
Current Classification and Management of Inflammatory Myopathies
Abstract
Inflammatory disorders of the skeletal muscle include polymyositis (PM), dermatomyositis (DM), (immune mediated) necrotizing myopathy (NM), overlap syndrome with myositis (overlap myositis, OM) including anti-synthetase syndrome (ASS), and inclusion body myositis (IBM). Whereas DM occurs in children and adults, all other forms of myositis mostly develop in middle aged individuals. Apart from a slowly progressive, chronic disease course in IBM, patients with myositis typically present with a subacute onset of weakness of arms and legs, often associated with pain and clearly elevated creatine kinase in the serum. PM, DM and most patients with NM and OM usually respond to immunosuppressive therapy, whereas IBM is largely refractory to treatment. The diagnosis of myositis requires careful and combinatorial assessment of (1) clinical symptoms including pattern of weakness and paraclinical tests such as MRI of the muscle and electromyography (EMG), (2) broad analysis of auto-antibodies associated with myositis, and (3) detailed histopathological work-up of a skeletal muscle biopsy. This review provides a comprehensive overview of the current classification, diagnostic pathway, treatment regimen and pathomechanistic understanding of myositis.
INTRODUCTION
Inflammatory myopathies (synonym: idiopathic inflammatory myopathy, IIM) – in short: myositis–are rare conditions that can affect multiple organs apart from muscle and often lead to a severe impairment of the quality of life [1, 2]. Diagnosis and treatment are often a challenge since several subspecialities are required for optimal care, including rheumatologist and/or neurologist, dermatologist, pulmonologist, cardiologist, physiotherapist etc. Except for classical dermatomyositis (DM), the diagnosis is mostly not straightforward and usually requires testing of auto-antibodies, histological evaluation of a skeletal muscle biopsy and further tests including muscle MRI and EMG. Novel diagnostic criteria have recently been established, but an update will be required (see below for details).
Major symptoms of myositis include an acute (within several days) or subacute (within several weeks) onset of weakness of arms and legs (see Table 1 for an overview of all symptoms in different subtypes of myositis). Typical complaints include problems in walking and climbing stairs or lifting an object above the head. Pain is often an accompanying symptom and, except for inclusion body myositis (IBM) cases, general laboratory assessment usually shows a profound upregulation of the creatine kinase (CK) by 10–50 fold and elevated liver enzymes as an indicator of muscle cell damage. As detailed below, it is of utmost importance to (1) make a reliable and quick diagnosis by combining clinical, antibody and biopsy results and (2) timely install appropriate treatment (Fig. 1 and see below for details). After an overview of the epidemiology of myositis, the following paragraphs will discuss each of the myositis forms including their auto-antibody findings, histopathology of the muscle and association with malignancies. The extramuscular organ manifestations, diagnostic criteria and their approach, pathophysiology and the current treatment modalities will be presented in separate paragraphs.
Table 1
Overview of the clinical presentation, auto-antibodies, muscle pathology and treatment in the main subtypes of myositis
| DM, JDM | NM | PM | OM, ASS | IBM | |
| Onset and disease course | Acute/ subacute onset; Short, benign or severe, chronic courses. | Acute/ subacute onset; Chronic, slow progression possible. | Acute/ subacute onset; Variable course. | Acute/ subacute onset; Mostly chronic course. | Slowly progressive; Always chronic. |
| Weakness, extramuscular symptoms | Amyopathic/ proximal tetraparesis±dysphagia; Specific skin- and organ manifestation; Malignancy in adults. | Proximal tetraparesis; Rarely extramuscular manifestation: heart, lung; Malignancy. | Proximal tetraparesis±dysphagia. No extramuscular manifestation. | Proximal tetraparesis; ASS: ILD, mechanic’s hands, arthritis, Raynaud’s syndrome. Other OM: Scleroderma, SLE | Long finger flexors, knee extensors, dysphagia. |
| CK level | Normal or ∼10–50 fold elevated | ∼10–50 fold elevated | ∼10–50 fold elevated | ∼10–50 fold elevated | Normal to 15 fold elevated |
| Auto-antibodies | Mi-2, MDA5 (ILD!), TIF-1γ (malignancy!), NXP2 (malignancy!), SAE | SRP, HMGCR (malignancy!) | Unspecific | ASS: Jo-1, PL-7, PL-12, HA, EJ, KS, Zo, OJ. Other OM: Ku, Ro/SS-A, SS-B, PM/Scl, U-snRNP | cN1A |
| Muscle pathology | Perimysial inflammation, perifascicular atrophy, MHC class I, complement on capillaries and/or sarcolemma, capillary loss. | Scattered necrosis; MHC class I, complement on capillaries and/or sarcolemma. | Endomysial CD8 + T cells | Perifascicular necrosis, MHC class I and II, complement on sarcolemma | Endomysial CD8 + T cells, MHC class I, amyloid, vacuoles, tubulofilaments, mitochondrial impairment (COX, paracr. inclusions) |
| Treatment and its response | Basic: GS, AZA/MTX/MMF; Skin &JDM: IVIG; Lung/ Escal.: RTX, CYC, IVIG, (CsA); Mostly good response except for malignancy or ILD. | Basic: GS, AZA/MTX/MMF; Lung/ Escal.: RTX, CYC, IVIG; Overall response good-moderate, but escalation often required. | Basic: GS, AZA/MTX/MMF; Escal.: RTX, CYC, IVIG; Mostly good response. | Basic: GS, AZA/MTX/MMF; Lung/ Escal.: RTX, CYC, IVIG, (CsA); Mostly good response except for malignancy or ILD. | No basic immuno-suppression; Probatory IVIG in selected patients justifiable; Severe dysphagia: local botulinum toxin or myotomy, percutaneous feeding tube. Usually refractory to treatment. |
EPIDEMIOLOGY
All forms of myositis are considered rare diseases: DM has a prevalence of ∼1–6 patients per 100,000 persons in the USA [3]. Overlap myositis (OM; synonym: overlap syndrome with myositis) presumably accounts for the largest group of the myositis forms with up to half of the cases, followed by DM with over one third of the cases [4, 5]. In a very recent large analysis of 3067 patients from the Euromyositis registry, DM was the most common disorder with 31% [6]. Necrotizing myopathy (NM, also termed immune mediated NM, IMNM) is thought to be the second largest group with ∼one fifth of the cases [4, 5]. The epidemiology of polymyositis (PM) is controversial, ranging from the largest fraction with ∼10 per 100,000 persons in the USA [3], 27% in the Euromyositis group [6] down to the rarest condition that should only be diagnosed per exclusion [5]. IBM is supposed to occur at a prevalence of up to 14 per million [7]. Precise epidemiological data are difficult to generate and previous publications should be considered with care since the diagnostic criteria have changed significantly during the last decades (see details below). Collectively, it is currently believed that OM, NM and DM make up 90% of the myositis cases [4]. In general, females are affected more often by myositis and a juvenile form of DM (JDM) is noted in children and adolescents.
CLINICAL PRESENTATION, AUTO-ANTIBODIES AND MUSCLE HISTOPATHOLOGY
Dermatomyositis (DM)
Patients with DM present with signs of inflammation of the skin such as a Gottron papules on the dorsal sides of the hands and fingers, a periorbital oedema, and erythema of the face (heliotrope rash), the anterior upper chest (V-sign) or the posterior neck (shawl sign). Periungal erythema and telangiectasia as well as cracked, thickened skin of the ventral and dorsal parts of the fingers and hands occur (“mechanic’s hands”), whereas the latter is also a typical feature of the anti-synthetase syndrome (ASS, see below) (Table 1). The muscle inflammation causes proximal weakness which can develop acutely (within several days) or subacutely (within several weeks up to a few months). The patients suffer from impaired walking and climbing stairs as well as lifting their arms and heavy objects. Pain can be present and laboratory workup usually displays a significant upregulation of muscle enzymes such as serum creatine kinase (CK) with 10–50 fold elevation.
Several variants of classical DM exist such as the amyopathic DM (ADM; synonym: “clinically amyopathic DM”, CADM) in appr. 20% of the cases, in which only skin manifestations are present but no weakness of the muscles and no elevation of the serum CK. However, patients with ADM often display an affection of the lungs in form of an interstitial lung disease (ILD), which indicates a severe phenotype and requires more aggressive treatment [8] (see below). Adermatopathic DM (“dermatomyositis sine dermatitis”) is an opposite syndrome, in which the weakness and histological signs are noted similar to DM but without inflammatory lesions of the skin [9]. However, it is much more likely that most of these patients are misclassified and in fact represent cases of anti-synthetase syndrome (ASS) or other forms of OM (see below). JDM affects children and usually presents with fever and skin rash. Calcinosis of the skin is common in JDM and swallowing abnormalities can occur in all severe forms of DM. Further details of extramuscular organ involvements are discussed below.
The most common and longest known auto-antibody associated with the classical form of DM is the Mi-2 antibody, which is found in up to 20% of the patients with DM [10] (see Table 2 for an overview of auto-antibodies in myositis). MDA5 antibodies (previous term: anti-clinically amyopathic dermatomyositis, CADM-140) were first described in 2009 [11] and are present in ∼10–30% of the DM patients, especially in cases with vasculitic skin lesions and a severe ILD with an increased mortality [12]. A common antibody in DM with a frequency of about one third of the cases is the anti-TIF-1α/β/γ antibody, which was first described in 2006/2007 [13, 14]. TIF-1 antibodies are strongly associated with malignancies in up to 75% of adult patients; in children, they present one of the most common antibodies and are associated with JDM [13], but not with tumors in this age group [15]. Another antibody with a strong tumor association is the NXP2 antibody (initially termed MJ antibody) [16] with a reported tumor rate of up to 37.5% (overview in [12]). NXP2 is also the second most common auto-antibody in children with a frequency of up to 22% [15] and can have a high degree of calcinosis [16]. A more recently identified antibody that is associated with ∼8% of the DM cases is SAE [17]. These patients often display an amyopathic disease course and present with dysphagia and a mild to moderate ILD [12].
Fig.1
Overview of the main items required for appropriate care for myositis.
