
Centronuclear myopathies under attack: A plethora of therapeutic targets
Abstract
Centronuclear myopathies are a group of congenital myopathies characterized by severe muscle weakness, genetic heterogeneity, and defects in the structural organization of muscle fibers. Their names are derived from the central position of nuclei on biopsies, while they are at the fiber periphery under normal conditions. No specific therapy exists yet for these debilitating diseases. Mutations in the myotubularin phosphoinositides phosphatase, the GTPase dynamin 2, or amphiphysin 2 have been identified to cause respectively X-linked centronuclear myopathies (also called myotubular myopathy) or autosomal dominant and recessive forms. Mutations in additional genes, as RYR1, TTN, SPEG or CACNA1S, were linked to phenotypes that can overlap with centronuclear myopathies. Numerous animal models of centronuclear myopathies have been studied over the last 15 years, ranging from invertebrate to large mammalian models. Their characterization led to a partial understanding of the pathomechanisms of these diseases and allowed the recent validation of therapeutic proof-of-concepts. Here, we review the different therapeutic strategies that have been tested so far for centronuclear myopathies, some of which may be translated to patients.
BACKGROUND
Centronuclear myopathies (CNM) are usually defined as congenital myopathies with abnormally positioned nuclei in the center of myofibers, in the absence of increased muscle regeneration commonly observed in dystrophies [1]. Of note, CNM are linked with a general disorganization of myofibers, with abnormal positioning of different organelles, internal membrane alterations and often myofibrillar misalignment and fiber shape defects.
Since the first clinical and histopathological description of a 12-year-old affected boy with myotubular myopathy in 1966 by Spiro et al. [2], remarkable advances have been made in understanding the physiopathology of CNM. A major step forward was the identification of the causative genetic anomalies. MTM1 was the first identified gene mutated in the most severe form of CNM: X-linked centronuclear or myotubular myopathy (XLMTM, CNMX, OMIM#310400) [3, 4]. This form is characterized clinically by a severe hypotonia and a generalized muscle weakness often leading to respiratory failure and swallowing difficulties, and usually correlated with a poor prognosis [5–7]. In the vast majority of cases, loss-of-function mutations cause a strong decrease of the MTM1 protein [8, 9], a phosphoinositides phosphatase implicated in multiple cellular processes such as endosomal trafficking [10–12], excitation-contraction coupling [13, 14], intermediate filament organization [15], neuromuscular junction (NMJ) structure [16, 17], satellite cell proliferation [18–20], and autophagy [21, 22]. Female carriers can display a wide spectrum of clinical and pathological involvement [23].
The autosomal dominant (ADCNM) form is mainly caused by mutations in the dynamin 2 (DNM2) gene (ADCNM; OMIM#160150) [24]. The severity ranges from severely affected infant to mildly affected adults [25]. In severe early onset cases, infants present with generalized muscle weakness, hypotonia with facial weakness in addition to ophthalmoplegia [26], while in the late-onset milder cases, young adult patients present with moderate muscle weakness leading to motor defects [27]. DNM2 is a GTPase mechanoenzyme involved in endocytosis [28], exocytosis [29, 30], intracellular membrane trafficking [31–33] and cytoskeletal organization [34–36]. In vitro studies showed that CNM mutations in DNM2 increase the GTPase activity and promote higher stability of polymers, suggesting a gain-of-function mechanism in ADCNM [37, 38]. This is supported by the appearance of a CNM-like phenotype in mice over-expressing wildtype DNM2 [39, 40].
Mutations in the BIN1 gene encoding amphiphysin 2 have been reported in CNM patients linked to two modes of inheritance: autosomal recessive [41, 42] and autosomal dominant [43] (OMIM#255200). A large variability in severity is observed in affected patients, however the main consistent presentation is muscular atrophy with diffuse muscle weakness [1, 44, 45]. BIN1 generates and maintains membrane curvature and tubulation through the BAR domain [46], while the SH3 domain is involved in recruitment of endocytotic proteins such as DNM2 [41, 47, 48]. Recessive mutations decrease the membrane tubulation properties of BIN1 [41]. Of note, BIN1 also binds phosphoinositides and MTM1 [46, 47, 49, 50], suggesting a common pathway linking MTM1, BIN1 and DNM2 [51].
The incidence of CNM is about 1-2 per 100,000 births, comprised of mutations in MTM1 (∼50%), DNM2 (∼15%) or BIN1 (∼3%), other implicated genes (∼12%), or CNM with unknown genetic basis (∼20%) [52]. Mutations in Ryanodine receptor 1 (RYR1) [53, 54], Titin (TTN) [55], SPEG [56], DHPR (CACNA1S) [57], ZAK [58], and potentially myotubularin-related protein 14 (MTMR14) [59] and CCDC78 [60], have been reported to cause congenital myopathies with a CNM-like phenotype. We concentrate here on CNM linked to MTM1, BIN1 or DNM2, as these forms present with highly similar pathogenesis and most translational research has focused on these forms in the recent years. We provide a summary of the different preclinical therapeutic developments that have been explored for XLMTM and autosomal forms of CNM.
INCREASING OUR UNDERSTANDING OF CNM THROUGH PHENOTYPING OF ANIMAL MODELS
Development of different animal models and their characterization greatly accelerated the understanding of the clinical, physiopathological and molecular aspects of CNM (Table 1). CNM, or their molecular defects, have been modelled and studied in cultured cells, yeast, drosophila, zebrafish, mice and dogs [51]. Moreover, they represent precious tools to test and validate the different therapeutic approaches preclinically before embarking on clinical trials.
Table 1
Comparison between the different therapeutic approaches tested on animal models of centronuclear myopathies
| Approach | Disease rescue | Products | Species &CNM form | Delivery | Positive outcomes | References | |||
|---|---|---|---|---|---|---|---|---|---|
| Lifespan | Body weight | Muscle mass and/or force | Muscle histology | ||||||
| Gene, RNA and protein replacement | |||||||||
| MTM1 gene delivery | Prevention and reversion | AAV-MTM1 | Mtm1KO mouse | Local (intramuscular) | NA | NA | ++ | +++ | Buj-Bello et al. 2008 |
| Systemic (intravenous) | +++ | +++ | +++ | +++ | Childers et al. 2014 | ||||
| AAV-MTM1 | XLMTM dog | Local (intramuscular) | NA | NA | +++ | +++ | Childers et al. 2014 | ||
| Systemic (intravenous) | +++ | +++ | +++ | +++ | Mack et al. 2017 | ||||
| Prevention | AAV-MTM1 dead phosphatase | Mtm1KO mouse | Local (intramuscular) | NA | NA | ++ | ++ | Amoasii et al. 2012 | |
| Myotubularin delivery | Prevention | 3E10Fv-myotubularin | Mtm1KI R69C mouse | Local (Intramuscular) | NA | NA | + | + | Lawlor et al. 2013 |
| DNM2 RNA trans-splicing | NA | AAV-5’ pre-trans-splicing nucleotides | WT mouse | Local (intramuscular) | NA | NA | NA | NA | Trochet et al. 2016 |
| Allele-specific DNM2 RNA silencing | Prevention | AAV-shRNA | Dnm2KI R465W mouse | Local (intramuscular) | NA | NA | +++ | +++ | Trochet et al. 2017 |
| Normalizing the disease pathway | |||||||||
| MTMR2 expression | Prevention | AAV-MTMR2 and isoforms | Mtm1KO mouse | Local (intramuscular) | NA | NA | ++ | ++ | Raess et al. 2017 |
| Prevention | AAV-MTMR2 | Mtm1KO mouse | Systemic (intravenous) | ++ | ++ | ++ | ++ | Danièle et al. 2018 | |
| PI3K inhibition | Prevention and reversion | NA | Mtm1KO mouse | Genetic cross Pik3c2bKO homozygous | +++ | +++ | +++ | +++ | Sabha et al. 2016 |
| Wortmannin; LY294002; PI-103 | mtm1KO zebrafish | Systemic (oral) | ++ | NA | ++ | ND | Sabha et al. 2016 | ||
| Wortmannin | Mtm1KO mouse | Systemic (oral or intraperitoneal) | ++ | – | ++ | + | Kutchukian et al. 2016; Sabha et al. 2016 | ||
| DNM2 reduction or normalization | Prevention and reversion | NA | Mtm1KO mouse | Genetic cross Dnm2KO heterozygous | +++ | +++ | +++ | +++ | Cowling et al. 2014 |
| antisense oligonucleotides | Mtm1KO mouse | Local (intramuscular) | NA | NA | +++ | +++ | Tasfaout et al. 2017 | ||
| Systemic (intraperitoneal) | +++ | +++ | +++ | +++ | Tasfaout et al. 2017 | ||||
| Prevention | AAV-shRNA | Mtm1KO mouse | Local (intramuscular) | NA | NA | +++ | +++ | Tasfaout et al. 2018 | |
| Prevention | NA | Bin1KO mouse | Genetic cross Dnm2KO heterozygous | +++ | ++ | +++ | +++ | Cowling et al. 2017 | |
| Treating the general muscle unbalance | |||||||||
| Autophagy activation | Prevention | mTOR inhibitors (RAD001, AZD8055) | Mtm1KO gt/y mouse | Systemic (oral) | ND | ND | + | ND | Fetalvero et al. 2013 |
| Myostatin inhibition | Prevention | ActRIIB-mFC | Mtm1KO mouse | Systemic (intraperetoneal) | + | + | + | + | Lawlor et al. 2011 |
| Mtm1KI R69C mouse | Systemic (intraperetoneal) | ND | – | + | + | Lawlor et al. 2014 | |||
| Prevention | AAV-PropD76A | Mtm1KO mouse | Local (Intramuscular) | NA | NA | – | ND | Mariot et al. 2017 | |
| Other potential approaches | |||||||||
| Acetylcholinesterase inhibition | Prevention | edrophonium | mtm1 morphant zebrafish | Systemic (oral) | ND | ND | ++ | ND | Robb et al. 2011 |
| dnm2 S619L zebrafish | Systemic (oral) | ND | ND | ++ | ND | Gibbs et al. 2013 | |||
| Reversion | pyridostigmine | XLMTM patient | Systemic (oral) | ND | ND | + | ND | Robb et al. 2011 | |
| DNM2-CNM patients | Systemic (oral) | ND | ND | + | ND | Gibbs et al. 2013 | |||
| Cell transplantation | Prevention | syngeneic WT myoblasts | Mtm1KI R69C mouse | Local (intramuscular) | ND | ND | + | ND | Lim et al. 2014 |
Notes: ND: no data; NA: not applicable. Most animal models are presented in Cowling et al. 2012. Extent of positive outcomes in animal models do not necessarily translate in human. In several set-up, only disease prevention was tested.
