
New Pathways and Therapeutic Targets in Autoimmune Myasthenia Gravis
Abstract
Acquired Myasthenia Gravis (MG) is a neuromuscular disease caused by autoantibodies against components of the neuromuscular junction. It is a prototype organ-specific autoimmune disease with well-defined antigenic targets mainly the nicotinic acetylcholine receptor (AChR). Patients suffer from fluctuating, fatigable muscle weakness that worsens with activity and improves with rest.
Various therapeutic strategies have been used over the years to alleviate MG symptoms. These strategies aim at improving the transmission of the nerve impulse to muscle or at lowering the immune system with steroids or immunosuppressant drugs. Nevertheless, MG remains a chronic disease and symptoms tend to persist in many patients, some being or becoming refractory over time. In this review, based on recent experimental data on MG or based on results from clinical trials for other autoimmune diseases, we explore new potential therapeutic approaches for MG patients, going from non-specific approaches with the use of stem cells with their anti-inflammatory and immunosuppressive properties to targeted therapies using monoclonal antibodies specific for cell-surface antigens or circulating molecules.
Abbreviations
AID: | Autoimmune disease |
AChR: | Acetylcholine receptor |
BAFF: | B-cell activating factor |
GC: | Germinal center |
EAMG: | Experimental autoimmune MG |
IFN: | Interferon |
Ig: | Immunoglobulin |
IL: | Interleukin |
IS: | Immunosuppressive |
IVIg: | Intravenous immunoglobulin |
KO: | Knockout |
LRP4: | Low-density lipoprotein-related protein 4 |
MG: | Myasthenia Gravis |
MuSK: | Muscle specific kinase |
NMJ: | Neuromuscular junction |
RA: | Rheumatoid arthritis |
SC: | Stem cell |
SLE: | Systemic lupus erythematosus |
Th cell: | T helper cell |
Tfh cell: | T follicular helper cell |
Treg cell: | Regulatory T cell |
INTRODUCTION
Autoimmune Myasthenia Gravis (MG) is a rare neuromuscular disorder characterized by a defective transmission of nerve impulses to muscles. This defect is caused by an autoimmune attack against components of the neuromuscular junction (NMJ) on the postsynaptic membrane of striated skeletal muscles. The characteristic feature of MG is a fluctuating fatigable muscle weakness which can range between mild forms affecting eye muscles only to severe generalized forms.
Autoantibody targets defining MG subtypes
A large majority of MG patients can be defined by the presence of specific autoantibodies (Table 1). Anti-acetylcholine receptor (AChR) antibodies have been described in the seventies [1]. Since 2001 [2] and 2011 [3], two new subgroups of patients have been characterized with autoantibodies against other components of the NMJ: respectively, the muscle-specific kinase (MuSK) and the low-density lipoprotein-related protein 4 (LRP4) [4].
Table 1
Classification of MG patients according to the nature of the autoantibodies
| Antigen target | AChR | Clustered-AChR | MuSK | LRP4 |
|---|---|---|---|---|
| First publication | Aharonov et al. [1] | Leite et al. [12] | Hoch et al. [2] | Higuchi et al. [3] |
| Frequency | ∼85% | ∼5% | ∼5% | ∼1–5% |
| Patients | EOMG: women > men LOMG: men > women | ? | Young women | Young women |
| Isotypes | IgG1, IgG3 mainly IgG2, IgG4 low levels | IgG1 | IgG4 mostly | IgG1, IgG2 mainly IgG3 low levels |
| Implication of complement | Yes | Yes | No | Yes |
| Correlation with severity | No | ? | Yes | ? |
| Thymic pathology | EOMG: follicular hyperplasia LOMG: thymoma | Mild follicular hyperplasia | No | ? |
MG with anti-AChR antibodies
The nicotinic AChR is composed of five protein subunits and is concentrated on muscle cells at the NMJ. The anti-AChR antibodies in one individual are composed of different subclasses of immunoglobulins (IgGs) and preferentially target a region on the extracellular side of the α-subunit of the AChR called the MIR (main immunogenic region) [5].
A number of mechanisms underlie the reduction of AChRs at the NMJ in MG patients [6]. However, the classical complement pathway is mainly involved in the pathogenesis of AChR-MG [6]. Formation of the membrane attack complex (MAC) causes severe structural injury of endplates, and lyses the postsynaptic membrane which ultimately disperses and depletes AChRs at the NMJ [7]. It is suggested that in some cases, the destruction of the NMJ accounts for irreversible, fixed symptoms which do not improve even with an optimal treatment.
Two main clinical subgroups of MG patients with anti-AChR antibodies can be distinguished: the ocular and generalized form. The ocular form, representing approximately 15% of patients, is diagnosed when symptoms of the disease are limited to ocular symptoms for at least 2 years. Patients with a generalized form of MG display a fluctuating weakness affecting in a variable, often asymmetric way the axial muscles, the limbs and the so-called “bulbar” and respiratory muscles. Furthermore, this form of myasthenia can be divided into two subgroups according to the age of onset.
• Early-onset AChR-MG (EOMG) (age of onset <45–50 years old): most EOMG patients present a high level of anti-AChR antibodies often associated with thymic follicular hyperplasia [8]. More than 80% of patients with follicular hyperplasia are women [9].
• Late-onset AChR-MG (LOMG) (age of onset >45–50 years old): this subgroup is frequently associated with the presence of a thymoma [10]. In recent years, a form of MG appearing mainly in males after 60 years of age but not associated with a thymoma has been described and is sometime defined as a very late-onset form of AChR-MG) [11].
Irrespective of their clinical presentation, response to treatment, and thymic abnormalities, some patients have anti-AChR antibodies that are not detectable by the classical immunoprecipitation assay, as their autoantibodies only recognize the AChR in its clustered configuration. In these patients, the anti-AChR antibodies are predominantly of the IgG1 isotype and can also activate complement [12] (Table 1).
MG with anti- MuSK antibodies
MuSK is a transmembrane tyrosine kinase that is expressed predominantly at the postsynaptic membrane of the NMJ. MuSK binds to LRP4 and transmits an agrin-mediated signal for the clustering of AChR [13]. MG with anti-MuSK antibodies corresponds to about 5% of the MG patients [2, 4] (Table 1). They typically have severe clinical symptoms involving the facial, bulbar and respiratory muscles; ocular symptoms are less prominent than in AChR-MG. Muscle atrophy is also more common in these patients [14], especially at the tongue level. No thymic pathology is observed [14, 15]. The mechanism of action of anti-MuSK antibodies is not yet clearly defined as autoantibodies are of the IgG4 subtype that does not activate the complement cascade. However, in mouse models, MuSK antibodies lead to a pre- and post-disorganization of the NMJ [16].