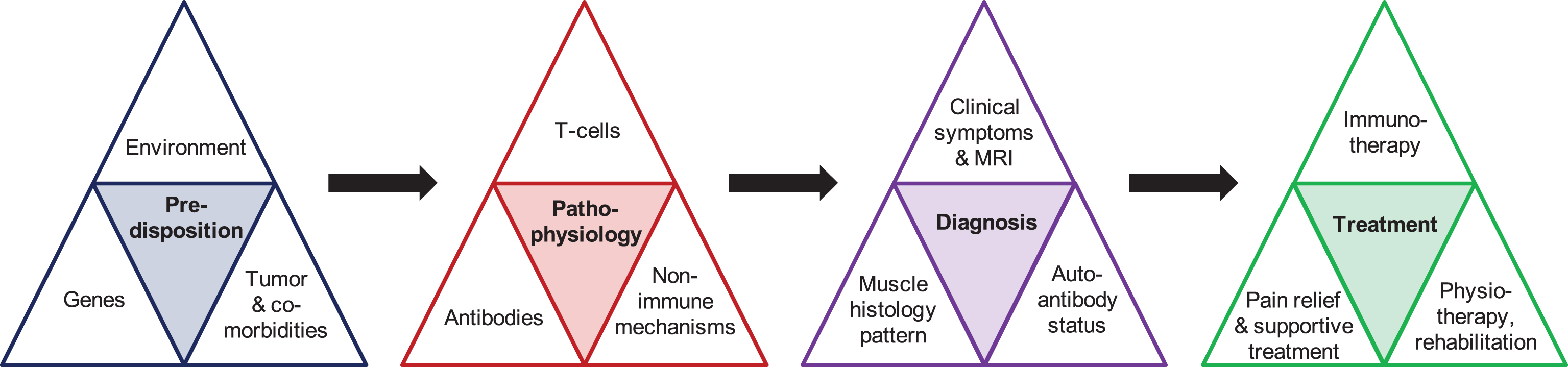
Table 2
Overview of auto-antibodies and their associated clinical features
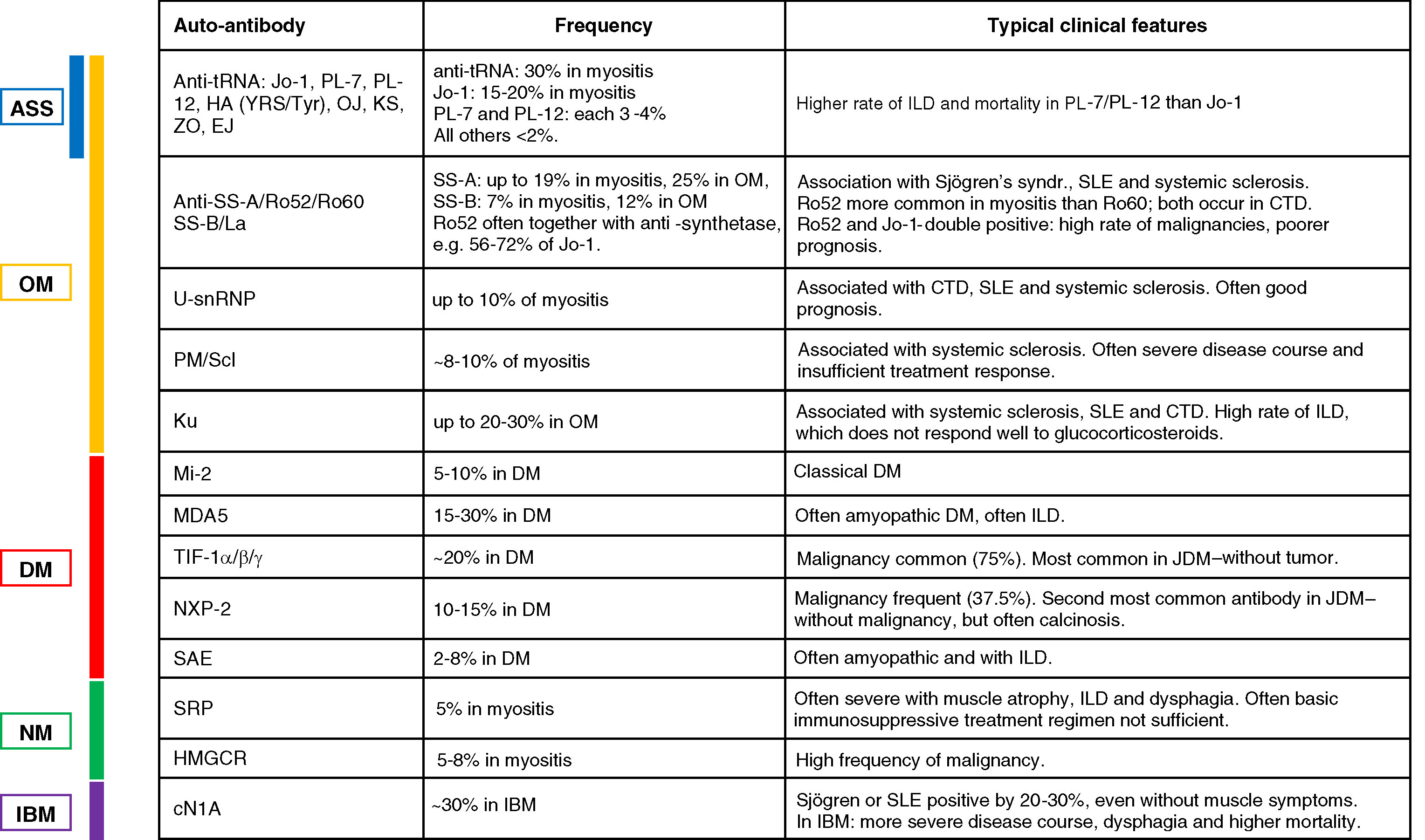 |
Histopathologic signs of DM consist of a perimysial inflammation, perifascicular atrophy and perifascicular elevation of MHC class 1, binding of complement to capillaries and the surface of the sarcolemma and reduction of capillaries [2, 18]. The typical pathological feature of perifascicular atrophy is more common in JDM than in DM. A recent report demonstrates that immunohistochemical staining for MxA protein is a more sensitive marker for DM than other classical pathological features including perifascicular atrophy [19], suggesting that this is a useful marker for histologic workup of suspected myositis.
Polymyositis (PM)
Polymyositis (PM) is by far the rarest form of myositis with an estimated portion of ∼5% of all cases [2, 4, 20] and some experts even question its existence [5, 21, 22]. A rash or other signs of inflammation of the skin do not occur in PM. The diagnosis of PM should be made by exclusion of all other types of myositis, and, in chronic courses of presumed PM, a muscular dystrophy should also be ruled out (see the diagnostic criteria section below).
Although several databases, reports and registry entries contain “PM cases” with the detection of one of the myositis antibodies depicted in Table 1, it is now generally well accepted that none of these is truly associated with the disease [4, 22, 23]. Rather, patients without a rash or other pathologic signs of the skin and the distribution of muscle weakness as in “PM”, with or without signs of an overlap syndrome (see below), could be re-classified either as adermatomyopathic DM or overlap myositis (OM, see below).
Histopathologic hallmarks of PM include an “invasion” of muscle fibers by presence of endomysial cytotoxic CD8 + T-cells and widespread upregulation of MHC class I [2, 18]. Importantly, invasion of muscle fibers by cytotoxic CD8 + T-cells is not unique for PM, but even more common in IBM [24–26] and potentially present in cases with DM or ASS [27] (see also below). Relevant signs of protein accumulation as in inclusion body myositis (see below) or increased connective tissue as in muscular dystrophy should be absent.
Necrotizing myopathy (NM)
Immune mediated necrotizing myopathy (IMNM; in short: necrotizing myopathy, NM; synonym: necrotizing autoimmune myopathy, NAM) leads to an acute or subacute proximal weakness of arms and legs. The disease course is often more rapid and more severe compared to DM and PM. The muscle enzymes are usually very high with a 20–50 fold elevated CK. Swallowing difficulties can occur and further organ manifestation is summarized below.
Two auto-antibodies have been shown to be associated with NM. Anti-SRP antibodies are expected in about 10 to 20% of patients with NM, although the rate of detection ranges largely (from 0% to 54%, summarized in [28]). Anti-SRP can be associated with a cardiomyopathy [29] and a severe disease course with muscular atrophy, interstitial lung disease (ILD) and dysphagia [30, 31]. The second auto-antibody that was identified in up to 60% of certain NM cohorts is anti-HMGCR [32]. A varying degree of 30–60% of these patients had prior exposure to statins [33, 34]. An association with malignancy has been shown to be higher in patients with anti-HMGCR and without an auto-antibody compared to patients with anti-SRP [35].
The histological picture in NM displays scattered necrotic myofibers of varying degree, moderate and mostly focal upregulation of MHC class I, particularly in areas with necrotic fibers, and binding of complement to the sarcolemma [2, 18, 21, 36]. Some inflammatory T-cells and other immune cells may be present around these focal spots, but there are no primary inflammatory lesions. Necrotic fibers typically display a secondary invasion by macrophages for clearance of the cell debris. Complement binding is present in capillaries and on the surface of muscle fibers. On electron microscopy, pipestem capillaries may occur [37], but no tubureticular inclusions in endothelial cells (which are present in DM).
Overlap Myositis (OM)
Overlap myositis (OM; synonym: overlap syndrome with myositis) is increasingly recognized as an individual form of myositis that itself is heterogeneous and presumably encompasses the largest subgroup of myositis [4]. The term OM has been already used since several years and relies on the identification of auto-antibodies associated with myositis [38] (see below for details). Patients with OM present with acute or subacute onset of weakness of arms and legs, similar to the aforementioned types of myositis. A profound elevation of muscle enzymes including CK is usually present (10–50 fold). OM can be diagnosed in conjunction with other collagen disorders such as Sjögren syndrome, systemic sclerosis or systemic lupus erythematosus (SLE). The most common condition within OM is the anti-synthetase syndrome (ASS), which can be considered a distinct subform of myositis. This consists of a typical collection of clinical symptoms including myositis, Raynaud’s phenomenon, arthritis, mechanic’s hands, interstitial lung disease (ILD) and presence of anti-transfer RNA synthetase auto-antibodies (see below for details).
The most common of the eight anti-synthetase antibodies is the Jo-1 antibody, which is observed in ∼30% myositis patients according to a recent meta-analysis [39]. Anti-PL-7 and anti-PL-12 are found in 3-4% of the cases and all other anti-synthetase antibodies are less common (<2% ): anti-EJ, anti-ZO, anti-OJ, anti-KS, anti-HA(YRS/Tyr). Two large subgroup analyses revealed a higher rate of ILD in patients with anti-PL-7 or anti-PL-12 compared to Jo-1 and this was accompanied by a higher mortality [40, 41]. Apart from the anti-synthetase antibodies, five other antibodies are associated with OM as well as with other connective tissue disorders. They can cause an identical clinical syndrome as ASS or some of the features, e.g. ILD and myositis plus the individual symptoms of the respective connective tissue disorder. The most common of these antibodies is anti-PM/Scl (targeting two subunits of 75 kDa and 100 kDa of a nuclear exosome complex), which is commonly associated with systemic sclerosis and present in up to 12% of myositis cases [39]. Anti-U-snRNP antibodies are observed in 3–8% of myositis patients and associated with mixed connective tissue disease (CTD). Whereas patients with anti-U-snRNP often have a good prognosis, anti-PM/Scl may indicate a more severe course and insufficient treatment response (reviewed in [23]). Anti-Ku antibodies are associated with systemic sclerosis, systemic lupus erythematodes (SLE) and other mixed and undifferentiated CTD. In myositis, they are overall rare with a frequency of 1–3%, but in overlap syndromes with myositis, they are present in up to 19% of the cases [39]. They have a high rate of ILD, which does not respond well to glucocorticosteroids, whereas the muscular symptoms usually respond [42]. Anti-SS-A/Ro52/Ro60 and anti-SS-B/La are usually present in Sjögren’s syndrome, SLE, and systemic sclerosis. They occur in patients with myositis at a frequency of 19% and 7% respectively, and in OM they are present in up to 25% and 12% of the cases [39]. SS-A has been shown to be present in 6% of the JDM patients. The anti-Ro52 subdomain is more common in patients with myositis than the Ro60, whereas both are equally present in CTD [23]. Anti-Ro52 is often present in conjunction with anti-synthetase antibodies, e.g. in 56–72% of anti-Jo-1 positive patients. These double-positive patients have a higher risk of malignancy and a poorer prognosis [43].
The histological picture encompasses a perifascicular necrosis and perifascicular binding of MHC class I and class II antibodies as well as complement binding to the sarcolemma in the same areas of the skeletal muscle [21, 44–46].
Inclusion Body Myositis (IBM)
The clinical presentation of inclusion body myositis is distinct from all other forms of myositis: The elevation of CK is much milder (up to 10–15 fold upregulation), the onset is much more asymmetric and may begin with unilateral affection of one leg or arm, e.g. the proximal leg, the forearm, or ventral calf; the progression is much slower than that of other forms of myositis, but continues relentlessly and leads to a profound muscle atrophy [47]. The typical pattern of muscle involvement includes weakness of the long finger flexors, the quadriceps, the tibialis anterior and, usually to a lesser extent, all other muscles of arms and legs. Swallowing problems (dysphagia) are common and may present as the initial symptom. The weakness often leads to injurious falls and the dysphagia may cause aspiration pneumonia, which explains a higher rate of mortality in these patients [48]. More male than female are affected (2 to 3 fold) and most patients are above 50 years of age when the first symptoms evolve, yet the diagnosis may be made even much later [49, 50]. Skin changes are not present.
The only antibody associated with IBM is cN1A (5NT1A / 5NTC1A, initially termed “Mup44”). It has been identified a few years ago [51, 52] and was initially thought to be present in more than half of the patients. More recent observations have demonstrated that in most cohorts the frequency is about 30% and only rarely as high as 60% and above; Importantly, in addition to other forms of myositis including DM, many other conditions such as Sjögren’s syndrome and SLE were also positive by ∼20–30%, even in absence of any muscle symptoms [53, 54]. This was recently corroborated in a Japanese cohort [55]. Recent data in reasonably sized IBM cohorts suggest that presence of cN1A is associated with a more severe disease course and dysphagia [56] and a higher mortality [57]. By contrast, in a group of 20 German patients, presence of cN1A did not correlate with the severity of dysphagia or muscle impairment [58].
The histological picture encompasses “invasion” of muscle fibers by endomysial cytotoxic CD8 + T-cells, widespread and moderate to strong upregulation of MHC class I, signs of protein accumulation by detection of amyloid (Congo red, thioflavin S, immunohistochemistry for p62 or TDP-43), detection of tubulofilaments on EM, vacuoles and signs of mitochondrial damage as evidenced by histochemical proof of COX-deficient muscle fibers, and paracristallin inclusions [2, 59]. However, signs of protein accumulation can be absent, even the “canonical” feature of rimmed vacuoles can be missing [25, 26].