Three different mouse lines were developed to mimic XLMTM. The Mtm1 knockout (KO) mice with a complete knockdown of the MTM1 protein present a progressive myopathy starting from around week 3 with a shortened lifespan to around 7-8 weeks of age [13, 61]. They exhibit similar features to XLMTM patients, including muscle atrophy and severe weakness, and muscle biopsies feature small rounded fibers with a high frequency of abnormal localization of nuclei and disorganized mitochondrial distribution. Ultrastructural analysis revealed dramatic decrease of normal triads [13], membrane invagination implicated in excitation coupling machinery, as well as sarcomere disorganization and enlargement of neuromuscular junctions [17, 62]. Additional studies have shown defects in mitochondrial dynamics, desmin aggregation and alteration in autophagy and ubiquitin-proteasome pathways [15, 63]. A knock-in (KI) mouse carrying the R69C mutation was generated to mimic a mutation identified in XLMTM patients with a mild phenotype [64]. The R69C mutation results in exon 4 skipping leading to premature termination of myotubularin translation. However, some full-length MTM1 protein was still expressed in this model, resulting in a milder phenotype compared to Mtm1KO mice, with a median lifespan of 66 weeks. This observation suggests that expression of a small amount of MTM1 may have significant clinical benefits in XLMTM. A third mouse model, the Mtm1gt/y, was generated using a gene trap strategy, and these mice survive for at least 41 weeks [22]. Mtm1gt/y mice present a milder phenotype than Mtm1KO mice with a similar histopathology including hypotrophic fibers with a mild increase in mislocalized organelles and internal nuclei, and autophagy defects. Overall these mouse models present with CNM phenotypes mimicking those observed in patients, making these models very attractive, and are therefore not surprisingly frequently used in preclinical research.
In addition to murine models, a zebrafish model of XLMTM was generated using morpholinos [14]. Injection of antisense oligonucleotides into embryos resulted in reduction of myotubularin expression resulting in a CNM phenotype including myofiber hypotrophy, centralization of nuclei, as well as mislocalization of organelles and NMJ defects [14, 16]. Triad defects were also noted in this model. These defects caused abnormal dorsal curvature with impairment in spontaneous movements and swimming capacities. More recently, a novel zebrafish model for XLMTM was created using zinc finger nucleases to delete 8-bp in exon 5 of the mtm1 gene causing a frameshift and premature stop codon [65]. In addition to a shortened survival of 7 days, mutants present a severe muscular phenotype similar to the morphant model, including reduced motor abilities, and morphological and muscle structural defects.
Finally, two naturally occurring canine models were identified for XLMTM: a Labrador Retriever dog and a Rottweiler dog with progressive myotubular myopathy harboring the N155K and Q384P mutations, respectively, in the MTM1 gene [66, 67]. The affected dogs develop a progressive myopathy and exhibit CNM features such as gait abnormalities with muscle weakness, and hypotrophic fibers with an increase in mis-positioned nuclei and organelles.
A third naturally occurring canine model was identified with a splicing mutation in BIN1 leading to a strong decrease in protein level and a rapidly progressive muscle atrophy coupled with exercise intolerance and a typical CNM histology [42]. This latter line represents a model for the recessive form of BIN1-CNM.
Concerning ADCNM due to DNM2 mutations, a KI mouse model harboring the most frequent ADCNM mutation R465W was generated [68]. While the homozygous KI mice present a neonatal lethality due to metabolic defect including hypoglycemia, increased liver glycogen content and hepatomegaly [69], the heterozygous KI mice are viable and have a normal lifespan, and develop a progressive myopathy with impairment of muscle contractile properties, atrophy and defects in mitochondria and reticular networks [68]. Similarly to Mtm1KO mice, alterations of autophagy were reported, in addition to defective clathrin-dependent endocytosis and calcium homeostasis [68–71]. As an alternative to germline mouse mutagenesis, adeno-associated virus (AAV) containing mutant DNM2-R465W were intramuscularly injected into WT mice and resulted in development of several CNM features such as fiber hypotrophy, histological abnormalities and muscle contraction defects, despite the endogenous expression of DNM2 [39]. In addition, overexpression of WT DNM2 in mouse, either with the same AAV transduction strategy or through transgenesis, also resulted in a CNM phenotype [39, 40]. These results suggest DNM2-CNM is not due to haploinsufficiency of DNM2, and support other in vitro observations suggesting that DNM2 mutations leading to CNM may be gain-of-function [37, 38]. Lastly, a more severe DNM2-CNM mutation, S619L, was modeled in zebrafish, resulting in severe weakness and motor defects with histopathological and NMJ abnormalities [72].
Overall, these animal models recapitulate many CNM features and have contributed to the identification of several pathomechanims. They all have strengths and limitations; the small size and high offspring number of drosophila and zebrafish make these models well adapted for preclinical work including drug screening, whereas the murine models allow in-depth analysis of the pathomechanisms in mammal. The canine models have similar body weight to affected children and are thus better suited for pharmacokinetic/pharmacodynamic modeling and validation of delivery methods. Based on their various strengths these animal models were used in several preclinical studies as discussed below.
GENE, RNA AND ENZYME REPLACEMENT STRATEGIES
MTM1 (Myotubularin) gene therapy
Gene therapy is a promising therapeutic approach for many monogenic disorders, especially with loss-of-function mechanism, which relies on the knowledge of the implicated genes but not on an in-depth understanding of the pathomechanisms. It was thus the first approach tested in CNM models [73]. Moreover, recent progress has greatly increased the prospects for successful gene transfer [74–76]. Among the different options, AAV vectors have emerged as effective and potentially safe candidates for certain indications such as neuromuscular or neurodegenerative diseases, hematologic disorders or heart defects [77, 78]. This is reflected by the increasing number of successful clinical trials that have driven the approval and marketing of different products based on AAV-gene delivery, such as alipogene tiparvovec (AAV1-LPLS447X) (commercial name: Glybera) for lipoprotein lipase deficiency (LPLD)[79, 80]. MTM1 gene replacement was first tested through intramuscular injection of Mtm1KO mice with AAV expressing the Mtm1 cDNA under the control of a CMV promoter, which resulted in the recovery of muscle mass and force and a normal histology [73]. Subsequently, AAV-Mtm1 with a desmin promoter was administered through loco-regional or systemic injections in both mice and dogs lacking MTM1 [81–83]. Significant improvements of survival and whole-body rescue were noted after a single injection, with increase of muscle force and mass as well as correction of histopathological hallmarks of CNM. An absence of obvious toxicity or immune response was noted, except histological alteration of cardiac tissue specifically in mice [81]. In treated dogs, amelioration of the phenotype was seen from 4 weeks post injection, and both survival and health status were greatly improved. This exciting proof-of-concept demonstrates the efficacy of gene therapy-based approaches in both small and large animals and indicates that postnatal gene replacement can reverse the XLMTM phenotype.
The selection of the best AAV serotype and promoter that controls the MTM1 expression is essential for effective and safe gene delivery. Although XLMTM patients exhibit mainly a muscular phenotype, MTM1 is ubiquitously expressed and might have other key roles in other tissues and organs. Tissue-tropism of AAV-Mtm1 should respect the natural tissue-distribution of MTM1 with similar expression level specific to each organ. The desmin promoter is highly active in skeletal and cardiac muscles after systemic delivery in mouse, but not in dog, resulting in massive overexpression of MTM1 in heart (about 700 fold) with an impact on cardiac histology [81]. The MTM1 promoter may be an ideal regulator of myotubularin expression in the different tissues. Furthermore, since MTM1 is barely expressed in XLMTM patient cells [8, 9], it may be necessary to ensure that delivering an “exogenous” protein will not trigger an immune response against the transgene. Also, as AAVs are present within cells mostly as non-integrated episomes, normal muscle turnover will likely lead to loss of transgene expression and reduce the efficacy; however this may be less the case for CNM than for the tested dystrophies as there are no signs of increased muscle regeneration in CNM [1]. Whether re-administration of these viral vectors several years after the initial infusion may be necessary, without eliciting immunoresponse, is unclear. A recent study in dog showed longterm expression of MTM1 four years after a single AAV-MTM1 isolated limb perfusion despite decrease in AAV vector copy number, correlating with near-normal respiratory and motor functions [82]. Finally, production of an effective dose of AAV vectors for human administration presents a manufacturing challenge and requires high titer solutions ranging from 1013 to 1014 vector genome per kg of body weight for effective gene delivery to at least vital affected muscles as shown in several ongoing clinical trials (NCT03199469, NCT03368742, NCT03375164). Production of these highly concentrated solutions relies on the use of costly methods that guarantee high-quality and reliable lots [84]. Importantly, a clinical trial delivering the AAV-MTM1 gene therapy to XLMTM patient was started by Audentes Therapeutics (www.audentestx.com, NCT03199469) and exciting positive preliminary results were very recently reported (www.audentestx.com). Significant improvements of neuromuscular and respiratory functions were detected, and the earliest treated patient has become ventilator independent 16 weeks post-injection.
Myotubularin replacement
Enzymotherapy or enzyme replacement is another approach that was tested to deliver the MTM1 protein to myofibers [85]. Preclinical research suggests a small amount of fully functional myotubularin is enough to significantly prevent or improve the disease. Indeed several female carriers are asymptomatic, and there is a tendency for a milder disease in patients with a residual protein level if the mutation is not in the catalytic site [8]. This is supported by animal models, such as the Mtm1KI R69C mouse that produces a small amount of myotubularin and is less affected than the Mtm1KO mouse [64]. For enzyme replacement, MTM1 was fused with a single chain fragment derived from a mouse monoclonal antibody (3E10Fv) [85]. 3E10Fv fusion proteins penetrate cells through interactions with the equilibrative nucleoside transporter 2 (ENT2), a plasma membrane receptor widely distributed in mammalian tissues with high abundance in skeletal muscle [86]. Interestingly, the phosphatase activity of the fused myotubularin-3E10Fv was similar to myotubularin alone suggesting that this fusion does not interfere with the enzymatic activity [85]. Intramuscular injection of 3E10Fv-MTM1 into Mtm1KO mice significantly improved the muscle function with an increase in muscle contraction that may be correlated to the increase in correctly positioned triads and T-tubules observed by electronic microscopy. However, no obvious improvement was seen in general muscle histology. Assessment of the beneficial effect of 3E10Fv-MTM1 was limited to intramuscular injections to date. Due to the generalized muscle defect in XLMTM disease, systemic delivery will be required and the efficacy of systemic delivery of 3E10Fv-MTM1 remains to be elucidated. In addition, the manufacturing of this active MTM1 protein for human delivery represents a challenge. The major advantage of this approach is the flexibility in dosing; however the expected short effect duration will necessitate a repeated re-administration, which may induce an immunoresponse against the 3E10Fv-MTM1 complex. Similar therapeutic strategies have been successfully employed especially in multisystemic lysosomal storage disorders including alglucosidase alfa (Myozyme) for Pompe disease, Agalsidase alfa (Replagal) or Agalsidase beta (Fabrazyme®) for Fabry disease, and Imiglucerase (Cerezyme®) for Gaucher disease [87–90]. These marketed enzymotherapies showed the feasibility of such approaches and may be used as a roadmap for a therapeutic development initiated by Valerion Therapeutics (www.valerion.com) based on myotubularin enzyme replacement for myotubular myopathy.