MG with anti-LRP4 antibodies
The LRP4 protein belongs to a family of proteins that has been recently identified as the receptor for the neural agrin that can activate MuSK [17]. Patients with anti-LRP4 antibodies were only described in 2011 [3, 18] (Table 1). A study has investigated a large number LRP4-MG patients [19]. The clinical data collected for LRP4-MG patients showed that at disease onset, symptoms are mild with some identified thymic changes (32% with follicular hyperplasia, none with thymoma). These patients are predominantly of the IgG1 and IgG2 subtypes that can activate the complement cascade. In the LRP4 induced-mouse model, alterations of the NMJ is observed [20]. Overall, the response of LRP4-MG patients to treatment seems more similar to that of AChR-MG than of MuSK-MG patients [19]. Unfortunately, so far, there are no routine lab tests to detect efficiently anti-LRP4 antibodies.
Alteration of the immune system in AChR-MG
Thymic abnormalities
The thymus provides a complex environment essential for the differentiation and generation of the T-cell repertoire: CD4 + and CD8 + T cells. In AChR-MG, histological abnormalities are very often found in the thymus [21]. In EOMG cases, 50–60% of the patients exhibit thymic hyperplasia of lympho-proliferative origin characterized by ectopic germinal centers (GCs) and 15% of the patients have a thymoma [22].
The hyperplastic thymus in AChR-MG displays numerous features normally only observed in secondary lymphoid organs [8, 23]. The hyperplastic thymus includes all the components of the anti-AChR response: AChR [24], B cells producing anti-AChR antibodies [25, 26], and anti-AChR autoreactive T cells [27]. The proportion of GCs correlates with the anti-AChR antibody titer [22, 28], which generally decreases after thymectomy [29]. All these observations support the role of the thymus in the pathogenesis of AChR-MG, and thymectomy is often advised for AChR-MG patients [30]. In contrast, the thymus of MuSK-MG patients shows no pathological changes, and beneficial effects of thymectomy have not been proven for this subgroup [14, 31]. There is still no clear information on the involvement of the thymus in LRP4-MG.
Imbalances of the immune system
The production of antibodies is dependent on CD4 + T cells, necessary to induce a subsequent B-cell response. Naive CD4 + T cells differentiate into different T helper (Th) cell subsets including Th1, Th2, Th17, T follicular helper (Tfh) cells, and regulatory T (Treg) cells. Both Th1 and Th2 cells are involved in MG pathogenic mechanisms, as demonstrated by type II interferon (IFN-II) and interleukin (IL)-4 production by peripheral blood cells [32, 33]. Tfh cells are a subset distinguishable from other Th cells by several criteria, including CXCR5 expression, location/migration (B-cell follicles), and function (B-cell help). MG patients showed a significantly higher frequency of Tfh in correlation with disease severity [34].
In recent years, autoimmunity has been reconsidered as being mainly controlled by the balance between inflammatory Th17 cells and Treg cells [35]. An increase in IL-17, a Th17 cytokine, has been described in the serum of AChR-MG patients but also in the thymus where it could be involved in GC development [36–38]. In AChR-MG patients defects have been observed about Treg cells that are clearly less functional [39, 40]. The altered immune regulatory function observed for T cells in MG patients is also due to effector T cells that are resistant to suppression [41].
Altogether, these data clearly suggest an imbalance between Treg cells and effector T cells (Th1, Th2, Tfh, and Th17) in MG patients. However, it is not clearly defined if these alterations of the immune system pre-exist in susceptible MG patients before the onset of the disease or if they are the consequence of the inflammatory environment.
Classical therapeutic approaches
Treating MG implies considering several parameters: the form of the disease (i.e. ocular, mild or severe generalized MG), the age of the patient, and the presence of other conditions (e.g. pregnancy, severe diabetes or arterial hypertension) or treatments that may lead to an absolute or relative contraindication of a potentially useful drug. Most patients will need a long-term treatment, sometimes for their whole life, but choosing the optimal treatment is not always easy, as reliable, controlled studies comparing the different treatment options are scarce, largely due to the vast heterogeneity of patients’ presentation and severity, and also to the differences observed in their management according to the “local culture” of their physicians. The “classical” treatments of MG include molecules alleviating the symptoms, drugs targeting the auto-immune mechanisms of the disease, and thymectomy. Beside the long-term management of the disease, severe myasthenic crises require particular treatments, including intravenous immunoglobulin (IVIg) infusions, plasmapheresis and non-specific intensive care unit management in case of respiratory involvement. One should also always remind that myasthenic symptoms can worsen (sometimes dramatically) due to potentially aggravating drugs, such as quinine, macrolides or beta-blockers.
Cholinesterase inhibitors
By inhibiting the action of acetylcholinesterase at the NMJ, these molecules increase the half-life of acetylcholine and optimize its potential interaction with AChR. They should be part of the first-line treatment of MG patients [42]. Pyridostigmine 60 mg is the main drug used in clinical practice throughout the world, and gives relief at a variable dose according to the patients’ symptoms. In some countries, ambemonium chloride 10 mg can be an alternative to pyridostigmine, especially in case of poor tolerance. The 180 mg extended release form of pyridostigmine is useful in patients who still have significant symptoms when they wake up. The drug intake should be prescribed late in the evening. Side-effects of cholinesterase inhibitors include muscle cramps and contractures, fasciculations, diarrhea. A cholinergic crisis with muscle weakness is possible in case of overdosage of the molecule and should be differentiated from myasthenic paralysis. Of note is the fact that anti-MuSK MG patients often responds poorly to cholinesterase inhibitors, and may even occasionally worsen under treatment.
Steroids
Steroids (or corticosteroids) work by decreasing inflammation and reducing the activity of the immune system. They are used in most patients who do not meet treatment objectives on pyridostigmine alone. The usual dose of prednisone prescribed in MG is 0.5 mg/kg/day in purely ocular forms, and 1 mg/kg/day in generalized forms. The tapering scheme of steroids varies, according to the patients’ response and local habits of the treating physicians. Three main problems may occur in practice: the patients may prove resistant or dependent on steroids and/or develop side-effects with a frequency increasing with treatment dose and duration. However, some patients reach long-standing, remission with no or minimal symptoms with a few milligrams of prednisone alone, which may be maintained over time if MG symptoms reappear when trying to stop the drug [42].