ASSOCIATION WITH MALIGNANCY AND EXTRAMUSCULAR MANIFESTATIONS
All forms of myositis except IBM have been shown to be associated with varying frequencies of malignancies with an increased risk by 2- to 7-fold compared to the general population [60–63]. As stated in the section on antibodies above, this risk of malignancy is particularly high in DM cases with anti-TIF-1 or anti-NXP2 antibodies as well as NM cases with anti-HMGCR antibodies. Patients without detectable presence of an auto-antibody may also suffer from cancer as evidenced in cases with NM (see above). The risk is highest within one year before and after the diagnosis of myositis and remains elevated within a time span of about 3 years. The types of malignancy in myositis include cancer of the lung, breast and ovary as well as lymphoma. All patients with newly diagnosed myositis should receive a CT-scan (or MRI) of the chest and abdomen, and in certain cases with high level of suspicion, possibly a PET-CT and tumor markers. Further tests such as ultrasound of the abdomen and gastroscopy or colonoscopy or referrals to specialists depend on the individual situation of each patient. Depending on the outcome of the tumor screening and the clinical course of the myositis, tumor screening should be repeated at least once a year for at least three years.
A range of extramuscular organs can be affected in myositis: lung, heart, joints, skin, kidneys and others. The skin is a commonly affected organ in myositis. The clinical presentation of skin manifestation in dermatomyositis with Gottron’s sign, heliotrope rash of the eye lids, shawl and V-sign have been explained in more detail above. Mechanics’ hands and Raynaud’s phenomenon have been mentioned in the ASS section above. In addition, in systemic sclerosis, the skin of hands and feet displays scars, ulcers and becomes thickened. Fingers and toes may display an impaired mobility (sclerodactyly) and the disease can progress to more proximal areas of arms and legs. Opening of the mouth can be impaired (microstomy, “tobacco pouch mouth”) and the face may appear like a mask. Telangiectasia and Raynaud’s phenomenon are common. Skin changes that occur in SLE include the typical “butterfly rash” (or malar rash) and can present as red, scaly patches of skin – so called discoid rash. A rash typically evolves in sun exposed skin. Further manifestations include hair loss, telangiectasia and calcinosis. Patients with suspected OM or detection of an auto-antibody of the anti-synthetase group or anti-Pm/Scl, anti-SS-A/Ro, anti-SS-B/La should be screened for any of the above mentioned skin changes.
Lung disease is a typical concomitant manifestation in myositis and the frequency and severity depends on the respective myositis subtype [64]. As mentioned and highlighted above, ILD is common in ASS [65]. The pattern of pulmonary manifestations includes pulmonary hypertension, serositis, and various degrees of ILD [64]. Ventilation can be additionally impaired by inflammation and weakness of the diaphragm and other muscles required for breathing. Symptoms of ILD include dyspnoea and cough, which can be dry and non-productive. Apart from all anti-synthetase antibodies, ILD in patients with myositis is associated with anti-U-snRNP, anti-PM/Scl, anti-Ku, anti-MDA5, and anti-SRP [39] (see the respective disease sections above and Table 2). Apart from malignancy, ILD is one of the most critical organ manifestation in myositis and severe ILD is clearly associated with an increased rate of mortality in myositis [66]. Therefore, patients should be asked for symptoms of lung involvement and pulmonary function tests should be performed in all cases. A (high-resolution) computed tomography (CT) of the chest should be routinely considered, depending on the presence of pulmonary symptoms, the overall severity of the myositis, the type of auto-antibody and other reasons for chest imaging such as screening for malignancy. Typical findings of an ILD include reticulation, linear and ground-glass opacity, peribronchovascular thickening, cystic spaces with thickened walls (“honeycombing”) and traction bronchiectasis [66]. The chest CT should be evaluated by a physician experienced in conditions with ILD and the clinical management of ILD should be conducted together with a pulmonary specialist in a multi-disciplinary fashion (see also below).
Inflammation of joints is common in connective tissue disease (CTD) and overlap syndromes with skin involvement, especially in ASS. Symptoms of arthritis include swelling, pain and stiffness of the joints. Typically, small hand joints are affected as well as wrist, hip, knee, and ankle. The diagnosis can be supported by changes on X-ray such as joint space narrowing and bone erosions.
Involvement of the heart is not uncommon in myositis and overlap syndromes, but it is often not recognized. The heart can be affected in form of a cardiomyopathy, serositis or pericarditis as well as conduction problems. Involvement of the heart is associated with an increased mortality [67]. Typical symptoms of heart involvement include dyspnoea, especially during exercise, and arrhythmia. Testing should include an ECG, potentially over 24 hours to detect arrhythmias, echocardiography, and, if available, MRI of the heart. Upon identification of heart problems, a cardiology specialist should be consulted for interdisciplinary treatment. Apart from symptomatic treatment for the heart, an intensified immunosuppression should be considered in these cases.
The gastrointestinal tract and liver can be affected in overlap syndromes and particularly impair function of the pharynx and oesophagus [68]. The symptoms range from a mild dysphagia with occasional coughing during eating to a severely impaired food passage with choking etc., possibly requiring a nasogastric or even a percutaneous feeding tube. Danger of unrecognized swallowing problems is the risk of aspiration with subsequent pneumonia or even death. Symptoms of dysphagia include repeated clearing of the throat during eating, coughing, a prolonged duration of food intake, feeling that “food gets stuck”, and choking. Importantly, patients may not report their symptoms without specific questions for it, e.g. by using specific swallowing questionnaires such as the Swal-Quol, Sydney swallowing questionnaire etc. Apart from speech therapy, patients can receive specific treatment such as local injections of botulinum toxin treatment for IBM (see section on IBM below for details). The diagnostic tests for evaluation of dysphagia include a fiber-optic endoscopic evaluation of swallowing. Patients with a confirmed swallowing abnormality should receive a fluoroscopy, an X-ray based assessment. In the future, real-time MRI may become available for general assessment of swallowing [58].
Other extramuscular manifestations are more rare in myositis, such as an involvement of the kidneys, which is seen in ∼60% of the patients with SLE (“Lupus nephritis”) with typical signs of renal dysfunction including oedema of the legs, proteinuria and haematuria. As a rare event, thromboangiopathic renal crisis can occur in systemic sclerosis in conjunction with initiation or high dose application of steroid treatment. Recently, such an event has also been demonstrated in an ASS case with anti-PL7 antibody [69].
CLASSIFICATION CRITERIA AND DIAGNOSTIC PARAMETERS
Most international experts agree that the current gold standard for a reliable diagnosis of myositis requires i) a robust clinical assessment including the pattern of weakness and paraclinical parameters such as MRI and CK etc., ii) a broad assessment of auto-antibodies, preferably a line-blot assay that covers all currently available auto-antibodies in myositis, iii) a muscle biopsy with detailed histological and immunohistological work-up that allows subtyping of the different pathological entities. This traditional concept [1, 2] is valid despite the recent publication of novel criteria for myositis that have been formally accepted by ACR and EULAR [70, 71]. These criteria have been developed by using a large, yet not sufficiently well characterized patient group of 976 patients with myositis. 12 clinical parameters are used and transformed into a numerical value: age, sex, pattern of weakness, signs of skin manifestations, laboratory features including Jo-1 antibody and 4 facultative biopsy parameters including pattern of inflammation, perifascicular atrophy and vacuoles. The total numerical score for each value is then used for classification into JDM, DM, ADM, IBM and PM – either with or without biopsy data–and each as “definite”, “probable” or “possible”. An easy to use web-calculator for this algorithm has been set up: http://www.imm.ki.se/biostatistics/calculators/iim/. The sensitivity and specificity of the new criteria have been compared to all relevant previous diagnostic criteria by Bohan & Peter [72], Tanimoto et al. [73], Targoff et al. [74], Dalakas and Hohlfeld [75], and ENMC Hoogendijk et al. [18]. The new criteria reached an overall sensitivity of 93% with biopsy and a specificity of 88% with biopsy and both was considerably lower without biopsy (87% sensitivity and 82% specificity). The sensitivity with a biopsy was comparable to the Bohan & Peter, Tanimoto and Targoff criteria and better than the Dalakas & Hohlfeld criteria, which were only 6% sensitive and the Hoogendijk criteria, which were 52% sensitive. The specificity was better than Bohan & Peter (55% ) and Tanimoto (31% ), comparable to Targoff (89% ) and lower than Dalakas & Hohlfeld (99% ) and ENMC Hoogendijk (97% ). The authors acknowledge that their new criteria have limitations and these are further corroborated by recent communications to the publication [76–78]: i) The group of patients is large, but a significant part of them has not been worked up in sufficient detail, as evidenced e.g. by the lack of biopsy results, ii) The entities NM and OM are not represented since these diagnoses were not sufficiently present – and it is highly likely that many patients within the Euromyositis registry are diagnosed as PM or DM in error, but actually suffer from OM or NM, iii) a broad antibody profile is lacking in many patients so that distinction of TIF-1, NXP-2, MDA-5, Mi-2, SRP, HMGCR etc. is not possible. The authors acknowledge that there is a need to validate the newly published diagnostic criteria, which they call “provisional”, and to revise them for inclusion of more auto-antibodies, more specific biopsy features, and inclusion of NM and OM as separate entities, since both belong to the spectrum of myositis subtypes as recently suggested [4].
None of the above mentioned myositis classifications include the diagnostic panel of auto-antibodies “myositis specific auto-antibodies” (MSA) and “myositis associated antibodies” (MAA). This distinction between MSA and MAA has been established several years ago and, due to recent findings of myositis in MAA cases and presence of MSA in cases without myositis, the arbitrary distinction between MSA and MAA appears to be outdated and currently rather obsolete. This is in line with the wording of recent summaries on myositis auto-antibodies by others [10, 22].
Initial criteria for IBM have relied strongly on histological criteria [79], whereas old ENMC criteria [80] and those from Dalakas [81] equally weighted clinical and histological features. The criteria for IBM have recently been redefined by an ENMC workshop [59] with suggestion of the clinical parameters that patients should be above 45 years, have a CK of less than 15-fold elevated, and clinical features of finger flexors weaker than shoulder abductors and/or quadriceps weaker than hip flexors. The histological parameters include signs of endomysial inflammation as well as rimmed vacuoles and signs of protein accumulation [47, 59]. The parameters were assessed in total and produced either “clinico-pathologically defined IBM”, “clinically defined IBM” or “probable IBM” [47, 59]. A combination of three of the relevant parameters of the new ENMC criteria appear to be ideally suited to diagnose IBM at 90% sensitivity and 96% specificity as evidenced in a computer-based, unbiased assessment of 371 patients (200 IBM vs. 171 other myopathies): (1) finger flexor weakness or quadriceps weakness, and (2) endomysial inflammation, and (3) either invasion of nonnecrotic muscle fibers or rimmed vacuoles [82].
In addition to the clinical parameters, which should be compared with those summarized in Table 1, patients should be assessed by paraclinical methods. Particularly MRI parameters such as inflammation, fibrosis and atrophy can be used to determine the (subclinical) pattern of disease activity [83, 84]. Ultrasound can help to distinguish a pattern of affected muscles [85] or at least help to identify a suitable muscle for biopsy [86, 87]. Apart from using MRI for diagnosis, it is a suitable tool to check for acute damage and current inflammation. A whole body MRI is now considered a state-of-the-art technique that helps to identify involvement of muscles even at a subclinical level [88, 89]. MRI is also useful for detection of muscle inflammation in juvenile myositis [90, 91]. In IBM, MRI can be used to identify the pattern of affected muscles, which has been shown to correlate well with other clinical parameters [92] and displays a high diagnostic accuracy and specificity for the disease [93]. Precise and broadly accepted scoring algorithms will help to improve the future use of MRI as an additional clinical follow-up item and further its use for clinical trials [94]. EMG changes are non-specific and do not help to differentiate between the different myositis subforms. Spontaneous discharges as seen in acute myositis can even give rise to an erroneous suspicion of motor neuron disease. An EMG can be useful to distinguish between acute changes in myositis and weakness due to steroid myopathy. In addition to the mere myositis diagnosis, paraclinical assessments should be performed in order to identify underlying conditions such as a malignancy or an organ manifestation such as interstitial lung disease (see each subgroup section for the respective diagnosis).
Pathomechanisms
A detailed summary of the underlying pathomechanisms of myositis is beyond the scope of this clinically oriented review. The mechanisms in myositis have been recently reviewed elsewhere: inflammatory mechanisms [95], overall/ mixed mechanisms [1, 96, 97], genetic factors [98], non-inflammatory mechanisms [99], myokines [100], innate immunity [101], and mechanisms in IBM [102].