RNA Trans-splicing for DNM2-related myopathy
DNM2-related CNM is a dominant form of CNM potentially due to gain-of-function mutations, and may not be a good target for conventional gene replacement. Reprogramming the Dnm2 RNA to replace the mutated part with a normal sequence was tested in the mouse through spliceosome-mediated RNA trans-splicing [91, 92]. In this specific example, the classical 3’ trans-splicing strategy led to toxic intermediate. However, the alternative usage of 5’ trans-splicing successfully allowed the detection of trans-splicing events at the mRNA and protein levels, both in vitro in fibroblasts and in vivo in WT murine tibialis muscle [91].
RNA trans-splicing was successful for different diseases including several myopathies in a pre-clinical setting [92–96]. This promising strategy appears still in its infancy and additional studies will have to improve the in vivo efficiency of the trans-splicing and the phenotypic rescue, while carefully investigating potential toxicity linked to the used pre-trans-splicing molecules. Also, these molecules have to be vectorized to be efficiently provided [97].
Allele-specific RNA silencing for DNM2-related myopathy
Another approach aiming to decrease the amount of the mutated DNM2 transcript consisted in silencing specifically the mutated RNA through the siRNA technology [98]. Allele-specific siRNA sequences were developed in order to specifically knock down the human or murine DNM2-mRNA harboring the p.R465W mutation without affecting the wild-type allele. Scanning the sequence encompassing the mutated site was necessary to identify the best siRNA molecules efficient in heterozygous Dnm2KI R465W mouse embryonic fibroblasts. The in vivo proof-of-concept was obtained in the Dnm2KI R465W mouse after intramuscular injection of AAV expressing the selected shRNA molecules. After 3 months, most CNM histological signs were partially or fully rescued, including muscle mass and specific force, fiber size and central accumulation of oxidative staining. A similar approach was validated in patient fibroblasts expressing the R465W mutation. The R465W RNA was strongly reduced while the WT transcript appeared unaffected. This was correlated with a normalization of endocytosis measured by transferrin uptake, while this is decreased in scramble-transfected cells. Overall, this allele-specific targeting approach appears very specific and bears promise for future pre-clinical development.
NORMALIZING THE DISEASE PATHWAY
MTMR2 expression
The MTM1 gene has many close homologs, which may provide an alternative therapeutic option. Indeed, 14 myotubularins have been identified in human [99, 100]. They express common protein domains including the PH-GRAM and coiled-coil that are implicated in phosphoinositide binding and in their oligomerization, respectively [4]. Of note, 6 myotubularins do not have an enzymatic activity and are named dead-phosphatases, while 8 display a phosphatase activity [4]. The myotubularin dead-phosphatases do however regulate their catalytically active homologs, either by modifying their subcellular localization or by potentially changing the allosteric activity [101]. MTM1 and MTMR2 share 65% of amino acid sequence identity and similar phosphatase activity [100, 102]. Of note, while MTM1 is mutated in XLMTM, MTMR2 mutations are linked to demyelinating Charcot-Marie-Tooth neuropathy (CMT4B1; OMIM 601382) [103]. Raess et al. studied the molecular basis for the functional difference between MTM1 and MTMR2 [104]. Two MTMR2 isoforms were previously identified [105]. The most studied and longest isoform has an additional N-terminal extension prior to the PH-GRAM domain, which is absent in the short MTMR2 isoform. Importantly, unlike the long isoform, the short MTMR2 isoform displays an MTM1-like activity in a yeast complementation system and was found to be downregulated in Mtm1KO muscle [104]. Upon intramuscular injection of AAV expressing the long MTMR2 isoform in the Mtm1KO mouse, a significant rescue of muscle phenotype was seen, however a better amelioration in muscle mass, force and histology were observed with the short isoform [104]. In addition, the elevated level of the MTM1 substrate phosphoinositol-3-phosphate (PtdIns3P) in Mtm1KO muscles was corrected upon this treatment. These observations suggest that the additional N-terminal extension in MTMR2 is responsible for the functional difference between MTM1 and MTMR2. More recently, intravenous administration of a single dose of the AAV9-Mtmr2 vector in the Mtm1KO mice was shown to improve the motor activity and muscle strength and prolonged survival throughout a 3-month study [106].
Thus, exogenous overexpression of short MTMR2 isoform or alternative splicing modulation of endogenous MTMR2 transcripts may be potential strategies to compensate for the loss of MTM1. This strategy could be applied as an approach to normalize the phosphoinositides level and treat XLMTM. While the targeting of an MTM1 homolog may avoid potential immune response compared to exogenous expression of the missing MTM1 protein, the rescue may be less efficient than using MTM1 itself, and the challenges for the delivery (e.g. AAV) remain as discussed above.
Rebalancing 3-phosphoinositides level
MTM1 is a PI 3-phosphatase that dephosphorylates PtdIns3P and PtdInsp(3,5)P2 into PtdIns and PtdIns5P respectively [107–109]. Different XLMTM animal models have shown an increase in PtdIns3P as a direct consequence of loss of MTM1 [14, 64, 65, 110]. These low-abundant phospholipids are key regulators of different cellular processes like membrane trafficking and intracellular signaling [111, 112]. Concerning PtdIns3P, several kinases were implicated in its generation including class II (PIK3C2) and class III (PIK3C3 or VPS34) PI 3-kinases. Reducing the elevated PtdIns3P level was investigated through downregulation or inhibition of these kinases. Based on the observation made in Drosophila melanogaster showing depletion of the class II PI 3-kinase Pi3K68D rescued the defects caused by mtm (ortholog of both MTM1 and MTMR2) deficiency [113], depletion of the class II PIK3C2B was achieved in Mtm1KO mice through the production of a double knockout Mtm1KO/muscle-specific Pikc2BKO [65]. PIK3C2B was potentially the best candidate to target as its total loss in mice is viable whereas loss of other PI 3-kinases including PIK3C3 leads to strong defects [114]. Genetic reduction of PIK3C2B before or after disease onset prevented or reverted the myopathy and extended the life expectancy, improved body weight and restored muscle force and histology. Surprisingly, genetic reduction of PIK3C3 in Mtm1KO mice (Mtm1KO/Pik3c3 heterozygous) worsened the CNM phenotype, suggesting that MTM1 and PIKC2B may control the same sub-pool of PtdIns3P and that blocking the phosphorylation activity of PIK3C2B, but not PIK3C3, would be a therapeutic strategy for XLMTM. To develop this approach further, the identification of drugs that can specifically target PIKC2B is required. In the mtm1 zebrafish model, inhibitors of PI 3-kinase class I or III, GDC-0941 or VPS34-IN1 respectively, did not improve the motor function and morphological defects. In the absence of specific class II inhibitors, pan PI 3-kinase inhibitors were tested, including PI-103, LYS294002 and wortmannin, and significant improvements of the survival, motor function and morphology were observed in the treated larvae [65]. In addition, treatment of Mtm1KO mice with wortmannin prolonged the survival, ameliorated the CNM phenotype and histology, and greatly improved Ca2 + release defects [65, 115].
A great advantage of this therapeutic strategy is that the target is an enzymatic activity and thus druggable. The therapeutic window, i.e. the range of doses which optimize between efficacy and toxicity, has to be defined and genetic studies suggested full depletion of PIK3C2B achieved a better rescue than partial depletion [65]. This successful strategy focused on targeting one function of MTM1, PtdIns3P dephosphorylation, but not PtdIns5P production or phosphoinositides-independent functions, suggesting increased in PtdIns3P is a main cause of XLMTM. Interestingly, a significant prevention of CNM phenotypes was also achieved by AAV-mediated expression of several phosphatase-dead MTM1 mutants in the Mtm1KO mouse [116] and confirmed in an independent study [63]. Thus targeting phosphoinositide-independent functions of MTM1 also appears promising as normalization of PtdIns3P level was not needed for the rescue of the intracellular organization of myofibers, including positioning of nuclei, mitochondria and triads, nor for muscle force, but seems important for triad shape and fiber size [116].
In conclusion, inhibiting specifically PIK3C2B in muscle represents a promising therapeutic approach in XLMTM; however development of specific PIK3C2B inhibitors is required, as alteration or inhibition of other PI 3-kinases is highly detrimental [117].
Normalizing or reducing DNM2 in several forms of CNM
As mutations in either MTM1 or DNM2 lead to CNM, it is probable that these proteins act in the same pathway for normal muscle maturation and/or maintenance [51]. In vitro experiments showed several CNM mutations increase the oligomerization and GTPase activity of DNM2 [37, 38], and in vivo over-expression of wildtype DNM2 in mice either through transgenesis or AAV injection produced a CNM-like phenotype [39, 40]. Moreover, it was realized that DNM2 protein level is increased in XLMTM patient muscles and in muscles from the Mtm1KO mouse [118]. Based on these results, and the rationale that DNM2 mutations in CNM are gain-of-function, the potential therapeutic effect of DNM2 downregulation was tested in Mtm1KO mice.
To test a negative epistasis between MTM1 and DNM2, a genetic reduction of DNM2 in Mtm1KO mice was first investigated in Mtm1KO/Dnm2 heterozygous KO animals. Unlike Mtm1KO mice, the double transgenic mice showed normal lifespan and whole-body weight, and greatly improved muscle histology, mass and force [118]. These results were confirmed recently using antisense oligonucleotides (ASO) targeting specifically Dnm2 transcript [119]. Systemic ASO delivery in pre-symptomatic Mtm1KO mice efficiently decreased DNM2 expression at RNA and protein levels and prevented the development of CNM. More importantly, post-symptomatic ASO systemic treatment stopped disease progression after a single injection and reverted CNM phenotypes within only 2 weeks. In this later experiment, the disease correction correlated with a normalized DNM2 expression, suggesting that decreasing DNM2 level to normal range is enough to restore muscle function in severely affected mice thereby reducing side effects related to DNM2 reduction below normal level.