Immunosuppressive (IS) drugs
They can be an alternative to steroids, especially in patients over 60 years of age, in steroid-resistant forms of MG. They are also used as a concomitant treatment which allows tapering the doses of prednisone in steroid-dependent patients with generalized forms of MG. Prescribing both steroids and IS drugs in the first place is a common habit in some centers, aiming at limiting the total long-term intake of steroids. Azathioprine, mycophenolate mofetil, cyclosporine, methotrexate and tacrolimus are currently considered as potentially useful in MG [42]. The first two drugs are generally used as first-line IS drugs, and all of them require an adequate monitoring of potential side-effects. Cyclophosphamide, a treatment used in chemotherapies but also suppressing the immune system, can also be occasionally proposed in non-responders [43].
One difficult question is the acceptable duration of treatment with IS drugs. There is currently no consensus on that point, and some patients remain heavily dependent, forbidding the discontinuation of IS drugs.
Rituximab
This anti-CD20 B-cell depleting monoclonal antibody has proved to be a major weapon in the fight against cases of MG which do not meet therapeutic goals with IS treatment. At the present time, it is thus a second-line treatment, all the more so that rare but severe complications (including progressive multifocal leukoencephalopathy) may potentially occur [44, 45]. However, it is questionable whether the risks of complications do not increase with the burden of immunosuppressant treatments preceding the introduction of Rituximab. Unfortunately, the data obtained on Rituximab over the last few years in MG are very heterogeneous in terms of treatment regimen, evaluation and patients’ previous management, and the best therapeutic scheme is not yet determined. A recent meta-analysis has shown that MuSK-MG patients, younger and mild to moderate cases seem to develop a better response to rituximab [46]. Independently of the antibody status, studies show a variable response rate, ranging from 50 to 100% [47]. The BeatMG (NCT02110706) and Rinomax (NCT02950155) trials, comparing Rituximab with placebo (unfortunately with different treatment schemes) should soon add new data to our knowledge.
Thymectomy
Thymectomy is obviously mandatory in patients with thymoma. In non-thymomatous patients, there is no evidence for an interest of surgery in MuSK-MG. In AChR-MG, thymectomy is proposed in order to limit the dose and duration of immunotherapy. The results of the MG thymectomy trial (MGTX) confirm the fact that transsternal thymectomy, associated with prednisone, allows a decrease in prednisone requirement in non-thymomatous patients when compared with prednisone alone, and a general alleviation of the disease burden over time [30]. The use of less invasive techniques, often proposed in clinical practice, is currently being evaluated and should add to the acceptability of this procedure, especially in younger patients [48, 49]. Surgery is never an emergency, and should be proposed in stable patients.
Intravenous immunoglobulin (IVIg) injections and plasmapheresis
They are used in cases of MG with life-threatening symptoms, mostly respiratory dysfunction or swallowing difficulties. Plasmapheresis consists in the unspecific removing of antibodies from the blood of patients, while IVIg treatment resides in injecting large amount of immunoglobulins prepared from the serum of different healthy donors. The therapeutic mechanisms of action of IVIg are complex but lead to potent immunomodulatory action. The choice of giving one treatment or the other depends on individual contra-indications (e.g. renal insufficiency for IVIg or sepsis for plasmapheresis), but also on the form of MG, plasma exchanges being probably more effective in MuSK-MG.
In clinical centers having a specific expertise with MG, refractory cases, which may represent about 10–15% of patients [47], can also be treated with chronic IVIg injections or plasmapheresis exchanges. The decision of using one of these approaches results from an individual assessment of the patient’s clinical situation and previous treatment.
Considering innovative therapeutic approaches
MG is a rare disease and it is not easy to gather the proper number of patients to set up clinical trials. Clinical trials for MG are thus often initiated on a limited number of patients with a severe and refractory disease. However, MG takes full use of research advances in other more common autoimmune diseases (AIDs) sharing similar dysfunctions of the immune system. Below, are listed new therapeutic approaches which could be effective for the treatment of MG patients. As indicated on Fig. 1, these therapeutic approaches can modulate the immune system at different levels or can target specific molecules. A few clinical trials are ongoing for some of these approaches as detailed on “clinicaltrial.gov.” and listed in Table 2.
Fig.1
Schematic representation of the cell cascade involved in the immune reaction leading to auto-antibody production in MG. The impact on this immune cascade of innovative therapeutic applications is indicated below.