Cellular immune mechanisms include cytotoxic CD8 + T-cells that make cell-to-cell contact with muscle fibers and exert their cytotoxic granules with perforin and granzyme B in direction of muscle fibers [103, 104]. Several reports have consistently shown that, in PM and IBM, these T-cells clonally expand within the muscle [105–107] and that individual clones can be tracked over years by T-cell receptor profiling and laser capture microdissection [108]. On the other hand, in PM and IBM, a clonal expansion of B cells has also been identified [109, 110], which supports the pathogenetic relevance of B-cells in these disorders. It is currently believed that, by local secretion of a range of pro-inflammatory cytokines and chemokines [95, 111, 112], muscle fibers can directly contribute to the inflammatory milieu in the muscle with attraction and local activation of immune cells that subsequently attack the muscle fibers [104]. Mediators of the innate immune system are also noted in myositis and expression of Toll-like-receptors is present on the sarcolemma of muscle fibers [113, 114].
Apart from inflammatory pathways, non-immune mechanisms are also operational in myositis muscle [99, 115], including ER-stress, NFκB-activation and free radicals such as NO. Such cell stress mechanisms are supposed to cause weakness of the skeletal muscle, even in absence of structural damage inflicted by cell lysis, protein aggregation or a cell death cascade [116]. In IBM, several lines of evidence suggest that there is a distinct interaction between inflammatory mechanisms, vacuolar transformation and accumulation of amyloid [117]. More recent evidence suggests that potential mediators in this network of events could be HMGB1, its receptor RAGE [118], intra-fiber production of NO by iNOS [119], or overexpression of the heat-shock protein αB-crystallin [120].
The HLA 8.1 haplotype has been identified to be a risk factor for myositis in a genome-wide analysis of 1710 patients with myositis vs. 4724 controls [121]. This was confirmed and explored in more detail by a single-nucleotide polymorphism (SNP) analysis via an immunochip assessment of 2566 myositis patients [122], which identified several variants within the HLA 8.1 that were associated with certain subsets of myositis including IBM [123]. Outside of the HLA molecules, the strongest association was observed with the PTPN22 gene in PM, but not in DM or other subtypes [122].
Autophagic activity has been associated with the accumulation of amyloid in IBM [124]. Recently, variants in the genes of VCP, SQSTM1 and FYCO1 were identified and the latter has been shown to be relevant for autophagic activity. This suggests that a variation in FYCO1 expression may contribute to the autophagic degeneration in IBM muscle [125].
It has been suggested for over two decades that myositis is associated with environmental factors [126]. Recent data have demonstrated that exposure to UV radiation [127, 128], infections [129] and various other environmental factors can have a protective or triggering role in myositis [130, 131]. It is hypothesized that environmental factors, in presence or absence of underlying genetic preconditions, can modulate immune mechanisms that either reduce inhibitory factors or increase stimulatory effects and that this triggers or aggravates an auto-inflammatory cascade [130].
DISEASE COURSE AND TREATMENT OF DM, PM, NM AND OM
Overall, a majority of the patients does respond reasonably well to the immunosuppressive treatment detailed below. Yet, it is important to point out that many patients continue to progress despite intense treatment efforts because the muscle inflammation cannot be sufficiently controlled. Another reason for persistent muscle weakness is a delayed start of the treatment, which can lead to an irreversible muscle damage with fibrosis or fat replacement. These elements of the course of myositis explain the profound burden of disease with an elevated morbidity and mortality in all subforms [132–134].
Glucocorticosteroids are the mainstay of the treatment for PM, DM, NM, and OM, whereas IBM is treated differently and will be reviewed separately below [1]. The rational for glucocorticosteroids relates to several old non-controlled trials and case series and is well accepted by all societies and medical subspecialities. Usually prednisolone is given orally at a dose of 0.5–1.0 mg/kg per day (see Fig. 2 for an overview of the treatment regimen). Particularly in acute and severe cases, the treatment can be initiated with an intravenous high dose pulse of 250–1000 mg per day for 3 to 5 days. The CK value should not be the sole parameter for decision of steroid tapering: steroids should be continued for about 4–12 weeks and a reduction can be considered once an obvious clinical improvement can be documented per muscle strength (MRC sum score) or subjective/objective assessment scales (see below). Tapering needs to be performed slowly by 10 mg every 1 or 2 weeks until 20 mg/day is reached. Subsequent tapering should be done in 2.5–5.0 mg steps every 1 or 2 weeks, depending on the course of the disease. A maintenance dose of ∼5 mg prednisolone per day is often necessary, at least for an interim phase or even long-term. Treatment with glucocorticosteroids on alternating days can be a useful alternative with less long-term side-effects and – at the same time–possibly an even higher efficacy [135]. The alternate day regimen was even associated with a higher long-term survival rate compared to daily application [136]. A third regimen of steroids in myositis is the monthly oral pulse treatment with 4 days of 40 mg/day dexamethasone [137]. This dosing achieved similar efficacy, and at the same time caused less side-effects. Potential long-term side-effects of steroids include osteoporosis, cataract, atrophy of the skin, diabetes mellitus, hypertension, mood swings, weight gain, and increased risk of infections. All patients receiving long-term steroid treatment (for several years) should receive 1000 mg/day calcium and 500 IU vitamin D. Long-term immunosuppression should be started in parallel with the steroid, unless only a very moderate disease course is present. Immunosuppressants include methotrexate (MTX), azathioprine (AZA) or mycophenolate mofetil (MMF). A Cochrane analysis compared all available clinical studies with these and other agents in myositis and could not identify any significant efficacy [138]. However, given a range of case series and expert experience and in view of the known pathogenesis of the disorders, it is international consensus to use glucocorticosteroids as well as immunosuppressants for treatment of myositis in an off-label fashion. MTX is given once a week at a dose of 5–20 mg (Fig. 2), followed by 5–10 mg folic acid on the following day. It can be applied orally or subcutaneously – the latter is usually better tolerated in view of gastrointestinal side-effects. Side-effects of MTX include leukopenia, elevated liver enzymes, an interstitial pneumonitis, and pulmonary fibrosis. Before use of AZA, it is advisable to assess the enzymatic activity of the thiopurinmethyltransferase (TPMT). If the activity is normal, AZA can be started at orally 50 mg/day for the first week and then increased every week up to 200 mg/day and more, given once a day or divided into three doses. In patients with reduced TPMT activity, an initial dose of 25 mg should be used and a much slower and careful dose increase should be performed. The optimal dose is distinguished by the absolute lymphocyte count, which should be between 600 and 1000 cells per μl. Main potential side effects of AZA are leukopenia, malignancy, liver damage and a range of gastrointestinal symptoms. The choice of the primary immunosuppressant depends on the co-medication, concomitant illnesses and personal experience with the drug by the physician. If both MTX and AZA are not well tolerated, MMF can be used and is usually started at 500 mg twice per day. The dose can be increased to 2.0 or 2.5 g or even 3.0 g/day, divided into three daily doses. Even if the lymphocyte count may be reduced quickly by all immunosuppressants, the biologic immunosuppressive effect of AZA, MMF and MTX can take several weeks before it starts: MTX and MMF may have a delay of up to 12 weeks and AZA can have a delay of up to 3 to 6 months (or rarely even up to one year) until clinically effective. A dose increase of AZA, MMF or MTX should be performed in biweekly intervals with regular monitoring of all relevant parameters of the blood including full blood count, liver and renal function tests.
![Overview of the basic and escalating treatment modalities in myositis (modified from [1]).](https://content.iospress.com:443/media/jnd/2018/5-2/jnd-5-2-jnd180308/jnd-5-jnd180308-g002.jpg)
In case the standard regimen with steroids and the above mentioned immunosuppressants is not tolerated or not sufficiently effective, two alternative options are available: oral ciclosporin or intravenous immunoglobulin G (IVIG). Ciclosporin (cyclosporine A, CsA) (or its modified drug tacrolimus) is an effective immunosuppressant that can be used either as replacement or in combination with other immunosuppressive drugs. Side-effects include gastrointestinal symptoms, hypertension, kidney disease, and malignancy. Dosing of CsA should be done according to therapeutic plasma levels, which should be checked regularly. Of note, most immunosuppressants are potentially embryotoxic or gonadotoxic (overview in [139]). In particular MTX, MMF and cyclophosphamide need to be avoided during pregnancy. AZA and CsA are considered less toxic and several reports have demonstrated a normal child development, but if possible it still appears advisable to avoid any immunosuppressive drug several months before conception (for female as well as for male!) and during pregnancy.
IVIG is another well established alternative or add-on treatment for myositis, which has been shown efficacious in several clinical studies and case series, particulary in DM and NM (recent overview in [140]. IVIG is applied at 1-2 g/kg per treatment cycle every 3 to 6 weeks (potentially every 8 weeks), usually over 1 to 2 or 3 days at a daily dose of 0.5–1.0 g/kg. The individual dose for each patient needs to be established over several treatment cycles. The total dose depends on the clinical effect of IVIG and the daily dose depends on how the drug is tolerated by the patient. Potential side effects are allergic reactions, headache, fever, thrombosis, and haemolysis. Many of the effects are associated with the dose and the infusion rate. Allergic reactions and the overall tolerability of IVIG often also depend on the specific formulation and may be abolished by changing the supplier. The therapeutic effect of IVIG is expected to be similar across all products.
If standard immunosuppression and IVIG are not sufficient, a treatment escalation to rituximab (RTX) or cyclophosphamide (CYC) should be considered. RTX has recently been tested in a trial with 200 juvenile and adult patients with myositis [141]. Although the primary end-point was not reached, presumably due to the study design, the overall response to RTX is generally interpreted as successful since a major part of the patients clearly improved during the trial [142].
In addition to the basic pharmacological treatment of DM, PM, NM, and OM as detailed above, several points should be considered for specific subcategories: a) in all of the patients, an initial and, depending on the type of myositis and auto-antibody status, also repeated screening of malignancy should be performed and treatment of the tumor should be prioritized vs. immunosuppression; b) in NM, the standard regimen is often not sufficient to achieve remission so that an add-on treatment with IVIG and/or escalation treatment with RTX is often required, particularly in patients with anti-SRP antibodies; c) involvement of the skin will require protection from sunlight/ UV exposure and add-on treatment with topical treatment, e.g. with glucocorticosteroids; IVIG has been shown efficacious regarding skin lesions in DM [145] and, particularly in juvenile DM, anti-malaria drugs (hydrochloroquine) can be useful [146]; d) the management of ILD should be performed interdisciplinary together with a pulmonologist and usually requires a more aggressive treatment (overview in [66, 146]) with a combination of high-dose glucocorticosteroids and an immunosuppressant such as AZA plus either RTX or CYC; milder forms may be sufficiently controlled by addition of CsA; MTX should be used with caution in patients with ILD since it may itself induce a pneumonitis; IVIG may be a temporary option during infection or an alternative/add-on treatment in patients with ILD and contraindications for immunosuppressive escalation therapy.
Apart from pharmacological treatment, patients with any form of myositis should receive physiotherapy from the beginning of the disease. Historic reports that suggest to avoid exercise during the acute phase are obsolete. Physiotherapy should include all aspects of rehabilitative medicine and non-fatiguing resistance exercise as long-term therapy of all patients until clinical symptoms are fully resolved [147].
TREATMENT OF IBM
No effective treatment is currently available for IBM [47]. In particular, the standard immunosuppressive regimen with glucocorticosteroids and immunosuppressants does not lead to an improvement of impaired muscle strength as evidenced by several clinical trials with glucocorticosteroids, MTX, CsA etc. (reviewed in [47]). Therefore, treatment with glucocorticosteroids or immunosuppressants is usually not recommended in IBM. Alemtuzumab did lead to a transient improvement in some patients within an unblinded proof-of-concept study [148]. In a post-hoc analysis, downmodulation of inflammatory markers was noted, but degenerative molecules remained unchanged [149]. However, due to the uncontrolled nature and small size of the study, the results need to be interpreted carefully and are not sufficient to justify a treatment with alemtuzumab in IBM – a clinical trial would be warranted to study the effect. Three placebo-controlled clinical trials assessed IVIG in IBM over 3 to 6 months: two of the studies observed a small increase of some of the outcomes including MRC scale and swallowing function [150–152] (overview in [47]). IVIG was further evaluated in several uncontrolled case-series, which showed an improvement of dysphagia and muscular weakness [153–155]. However, all clinical IVIG trials missed their primary outcome, which most likely is explained by an insufficient duration of the studies (3 months in 2 studies and 6 months in 1 study) so that a reliable conclusion for the use of IVIG in IBM cannot be made [140]. In view of the positive case series in this devastating disease that often is associated with severe dysphagia, aspiration pneumonia and an increased mortality [48], a probatory treatment with IVIG for 6 months with a dose of 1-2 g/kg every 6–8 weeks appears to be justifiable in such patients [47, 156, 157], yet its use may be restricted by national reimbursement policies. An international group of experts does currently establish the standard of care for IBM including all pharmacological and non-pharmacological treatments [158].