These observations make the reduction/normalization of DNM2 by ASO injection an attractive therapeutic approach. Indeed, injection and dosing regime can be easily adapted during the treatment. In addition, many clinical trials using ASO have been made with some ASO-based drugs approved for use in clinic, such as fomivirsen upon local administration in Cytomegalovirus retinitis, or systemic delivery of mipomersen for familial hypercholesterolemia, and more recently nusinersen and eteplirsen for spinal muscular atrophy and Duchenne muscular dystrophy, respectively [120]. Nevertheless, repeated injections are needed and certain ASO chemistries have been reported to preferentially distribute into liver and kidney potentially causing toxicity [121–124]. Thus, translation of this approach into XLMTM patients will require the development of efficient and safe human ASO candidates that allow a preferential targeting of skeletal muscle. Also, DNM2 has important roles in cellular organization in different tissues; therefore determining the benefit:risk ratio and to which extent DNM2 can and needs to be downregulated is a main point being investigated by Dynacure (www.dynacure.fr) which is developing clinical candidates for DNM2 targeting.
The concept of “cross therapy” in CNM, i.e. targeting one CNM gene (DNM2) to rescue the myopathy due to mutations in another CNM gene (MTM1 as above), has been recently further developed by showing that DNM2 downregulation can also rescue BIN1-related CNM [125]. While Bin1KO mice die at birth from a skeletal muscle defect, Bin1KO mice with reduced level of DNM2 (Bin1KO/Dnm2 heterozygous KO) survive at least 18 months, and have normal muscle force and intracellular organization of muscle fibers. Modulating DNM2 level thus represents a first therapeutic concept for patients with BIN1 defects.
Taken together, these results validate the epistasis between MTM1, BIN1 and DNM2. Of note, DNM2 targeting appears to be the first therapeutic strategy targeting several forms of CNM, suggesting a single ASO therapy may thus be sufficient to target a wide range of CNM mutations.
TREATING THE GENERAL MUSCLE IMBALANCE
XLMTM is characterized by severe muscle atrophy with small muscle fibers in Human and animal models [1, 14, 61, 66]. Moreover, a correlation between myofiber size, MTM1 mutation and clinical outcome has been documented by comparing histology of XLMTM infant muscle biopsies and prognosis [126]. Other data on murine and canine models suggest that a very useful histological indicator of disease improvement is normalization of myofiber size together with organelle distribution. These observations support that therapeutic strategies increasing fiber size and/or muscle bulk together with myofibers intracellular organization may be beneficial. The regulation of muscle mass and fiber size is reflected by the balance between protein synthesis and degradation, which are controlled by several intracellular signaling pathways. Major pathways implicated to date are the IGF1-AKT-mTOR pathway leading to muscle hypertrophy mainly through increased protein synthesis, the myostatin-Smad2/3 pathway which is a negative regulator of protein synthesis, and the protein degradation pathways, autophagy and the ubiquitin-proteasome system [127]. These pathways represent interesting targets that were investigated mainly for XLMTM.
Targeting cellular homeostasis: Autophagy and ubiquitin-proteasome system
The ubiquitin-proteasome and autophagy-lysosomal pathways play a vital role in cellular homeostasis by degrading damaged organelles and unnecessary or dysfunctional components such as proteins, and were shown essential for normal muscle growth and maintenance [128]. Alterations of these cellular processes have been reported in different Mtm1-deficient animals [15, 21, 22, 63]. Analysis of muscles from Mtm1KO mice revealed an accumulation of ubiquitinated proteins and defective mitochondria as well as a low conversion rate of LC3I to LC3II suggesting a blockade of the late stage of autophagy [15, 22]. In addition, misfolding and aggregation of intermediate filaments, such as desmin, were also noted [15]. Loss of MTM1 was associated with hyperactivation of the IGF1R/AKT/mTOR pathway reflected by an hyperphosphorylation of S6K1Thr389, S6ser240/244 and 4EBP1Thr37/46 and an increase of the autophagy induction marker phospho-ULK1Ser757 [19, 22]. On the other hand, hypophosphorylation of FoxO3aser318/321, the transcription factor implicated in muscle protein degradation, was noted in MTM1-deficient muscles resulting in increased transcription of atrogenes such as Atrogin1, Murf1, Nedd4 and ZNF2016 [21]. Moreover, a molecular interaction was characterized between MTM1 and UBQLN2, an ubiquilin protein regulating the degradation of ubiquitinated proteins by the proteasome [63]. Taken together, these results indicate that MTM1-deficiency results in defects in cellular clearance pathways and suggest that activating these degradation processes may normalize homeostasis of the muscle fibers.
Pharmacological inhibition of mTOR, a major negative regulator of autophagy, using the allosteric inhibitor RAD001 or the ATP-competitive inhibitor AZD8055 remarkably normalized autophagy markers by lowering p62 and increasing LC3I/LC3II conversion [22]. Moreover, AZD8055 treatment in MTM1-deficient mice had positive effect on muscle mass and reduced the abnormal accumulation of desmin intermediate filament. These encouraging results highlight the implication of MTM1 in autophagy-lysosome and ubiquitin-proteasome pathways and propose autophagy normalization as a therapeutic target for XLMTM. As drugs affecting these pathways are in clinical development for different diseases [129], it may benefit the field of centronuclear myopathies.
It is worth noting that DNM2 was also reported to be implicated in autophagy [68, 69]. Liver, skeletal muscle and fibroblasts of Dnm2 R465W KI mice presented a defect in autophagosome maturation due to a decrease in the autophagic flux [69]. As these data were mainly obtained on cells and mice homozygous for this specific DNM2 mutation, while DNM2-CNM is linked to different heterozygous mutations, the importance of autophagy in the DNM2-CNM forms remains to be confirmed. To this end it was shown that fasting in heterozygous animals induced an increase in LC3-II, p62 and autophagosomes in muscle [69]. These findings suggest a common molecular defect between XLMTM and ADCNM, and broaden the therapeutic interest of the autophagy normalization approach.
Targeting muscle growth pathways with Activin Receptor type IIB inhibition
Myostatin, a member of the TGFβ (transforming growth factor beta) family, is a highly conserved negative regulator of skeletal muscle bulk [130]. Furthermore, natural deficiency or pharmacological inhibition of the myostatin pathway has been reported to stimulate muscle growth by increasing myofiber size and muscle mass [131–135], which makes anti-myostatin based approaches very attractive to different muscle wasting conditions including Duchenne muscular dystrophy [136], amyotrophic lateral sclerosis [137], sarcopenia [138] and cancer cachexia [139, 140]. Indeed, various myostatin inhibitor approaches have been identified and tested in preclinical or clinical trials with varying efficacies, e.g. gene delivery of follistatin and homologues [135], myostatin propeptide [141, 142], myostatin antibodies [136] and more recently a soluble form of myostatin receptors [143, 144]. In Mtm1KO mice, administration of a soluble form of the extracellular region of the myostatin receptor (ActRIIB) fused to the Fc portion of human IgG resulted in a transient increase in muscle mass and force and myofiber size, that was reflected clinically by a significant albeit modest extension of lifespan [145], whereas no clinical improvements were noted following ActRIIB injection in the less severely affected Mtm1KI R69C mice apart from hypertrophy of gastrocnemius muscle [19]. Histology analysis of skeletal muscles of both treated Mtm1KO or Mtm1KI R69C mice showed that the hypertrophy was restricted to myofiber type 2b, suggesting that inhibiting myostatin has an effect mainly on fibers with low oxidative capacities. However, in XLMTM cases, muscle biopsies are characterized by predominance of highly oxidative type 1 fibers [1], which may compromise the efficacy of this strategy in XLMTM patients. An explanation for the modest benefit of downregulating the myostatin pathway in the XLMTM mice models is that, in the Mtm1KO at least, there is a strong decrease of myostatin and a corresponding increase of the myostatin antagonist follistatin, supporting that the myostatin pathway is already turn down in the disease and thus is not available for further inhibition [146]. Moreover, despite the substantial increase in myofiber type 2b size, no improvement of the excitation-contraction coupling related defects was noted, supporting myostatin inhibition as a potential additive strategy for increasing muscle bulk in combination with another strategy improving excitation-contraction coupling. In addition to its skeletal muscle expression, myostatin is also expressed at much lower level in the heart and adipose tissue. Myostatin null mice exhibit cardiac hypertrophy with changes in structure and contractile parameters [147]. Consequently, the benefit:risk balance for an anti-myostatin approach in XLMTM should be considered before application on patients.
OTHER POTENTIAL THERAPEUTIC APPROACHES
Acetylcholinesterase inhibition
Defects of NMJ have been documented in XLMTM patients and animal models of myotubular myopathy [16, 17, 148]. This chemical synapse is where a motoneuron activates muscle contraction upon the arrival of an action potential at the presynaptic neuron leading to a release of neurotransmitters. Its function is defective in congenital myasthenic syndromes (CMS) that share some clinical features with CNM. Previous studies described structural and functional defects of NMJ in some patients with CNM without genetic diagnosis at that time [149, 150]. Using electron microscopy, muscle biopsies from CNM affected neonates showed shallow primary synaptic clefts with few large synaptic vesicles indicating a poor NMJ transmission. Furthermore, similar defects have been observed in a zebrafish model of XLMTM (mtm1 hypomorphic morpholino model) or ADCNM transiently overexpressing the most severe DNM2 S619L mutation [16, 151]. In fact, these zebrafish models exhibit an abnormal distribution of acetylcholine receptors that was associated with a decrease in swimming capacities, while treatment with edrophonium, an acetylcholinesterase inhibitor, dramatically enhanced both spontaneous and touch evoked movement. Finally, additional studies confirmed the NMJ defect in two mouse models of XLMTM, Mtm1KO and Mtm1KI R69C [17, 148]. In Mtm1KO, through either intravital imaging or examination of muscle sections, NMJs appear larger and ultrastructural examination revealed simplification of post-junctional folds with presence of many vacuolar structures, resembling NMJ ultrastructure described in XLMTM patients. In all, these findings suggest a contribution of NMJ defects in the muscle weakness of CNM patients and a role of two CNM-causing genes (MTM1 and DNM2) in the regulation of NMJ. Whether NMJ defects are a primary defect of the nerve or secondary to muscle alteration is not yet known. Importantly, a significant clinical improvement was noted in a confirmed case with XLMTM following treatment with pyridostigmine, an acetylcholinesterase inhibitor which increases acetylcholine half-life within the synapse and is commonly used to treat myasthenia gravis and CMS [16]. Thus, acetylcholinesterase inhibitors may represent an effective therapy to treat symptoms for patients with CNM.