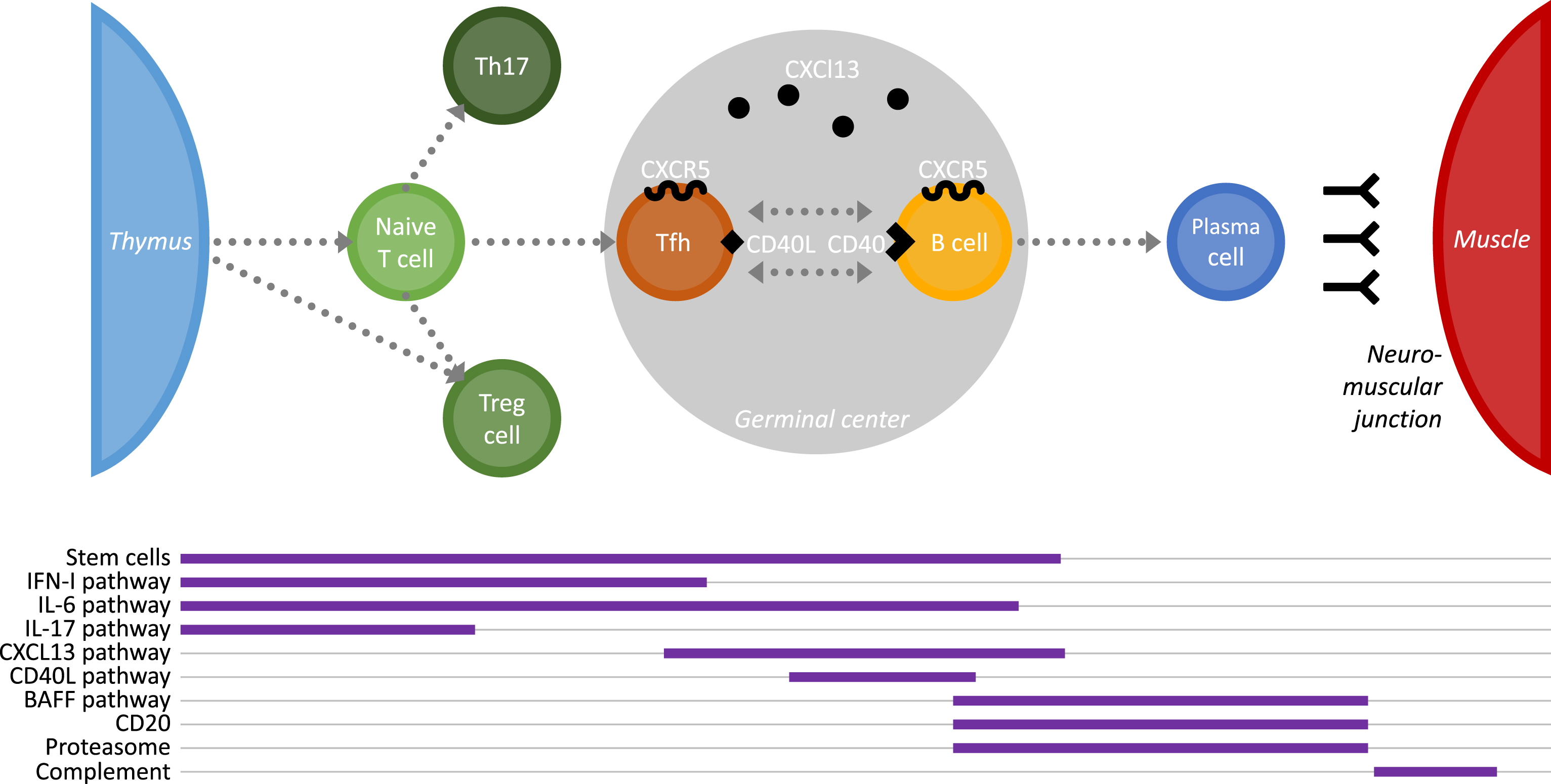
Table 2
List of potential future therapeutic approaches for MG patients
| Target | Function | Therapeutics | Ongoing clinical trial for myasthenia gravis |
|---|---|---|---|
| Undefined | Immuno-regulation of the immune system | - Hematopoietic SCs | - 7 patients (NCT00424489) |
| - Mesenchymal SCs | - No | ||
| CD20 | Deplete B cells | - Rituximab | - Approved |
| CD40L pathway | Inhibit B-cell activation | - CFZ 533 (Novartis) | - Phase II (NCT02565576) |
| - Bleselumab (Astellas Pharma) | - No | ||
| BAFF pathway | Inhibit B-cell development and survival | - Belimumab (GSK) | - Phase II (NCT01480596) |
| - Blisibimod (Anthera Pharmaceuticals) | - No | ||
| - Atacicept (Merck Serono) | - No | ||
| CXCL13 pathway | Inhibit B- and Tfh-cell migration and GC development | - SAR113244 (Sanofi) | - No |
| - VV5 (Vaccinex) | - No | ||
| Th17 pathway | Inhibit pro-inflammatory Th17 cells | - Secukinumab (Novartis) | - No |
| - Ixekizumab (Eli Lilly &Co) | - No | ||
| - Bimekizumab (UCB) | - No | ||
| - Brodalumab (AstraZeneca) | - No | ||
| - Ustekinumab (Janssen) | - No | ||
| IFN-I pathway | Inhibit IFN-I inflammatory effects | - Rontalizumab (Genentech) | - No |
| - Sifalimumab (MedImmune) | - No | ||
| - Anifrolumab (MedImmune) | - No | ||
| - Ruxolitinib (Novartis) | - No | ||
| - Tofacitinib (Pfizer) | - No | ||
| - Baracitinib (Eli Lilly &Co/Incyte Corporation) | - No | ||
| IL-6 pathway | Inhibit IL-6 pro-inflammatory effects | - Sirukumab (Johnson &Johnson’s) | - No |
| - Siltuximab (Janssen) | - No | ||
| - Tocilizumab (Hoffmann-La Roche) | - No | ||
| Proteasome | Deplete antibody producing B cells | - Bortezomib (Millennium Pharmaceuticals) | - Phase II (NCT02102594) |
| Complement | Diminish the NMJ destruction | - Eculizimab (Alexion Pharmaceuticals) | - Approved (USA) |
Stem cell therapies to rescue the immune system
Stem cell-based therapies have been considered for years as a promising approach to treat chronic diseases. Different stem cell (SC) sources can be considered such as hematopoietic SCs or mesenchymal SCs. Hematopoietic SCs have been among the first used in the clinic, mainly to treat blood disorders and restore hematopoietic function after radiation and chemotherapy. Afterwards, they have also been considered to treat other diseases, such as AIDs [50]. In 2016, seven patients with refractory and severe MG who received autologous hematopoietic SC transplants have experienced long-term remission, and remained symptom and treatment free for many years. This therapeutic approach can only be applied to severe and treatment-resistant patients, as the procedure is not without risk [51]. Mesenchymal SCs are distinct from the hematopoietic SCs and have the capacity to differentiate into multiple cell types including adipocytes, myocytes, but not in the hematopoietic cells. Mesenchymal SCs display regenerative and trophic features, as well as anti-inflammatory and immunosuppressive properties.
Studies have reported the beneficial effects of mesenchymal SCs in experimental autoimmune MG (EAMG) models using either syngeneic mesenchymal SCs or human xenogeneic mesenchymal SCs [52, 53]. Using a new humanized mouse model of MG, Sudres et al. have also demonstrated that human mesenchymal SCs, initially conditioned in vitro, were efficient in alleviating the disease. They observed that conditioned mesenchymal SCs act by several mechanisms, including reduction in the expression of costimulatory molecules and proliferation, and activation of complement regulator components [54]. Mesenchymal SC-based therapy has not yet been tested in MG patients and the mechanisms by which mesenchymal SCs exert their regulatory effects in other AIDs remain largely unknown.
Targeting the CD40L pathway to inhibit B-cell activation
CD40 is expressed on antigen-presenting cells, such as macrophages, dendritic cells and B cells, while its ligand CD40L is expressed on activated T cells. CD40L induces activation of B cells, immunoglobulin class switch, plasma cell differentiation, as well as GC formation. CD40-CD40L interactions were shown to be essential for the development of several AIDs in experimental models, including MG. Indeed in the EAMG model, CD40L knockout (KO) mice are resistant to the induction of the disease and display diminished Th1 and Th2 responses as well as severely impaired T-cell dependent AChR-reactive B-cell responses [55]. Moreover the use of an anti-CD40L antibody is an effective treatment to suppress EAMG even when it is given during the chronic stage of disease [56].
Novartis has developed, a fully human IGg1 anti-CD40 monoclonal antibody (CFZ533) that inhibits B-cell activation without B-cell depletion, for diverse autoimmune conditions. A phase II clinical trial is ongoing to evaluate the efficiency of CFZ533 in patients with generalized MG. Other monoclonal antibodies against CD40 have been developed, such as Bleselumab (Astellas Pharma) but it has not yet been evaluated in clinical trial for AIDs.