About 2/3 of the patients suffer from varying degrees of impaired swallowing, sometimes even as the sole symptom [2, 47]. Cause of the swallowing is a functional stenosis of the upper oesophagus sphincter, which can be detected by videofluoroscopy [159] or real-time MRI (the latter in a research setting) [58]. Local treatment possibilities include cricopharyngeal myotomy, pharyngoesophageal balloon dilatation and repeated injections of botulinum toxin into the upper oesophageal sphincter [47, 157]. Since recent data suggest an impaired laryngeal excursion as cause of dysphagia in IBM [160], a beneficial effect of balloon dilatation should not be expected and our own interpretation is that the dilatation may even cause additional muscle damage in the pharynx/oesophagus. The cricopharyngeal myotomy is an effective technique, but it is irreversible and patients may later suffer from reflux or other swallowing problems. Local injections of botulinum toxin have been demonstrated to be effective in IBM in several case series [161–164] and our own experience also supports this treatment (unpublished observations). Once dysphagia has evolved, it is likely to persist and worsen. Regular screening for aspiration including a fiberoptic endoscopic evaluation of swallowing (FEES) is advisable in order to identify dysphagia and prevent aspiration pneumonia or even risk of death, which is more common in IBM patients with severe dysphagia [48]. Weight management should be conducted to avoid cachexia and if other treatment options fail, a percutaneus feeding tube should be considered to avoid aspiration and assure sufficient calorie supply.
ASSESSMENTS OF THE DISEASE COURSE AND OVERVIEW OF RELEVANT CLINICAL TRIALS
In clinical practice just as much as in clinical studies, clinical scales are required that allow a quick and reliable comparison of two visits of the patient (see [165] for a recent overview). This is indispensable for a consistent evaluation of the treatment and decisions of dose escalation including the switch to a more aggressive drug. The International Myositis Assessment and Clinical Studies Group (IMACS) has developed highly relevant assessment tools that provide appropriate clinical measures: six items were contained in the core set of disease activity including physicians’ and patients’ global activity assessment, muscle strength (manual muscle testing), functional disability, muscular enzymes and extramuscular disease activity [166, 167]. The extra muscular disease activity is an important component of the core set since it reflects tissue damage of joints, lung, skin, heart, or the gastrointestinal system [168]. Recently, the core set measures were transformed into a numeric scale and weighted depending on their impact on the final score; this composite score was officially accredited as “response criteria” by the American College of Rheumatology (ACR) and the European League Against Rheumatology (EULAR) for adult myositis [169, 170] as well as for juvenile myositis [171, 172]. Manual muscle testing of a group of muscles (MMT-6 or MMT-8) is an important component of this longitudinal assessment in myositis and should be performed by the physician at each clinical visit. In order to capture the endurance component in addition to the short maximal strength, the functional index and its second version have been developed for myositis [173] and it has been demonstrated that a 2-minute walk test is a reliable measure in patients with IBM [174]. Patient reported outcome (PRO) measures such as the health assessment questionnaire (HAQ) are an essential component of long-term observation of myositis patients. It is important to note the degree of pain, fatigue, and severe, often nearly ubiquitous pruritus as in DM and morphological affection of the skin – the latter by using the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) [167, 175]. Two very similar, useful and strongly correlating PRO’s have been developed for IBM: the inclusion body myositis functional rating scale (IBM-FRS) [176] and the sporadic inclusion body myositis physical functional assessment (sIFA) [177]. Another tool that was developed specifically to capture functional and endurance aspects in patients with a myopathy including myositis is the adult myopathy assessment tool (AMAT) [178]. In an international effort, PRO’s for myositis are currently assessed with the aim of making them more precise and reliable [179]. Particularly in patients with dysphagia, it is important to also include a scale that captures symptoms of swallowing, e.g. the Sydney Swallowing Questionnaire (SSQ) [180].
Regarding the number of major clinical trials in the past decades, inflammatory myopathies (and neuromuscular disorders in general) have been neglected compared to other disease areas. This has changed tremendously in recent years and it is expected that neuromuscular disorders will stay in the focus of interest for the coming years. Several clinical trials in DM and/or PM have been conducted recently or are currently ongoing. The ideal classification that can be used as an easy and reliable inclusion criterion for clinical trials is still not existent. The recently published classification criteria [70, 71] have inherent problems as discussed above and, therefore, do not appear to be useful without additional diagnostic items such as the antibody status. It is beyond the scope of this review to discuss all of the recent trials and only a small selection can be discussed here (for a broader recent overview see [181]): Belimumab is an inhibitor of the B cell activation factor (BAFF) and is studied for the maintenance of DM/PM in a placebo-controlled phase 2/3 trial (NCT02347891 on clinicaltrials.gov). A placebo-controlled phase 2 trial with tocilizumab, an IL-6 receptor blocker, is ongoing (NCT02043548). A placebo-controlled phase 3 trial in all subforms of myositis with Abatacept, a CTLA-4 fusion protein that acts as co-stimulation inhibitor for T-cells, was started in 2017 (NCT02971683). A placebo-controlled phase 3 trial in DM with the IVIG Octagam 10% was started in 2017 (NCT02728752). Basiliximab, a novel calcineurin inhibitor, is currently studied as an add-on drug in an open label phase 2 trial in patients with ILD in ADM (NCT03192657). IFN-Kinoid is an immunizing drug that induces generation of neutralizing anti-IFN-α antibodies and is currently studied in a single-placebo-controlled phase 2 study in DM (NCT02980198). The clinical trials on DM and PM with BAF321, a sphingosine 1-phosphate (S1P) receptor modulator that inhibits lymphocyte migration out of lymph nodes, have been terminated. TNF-α blockers should be rather avoided in myositis since worsening has been described [182, 183].
A recent phase 2b/3 study with bimagrumab, a monoclonal antibody that acts via the myostatin-mediated induction of muscle growth [184], has failed its primary endpoint 6-minute walk test, but the full data have not yet been published (NCT01925209). In a recently completed double-blind, proof-of-concept study (phase 2b), 44 patients with IBM were treated with oral 2 mg/day rapamycin vs. placebo for one year [185]. Although the quadriceps strength as primary outcome measure was not improved, several clinical parameters including the 6-minute-walk test and a functional composite index displayed a significant positive treatment effect. This is the first clinical study in IBM with a sufficient duration that shows a significant and clinically meaningful improvement, which supports the conduction of a larger trial with rapamycin in the future. Another recent concept study was conducted with arimoclomol, which was effective in the valosin-containing protein (VCP) mouse model, muscle cell cultures and was well tolerated in a small, double-blind, placebo-controlled phase 2 trial in IBM over 4 months with additional 8 months clinical assessments [186]. A large, double-blind, placebo-controlled phase 2 trial has recently started in 150 IBM patients who will receive 400 mg arimoclomol three times per day for one year (NCT02753530). Further recent clinical studies and potential drug targets for future efforts have been reviewed by us before [47, 156].
CONCLUSIONS
Inflammatory myopathies are heterogeneous disorders that, apart from an often severe and acute inflammation of the muscle, can affect several extramuscular organs. The main subtypes of myositis include DM, PM, NM, IBM, OM, and ASS. The classification and management of the different subforms usually requires attention by a multi-disciplinary team that includes expertise from several specialities: rheumatology, neurology, dermatology, neuropathology, pulmonology and others. The diagnosis is based upon a combined assessment of i) the clinical symptoms and course of the disease including muscle MRI and screening for extramuscular organ manifestation such as ILD, ii) a full screening of auto-antibodies associated with myositis including anti-synthetase antibodies, Mi-2, Jo-1, TIF-1, NXP-2, SRP, HMGCR, cN1A and others, iii) a detailed histopathological workup of a muscle biopsy including immunohistochemical subtyping of immune cells and staining for complement and MHC class 1 and 2. The basic treatment of all myositis subforms except for IBM includes glucocorticosteroids for several weeks to months (and possibly longer) and often an immunosuppression by AZA or MTX. Add-on treatment or escalation therapy may include IVIG, CYC and RTX. In IBM, a probatory treatment with IVIG may be justifiable in selected patients. All patients with myositis should receive long-term physiotherapy and regular rehabilitation. All treatments need to be monitored closely by use of suitable scales that depict the current status of the patient. Modification of the treatment regimen needs to take into account the overall disease course as well as extramuscular manifestations. Future improvements should aim to advance the therapeutic escalation scheme and continue to explore the use of targeted treatments including the use of biologicals as in several ongoing clinical trials. Improvement of the diagnostic criteria that incorporate clinical, histological and auto-antibody data is required in order to provide a reliable diagnosis and allow an effective treatment across all subforms of myositis.
CONFLICT OF INTEREST
JS has received honoraria, research grants or reimbursements for travel from Bayer, Biogen, BioMarin, Biotest, CSL Behring, Grifols, Hormosan, Novartis, and Octapharma.