Cell transplantation or cell therapy
Cell transplantation has been tested in the clinic for many diseases with variable positive outcomes [152]. At present, the most successful trials have been achieved in hematological diseases using bone marrow cells which are relatively easy to harvest, manipulate and replace, and have good safety profiles and obvious selective advantages [153, 154]. In contrast, due to the wide tissue distribution and slow cellular turnover of muscle fibers, stem cell transplantation showed very limited beneficial effects when used in muscle disorders [155]. In this field, the majority of trials focused on Duchenne muscular dystrophy which is characterized by myofiber degeneration/regeneration and satellite cell pool exhaustion [156–159]. Albeit XLMTM is not a dystrophy, the number of satellite cells in three different muscles of XLMTM patients is significantly lower than controls [20]. The Mtm1KO mice also present a decrease in satellite cells [18, 19]. In addition, Mtm1KO muscle cells displayed decreased proliferation, increased apoptosis and poor cell engraftment indicating an inability of satellite cells to “rebuild” damaged skeletal muscle [18] and suggesting cell replacement as a potential therapeutic option. In the Mtm1KI R69C mice, a model of mild XLMTM, injection of syngeneic skeletal muscle-derived WT myoblasts into the gastrocnemius resulted in moderate improvements in skeletal muscle mass and force [160]. This encouraging, albeit limited, positive effect demonstrated the potency of this approach in XLMTM disease.
Furthermore, tremendous progress has been achieved in the isolation of muscle satellite cells and understanding the molecular and cellular mechanisms that control their activity [156, 161–164]. Combination of autologous cell therapy with other approaches like ex vivo genome editing or ex vivo gene replacement may present a safe and efficient way to achieve a successful cell therapy-based approach, while avoiding immunoreaction to exogenous cells in case of allotransplantation. Interestingly, the myostatin inhibition-based approach described above showed a significant increase of satellite cells activation in XLMTM animals [19]. Importantly, myostatin inhibition increased the efficacy of myoblast transplantation in other myopathy models [165], indicating that inhibition of myostatin could potentiate myoblast transfer. Nonetheless, systemic delivery of stem cells to repopulate most affected muscles represents the major limitation of this approach due to the wide distribution required to target this tissue. Overall, this approach has a promising future and encouraging perspectives in XLMTM disease assuming methodological bottlenecks are solved.
CONCLUSIONS
Substantial advances have been achieved in the recent years in our understanding of the molecular pathomechanisms causing CNM, and have allowed the emergence of therapeutic strategies targeting different cellular defects (Fig. 1) and the creation of several companies dedicated to developing these approaches to clinic.
Fig.1
Conceptual figure depicting the different cellular defects reported in X-linked and/or autosomal CNM (in red) and indicating the various therapeutic strategies tested (in green). CNM fibers can be rounder and smaller, especially for the X-linked form. NMJ: neuromuscular junction.
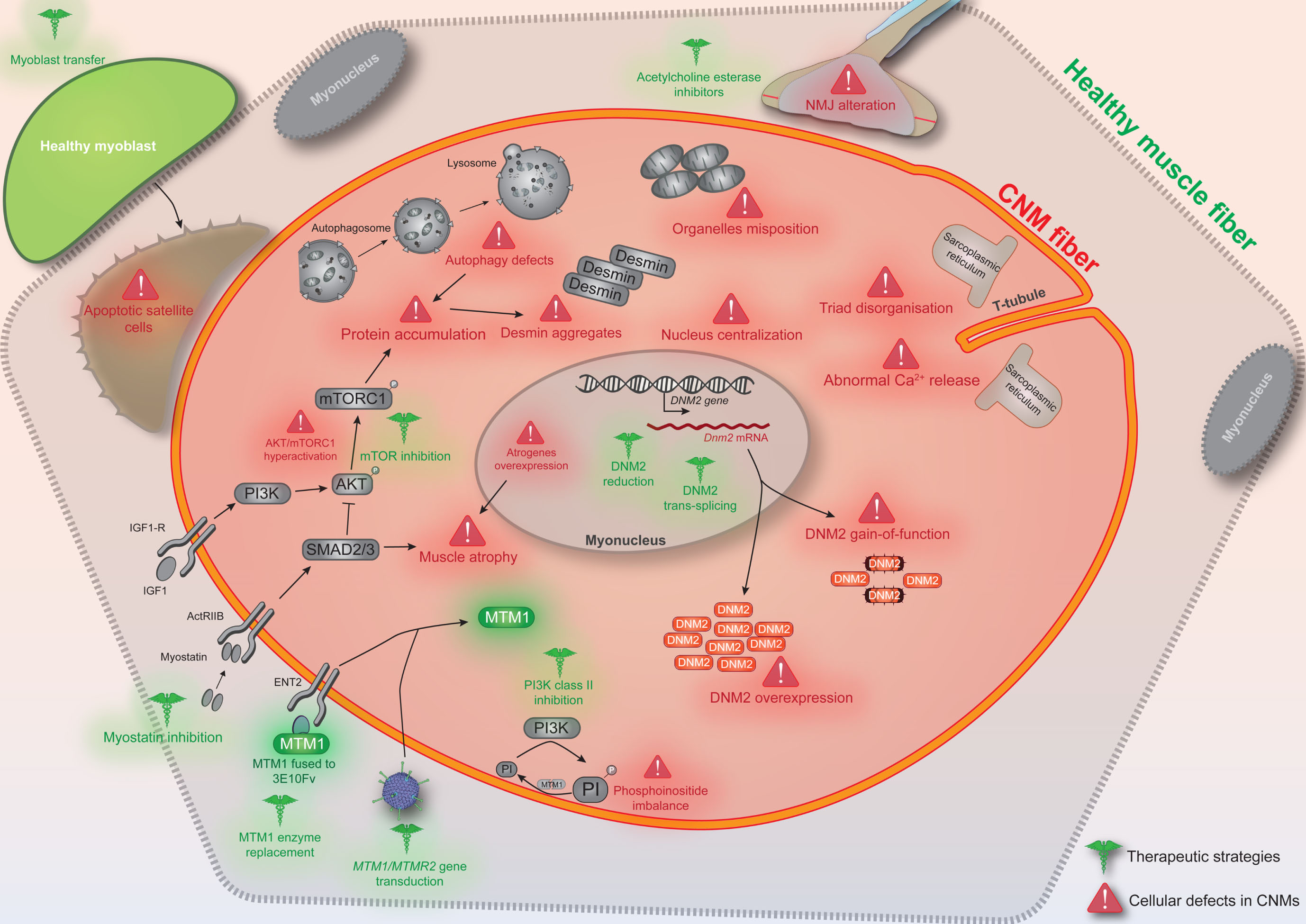
However, while the overlap observed in clinical features, histology and several cellular defects between different CNM forms suggests a common pathway where the different mutated proteins may be involved, the functional interplay between these proteins and their links with the different cellular abnormalities are not well understood. Moreover, a significant number of individuals with CNM-like phenotype remains without genetic diagnosis, suggesting the implication of other genes. Identification of novel CNM genes and pathomechanisms will certainly contribute to the characterization of additional defective cellular pathways that represent new therapeutic targets.
Each of the therapeutic approaches has its own pros and cons (summarized in Table 2). Current bottlenecks for translation of therapeutic concepts achieved in cellular and animal models need to be overcome, such as increasing efficacy, decreasing on-target toxicity and off-target effects, benefit sustainability, tissue specific targeting, and vector/drug production and delivery. A first clinical trial has been started in 2017 for MTM1 gene therapy, underlining that myotubular and centronuclear myopathies are now under attack. The plethora of therapeutic targets recently identified should pave the way to future combinatorial therapies for the benefit of patients.
Table 2
Pros and Cons of the potential therapeutic approaches for centronuclear myopathies
| Approach | Products | Pros | Cons |
|---|---|---|---|
| Gene, RNA and protein replacement | |||
| MTM1 gene delivery | AAV-MTM1; AAV-MTM1 dead phosphatase | Single injection; longterm expression; partial organ-specific delivery | Not fine-tunable; no treatment interruption; immunity to AAV; immunity to MTM1 |
| Myotubularin delivery | 3E10Fv-myotubularin | Fine-tunable; possible treatment interruption | Re-administration needed; immunity to MTM1 or 3E10Fv |
| DNM2 RNA trans-splicing | AAV-5’ pre-trans-splicing nucleotides | Single injection; longterm expression | Requires DNA delivery (naked, liposome, viral-based); potential toxicity of pre-trans-splicing molecules |
| Allele-specific DNM2 RNA silencing | AAV-shRNA | Single injection; allele-specificity decreasing on-target toxicity | Late treatment appeared less efficient for reversion; immunity to AAV |
| Normalizing the disease pathway | |||
| MTMR2 expression | AAV-MTMR2 | Single injection; longterm expression; partial organ-specific delivery | Not fine-tunable; no treatment interruption; immunity to AAV |
| PI3K inhibition | Wortmannin; LY294002; PI-103 | Fine-tunable; possible treatment interruption | Re-administration needed; poor organ specificity; on-target toxicity (lack of specific PIK3C2B inhibitor) |
| DNM2 reduction or normalization | antisense oligonucleotides | Fine-tunable; possible treatment interruption; sequence substitution | Re-administration needed; poor organ specificity; on-target toxicity |
| AAV-shRNA | Single injection; longterm expression | Not fine-tunable; no treatment interruption; immunity to AAV; on-target toxicity | |
| Treating the general muscle unbalance | |||
| Autophagy activation | mTOR inhibitors (RAD001, AZD8055) | Fine-tunable; possible treatment interruption | Re-administration needed; poor organ specificity; on-target toxicity |
| Myostatin inhibition | ActRIIB-mFC | Fine-tunable; possible treatment interruption; in clinical use | Re-administration needed; poor organ specificity; on-target toxicity; good histological but poor functional improvements in XLMTM model |
| Other potential approaches | |||
| Acetylcholinesterase inhibition | pyridostigmine; edrophonium | Fine-tunable; possible treatment interruption; in clinical use for myopathies (FDA approved) | Symptomatic treatment |
| Cell transplantation | syngeneic WT myoblasts | Ex vivo cell manipulation possible | Re-administration needed; systemic delivery difficult; longterm effect unknown |
Notes: Extent of positive outcomes in animal models do not necessarily translate in human. Pros and Cons and mode of delivery may change depending on results in pre-clinical development and methodological updates.
LIST OF ABBREVIATIONS
XLMTM: X-linked myotubular or centronuclear myopathy
CNM: centronuclear myopathy
ADCNM:autosomal dominant centronuclear myopathy
MTM1: myotubularin
BIN1: amphiphysin 2
DNM2: dynamin 2
NMJ: neuromuscular junction
PtdIns3P: phosphoinositide 3-monophosphate
PH-GRAM: pleckstrin homology- glucosyltransferases, Rab-like GTPase activators and myotubularins
ASO: antisense oligonucleotide
CMS: congenital myasthenic syndrome
DECLARATIONS
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Availability of data and material
Not applicable
Competing interests
H.T., J.L. and B.S.C. are inventors of a patent on targeting DNM2 for the treatment of centronuclear myopathies. J.L. and B.S.C. are scientific advisors for Dynacure.