Antibodies targeting CD40L have also been developed and were promising in experimental models. They have been evaluated in clinical trials for multiple sclerosis and Crohn’s disease but halted due to thromboembolic events in treatedsubjects [57].
Targeting the BAFF pathway to inhibit B cells
B-cell activating factor (BAFF) is playing a role in B-cell development and survival. An excess of BAFF can favor the proliferation and survival of autoreactive B cells [58]. Three functional receptors for BAFF have been identified BAFF-R, TACI and BCMA. Different independent studies have shown that circulating BAFF levels were higher in AChR-MG patients compared to healthy controls [59–62]. Moreover, BAFF is also overexpressed in the thymus of MG patients [63]. A polymorphism has been observed in BAFF in a large cohort of MG patients that could explained its higher expression in MG [64].
Belimumab (Benlystatrademark), commercialized now by GSK, is a monoclonal antibody which neutralizes soluble BAFF. Its use has been approved in different countries for the treatment of systemic lupus erythematosus (SLE) with mixed results [65]. In MG, a phase II study investigating Belimumab effects in both AChR-MG and MuSK-MG patients has been completed and results are expected. Blisibimod (Anthera Pharmaceuticals) is an inhibitor of both soluble and membrane-bound BAFF. A clinical trial was set up for SLE patients but failed to demonstrate meaningful improvement in patients’ disease activity [66]. APRIL is another molecule of the same family that can also bind TACI and BCMA [58]. Other selective targeted therapies could thus be considered for all these molecules, such Atacicept (developed by ZymoGenetics and handles by Merck Serono), a recombinant fusion protein that blocks activation of TACI by APRIL and BAFF.
Targeting the CXCL13 signalization to inhibit B-cell and Thf migration and GC development
The chemokine CXCL13 and its receptor CXCR5 that is expressed mainly on B cells and follicular T cells, play a key role in cell migration towards secondary lymphoid organs or inflammatory organs, and the development of a GC reactions. In EOMG patients, CXCL13 is overexpressed in the thymus leading to the abnormal B-cell recruitment [67] and a higher frequency of circulating follicular T cells was observed in the peripheral blood of MG patients [68]. Therapies aiming at blocking CXCL13 and CXCR5 interaction are thus of great interest. An anti-CXCR5 monoclonal antibody (SAR113244) was initially proposed by Sanofi for SLE patients but the clinical trial has not been pursued. Recently, a human monoclonal anti-CXCL13 antibody has been developed (VX5-Vaccinex) and tested in several experimental models for different AIDs with very encouraging results [69]. This new observation supports the potential interest of this therapeutic approach.
Targeting IL-17 pathway to inhibit pro-inflammatory Th17-cell effects
In recent years, the development of AIDs has been linked to the imbalance between Treg cells and pro-inflammatory Th17 cells, in favor of these lasts. Treg cells possess immunomodulatory properties indispensable to terminate a normal immune reaction, maintain peripheral tolerance to self-antigens and avoid autoimmunity. In contrast, Th17 cells are highly inflammatory. They are defined by the production of different IL-17 cytokines that are involved in inflammation and AIDs. Th17 cells differentiate from naïve T cells upon stimulation with IL-21 and Il-23.
The overexpression of cytokines of the IL-17 family has been observed in T cells from MG thymuses together with the up-regulation of IL-23 receptor expression [41]. Elevated levels of IL-17A has been demonstrated in the serum of EOMG patients in correlation with disease severity [37]. The implication of Th17 cells has also been demonstrated in the EAMG mouse model. Indeed, IL-17 KO mice develop fewer EAMG symptoms [70, 71].
Different monoclonal antibodies have been developed against IL-17A, such as Secukinumab (CosentyxTM, Novartis), Ixekizumab (TalzTM, Eli Lilly & Co), that seem promising for the treatment of psoriasis and psoriasis arthritic [72]. Bimekizumab (UCB) is another monoclonal antibody against both IL-17A and IL-17F that demonstrated positive results in early development in patients with psoriatic arthritis [73].
IL-17 inhibition can be also achieved by blocking the IL-17 receptor with Brodalumab a human IgG2 monoclonal antibody. Brodalumab (AstraZeneca) is licensed for the treatment of psoriasis. Of note, the blockade of the different IL-17 cytokines or their receptor can lead to different therapeutic responses and both Secukinumab and Brodalumab give mixed responses for rheumatoid arthritis (RA) and even worsen Crohn’s disease [74]. Other therapeutic approaches aiming at blocking Th17 cell differentiation can also be envisaged with the use of molecules targeting IL-23. For example Ustekinumab (StelaraTM, Janssen) is an antibody against the p40 protein subunit present in IL-12 and IL-23. It is licensed again for the treatment of psoriasis, psoriasis arthritic and also Crohn’s disease. The efficacy in RA is not yet clearly demonstrated and a clinical trial is ongoing for SLE.
Targeting the IFN-I signaling pathway to switch off chronic inflammation
Type I IFN (IFN-I) plays a central role to set up a balanced immune response to pathogens. The IFN-I family comprises different subtypes, such as IFN-β and several IFN-α. They all signal via a common heterodimeric receptor, composed of IFNAR1 and IFNAR2, which relies on associated kinases (JAK1 and TYK2) to phosphorylate STAT transcription factors and induce an IFN-I signalization [75].
In thymoma-associated MG patients, IFN-I subtypes are overexpressed in thymoma and patients display high levels of anti-IFN-I autoantibodies [76]. In EOMG, IFN-β is also overexpressed in the thymus and could be the orchestrator of thymic changes associated with MG [63]. IFN-β is overexpressed by thymic epithelial cells even long after the disease onset. These observations suggest that targeting the IFN-I signaling pathway could shut down the chronic inflammation characteristic of many AIDs. In this way, different therapeutic approaches are investigated but not yet in MG. The use of antibodies against anti-IFN-α (Rontalizumab (Genentech), Sifalimumab (MedImmune)) or against the IFNAR1 (Anifrolumab (MedImmune)) is under consideration for SLE but also dermatomyositis and polymyositis [77].
Drugs that block JAK/STAT signalization could also interfere in a non-specific way with various cytokine signalization, such as IL-6. Small molecules inhibiting JAKs (Jakinibs) are proposed for the treatment of RA: Ruxolitinib (JakaviTM, Novartis) and Baracitinib (Eli Lilly & Co/Incyte Corporation) for JAK1 and JAK2, or Tofacitinib (XeljanzTM, Pfizer) for JAK1, JAK3 and to a lesser extend JAK2 [78].