REFERENCES
[1] | Carstens PO , Schmidt J Diagnosis, pathogenesis and treatment of myositis: Recent advances. Clin Exp Immunol. (2014) ;175: (3):349–58. |
[2] | Dalakas MCInflammatory muscle diseases. N Engl J Med. (2015) ;372: (18):1734–47. |
[3] | Furst DE , Amato AA , Iorga SR , Gajria K , Fernandes AW Epidemiology of adult idiopathic inflammatory myopathies in a U. S. managed care plan. Muscle Nerve. (2012) ;45: (5):676–83. |
[4] | Senecal JL , Raynauld JP , Troyanov Y Editorial: A New Classification of Adult Autoimmune Myositis. Arthritis Rheumatol. (2017) ;69: (5):878–84. |
[5] | van der Meulen MFG , Bronner IM , Hoogendijk JE , Burger H , van Venrooij WJ , Voskuyl AE , et al. Polymyositis - An overdiagnosed entity. Neurology. (2003) ;61: (3):316–21. |
[6] | Lilleker JB , Vencovsky J , Wang G , Wedderburn LR , Diederichsen LP , Schmidt J , et al. The EuroMyositis registry: An internationalcollaborative tool to facilitate myositis research. Ann Rheum Dis. (2018) ;77: (1):30–9. |
[7] | Needham M , Corbett A , Day T , Christiansen F , Fabian V , Mastaglia FL Prevalence of sporadic inclusion bodymyositis and factors contributing to delayed diagnosis. J Clin Neurosci. (2008) ;15: (12):1350–3. |
[8] | Sun Y , Liu Y , Yan B , Shi G Interstitial lung disease in clinically amyopathic dermatomyositis (CADM) patients: A retrospective study of 41 Chinese Han patients. Rheumatol Int. (2013) ;33: (5):1295–302. |
[9] | Iaccarino L , Ghirardello A , Bettio S , Zen M , Gatto M , Punzi L , et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun. (2014) ;48-49: :122–7. |
[10] | Gunawardena HThe Clinical Features of Myositis-Associated Autoantibodies: A Review. Clin Rev Allergy Immunol. (2017) ;52: (1):45–57. |
[11] | Sato S , Hirakata M , Kuwana M , Suwa A , Inada S , Mimori T , et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanesepatients with clinically amyopathic dermatomyositis. Arthritis Rheum. (2005) ;52: (5):1571–6. |
[12] | Fujimoto M , Watanabe R , Ishitsuka Y , Okiyama N Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol. (2016) ;28: (6):636–44. |
[13] | Targoff IN , Mamyrova G , Trieu EP , Perurena O , Koneru B , O’Hanlon TP , et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. (2006) ;54: (11):3682–9. |
[14] | Kaji K , Fujimoto M , Hasegawa M , Kondo M , Saito Y , Komura K , et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: An association with malignancy. Rheumatology (Oxford). (2007) ;46: (1):25–8. |
[15] | Rider LG , Shah M , Mamyrova G , Huber AM , Rice MM , Targoff IN , et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). (2013) ;92: (4):223–43. |
[16] | Gunawardena H , Wedderburn LR , Chinoy H , Betteridge ZE , North J , Ollier WE , et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. (2009) ;60: (6):1807–14. |
[17] | Betteridge ZE , Gunawardena H , Chinoy H , North J , Ollier WE , CooperRG, et al. Clinical and human leucocyte antigen class II haplotypeassociations of autoantibodies to small ubiquitin-like modifierenzyme, a dermatomyositis-specific autoantigen target, in UKCaucasian adult-onset myositis. Ann Rheum Dis. (2009) ;68: (10):1621–5. |
[18] | Hoogendijk JE , Amato AA , Lecky BR , Choy EH , Lundberg IE , Rose MR , et al. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October Naarden, The Netherlands. Neuromuscul Disord. (2004) ;14: (5):337–45. |
[19] | Uruha A , Nishikawa A , Tsuburaya RS , Hamanaka K , Kuwana M , Watanabe Y , et al. Sarcoplasmic MxA expression: A valuable marker of dermatomyositis. Neurology. (2017) ;88: (5):493–500. |
[20] | Malik A , Hayat G , Kalia JS , Guzman MA Idiopathic Inflammatory Myopathies: Clinical Approach and Management. Front Neurol. (2016) ;7: :64. |
[21] | Allenbach Y , Benveniste O , Goebel HH , Stenzel W Integrated classification of inflammatory myopathies. Neuropathol Appl Neurobiol. (2017) ;43: (1):62–81. |
[22] | Benveniste O , Stenzel W , Allenbach Y Advances in serological diagnostics of inflammatory myopathies. Curr Opin Neurol. (2016) ;29: (5):662–73. |
[23] | Betteridge Z , McHugh N Myositis-specific autoantibodies: An important tool to support diagnosis of myositis. J Intern Med. (2016) ;280: (1):8–23. |
[24] | van de Vlekkert J , Hoogendijk JE , de Visser M Myositis with endomysial cell invasion indicates inclusion body myositis even if other criteria are not fulfilled. Neuromuscul Disord. (2015) ;25: (6):451–6. |
[25] | Ikenaga C , Kubota A , Kadoya M , Taira K , Uchio N , Hida A , et al. Clinicopathologic features of myositis patients with CD8-MHC-1 complex pathology. Neurology. (2017) ;89: (10):1060–8. |
[26] | Chahin N , Engel AG Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology. (2008) ;70: (6):418–24. |
[27] | Pinal-Fernandez I , Casciola-Rosen LA , Christopher-Stine L , Corse AM , Mammen AL The Prevalence of Individual Histopathologic Features Varies according to Autoantibody Status in Muscle Biopsies from Patients with Dermatomyositis. J Rheumatol. (2015) ;42: (8):1448–54. |
[28] | Satoh M , Tanaka S , Ceribelli A , Calise SJ , Chan EK A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin Rev Allergy Immunol. (2017) ;52: (1):1–19. |
[29] | Miller T , Al-Lozi MT , Lopate G , Pestronk A Myopathy with antibodies to the signal recognition particle: Clinical and pathological features. J Neurol Neurosurg Psychiatry. (2002) ;73: (4):420–8. |
[30] | Targoff IN , Johnson AE , Miller FW Antibody to signal recognition particle in polymyositis. Arthritis Rheum. (1990) ;33: (9):1361–70. |
[31] | Suzuki S , Nishikawa A , Kuwana M , Nishimura H , Watanabe Y , Nakahara J , et al. Inflammatory myopathy withanti-signal recognition particle antibodies: Case series of 100 patients. Orphanet J Rare Dis. (2015) ;10: :61. |
[32] | Christopher-Stine L , Casciola-Rosen LA , Hong G , Chung T , Corse AM , Mammen AL A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. (2010) ;62: (9):2757–66. |
[33] | Mammen AL , Chung T , Christopher-Stine L , Rosen P , Rosen A , Doering KR , et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. (2011) ;63: (3):713–21. |
[34] | Allenbach Y , Drouot L , Rigolet A , Charuel JL , Jouen F , Romero NB , et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: Inconstant exposure to statin. Medicine (Baltimore). (2014) ;93: (3):150–7. |
[35] | Allenbach Y , Keraen J , Bouvier AM , Jooste V , Champtiaux N , Hervier B , et al. High risk of cancer in autoimmune necrotizing myopathies: Usefulness of myositis specific antibody. Brain. (2016) ;139: (Pt 8):2131–5. |
[36] | Stenzel W , Goebel HH , Aronica E Review: Immune-mediated necrotizingmyopathies–a heterogeneous group of diseases with specificmyopathological features. Neuropathol Appl Neurobiol. (2012) ;38: (7):632–46. |
[37] | Schroder NW , Goebel HH , Brandis A , Ladhoff AM , Heppner FL , StenzelWPipestem capillaries in necrotizing myopathy revisited. Neuromuscul Disord. (2013) ;23: (1):66–74. |
[38] | Troyanov Y , Targoff IN , Tremblay JL , Goulet JR , Raymond Y , Senecal JL Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: Analysis of 100 French Canadian patients. Medicine (Baltimore). (2005) ;84: (4):231–49. |
[39] | Lega JC , Fabien N , Reynaud Q , Durieu I , Durupt S , Dutertre M , et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: A meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. (2014) ;13: (9):883–91. |
[40] | Hervier B , Devilliers H , Stanciu R , Meyer A , Uzunhan Y , Masseau A , et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: Phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev. (2012) ;12: (2):210–7. |
[41] | Aggarwal R , Cassidy E , Fertig N , Koontz DC , Lucas M , Ascherman DP , et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. (2014) ;73: (1):227–32. |
[42] | Rigolet A , Musset L , Dubourg O , Maisonobe T , Grenier P , Charuel JL , et al. Inflammatory myopathies with anti-Ku antibodies: A prognosis dependent on associated lung disease. Medicine (Baltimore). (2012) ;91: (2):95–102. |
[43] | Marie I , Hatron PY , Dominique S , Cherin P , Mouthon L , Menard JF , et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum. (2012) ;41: (6):890–9. |
[44] | Mescam-Mancini L , Allenbach Y , Hervier B , Devilliers H , Mariampillay K , Dubourg O , et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain. (2015) ;138: (Pt 9):2485–92. |
[45] | Stenzel W , Preusse C , Allenbach Y , Pehl D , Junckerstorff R , Heppner FL , et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology. (2015) ;84: (13):1346–54. |
[46] | Uruha A , Suzuki S , Suzuki N , Nishino I Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain. (2016) ;139: (Pt 9):e50. |
[47] | Schmidt K , Schmidt J Inclusion body myositis: Advancements in diagnosis, pathomechanisms, and treatment. Curr Opin Rheumatol. (2017) ;29: (6):632–8. |
[48] | Price MA , Barghout V , Benveniste O , Christopher-Stine L , Corbett A , de Visser M , et al. Mortality and Causes of Death in Patients with Sporadic Inclusion Body Myositis: Survey Study Based on the Clinical Experience of Specialists in Australia, Europe and the USA. J Neuromuscul Dis. (2016) ;3: (1):67–75. |
[49] | Cox FM , Titulaer MJ , Sont JK , Wintzen AR , Verschuuren JJ , Badrising UA A 12-year follow-up in sporadic inclusion body myositis: An end stage with major disabilities. Brain. (2011) ;134: (Pt 11):3167–75. |
[50] | Benveniste O , Guiguet M , Freebody J , Dubourg O , Squier W , Maisonobe T , et al. Long-term observational study of sporadic inclusion body myositis. Brain. (2011) ;134: (Pt 11):3176–84. |
[51] | Pluk H , van Hoeve BJ , van Dooren SH , Stammen-Vogelzangs J , van der Heijden A , Schelhaas HJ , et al. Autoantibodies to cytosolic 5’-nucleotidase 1A in inclusion body myositis. Ann Neurol. (2013) ;73: (3):397–407. |
[52] | Larman HB , Salajegheh M , Nazareno R , Lam T , Sauld J , Steen H , et al. Cytosolic 5’-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. (2013) ;73: (3):408–18. |
[53] | Lloyd TE , Christopher-Stine L , Pinal-Fernandez I , Tiniakou E , Petri M , Baer A , et al. Cytosolic 5’-Nucleotidase 1A As a Target of Circulating Autoantibodies in Autoimmune Diseases. Arthritis Care Res (Hoboken). (2016) ;68: (1):66–71. |
[54] | Herbert MK , Stammen-Vogelzangs J , Verbeek MM , Rietveld A , Lundberg IE , Chinoy H , et al. Disease specificity of autoantibodies to cytosolic 5’-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis. (2016) ;75: (4):696–701. |
[55] | Muro Y , Nakanishi H , Katsuno M , Kono M , Akiyama M Prevalence of anti-NT5C1A antibodies in Japanese patients withautoimmune rheumatic diseases in comparison with other patient cohorts. Clin Chim Acta. (2017) ;472: :1–4. |
[56] | Goyal NA , Cash TM , Alam U , Enam S , Tierney P , Araujo N , et al. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatry. (2016) ;87: (4):373–8. |
[57] | Lilleker JB , Rietveld A , Pye SR , Mariampillai K , Benveniste O , Peeters MT , et al. Cytosolic 5’-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis. (2017) ;76: (5):862–8. |
[58] | Olthoff A , Carstens PO , Zhang S , von FE , Friede T , Lotz J , et al. Evaluation of dysphagia by novel real-time MRI. Neurology. (2016) ;87: (20):2132–8. |
[59] | Rose MR188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December Naarden, The Netherlands. Neuromuscul Disord. (2013) ;23: (12):1044–55. |
[60] | Zahr ZA , Baer AN Malignancy in myositis. Curr Rheumatol Re. (2011) ;13: (3):208–15. |
[61] | Selva-O’Callaghan A , Trallero-Araguas E , Grau-Junyent JM , Labrador-Horrillo M Malignancy and myositis: Novel autoantibodies and new insights. Curr Opin Rheumatol. (2010) ;22: (6):627–32. |
[62] | Qiang JK , Kim WB , Baibergenova A , Alhusayen R Risk of Malignancy in Dermatomyositis and Polymyositis. J CutanMed Surg. (2017) ;21: (2):131–6. |
[63] | Olazagasti JM , Baez PJ , Wetter DA , Ernste FC Cancer risk in dermatomyositis: A meta-analysis of cohort studies. Am J Clin Dermatol. (2015) ;16: (2):89–98. |
[64] | Lega JC , Reynaud Q , Belot A , Fabien N , Durieu I , Cottin V Idiopathic inflammatory myopathies and the lung. Eur Respir Rev. (2015) ;24: (136):216–38. |