Funding
Not applicable
Authors’ contributions
H.T wrote the manuscript draft with editions by B.S.C and J.L.
ACKNOWLEDGMENTS
H.T. was supported by a MRT fellowship, and B.S.C and J.L. by INSERM.
REFERENCES
[1] | Romero NB. Centronuclear myopathies: A widening concept. Neuromuscul Disord. (2010) ;20: (4):223–8. |
[2] | Spiro AJ , Shy GM , Gonatas NK. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. (1966) ;14: (1):1–14. |
[3] | Laporte J , Hu LJ , Kretz C , Mandel JL , Kioschis P , Coy JF , et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. (1996) ;13: (2):175–82. |
[4] | Raess MA , Friant S , Cowling BS , Laporte J. WANTED - Dead or alive: Myotubularins, a large disease-associated protein family. Adv Biol Regul. (2017) ;63: :49–58. |
[5] | Jungbluth H , Wallgren-Pettersson C , Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. (2008) ;3: :26. |
[6] | Herman GE , Finegold M , Zhao W , de Gouyon B , Metzenberg A. Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr. (1999) ;134: (2):206–14. |
[7] | Amburgey K , Tsuchiya E , de Chastonay S , Glueck M , Alverez R , Nguyen CT , et al. A natural history study of X-linked myotubular myopathy. Neurology. (2017) ;89: (13):1355–64. |
[8] | Laporte J , Kress W , Mandel JL. Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann Neurol. (2001) ;50: (1):42–6. |
[9] | Tosch V , Vasli N , Kretz C , Nicot AS , Gasnier C , Dondaine N , et al. Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul Disord. (2010) ;20: (6):375–81. |
[10] | Tsujita K , Itoh T , Ijuin T , Yamamoto A , Shisheva A , Laporte J , et al. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. (2004) ;279: (14):13817–24. |
[11] | Cao C , Laporte J , Backer JM , Wandinger-Ness A , Stein MP. Myotubularin lipid phosphatase binds the hVPS15/hVPS34 lipid kinase complex on endosomes. Traffic. (2007) ;8: (8):1052–67. |
[12] | Ketel K , Krauss M , Nicot AS , Puchkov D , Wieffer M , Muller R , et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature. (2016) ;529: (7586):408–12. |
[13] | Al-Qusairi L , Weiss N , Toussaint A , Berbey C , Messaddeq N , Kretz C , et al. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U S A. (2009) ;106: (44):18763–8. |
[14] | Dowling JJ , Vreede AP , Low SE , Gibbs EM , Kuwada JY , Bonnemann CG , et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. (2009) ;5: (2):e1000372. |
[15] | Hnia K , Tronchere H , Tomczak KK , Amoasii L , Schultz P , Beggs AH , et al. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J Clin Invest. (2011) ;121: (1):70–85. |
[16] | Robb SA , Sewry CA , Dowling JJ , Feng L , Cullup T , Lillis S , et al. Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul Disord. (2011) ;21: (6):379–86. |
[17] | Dowling JJ , Joubert R , Low SE , Durban AN , Messaddeq N , Li X , et al. Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis Model Mech. (2012) ;5: (6):852–9. |
[18] | Lawlor MW , Alexander MS , Viola MG , Meng H , Joubert R , Gupta V , et al. Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am J Pathol. (2012) ;181: (3):961–8. |
[19] | Lawlor MW , Viola MG , Meng H , Edelstein RV , Liu F , Yan K , et al. Differential muscle hypertrophy is associated with satellite cell numbers and Akt pathway activation following activin type IIB receptor inhibition in Mtm1 R69C mice. Am J Pathol. (2014) ;184: (6):1831–42. |
[20] | Shichiji M , Biancalana V , Fardeau M , Hogrel JY , Osawa M , Laporte J , et al. Extensive morphological and immunohistochemical characterization in myotubular myopathy. Brain Behav. (2013) ;3: (4):476–86. |
[21] | Al-Qusairi L , Prokic I , Amoasii L , Kretz C , Messaddeq N , Mandel JL , et al. Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. FASEB J. (2013) ;27: (8):3384–94. |
[22] | Fetalvero KM , Yu Y , Goetschkes M , Liang G , Valdez RA , Gould T , et al. Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol Cell Biol. (2013) ;33: (1):98–110. |
[23] | Biancalana V , Scheidecker S , Miguet M , Laquerriere A , Romero NB , Stojkovic T , et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: Delineating diagnostic clues. Acta Neuropathol. (2017) ;134: (6):889–904. |
[24] | Bitoun M , Maugenre S , Jeannet PY , Lacene E , Ferrer X , Laforet P , et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. (2005) ;37: (11):1207–9. |
[25] | Bohm J , Biancalana V , Dechene ET , Bitoun M , Pierson CR , Schaefer E , et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat. (2012) ;33: (6):949–59. |
[26] | Bitoun M , Bevilacqua JA , Prudhon B , Maugenre S , Taratuto AL , Monges S , et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. (2007) ;62: (6):666–70. |
[27] | Susman RD , Quijano-Roy S , Yang N , Webster R , Clarke NF , Dowling J , et al. Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul Disord. (2010) ;20: (4):229–37. |
[28] | Warnock DE , Baba T , Schmid SL . Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol Biol Cell. (1997) ;8: (12):2553–62. |
[29] | Di A , Nelson DJ , Bindokas V , Brown ME , Libunao F , Palfrey HC . Dynamin regulates focal exocytosis in phagocytosing macrophages. Mol Biol Cell. (2003) ;14: (5):2016–28. |
[30] | Arneson LN , Segovis CM , Gomez TS , Schoon RA , Dick CJ , Lou Z , et al. Dynamin 2 regulates granule exocytosis during NK cell-mediated cytotoxicity. J Immunol. (2008) ;181: (10):6995–7001. |
[31] | Maier O , Knoblich M , Westermann P . Dynamin II binds to the trans-Golgi network. Biochem Biophys Res Commun. (1996) ;223: (2):229–33. |
[32] | Jones SM , Howell KE , Henley JR , Cao H , McNiven MA . Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science. (1998) ;279: : 573–7. |
[33] | Kreitzer G , Marmorstein A , Okamoto P , Vallee R , Rodriguez-Boulan E . Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat Cell Biol. (2000) ;2: (2):125–7. |
[34] | Gu C , Yaddanapudi S , Weins A , Osborn T , Reiser J , Pollak M , et al. Direct dynamin-actin interactions regulate the actin cytoskeleton. Embo J. (2010) ;29: (21):3593–606. |
[35] | McNiven MA , Kim L , Krueger EW , Orth JD , Cao H , Wong TW . Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol. (2000) ;151: (1):187–98. |
[36] | Thompson HM , Cao H , Chen J , Euteneuer U , McNiven MA . Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nat Cell Biol. (2004) ;6: (4):335–42. |
[37] | Wang L , Barylko B , Byers C , Ross JA , Jameson DM , Albanesi JP . Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J Biol Chem. (2010) ;285: (30):22753–7. |
[38] | Kenniston JA , Lemmon MA . Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. (2010) ;29: (18):3054–67. |
[39] | Cowling BS , Toussaint A , Amoasii L , Koebel P , Ferry A , Davignon L , et al. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am J Pathol. (2011) ;178: (5):2224–35. |
[40] | Liu N , Bezprozvannaya S , Shelton JM , Frisard MI , Hulver MW , McMillan RP , et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J Clin Invest. (2011) ;121: (8):3258–68. |
[41] | Nicot AS , Toussaint A , Tosch V , Kretz C , Wallgren-Pettersson C , Iwarsson E , et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. (2007) ;39: (9):1134–9. |
[42] | Bohm J , Vasli N , Maurer M , Cowling B , Shelton GD , Kress W , et al. Altered Splicing of the BIN1 Muscle-Specific Exon in Humans and Dogs with Highly Progressive Centronuclear Myopathy. PLoS Genet. (2013) ;9: (6):e1003430. |
[43] | Bohm J , Biancalana V , Malfatti E , Dondaine N , Koch C , Vasli N , et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. (2014) ;137: (Pt 12):3160–70. |
[44] | Bohm J , Yis U , Ortac R , Cakmakci H , Kurul SH , Dirik E , et al. Case report of intrafamilial variability in autosomal recessive centronuclear myopathy associated to a novel BIN1 stop mutation. Orphanet J Rare Dis. (2010) ;5: : 35. |
[45] | Claeys KG , Maisonobe T , Bohm J , Laporte J , Hezode M , Romero NB , et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. (2010) ;74: (6):519–21. |
[46] | Lee E , Marcucci M , Daniell L , Pypaert M , Weisz OA , Ochoa GC , et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. (2002) :297: (5584):1193–6. |
[47] | Kojima C , Hashimoto A , Yabuta I , Hirose M , Hashimoto S , Kanaho Y , et al. Regulation of Bin1 SH3 domain binding by phosphoinositides. EMBO J. (2004) ;23: (22):4413–22. |
[48] | Owen DJ , Wigge P , Vallis Y , Moore JD , Evans PR , McMahon HT . Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. Embo J. (1998) ;17: (18):5273–85. |
[49] | Royer B , Hnia K , Gavriilidis C , Tronchere H , Tosch V , Laporte J . The myotubularin-amphiphysin 2 complex in membrane tubulation and centronuclear myopathies. EMBO Re. (2013) ;14: (10):907–15. |
[50] | Fugier C , Klein AF , Hammer C , Vassilopoulos S , Ivarsson Y , Toussaint A , et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. (2011) ;17: (6):720–5. |
[51] | Cowling BS , Toussaint A , Muller J , Laporte J . Defective membrane remodeling in neuromuscular diseases: Insights from animal models. PLoS Genet. (2012) ;8: (4):e1002595. |
[52] | Biancalana V , Beggs AH , Das S , Jungbluth H , Kress W , Nishino I , et al. Clinical utility gene card for: Centronuclear and myotubular myopathies. Eur J Hum Genet. (2012) ) 20: (10). |
[53] | Wilmshurst JM , Lillis S , Zhou H , Pillay K , Henderson H , Kress W , et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. (2010) ;68: (5):717–26. |
[54] | Bevilacqua JA , Monnier N , Bitoun M , Eymard B , Ferreiro A , Monges S , et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol Appl Neurobiol. (2011) ;37: (3):271–84. |
[55] | Ceyhan-Birsoy O , Agrawal PB , Hidalgo C , Schmitz-Abe K , DeChene ET , Swanson LC , et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. (2013) ;81: (14):1205–14. |
[56] | Agrawal PB , Pierson CR , Joshi M , Liu X , Ravenscroft G , Moghadaszadeh B , et al. SPEG Interacts with Myotubularin, and Its Deficiency Causes Centronuclear Myopathy with Dilated Cardiomyopathy. Am J Hum Genet. (2014) ;95: (2):218–26. |