Targeting the IL-6 pathway to overcome its pro-inflammatory effects
IL-6 is a pleiotropic cytokine that has context-dependent pro- and anti-inflammatory properties. IL-6 is produced by various cell types and not only immune cells. IL-6 actively stimulates T lymphocytes and favors the switch of Treg cells towards a pathogenic Th17 cell phenotype. IL-6 also induces B-cell differentiation into antibody-secreting cells and promotes the survival of long-lived plasma cells [79].
IL-6 expression is increased in different cell types in MG patients: in vitro, IL-6 mRNA expression is increased in peripheral blood mononuclear cells from MG patients in response to AChR [80], increased expression of IL-6 has also been observed in thymic epithelial cells from MG patients [81] and IL-6 is expressed at a higher level in MG than control muscles [82]. IL-6 KO mice are resistant to EAMG and this is associated with a significant reduction in GC formation in the spleen and a decrease in serum complement C3 levels [83]. Moreover, antibodies against IL-6 suppress EAMG in the rat model when administrated either during the acute or the chronic phase of disease. Suppression of EAMG is accompanied by a decrease in anti-AChR antibodies and by a reduced number of B cells [84].
Compounds have been developed that target IL-6, its receptors or downstream signaling molecules. Monoclonal antibodies against IL-6 have been designed for other AIDs: Sirukumab (Johnson & Johnson’s) for RA [85], Siltuximab (Janssen) for the treatment of neoplastic diseases, and Tocilizumab (ActemraTM, Hoffmann-La Roche) for RA and systemic juvenile idiopathic arthritis [86]. None has been considered so far for MG patients.
Targeting the proteasome to reduce antibody-producing cells
Bortezomib (VelcadeTM, Millennium Pharmaceuticals) is an inhibitor of the proteasome. The proteasome is a large complex of proteases involved in the degradation of abnormal or unneeded proteins. The immunoproteasome is formed by the replacement of certain subunits of the proteasome and is abundantly expressed in cells of hematopoietic origin. It actively participates in the degradation of intracellular proteins for presentation of antigens by the major histocompatibility complex molecules [87].
Bortezomib was initially used as an anti-cancer drug in particular in hematological malignancies. However, Bortezomib has been reported to reduce autoantibody titers improving clinical condition in a SLE mouse model [88]. Since Bortezomib has been efficiently used in other experimental autoimmune models. It depletes both short-lived and long-lived plasma cells that usually survive the standard immunosuppressant treatments. In the mouse EAMG model, Bortezomib efficiently reduces the rise of anti-AChR autoantibodies, prevents ultrastructural damage of the postsynaptic membrane, improves neuromuscular transmission, and decreases MG symptoms [89]. It was also demonstrated that in vitro Bortezomib can kill long-lived plasma cells derived from the thymus of EOMG patients [90]. A selective inhibitor of the immunoproteasome (ONX-0914) has also proved to be efficient in the classical EAMG model ameliorating disease severity [91]. A clinical trial is ongoing to investigate the effect of Bortezomib in patients with refractory MG, SLE or RA.
Complement inhibition to attenuate the consequences of the autoantibody attack
Binding and activation of complement at the NMJ is the predominant pathogenic mechanism of anti-AChR antibodies [92]. In the MG thymus, the activation of the complement cascade has also been observed on myoid cells and on thymic epithelial cells [93]. Numerous studies on EAMG models have clearly shown that inhibition of complement pathways effectively and specifically diminish the NMJ destruction induced by anti-AChR antibodies. Changes in complement concentration can influence the severity of MG: (1) Experiments in rats showed that blocking or depletion of complement protects animals from developing EAMG [94], (2) antibodies that block complement component C6 or soluble complement receptor 1 protect rodents from EAMG [95], (3) an inhibitor of C5 reduces symptoms in the rat EAMG model [96], and (4) Mice deficient for classical complement pathway factors are resistant to antibody and complement mediated EAMG [97]. Moreover, increased complement consumption was detected in MG patients with high AChR antibody concentrations [98].
Altogether, these data suggest that molecules aiming at decreasing complement deposition at the NMJ could represent a therapeutic approach of major interest [99]. Nevertheless, only Eculizimab (SolirisTM, Alexion Pharmaceuticals) but no other molecules aiming at inhibiting the complement cascade activation have been considered for MG patients. Eculizumab is a humanized monoclonal antibody that inhibits the cleavage of the complement protein C5 acting at the terminal complement activation cascade. An initial phase II pilot study showed clinically improvements in severe and refractory of AChR-MG patients [100]. Next, the REGAIN phase III trial has compared this drug to placebo. The primary endpoint based on the measure of a change in MG activities of daily living was not reached due to the use of prespecified worst-rank criteria. However, secondary end points and sensitivity analyses suggested efficacy of Eculizumab leading to a reduction in disease severity [101]. After this clinical trial, in October 2017, the US Food and drug administration has extended the indication for this molecule as a potential treatment for patients with refractory and generalized AChR-MG. Nevertheless, as C5 terminal complement activation is necessary in the defense against encapsulated bacterial infections, meningococcal vaccines and antimicrobial prophylaxis must be considered to reduce the risk of meningococcal disease [102].
Conclusion
Various therapeutic strategies have been developed for MG patients, but MG is still a chronic disease and most patients will need treatments for the rest of their life. Our current strategies have some limitations, mainly due to the potential toxicity of the drugs over time, and to the fact that some patients are or become refractory to their treatment. There is a real need for new therapeutic approaches that could be envisaged earlier in the course of the disease and not only in refractory forms, often after complications linked to the burden of immunosuppressant treatments.
Conflict of interest
There is no conflict of interest to declare pertaining to this study.
ACKNOWLEDGMENTS
We thank “Association Française contre les Myopathies” (AFM) for its support.