[65] | Hallowell RW , Danoff SK Interstitial lung disease associated with the idiopathic inflammatory myopathies and the antisynthetase syndrome: Recent advances. Curr Opin Rheumatol. (2014) ;26: (6):684–9. |
[66] | Morisset J , Johnson C , Rich E , Collard HR , Lee JS Management of Myositis-Related Interstitial Lung Disease. Chest. (2016) ;150: (5):1118–28. |
[67] | Schwartz T , Diederichsen LP , Lundberg IE , Sjaastad I , Sanner H Cardiac involvement in adult and juvenile idiopathic inflammatory myopathies. RMD Open. (2016) ;2: (2):e000291. |
[68] | Gadiparthi C , Hans A , Potts K , Ismail MK Gastrointestinal and Hepatic Disease in the Inflammatory Myopathies. Rheum Dis Clin North Am. (2018) ;44: (1):113–29. |
[69] | Hermans MAW , Miedema JR , Verdijk RM , van Daele PLA Scleroderma-like renal crisis in a patient with anti-threonyl-tRNA synthetase-associated antisynthetase syndrome. Rheumatology (Oxford). (2018) )4831053. |
[70] | Lundberg IE , Tjarnlund A , Bottai M , Werth VP , Pilkington C , de VM , et al. European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Arthritis Rheumatol. (2017) ;69: (12):2271–82. |
[71] | Lundberg IE , Tjarnlund A , Bottai M , Werth VP , Pilkington C , Visser M , et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. (2017) ;76: (12):1955–64. |
[72] | Bohan A , Peter JB Polymyositis and dermatomyositis (first of two parts). N Engl J Med. (1975) ;292: (7):344–7. |
[73] | Tanimoto K , Nakano K , Kano S , Mori S , Ueki H , Nishitani H , et al. Classification criteria for polymyositis and dermatomyositis. JRheumatol. (1995) ;22: (4):668–74. |
[74] | Targoff IN , Miller FW , Medsger TA Jr , Oddis CV Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol. (1997) ;9: (6):527–35. |
[75] | Dalakas MC , Hohlfeld R Polymyositis and dermatomyositis. Lancet. (2003) )362: (9388)971–82. |
[76] | Hocevar A , Rotar Z , Krosel M , Cucnik S , Tomsic M Performance of the 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies in clinical practice. Ann Rheum Dis. (2017) )annrheumdis-212774. |
[77] | Lundberg IE , Bottai M , Tjarnlund A Response to: ‘Performance of the 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies in clinical practice’ by Hocevar et al. Ann Rheum Dis. (2018) )annrheumdis-212786. |
[78] | Lundberg IE , Tjarnlund A Response to: ’2017 EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups: Little emphasis on autoantibodies, why?’ by Malaviya. Ann Rheum Dis.. (2017) ;annrheumdis-212709. |
[79] | Griggs RC , Askanas V , DiMauro S , Engel A , Karpati G , Mendell JR , et al. Inclusion body myositis and myopathies. Ann Neurol. (1995) ;38: (5):705–13. |
[80] | Badrising UA , Maat-Schieman M , van Duinen SG , Breedveld F , van Doorn P , van Engelen B , et al. Epidemiology of inclusion body myositis in the Netherlands: A nationwide study. Neurology. (2000) ;55: (9):1385–7. |
[81] | Dalakas MCSporadic inclusion body myositis-diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol. (2006) ;2: (8):437–47. |
[82] | Lloyd TE , Mammen AL , Amato AA , Weiss MD , Needham M , Greenberg SA Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. (2014) ;83: (5):426–33. |
[83] | Day J , Patel S , Limaye V The role of magnetic resonance imaging techniques in evaluation and management of the idiopathic inflammatory myopathies. Semin Arthritis Rheum. (2017) ;46: (5):642–9. |
[84] | Pipitone NValue of MRI in diagnostics and evaluation of myositis. Curr Opin Rheumatol. (2016) ;28: (6):625–30. |
[85] | Burlina P , Billings S , Joshi N , Albayda J Automated diagnosis of myositis from muscle ultrasound: Exploring theuse of machine learning and deep learning methods. PLoS One. (2017) ;12: (8):e0184059. |
[86] | Adler RS , Garofalo G Ultrasound in the evaluation of the inflammatory myopathies. Curr Rheumatol Re. (2009) ;11: (4):302–8. |
[87] | Weber MAUltrasound in the inflammatory myopathies. Ann N Y Acad Sci. (2009) ;1154: :159–70. |
[88] | Huang ZG , Gao BX , Chen H , Yang MX , Chen XL , Yan R , et al. An efficacy analysis of whole-body magnetic resonance imaging in the diagnosis and follow-up of polymyositis and dermatomyositis. PLoS One. (2017) ;12: (7):e0181069. |
[89] | Elessawy SS , Abdelsalam EM , Abdel RE , Tharwat S Whole-body MRI for full assessment and characterization of diffuse inflammatory myopathy. Acta Radiol Open. (2016) ;5: (9):2058460116668216. |
[90] | Castro TC , Lederman H , Terreri MT , Caldana WI , Zanoteli E , Hilario MO Whole-body magnetic resonance imaging in the assessment of muscular involvement in juvenile dermatomyositis/polymyositis patients. Scand J Rheumatol. (2014) ;43: (4):329–33. |
[91] | Malattia C , Damasio MB , Madeo A , Pistorio A , Providenti A , Pederzoli S , et al. Whole-body MRI in the assessment of disease activity in juvenile dermatomyositis. Ann Rheum Dis. (2014) ;73: (6):1083–90. |
[92] | Guimaraes JB , Zanoteli E , Link TM , de Camargo LV , Facchetti L , Nardo L , et al. Sporadic Inclusion Body Myositis: MRI Findings and Correlation With Clinical and Functional Parameters. AJR Am J Roentgenol. (2017) ;209: (6):1340–7. |
[93] | Tasca G , Monforte M , De FC , Kley RA , Ricci E , Mirabella M Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve. (2015) ;52: (6):956–62. |
[94] | Kubinova K , Mann H , Vencovsky J MRI scoring methods used in evaluation of muscle involvement in patients with idiopathic inflammatory myopathies. Curr Opin Rheumatol. (2017) ;29: (6):623–31. |
[95] | De Paepe B , Zschuntzsch J Scanning for Therapeutic Targets within the Cytokine Network of Idiopathic Inflammatory Myopathies. Int J Mol Sci. (2015) ;16: (8):18683–713. |
[96] | Ceribelli A , De SM , Isailovic N , Gershwin ME , Selmi C The Immune Response and the Pathogenesis of Idiopathic Inflammatory Myositis: A Critical Review. Clin Rev Allergy Immunol. (2017) ;52: (1):58–70. |
[97] | Schmidt J , Dalakas MC Pathomechanisms of inflammatory myopathies: Recent advances and implications for diagnosis and therapies. Expert Opin Med Diagn. (2010) ;4: (3):241–50. |
[98] | Rothwell S , Lamb JA , Chinoy H New developments in genetics of myositis. Curr Opin Rheumatol. (2016) ;28: (6):651–6. |
[99] | Manole E , Bastian AE , Butoianu N , Goebel HH Myositis non-inflammatory mechanisms: An up-dated review. J Immunoassay Immunochem. (2017) ;38: (2):115–26. |
[100] | Lightfoot AP , Cooper RG The role of myokines in muscle health and disease. Curr Opin Rheumatol. (2016) ;28: (6):661–6. |
[101] | Day J , Otto S , Proudman S , Hayball JD , Limaye V Dysregulated innate immune function in the aetiopathogenesis of idiopathic inflammatory myopathies. Autoimmun Rev. (2017) ;16: (1):87–95. |
[102] | Keller CW , Schmidt J , Lunemann JD Immune and myodegenerative pathomechanisms in inclusion body myositis. Ann Clin Transl Neurol. (2017) ;4: (6):422–45. |
[103] | Goebels N , Michaelis D , Engelhardt M , Huber S , Bender A , Pongratz D , et al. Differential expression of perforin in muscle-infiltrating Tcells in polymyositis and dermatomyositis. J Clin Invest. (1996) ;97: (12):2905–10. |
[104] | Schmidt J , Rakocevic G , Raju R , Dalakas MC Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: Significance for CD8(+) T cell cytotoxicity. Brain. (2004) ;127: :1182–90. |
[105] | Lindberg C , Oldfors A , Tarkowski A Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur J Immunol. (1994) ;24: (11):2659–63. |
[106] | Benveniste O , Herson S , Salomon B , Dimitri D , Trebeden-Negre H , Jean L , et al. Long-term persistence of clonally expanded T cells in patients with polymyositis. Ann Neurol. (2004) ;56: (6):867–72. |
[107] | Amemiya K , Granger RP , Dalakas MC Clonal restriction of T-cellreceptor expression by infiltrating lymphocytes in inclusion bodymyositis persists over time. Studies in repeated muscle biopsies.Brain. (2000) ;123: (Pt 10):2030–9. |
[108] | Hofbauer M , Wiesener S , Babbe H , Roers A , Wekerle H , Dornmair K , et al. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci U S A. (2003) ;100: (7):4090–5. |
[109] | Bradshaw EM , Orihuela A , McArdel SL , Salajegheh M , Amato AA , Hafler DA , et al. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol. (2007) ;178: (1):547–56. |
[110] | Greenberg SA , Bradshaw EM , Pinkus JL , Pinkus GS , Burleson T , Due B , et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. (2005) ;65: (11):1782–7. |
[111] | De Paepe B , Creus KK , De Bleecker JL Role of cytokines and chemokines in idiopathic inflammatory myopathies. Curr Opin Rheumatol. (2009) ;21: (6):610–6. |
[112] | Moran EM , Mastaglia FL Cytokines in immune-mediated inflammatory myopathies: Cellular sources, multiple actions and therapeutic implications. Clin Exp Immunol. (2014) ;178: (3):405–15. |
[113] | Tournadre A , Lenief V , Miossec P Expression of Toll-like receptor 3 and Toll-like receptor 7 in muscle is characteristic of inflammatory myopathy and is differentially regulated by Th1 and Th17 cytokines. Arthritis Rheum. (2010) ;62: (7):2144–51. |
[114] | Cappelletti C , Baggi F , Zolezzi F , Biancolini D , Beretta O , Severa M , et al. Type I interferon and Toll-like receptor expression characterizes inflammatory myopathies. Neurology. (2011) ;76: (24):2079–88. |
[115] | Coley W , Rayavarapu S , Nagaraju K Role of non-immune mechanisms of muscle damage in idiopathic inflammatory myopathies. Arthritis Res Ther. (2012) ;14: (2):209. |
[116] | Lightfoot AP , Nagaraju K , McArdle A , Cooper RG Understanding the origin of non-immune cell-mediated weakness in the idiopathic inflammatory myopathies - potential role of ER stress pathways. Curr Opin Rheumatol. (2015) ;27: (6):580–5. |
[117] | Schmidt J , Barthel K , Wrede A , Salajegheh M , Bahr M , Dalakas MC Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain. (2008) ;131: (Pt 5):1228–40. |
[118] | Muth IE , Zschuntzsch J , Kleinschnitz K , Wrede A , Gerhardt E , Balcarek P , et al. HMGB1 and RAGE in skeletal muscle inflammation: Implications for protein accumulation in inclusion body myositis. Exp Neurol. (2015) ;271: :189–97. |
[119] | Schmidt J , Barthel K , Zschuntzsch J , Muth IE , Swindle EJ , Hombach A , et al. Nitric oxide stress in sporadic inclusion body myositis muscle fibres: Inhibition of inducible nitric oxide synthase prevents interleukin-1beta-induced accumulation of beta-amyloid and cell death. Brain. (2012) ;135: (Pt 4):1102–14. |
[120] | Muth IE , Barthel K , Bahr M , Dalakas MC , Schmidt J Proinflammatory cell stress in sporadic inclusion body myositis muscle: Overexpression of alphaB-crystallin is associated with amyloid precursor protein and accumulation of beta-amyloid. J Neurol Neurosurg Psychiatry. (2009) ;80: (12):1344–9. |
[121] | Miller FW , Chen W , O’Hanlon TP , Cooper RG , Vencovsky J , Rider LG , et al. Genome-wide association study identifies HLA 8. 1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. (2015) ;16: (7):470–80. |
[122] | Rothwell S , Cooper RG , Lundberg IE , Miller FW , Gregersen PK , Bowes J , et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis. (2016) ;75: (8):1558–66. |
[123] | Rothwell S , Cooper RG , Lundberg IE , Gregersen PK , Hanna MG , MachadoPM, et al. Immune-Array Analysis in Sporadic Inclusion Body MyositisReveals HLA-DRB1 Amino Acid Heterogeneity Across the MyositisSpectrum. Arthritis Rheumatol. (2017) ;69: (5):1090–9. |
[124] | Lunemann JD , Schmidt J , Schmid D , Barthel K , Wrede A , Dalakas MC , et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. (2007) ;61: (5):476–83. |
[125] | Guttsches AK , Brady S , Krause K , Maerkens A , Uszkoreit J , Eisenacher M , et al. Proteomics of rimmed vacuoles define new risk allele in inclusion body myositis. Ann Neurol. (2017) ;81: (2):227–39. |
[126] | Leff RL , Burgess SH , Miller FW , Love LA , Targoff IN , Dalakas MC , et al. Distinct seasonal patterns in the onset of adult idiopathic inflammatory myopathy in patients with anti-Jo-1 and anti-signal recognition particle autoantibodies. Arthritis Rheum. (1991) ;34: (11):1391–6. |
[127] | Shah M , Targoff IN , Rice MM , Miller FW , Rider LG Brief report: Ultraviolet radiation exposure is associated with clinical and autoantibody phenotypes in juvenile myositis. Arthritis Rheum. (2013) ;65: (7):1934–41. |