[57] | Schartner V , Romero NB , Donkervoort S , Treves S , Munot P , Pierson TM , et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. (2017) ;133: (4):517–33. |
[58] | Vasli N , Harris E , Karamchandani J , Bareke E , Majewski J , Romero NB , et al. Recessive mutations in the kinase ZAK cause a congenital myopathy with fibre type disproportion. Brain. (2017) ;140: (Pt 1):37–48. |
[59] | Tosch V , Rohde HM , Tronchere H , Zanoteli E , Monroy N , Kretz C , et al. A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet. (2006) ;15: (21):3098–106. |
[60] | Majczenko K , Davidson AE , Camelo-Piragua S , Agrawal PB , Manfready RA , Li X , et al. Dominant mutation of CCDC78 in a unique congenital myopathy with prominent internal nuclei and atypical cores. Am J Hum Genet. (2012) ;91: (2):365–71. |
[61] | Buj-Bello A , Laugel V , Messaddeq N , Zahreddine H , Laporte J , Pellissier JF , et al. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A. (2002) ;99: (23):15060–5. |
[62] | Toussaint A , Cowling BS , Hnia K , Mohr M , Oldfors A , Schwab Y , et al. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. (2011) ;121: (2):253–66. |
[63] | Gavriilidis C , Laredj L , Solinhac R , Messaddeq N , Viaud J , Laporte J , et al. The MTM1-UBQLN2-HSPs complex mediate degradation of misfolded intermediate filaments in skeletal muscle. Nature Cell Biol. (2018) ;20: (2):198–210. |
[64] | Pierson CR , Dulin-Smith AN , Durban AN , Marshall ML , Marshall JT , Snyder AD , et al. Modeling the human MTM1 R69C mutation in murine Mtm1 results in exon 4 skipping and a less severe myotubular myopathy phenotype. Hum Mol Genet. (2012) ;21: (4):811–25. |
[65] | Sabha N , Volpatti JR , Gonorazky H , Reifler A , Davidson AE , Li X , et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J Clin Invest. (2016) ;126: (9):3613–25. |
[66] | Beggs AH , Bohm J , Snead E , Kozlowski M , Maurer M , Minor K , et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc Natl Acad Sci U S A. (2010) ;107: (33):14697–702. |
[67] | Shelton GD , Rider BE , Child G , Tzannes S , Guo LT , Moghadaszadeh B , et al. X-linked myotubular myopathy in Rottweiler dogs is caused by a missense mutation in Exon 11 of the MTM1 gene. Skelet Muscle. (2015) ;5: (1):1. |
[68] | Durieux AC , Vignaud A , Prudhon B , Viou MT , Beuvin M , Vassilopoulos S , et al. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet. (2010) ;19: (24):4820–36. |
[69] | Durieux AC , Vassilopoulos S , Laine J , Fraysse B , Brinas L , Prudhon B , et al. A centronuclear myopathy–dynamin 2 mutation impairs autophagy in mice. Traffic. (2012) ;13: (6):869–79. |
[70] | Fraysse B , Guicheney P , Bitoun M . Calcium homeostasis alterations in a mouse model of the Dynamin 2-related centronuclear myopathy. Biol Open. (2016) ;5: (11):1691–6. |
[71] | Kutchukian C , Szentesi P , Allard B , Trochet D , Beuvin M , Berthier C , et al. Impaired excitation-contraction coupling in muscle fibres from the dynamin2(R465W) mouse model of centronuclear myopathy. J Physiol. (2017) ;595: (24):7369–82. |
[72] | Gibbs EM , Davidson AE , Telfer WR , Feldman EL , Dowling JJ . The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis Model Mech. (2014) ;7: (1):157–61. |
[73] | Buj-Bello A , Fougerousse F , Schwab Y , Messaddeq N , Spehner D , Pierson CR , et al. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum Mol Genet. (2008) ;17: (14):2132–43. |
[74] | Aiuti A , Cattaneo F , Galimberti S , Benninghoff U , Cassani B , Callegaro L , et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. (2009) ;360: (5):447–58. |
[75] | Bainbridge JW , Smith AJ , Barker SS , Robbie S , Henderson R , Balaggan K , et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. (2008) ;358: (21):2231–9. |
[76] | Nathwani AC , Tuddenham EG , Rangarajan S , Rosales C , McIntosh J , Linch DC , et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. (2011) ;365: (25):2357–65. |
[77] | Marks WJ, Jr , Ostrem JL , Verhagen L , Starr PA , Larson PS , Bakay RA , et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol. (2008) ;7: (5):400–8. |
[78] | Worgall S , Sondhi D , Hackett NR , Kosofsky B , Kekatpure MV , Neyzi N , et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. (2008) ;19: (5):463–74. |
[79] | Carpentier AC , Frisch F , Labbe SM , Gagnon R , de Wal J , Greentree S , et al. Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab. (2012) ;97: (5):1635–44. |
[80] | Gaudet D , Methot J , Dery S , Brisson D , Essiembre C , Tremblay G , et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: An open-label trial. Gene Ther. (2013) ;20: (4):361–9. |
[81] | Childers MK , Joubert R , Poulard K , Moal C , Grange RW , Doering JA , et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med. (2014) ;6: (220):220ra10. |
[82] | Elverman M , Goddard MA , Mack D , Snyder JM , Lawlor MW , Meng H , et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve. (2017) ;56: (5):943–53. |
[83] | Mack DL , Poulard K , Goddard MA , Latournerie V , Snyder JM , Grange RW , et al. Systemic AAV8-Mediated Gene Therapy Drives Whole-Body Correction of Myotubular Myopathy in Dogs. Mol Ther. (2017) ;25: (4):839–54. |
[84] | Kotin RM , Snyder RO . Manufacturing Clinical Grade Recombinant Adeno-Associated Virus Using Invertebrate Cell Lines. Hum Gene Ther. (2017) ;28: (4):350–60. |
[85] | Lawlor MW , Armstrong D , Viola MG , Widrick JJ , Meng H , Grange RW , et al. Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum Mol Genet. (2013) ;22: (8):1525–38. |
[86] | Crawford CR , Patel DH , Naeve C , Belt JA . Cloning of the human equilibrative, nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. J Biol Chem. (1998) ;273: (9):5288–93. |
[87] | Eng CM , Guffon N , Wilcox WR , Germain DP , Lee P , Waldek S , et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. (2001) ;345: (1):9–16. |
[88] | Brady RO , Pentchev PG , Gal AE , Hibbert SR , Dekaban AS . Replacement therapy for inherited enzyme deficiency Use of purified glucocerebrosidase in Gaucher’s disease. N Engl J Med. (1974) ;291: (19):989–93. |
[89] | Van den Hout H , Reuser AJ , Vulto AG , Loonen MC , Cromme-Dijkhuis A , Van der Ploeg AT . Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. (2000) ;356: (9227):397–8. |
[90] | Van den Hout JM , Kamphoven JH , Winkel LP , Arts WF , De Klerk JB , Loonen MC , et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. (2004) ;113: (5):e448–57. |
[91] | Trochet D , Prudhon B , Jollet A , Lorain S , Bitoun M . Reprogramming the Dynamin 2 mRNA by Spliceosome-mediated RNA Trans-splicing. Mol Ther Nucleic Acids. (2016) ;5: (9):e362. |
[92] | Berger A , Maire S , Gaillard MC , Sahel JA , Hantraye P , Bemelmans AP . mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip Rev RNA. (2016) ;7: (4):487–98. |
[93] | Philippi S , Lorain S , Beley C , Peccate C , Precigout G , Spuler S , et al. Dysferlin rescue by spliceosome-mediated pre-mRNA trans-splicing targeting introns harbouring weakly defined 3’ splice sites. Hum Mol Genet. (2015) ;24: (14):4049–60. |
[94] | Monjaret F , Bourg N , Suel L , Roudaut C , Le Roy F , Richard I , et al. Cis-splicing and translation of the pre-trans-splicing molecule combine with efficiency in spliceosome-mediated RNA trans-splicing. Mol Ther. (2014) ;22: (6):1176–87. |
[95] | Mansfield SG , Kole J , Puttaraju M , Yang CC , Garcia-Blanco MA , Cohn JA , et al. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Ther. (2000) ;7: (22):1885–95. |
[96] | Rodriguez-Martin T , Anthony K , Garcia-Blanco MA , Mansfield SG , Anderton BH , Gallo JM . Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum Mol Genet. (2009) ;18: (17):3266–73. |
[97] | Wally V , Murauer EM , Bauer JW . Spliceosome-mediated trans-splicing: The therapeutic cut and paste. J Invest Dermatol. (2012) ;132: (8):1959–66. |
[98] | Trochet D , Prudhon B , Beuvin M , Peccate C , Lorain S , Julien L , et al. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. EMBO Mol Med.2017. |
[99] | Laporte J , Bedez F , Bolino A , Mandel JL . Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Hum Mol Genet. (2003) ; 12: Spec No 2: R285–92. |
[100] | Laporte J , Liaubet L , Blondeau F , Tronchere H , Mandel JL , Payrastre B . Functional redundancy in the myotubularin family. Biochem Biophys Res Commun. (2002) ;291: (2):305–12. |
[101] | Nandurkar HH , Caldwell KK , Whisstock JC , Layton MJ , Gaudet EA , Norris FA , et al. Characterization of an adapter subunit to a phosphatidylinositol (3)P 3- phosphatase: Identification of a myotubularin-related protein lacking catalytic activity. Proc Natl Acad Sci U S A. (2001) ;98: (17):9499–504. |
[102] | Berger P , Bonneick S , Willi S , Wymann M , Suter U . Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot-Marie-Tooth disease type 4B1. Hum Mol Genet. (2002) ;11: (13):1569–79. |
[103] | Bolino A , Muglia M , Conforti FL , LeGuern E , Salih MA , Georgiou DM , et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. (2000) ;25: (1):17–9. |
[104] | Raess MA , Cowling BS , Bertazzi DL , Kretz C , Rinaldi B , Xuereb JM , et al. Expression of the neuropathy-associated MTMR2 gene rescues MTM1-associated myopathy. Hum Mol Genet. (2017) ;26: (19):3736–48. |
[105] | Bolino A , Marigo V , Ferrera F , Loader J , Romio L , Leoni A , et al. Molecular characterization and expression analysis of Mtmr2, mouse homologue of MTMR2, the Myotubularin-related 2 gene, mutated in CMT4B. Gene. (2002) ;283: (1-2):17–26. |
[106] | Daniele N , Moal C , Julien L , Marinello M , Jamet T , Martin S , et al. Intravenous Administration of a MTMR2-Encoding AAV Vector Ameliorates the Phenotype of Myotubular Myopathy in Mice. J Neuropathol Exp Neurol. (2018) ;77: (4):282–95. |
[107] | Blondeau F , Laporte J , Bodin S , Superti-Furga G , Payrastre B , Mandel JL . Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. (2000) ;9: (15):2223–9. |