REFERENCES
[1] | Aharonov A , Abramsky O , Tarrab-Hazdai R , Fuchs S .Humoral antibodies to acetylcholine receptor in patients with myasthenia gravis. Lancet. (1975) ;2: (7930):340–2. |
[2] | Hoch W , McConville J , Helms S , Newsom-Davis J , Melms A , Vincent A .Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. (2001) ;7: (3):365–68. |
[3] | Higuchi O , Hamuro J , Motomura M , Yamanashi Y .Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. (2011) ;69: (2):418–22. |
[4] | Berrih-Aknin S , Frenkian-Cuvelier M , Eymard B .Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun. (2014) ;48-49: :143–8. |
[5] | Tzartos S , Hochschwender S , Vasquez P , Lindstrom J .Passive transfer of experimental autoimmune myasthenia gravis by monoclonal antibodies to the main immunogenic region of the acetylcholine receptor. J Neuroimmunol. (1987) ;15: (2):185–94. |
[6] | Le Panse R , Berrih-Aknin S .Autoimmune myasthenia gravis: Autoantibody mechanisms and new developments on immune regulation. Curr Opin Neurol. (2013) ;26: (5):569–76. |
[7] | Nakano S , Engel AG .Myasthenia gravis: Quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. (1993) ;43: (6):1167–72. |
[8] | Berrih-Aknin S , Ruhlmann N , Bismuth J , Cizeron-Clairac G , Zelman E , Shachar I , et al. CCL21 overexpressed on lymphatic vessels drives thymic hyperplasia in myasthenia. Ann Neurol. (2009) ;66: (4):521–31. |
[9] | Eymard B , Berrih-Aknin S .Role of the thymus in the physiopathology of myasthenia. Revue Neurologique. (1995) ;151: (1):6–15. |
[10] | Romi F , Skeie GO , Aarli JA , Gilhus NE .Muscle autoantibodies in subgroups of myasthenia gravis patients. J Neurol. (2000) ;247: (5):369–75. |
[11] | Aragones JM , Bolibar I , Bonfill X , Bufill E , Mummany A , Alonso F , et al. Myasthenia gravis: A higher than expected incidence in the elderly. Neurology. (2003) ;60: (6):1024–6. |
[12] | Leite MI , Jacob S , Viegas S , Cossins J , Clover L , Morgan BP , et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain. (2008) ;131: (Pt 7):1940–52. |
[13] | Lin W , Burgess RW , Dominguez B , Pfaff SL , Sanes JR , Lee KF .Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. . (2001) ;410: (6832):1057–64. |
[14] | Evoli A , Tonali PA , Padua L , Monaco ML , Scuderi F , Batocchi AP , et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. (2003) ;126: (Pt 10):2304–11. |
[15] | Leite MI , Strobel P , Jones M , Micklem K , Moritz R , Gold R , et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Annals of Neurology. (2005) ;57: (3):444–8. |
[16] | Mori S , Kubo S , Akiyoshi T , Yamada S , Miyazaki T , Hotta H , et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol. (2012) ;180: (2):798–810. |
[17] | Kim N , Stiegler AL , Cameron TO , Hallock PT , Gomez AM , Huang JH , et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. (2008) ;135: (2):334–42. |
[18] | Zhang B , Tzartos JS , Belimezi M , Ragheb S , Bealmear B , Lewis RA , et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. (2012) ;69: (4):445–51. |
[19] | Zisimopoulou P , Evangelakou P , Tzartos J , Lazaridis K , Zouvelou V , Mantegazza R , et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun. (2014) ;52: :139–45. |
[20] | Shen C , Lu Y , Zhang B , Figueiredo D , Bean J , Jung J , et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest. (2013) ;123: (12):5190–202. |
[21] | Levinson AI , Wheatley LM .The thymus and the pathogenesis of myasthenia gravis. Clin Immunol Immunopathol. (1996) ;78: (1):1–5. |
[22] | Berrih-Aknin S , Morel E , Raimond F , Safar D , Gaud C , Binet J , et al. The role of the thymus in myasthenia gravis: Immunohistological and immunological studies in 115 cases. Ann N Y Acad Sci. (1987) ;505: :50–70. |
[23] | Weiss JM , Cufi P , Bismuth J , Eymard B , Fadel E , Berrih-Aknin S , et al. SDF-1/CXCL12 recruits B cells and antigen-presenting cells to the thymus of autoimmune myasthenia gravis patients. Immunobiology. (2013) ;218: (3):373–81. |
[24] | Wakkach A , Guyon T , Bruand C , Tzartos S , Cohen-Kaminsky S , Berrih-Aknin S .Expression of acetylcholine receptor genes in human thymic epithelial cells: Implications for myasthenia gravis. J Immunol. (1996) ;157: (8):3752–60. |
[25] | Safar D , Berrih-Aknin S , Morel E .In vitro anti-acetylcholine receptor antibody synthesis by myasthenia gravis patient lymphocytes: Correlations with thymic histology and thymic epithelial-cell interactions. J Clin Immunol. (1987) ;7: (3):225–34. |
[26] | Leprince C , Cohen-Kaminsky S , Berrih-Aknin S , Vernet-Der Garabedian B , Treton D , Galanaud P , et al. Thymic B cells from myasthenia gravis patients are activated B cells. Phenotypic and functional analysis. J Immunol. (1990) ;145: (7):2115–22. |
[27] | Melms A , Schalke BC , Kirchner T , Muller-Hermelink HK , Albert E , Wekerle H .Thymus in myasthenia gravis. Isolation of T-lymphocyte lines specific for the nicotinic acetylcholine receptor from thymuses of myasthenic patients. J Clin Invest. (1988) ;81: (3):902–8. |
[28] | Berrih S , Morel E , Gaud C , Raimond F , Le Brigand H , Bach JF .Anti-AChR antibodies, thymic histology, and T cell subsets in myasthenia gravis. Neurology. (1984) ;34: (1):66–71. |
[29] | Kuks JB , Oosterhuis HJ , Limburg PC , The TH .Anti-acetylcholine receptor antibodies decrease after thymectomy in patients with myasthenia gravis. Clinical correlations. J Autoimmun. (1991) ;4: (2):197–211. |
[30] | Wolfe GI , Kaminski HJ , Aban IB , Minisman G , Kuo HC , Marx A , et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. (2016) ;375: (6):511–22. |
[31] | Ponseti JM , Caritg N , Gamez J , Lopez-Cano M , Vilallonga R , Armengol M .A comparison of long-term post-thymectomy outcome of anti-AChR-positive, anti-AChR-negative and anti-MuSK-positive patients with non-thymomatous myasthenia gravis. Expert Opinion on Biological Therapy. (2009) ;9: (1):1–8. |