[128] | Love LA , Weinberg CR , McConnaughey DR , Oddis CV , Medsger TAJr , Reveille JD , et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. (2009) ;60: (8):2499–504. |
[129] | Uruha A , Noguchi S , Hayashi YK , Tsuburaya RS , Yonekawa T , Nonaka I , et al. Hepatitis C virus infection in inclusion body myositis: A case-control study. Neurology. (2016) ;86: (3):211–7. |
[130] | Miller FWEnvironmental agents and autoimmune diseases. Adv Exp Med Biol. (2011) ;711: :61–81. |
[131] | Mamyrova G , Rider LG , Ehrlich A , Jones O , Pachman LM , Nickeson R , et al. Environmental factors associated with disease flare in juvenile and adult dermatomyositis. Rheumatology (Oxford). (2017) ;56: (8):1342–7. |
[132] | Marie IMorbidity and mortality in adult polymyositis anddermatomyositis. Curr Rheumatol Rep. (2012) ;14: (3):275–85. |
[133] | Bronner IM , van der Meulen MF , de VM , Kalmijn S , van Venrooij WJ , Voskuyl AE , et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis. (2006) ;65: (11):1456–61. |
[134] | van de Vlekkert J , Hoogendijk JE , de Visser M Long-term follow-up of 62 patients with myositis. J Neurol. (2014) ;261: (5):992–8. |
[135] | Uchino M , Araki S , Yoshida O , Uekawa K , Nagata J High single-dose alternate-day corticosteroid regimens in treatment of polymyositis. J Neurol. (1985) ;232: (3):175–8. |
[136] | Uchino M , Yamashita S , Uchino K , Hara A , Koide T , Suga T , et al. Long-term outcome of polymyositis treated with high single-dose alternate-day prednisolone therapy. Eur Neurol. (2012) ;68: (2):117–21. |
[137] | van de Vlekkert J , Hoogendijk JE , de Haan RJ , Algra A , van der Tweel I , van der Pol WL , et al. Oral dexamethasone pulse therapy versus daily prednisolone in sub-acute onset myositis, a randomised clinical trial. Neuromuscul Disord. (2010) ;20: (6):382–9. |
[138] | Gordon PA , Winer JB , Hoogendijk JE , Choy EH Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev.. (2012) )8CD003643. |
[139] | Leroy C , Rigot JM , Leroy M , Decanter C , Le MK , Parent AS , et al. Immunosuppressive drugs and fertility. Orphanet J Rare Dis. (2015) ;10: :136. |
[140] | Hoa SAT , Hudson M Critical review of the role of intravenous immunoglobulins in idiopathic inflammatory myopathies. Semin Arthritis Rheum. (2017) ;46: (4):488–508. |
[141] | Oddis CV , Reed AM , Aggarwal R , Rider LG , Ascherman DP , Levesque MC , et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum. (2013) ;65: (2):314–24. |
[142] | de Visser MThe efficacy of rituximab in refractory myositis: The jury is still out. Arthritis Rheum. (2013) ;65: (2):303–6. |
[143] | Pinal-Fernandez I , Parks C , Werner JL , Albayda J , Paik J , Danoff SK , et al. Longitudinal Course of Disease in a Large Cohort of Myositis Patients With Autoantibodies Recognizing the Signal Recognition Particle. Arthritis Care Res (Hoboken). (2017) ;69: (2):263–70. |
[144] | Werner JL , Christopher-Stine L , Ghazarian SR , Pak KS , Kus JE , Daya NR , et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. (2012) ;64: (12):4087–93. |
[145] | Dalakas MC , Illa I , Dambrosia JM , Soueidan SA , Stein DP , Otero C , etalA controlled trial of high-dose intravenous immune globulininfusions as treatment for dermatomyositis. N Engl J Med. (1993) ;329: (27):1993–2000. |
[146] | Cavagna L , Monti S , Caporali R , Gatto M , Iaccarino L , Doria A How I treat idiopathic patients with inflammatory myopathies in the clinical practice. Autoimmun Rev. (2017) ;16: (10):999–1007. |
[147] | Alexanderson HPhysical exercise as a treatment for adult and juvenile myositis. J Intern Med. (2016) ;280: (1):75–96. |
[148] | Dalakas MC , Rakocevic G , Schmidt J , Salajegheh M , McElroy B , Harris-Love MO , et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain. (2009) ;132: (Pt 6):1536–44. |
[149] | Schmidt K , Kleinschnitz K , Rakocevic G , Dalakas MC , Schmidt J Molecular treatment effects of alemtuzumab in skeletal muscles of patients with IBM. BMC Neurol. (2016) ;16: :48. |
[150] | Dalakas MC , Sonies B , Dambrosia J , Sekul E , Cupler E , Sivakumar K Treatment of inclusion-body myositis with IVIg: A double-blind, placebo- controlled study. Neurology. (1997) ;48: (3):712–6. |
[151] | Walter MC , Lochmuller H , Toepfer M , Schlotter B , Reilich P , Schroder M , et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: A double-blind, placebo-controlled study. J Neurol. (2000) ;247: (1):22–8. |
[152] | Dalakas MC , Koffman B , Fujii M , Spector S , Sivakumar K , Cupler E A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology. (2001) ;56: (3):323–7. |
[153] | Dobloug C , Walle-Hansen R , Gran JT , Molberg O Long-term follow-up of sporadic inclusion body myositis treated with intravenous immunoglobul A retrospective study of 16 patients Clin Exp Rheumatol. (2012) ;30: (6):838–42. |
[154] | Cherin P , Pelletier S , Teixeira A , Laforet P , Genereau T , Simon A , et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: An open study with thirty-five adult patients. Arthritis Rheum. (2002) ;46: (2):467–74. |
[155] | Cherin P , Delain JC , de JC , Crave JC Subcutaneous Immunoglobulin Use in Inclusion Body Myositis: A Review of 6 Cases. Case Rep Neurol. (2015) ;7: (3):227–32. |
[156] | Breithaupt M , Schmidt J Treatment strategies for inclusion body myositis. Expert Opin Orphan Drugs. (2014) ;2: (12):1255–65. |
[157] | Breithaupt M , Schmidt J Update on treatment of inclusion body myositis. Curr Rheumatol Rep. (2013) ;15: (5):329. |
[158] | Jones KL , Sejersen T , Amato AA , Hilton-Jones D , Schmidt J , Wallace AC , et al. A protocol to develop clinical guidelines for inclusion-body myositis. Muscle Nerve. (2016) ;53: (4):503–7. |
[159] | Cox FM , Verschuuren JJ , Verbist BM , Niks EH , Wintzen AR , Badrising UA Detecting dysphagia in inclusion body myositis. J Neurol. (2009) ;256: (12):2009–13. |
[160] | Langdon PC , Mulcahy K , Shepherd KL , Low VH , Mastaglia FL Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia. (2012) ;27: (3):408–17. |
[161] | Oh TH , Brumfield KA , Hoskin TL , Kasperbauer JL , Basford JR . Dysphagia in inclusion body myositis: Clinical features, management, and clinical outcome. Am J Phys Med Rehabil. (2008) ;87: (11):883–9. |
[162] | Liu LW , Tarnopolsky M , Armstrong D Injection of botulinum toxin A to the upper esophageal sphincter for oropharyngeal dysphagia in two patients with inclusion body myositis. Can J Gastroenterol. (2004) ;18: (6):397–9. |
[163] | Schrey A , Airas L , Jokela M , Pulkkinen J Botulinum toxin alleviates dysphagia of patients with inclusion body myositis. J Neurol Sci. (2017) ;380: :142–7. |
[164] | Di Pede C , Masiero S , Bonsangue V , Ragona RM , Del Felice A Botulinum toxin and rehabilitation treatment in inclusion body myositis for severe oropharyngeal dysphagia. Neurol Sci. (2016) ;37: (10):1743–5. |
[165] | Espinosa-Ortega HF , Moreno-Ramirez M , Alexanderson H Novel insights of disability assessment in adult myositis. Curr Opin Rheumatol. (2017) ;29: (6):591–7. |
[166] | Miller FW , Rider LG , Chung YL , Cooper R , Danko K , Farewell V , et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford). (2001) ;40: (11):1262–73. |
[167] | Rider LG , Werth VP , Huber AM , Alexanderson H , Rao AP , Ruperto N , et al. Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis: Physician and Patient/Parent Global Activity, Manual Muscle Testing (MMT), Health Assessment Questionnaire (HAQ)/Childhood Health Assessment Questionnaire (C-HAQ), Childhood Myositis Assessment Scale (CMAS), Myositis Disease Activity Assessment Tool (MDAAT), Disease Activity Score (DAS), Short Form 36 (SF-36), Child Health Questionnaire (CHQ), physician global damage, Myositis Damage Index (MDI), Quantitative Muscle Testing (QMT), Myositis Functional Index-2 (FI-2), Myositis Activities Profile (MAP), Inclusion Body Myositis Functional Rating Scale (IBMFRS), Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), Cutaneous Assessment Tool (CAT), Dermatomyositis Skin Severity Index (DSSI), Skindex, and Dermatology Life Quality Index (DLQI). Arthritis Care Res (Hoboken). (2011) ;63: (Suppl 11):S118–S157. |
[168] | Rider LG , Lachenbruch PA , Monroe JB , Ravelli A , Cabalar I , Feldman BM , et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis Rheum. (2009) ;60: (11):3425–35. |
[169] | Aggarwal R , Rider LG , Ruperto N , Bayat N , Erman B , Feldman BM , et al. American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. (2017) ;76: (5):792–801. |
[170] | Aggarwal R , Rider LG , Ruperto N , Bayat N , Erman B , Feldman BM , et al. American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Adult Dermatomyositis and Polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. (2017) ;69: (5):898–910. |
[171] | Rider LG , Aggarwal R , Pistorio A , Bayat N , Erman B , Feldman BM , et al. 2016 American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Juvenile Dermatomyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. (2017) ;76: (5):782–91. |
[172] | Rider LG , Aggarwal R , Pistorio A , Bayat N , Erman B , Feldman BM , et al. 2016 American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Juvenile Dermatomyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. (2017) ;69: (5):911–23. |
[173] | Alexanderson H , Broman L , Tollback A , Josefson A , Lundberg IE , Stenstrom CH Functional index- Validity and reliability of a disease-specific measure of impairment in patients with polymyositis and dermatomyositis. Arthritis Rheum. (2006) ;55: (1):114–22. |
[174] | Alfano LN , Lowes LP , Dvorchik I , Yin H , Maus EG , Flanigan KM , et al. The 2-min walk test is sufficient for evaluating walking abilities in sporadic inclusion body myositis. Neuromuscul Disord. (2014) ;24: (3):222–6. |
[175] | Chansky PB , Olazagasti JM , Feng R , Werth VP Cutaneous dermatomyositis disease course followed over time using the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI). J Am Acad Dermatol. (2017) ;pii:S0190-9622(17):32578–1. |
[176] | Jackson CE , Barohn RJ , Gronseth G , Pandya S , Herbelin L Inclusion body myositis functional rating scale: A reliable and valid measure of disease severity. Muscle Nerve. (2008) ;37: (4):473–6. |
[177] | DeMuro C , Lewis S , Lowes L , Alfano L , Tseng B , Gnanasakthy A Development of the sporadic inclusion body myositis physical functioning assessment. Muscle Nerve. (2016) ;54: (4):653–7. |
[178] | Harris-Love MO , Joe G , Davenport TE , Koziol D , Abbett RK , Shrader JA , et al. Reliability of the adult myopathy assessment tool in individuals with myositis. Arthritis Care Res (Hoboken). (2015) ;67: (4):563–70. |
[179] | Park JK , Mecoli CA , Alexanderson H , Regardt M , Christopher-Stine L , Dominguez MC , et al. Advancing the Development of Patient-reported Outcomes for Adult Myositis at OMERACT 2016: An International Delphi Study. J Rheumatol. (2017) ;44: (11):1683–7. |
[180] | Wallace KL , Middleton S , Cook IJ Development and validation of a self-report symptom inventory to assess the severity of oral-pharyngeal dysphagia. Gastroenterology. (2000) ;118: (4):678–87. |
[181] | Leclair V , Lundberg IE Recent clinical trials in idiopathic inflammatory myopathies. Curr Opin Rheumatol. (2017) ;29: (6):652–9. |
[182] | Brunasso AM , Aberer W , Massone C New onset of dermatomyositis/polymyositis during anti-TNF-alpha therapies: A systematic literature review. ScientificWorldJournal. (2014) ;179180. |
[183] | Arandomized, pilot trial of etanercept in dermatomyositis. Ann Neurol. (2011) ;70: (3):427–36. |
[184] | Amato AA , Sivakumar K , Goyal N , David WS , Salajegheh M , Praestgaard J , et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. (2014) ;83: (24):2239–46. |
[185] | Benveniste O , Hogrel JY , Annoussamy M , Bachasson D , Rigolet A , Servais L , et al. Rapamycin Vs. Placebo for the Treatment of Inclusion Body Myositis: Improvement of the 6 Min Walking Distance, a Functional Scale, the FVC and Muscle Quantitative MRI. Arthritis Rheumatol.. (2018) ;69: (Suppl 10)(Abstract). |
[186] | Ahmed M , Machado PM , Miller A , Spicer C , Herbelin L , He J , et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med. (2016) ;8: (331):331ra41. |