[108] | Taylor GS , Maehama T , Dixon JE . Inaugural article: Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. (2000) ;97: (16):8910–5. |
[109] | Tronchere H , Laporte J , Pendaries C , Chaussade C , Liaubet L , Pirola L , et al. Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J Biol Chem. (2004) ;279: (8):7304–12. |
[110] | Amoasii L , Hnia K , Chicanne G , Brech A , Cowling BS , Muller MM , et al. Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo. J Cell Sci. (2013) ;126: (Pt 8):1806–19. |
[111] | Di Paolo G , De Camilli P . Phosphoinositides in cell regulation and membrane dynamics. Nature. (2006) ;443: (7112):651–7. |
[112] | Krauss M , Haucke V . Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep. (2007) ;8: (3):241–6. |
[113] | Velichkova M , Juan J , Kadandale P , Jean S , Ribeiro I , Raman V , et al. Drosophila Mtm and class II PI3K coregulate a PI(3)P pool with cortical and endolysosomal functions. J Cell Biol. (2010) ;190: (3):407–25. |
[114] | Reifler A , Li X , Archambeau AJ , McDade JR , Sabha N , Michele DE , et al. Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am J Pathol. (2014) ;184: (6):1819–30. |
[115] | Kutchukian C , Lo Scrudato M , Tourneur Y , Poulard K , Vignaud A , Berthier C , et al. Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice. Proc Natl Acad Sci U S A. (2016) ;113: (50):14432–7. |
[116] | Amoasii L , Bertazzi DL , Tronchere H , Hnia K , Chicanne G , Rinaldi B , et al. Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS Genet. (2012) ;8: (10):e1002965. |
[117] | Vanhaesebroeck B , Guillermet-Guibert J , Graupera M , Bilanges B . The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. (2010) ;11: (5):329–41. |
[118] | Cowling BS , Chevremont T , Prokic I , Kretz C , Ferry A , Coirault C , et al. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest. (2014) ;124: (3):1350–63. |
[119] | Tasfaout H , Buono S , Guo S , Kretz C , Messaddeq N , Booten S , et al. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat Commun. (2017) ;8: : 15661. |
[120] | Stein CA , Castanotto D . FDA-Approved Oligonucleotide Therapies in Mol Ther (2017) ;25: (5):1069–75. |
[121] | Swayze EE , Siwkowski AM , Wancewicz EV , Migawa MT , Wyrzykiewicz TK , Hung G , et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. (2007) ;35: (2):687–700. |
[122] | Burdick AD , Sciabola S , Mantena SR , Hollingshead BD , Stanton R , Warneke JA , et al. Sequence motifs associated with hepatotoxicity of locked nucleic acid–modified antisense oligonucleotides. Nucleic Acids Res. (2014) ;42: (8):4882–91. |
[123] | van Poelgeest EP , Swart RM , Betjes MG , Moerland M , Weening JJ , Tessier Y , et al. Acute kidney injury during therapy with an antisense oligonucleotide directed against PCSK9. Am J Kidney Dis. (2013) ;62: (4):796–800. |
[124] | Herrington WG , Talbot DC , Lahn MM , Brandt JT , Callies S , Nagle R , et al. Association of long-term administration of the survivin mRNA-targeted antisense oligonucleotide LY308 with reversible kidney injury in a patient with metastatic melanoma. Am J Kidney Dis. (2011) ;57: (2):300–3. |
[125] | Cowling BS , Prokic I , Tasfaout H , Rabai A , Humbert F , Rinaldi B , et al. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J Clin Invest. (2017) . |
[126] | Pierson CR , Agrawal PB , Blasko J , Beggs AH . Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul Disord. (2007) ;17: (7):562–8. |
[127] | Schiaffino S , Dyar KA , Ciciliot S , Blaauw B , Sandri M . Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. (2013) ;280: (17):4294–314. |
[128] | Lilienbaum A . Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol. (2013) ;4: (1):1–26. |
[129] | Xie J , Wang X , Proud CG . mTOR inhibitors in cancer therapy. FRes. (2016) ;5. |
[130] | McPherron AC , Lawler AM , Lee SJ . Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. (1997) ;387: (6628):83–90. |
[131] | Clop A , Marcq F , Takeda H , Pirottin D , Tordoir X , Bibe B , et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects mus1524 cularity in sheep. Nat Genet. (2006) ;38: (7):813–8. |
[132] | McPherron AC , Lee SJ . Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. (1997) ;94: (23):12457–61. |
[133] | Mosher DS , Quignon P , Bustamante CD , Sutter NB , Mellersh CS , Parker HG , et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. (2007) ;3: (5):e79. |
[134] | Schuelke M , Wagner KR , Stolz LE , Hubner C , Riebel T , Komen W , et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. (2004) ;350: (26):2682–8. |
[135] | Haidet AM , Rizo L , Handy C , Umapathi P , Eagle A , Shilling C , et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A. (2008) ;105: (11):4318–22. |
[136] | Bogdanovich S , Krag TO , Barton ER , Morris LD , Whittemore LA , Ahima RS , et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. (2002) ;420: (6914):418–21. |
[137] | Holzbaur EL , Howland DS , Weber N , Wallace K , She Y , Kwak S , et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis. (2006) ;23: (3):697–707. |
[138] | Siriett V , Salerno MS , Berry C , Nicholas G , Bower R , Kambadur R , et al. Antagonism of myostatin enhances muscle regeneration during sarcopenia. Mol Ther. (2007) ;15: (8):1463–70. |
[139] | Benny Klimek ME , Aydogdu T , Link MJ , Pons M , Koniaris LG , Zimmers TA . Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun. (2010) ;391: (3):1548–54. |
[140] | Zhou X , Wang JL , Lu J , Song Y , Kwak KS , Jiao Q , et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. (2010) ;142: (4):531–43. |
[141] | Thies RS , Chen T , Davies MV , Tomkinson KN , Pearson AA , Shakey QA , et al. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. (2001) ;18: (4):251–9. |
[142] | Hill JJ , Davies MV , Pearson AA , Wang JH , Hewick RM , Wolfman NM , et al. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. (2002) ;277: (43):40735–41. |
[143] | Cadena SM , Tomkinson KN , Monnell TE , Spaits MS , Kumar R , Underwood KW , et al. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. (2010) ;109: (3):635–42. |
[144] | Morrison BM , Lachey JL , Warsing LC , Ting BL , Pullen AE , Underwood KW , et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. (2009) ;217: (2):258–68. |
[145] | Lawlor MW , Read BP , Edelstein R , Yang N , Pierson CR , Stein MJ , et al. Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am J Pathol. (2011) ;178: (2):784–93. |
[146] | Mariot V , Joubert R , Hourde C , Feasson L , Hanna M , Muntoni F , et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun. (2017) ;8: (1):1859. |
[147] | Rodgers BD , Interlichia JP , Garikipati DK , Mamidi R , Chandra M , Nelson OL , et al. Myostatin represses physiological hypertrophy of the heart and excitation-contraction coupling. J Physiol. (2009) ;587: (Pt 20):4873–86. |
[148] | Mercier L , Bohm J , Fekonja N , Allio G , Lutz Y , Koch M , et al. In vivo imaging of skeletal muscle in mice highlights muscle defects in a model of myotubular myopathy. Intravital. (2016) ;5: (1):e1168553. |
[149] | Ambler MW , Neave C , Singer DB . X-linked recessive myotubular myopathy: II Muscle morphology and human myogenesis. Hum Pathol. (1984) ;15: : 1107–20. |
[150] | Fidzianska A , Goebel HH . Aberrant arrested in maturation neuromuscular junctions in centronuclear myopathy. J Neurol Sci. (1994) ;124: (1):83–8. |
[151] | Gibbs EM , Clarke NF , Rose K , Oates EC , Webster R , Feldman EL , et al. Neuromuscular junction abnormalities in DNM2-related centronuclear myopathy. J Mol Med (Berl). (2013) ;91: (6):727–37. |
[152] | Trounson A , McDonald C . Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell. (2015) ;17: (1):11–22. |
[153] | Copelan EA . Hematopoietic stem-cell transplantation. N Engl J Med. (2006) ;354: (17):1813–26. |
[154] | Passweg JR , Halter J , Bucher C , Gerull S , Heim D , Rovo A , et al. Hematopoietic stem cell transplantation: A review and recommendations for follow-up care for the general practitioner. Swiss Med Wkly. (2012) ;142: : w13696. |
[155] | Skuk D , Tremblay JP . Clarifying misconceptions about myoblast transplantation in myology. Mol Ther. (2014) ;22: (5):897–8. |
[156] | Cerletti M , Jurga S , Witczak CA , Hirshman MF , Shadrach JL , Goodyear LJ , et al. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell. (2008) ;134: (1):37–47. |
[157] | Gussoni E , Blau HM , Kunkel LM . The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat Med. (1997) ;3: (9):970–7. |
[158] | Mendell JR , Kissel JT , Amato AA , King W , Signore L , Prior TW , et al. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med. (1995) ;333: (13):832–8. |
[159] | Tremblay JP , Malouin F , Roy R , Huard J , Bouchard JP , Satoh A , et al. Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplant. (1993) ;2: (2):99–112. |
[160] | Lim HJ , Joo S , Oh SH , Jackson JD , Eckman DM , Bledsoe TM , et al. Syngeneic Myoblast Transplantation Improves Muscle Function in a Murine Model of X-Linked Myotubular Myopathy. Cell Transplant. (2015) ;24: (9):1887–900. |
[161] | Alessandri G , Pagano S , Bez A , Benetti A , Pozzi S , Iannolo G , et al. Isolation and culture of human muscle-derived stem cells able to differentiate into myogenic and neurogenic cell lineages. Lancet. (2004) ;364: (9448):1872–83. |
[162] | Conboy IM , Rando TA . The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. (2002) ;3: (3):397–409. |
[163] | Conboy IM , Conboy MJ , Smythe GM , Rando TA . Notch-mediated restoration of regenerative potential to aged muscle. Science. (2003) ;302: (5650):1575–7. |
[164] | Sherwood RI , Christensen JL , Conboy IM , Conboy MJ , Rando TA , Weissman IL , et al. Isolation of adult mouse myogenic progenitors: Functional heterogeneity of cells within and engrafting skeletal muscle. Cell. (2004) ;119: (4):543–54. |
[165] | Fakhfakh R , Lee SJ , Tremblay JP . Administration of a soluble activin type IIB receptor promotes the transplantation of human myoblasts in dystrophic mice. Cell Transplant. (2012) ;21: (7):1419–30. |


