, Austria; (2008) . Available from: URL: http://www.R-project.org |
[15] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) ;82: (6):883–91. |
[16] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , Vivo DC de , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet. (2016) ;388: (10063):3017–26. |
[17] | Foust KD , Wang X , McGovern VL , Braun L , Bevan AK , Haidet AM , et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. (2010) ;28: (3):271–4. |
[18] | Hua Y , Sahashi K , Rigo F , Hung G , Horev G , Bennett CF , et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. (2011) ;478: (7367):123–6. |
[19] | Kariya S , Obis T , Garone C , Akay T , Sera F , Iwata S , et al. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest. (2014) ;124: (2):785–800. |
[20] | Chiriboga CA . Nusinersen for the treatment of spinal muscular atrophy. Expert Rev Neurother. (2017) ;17: (10):955–62. |
[21] | Taylor JL , Lee FK , Yazdanpanah GK , Staropoli JF , Liu M , Carulli JP , et al. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin Chem. (2015) ;61: (2):412–9. |
[22] | Dobrowolski SF , Pham HT , Downes FP , Prior TW , Naylor EW , Swoboda KJ . Newborn screening for spinal muscular atrophy by calibrated short-amplicon melt profiling. Clin Chem. (2012) ;58: (6):1033–9. |
[23] | Prior TW , Swoboda KJ , Scott HD , Hejmanowski AQ . Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. (2004) ;130A: (3):307–10. |
[24] | Mailman MD , Heinz JW , Papp AC , Snyder PJ , Sedra MS , Wirth B , et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. (2002) ;4: (1):20–6. |
[25] | Calucho M , Bernal S , Alías L , March F , Venceslá A , Rodríguez-Álvarez FJ , et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of reported cases. Neuromuscul Disord. (2018) ;28: (3):208–15. |