[32] | Yi Q , Ahlberg R , Pirskanen R , Lefvert AK .Acetylcholine receptor-reactive T cells in myasthenia gravis: Evidence for the involvement of different subpopulations of T helper cells. J Neuroimmunol. (1994) ;50: (2):177–86. |
[33] | Link J , Navikas V , Yu M , Fredrikson S , Osterman PO , Link H .Augmented interferon-gamma, interleukin-4 and transforming growth factor-beta mRNA expression in blood mononuclear cells in myasthenia gravis. J Neuroimmunol. (1994) ;51: (2):185–92. |
[34] | Saito R , Onodera H , Tago H , Suzuki Y , Shimizu M , Matsumura Y , et al. Altered expression of chemokine receptor CXCR5 on T cells of myasthenia gravis patients. J Neuroimmunol. (2005) ;170: (1-2):172–8. |
[35] | Park H , Li Z , Yang XO , Chang SH , Nurieva R , Wang YH , et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) ;6: (11):1133–41. |
[36] | Gradolatto A , Nazzal D , Truffault F , Bismuth J , Fadel E , Foti M , et al. Both Treg cells and Tconv cells are defective in the Myasthenia gravis thymus: Roles of IL-17 and TNF-alpha. J Autoimmun. (2014) ;52: :53–63. |
[37] | Xie Y , Li HF , Jiang B , Li Y , Kaminski HJ , Kusner LL .Elevated plasma interleukin-17A in a subgroup of Myasthenia Gravis patients. Cytokine. (2016) ;78: :44–6. |
[38] | Peters A , Pitcher LA , Sullivan JM , Mitsdoerffer M , Acton SE , Franz B , et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) ;35: (6):986–96. |
[39] | Balandina A , Lecart S , Dartevelle P , Saoudi A , Berrih-Aknin S . Functional defect of regulatory CD4(+)CD25+T cells in the thymus of patients with autoimmune myasthenia gravis. Blood. (2005) ;105: (2):735–41. |
[40] | Thiruppathi M , Rowin J , Ganesh B , Sheng JR , Prabhakar BS , Meriggioli MN . Impaired regulatory function in circulating CD4(+)CD25(high)CD127(low/-) T cells in patients with myasthenia gravis. Clin Immunol. (2012) ;145: (3):209–23. |
[41] | Gradolatto A , Nazzal D , Truffault F , Bismuth J , Fadel E , Foti M , et al. Both Treg cells and Tconv cells are defective in the Myasthenia gravis thymus: Roles of IL-17 and TNF-alpha. J Autoimmun. (2014) ;52: :53–63. |
[42] | Sanders DB , Wolfe GI , Benatar M , Evoli A , Gilhus NE , Illa I , et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology. (2016) ;87: (4):419–25. |
[43] | Dezern AE , Styler MJ , Drachman DB , Hummers LK , Jones RJ , Brodsky RA .Repeated treatment with high dose cyclophosphamide for severe autoimmune diseases. American Journal of Blood Research. (2013) ;3: (1):84–90. |
[44] | Kanth KM , Solorzano GE , Goldman MD .PML in a patient with myasthenia gravis treated with multiple immunosuppressing agents. Neurology Clinical Practice. (2016) ;6: (2):e17–e19. |
[45] | Robeson KR , Kumar A , Keung B , DiCapua DB , Grodinsky E , Patwa HS , et al. Durability of the rituximab response in acetylcholine receptor autoantibody-positive myasthenia gravis. JAMA Neurology. (2017) ;74: (1):60–66. |
[46] | Tandan R , Hehir MK 2nd , Waheed W , Howard DB . Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve. (2017) ;56: (2):185–96. |
[47] | Evoli A .Myasthenia gravis: New developments in research and treatment. Curr Opin Neurol. (2017) ;30: (5):464–70. |
[48] | Siwachat S , Tantraworasin A , Lapisatepun W , Ruengorn C , Taioli E , Saeteng S .Comparative clinical outcomes after thymectomy for myasthenia gravis: Thoracoscopic versus trans-sternal approach. Asian J Surg. (2018) ;41: (1):77–85. |
[49] | Keijzers M , de Baets M , Hochstenbag M , Abdul-Hamid M , Zur Hausen A , van der Linden M , et al. Robotic thymectomy in patients with myasthenia gravis: Neurological and surgical outcomes. Eur J Cardiothorac Surg. (2015) ;48: (1):40–5. |
[50] | Zeher M , Papp G , Nakken B , Szodoray P .Hematopoietic stem cell transplantation in autoimmune disorders: From immune-regulatory processes to clinical implications. Autoimmun Rev. (2017) ;16: (8):817–25. |
[51] | Bryant A , Atkins H , Pringle CE , Allan D , Anstee G , Bence-Bruckler I , et al. Myasthenia gravis treated with autologous hematopoietic stem cell transplantation. JAMA Neurology. (2016) ;73: (6):652–8. |
[52] | Kong QF , Sun B , Wang GY , Zhai DX , Mu LL , Wang DD , et al. BM stromal cells ameliorate experimental autoimmune myasthenia gravis by altering the balance of Th cells through the secretion of IDO. Eur J Immunol. (2009) ;39: (3):800–9. |
[53] | Yu J , Zheng C , Ren X , Li J , Liu M , Zhang L , et al. Intravenous administration of bone marrow mesenchymal stem cells benefits experimental autoimmune myasthenia gravis mice through an immunomodulatory action. Scand J Immunol. (2010) ;72: (3):242–9. |
[54] | Sudres M , Maurer M , Robinet M , Bismuth J , Truffault F , Girard D , et al. Preconditioned mesenchymal stem cells treat myasthenia gravis in a humanized preclinical model. JCI Insight. (2017) ;2: (7):e89665. |
[55] | Shi FD , He B , Li H , Matusevicius D , Link H , Ljunggren HG .Differential requirements for CD28 and CD40 ligand in the induction of experimental autoimmune myasthenia gravis. Eur J Immunol. (1998) ;28: (11):3587–93. |
[56] | Im SH , Barchan D , Maiti PK , Fuchs S , Souroujon MC .Blockade of CD40 ligand suppresses chronic experimental myasthenia gravis by down-regulation of Th1 differentiation and up-regulation of CTLA-4. J Immunol. (2001) ;166: (11):6893–8. |
[57] | Sidiropoulos PI , Boumpas DT .Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. LUPUS. (2004) ;13: (5):391–7. |
[58] | Ragheb S , Lisak RP .B-cell-activating factor and autoimmune myasthenia gravis. Autoimmune Diseases. (2011) ;939520. |
[59] | Kim JY , Yang Y , Moon JS , Lee EY , So SH , Lee HS , et al. Serum BAFF expression in patients with myasthenia gravis. J Neuroimmunol. (2008) ;199: (1-2):151–4. |
[60] | Ragheb S , Lisak R , Lewis R , Van Stavern G , Gonzales F , Simon K .A potential role for B-cell activating factor in the pathogenesis of autoimmune myasthenia gravis. Arch Neurol. (2008) ;65: (10):1358–62. |
[61] | Scuderi F , Alboini PE , Bartoccioni E , Evoli A .BAFF serum levels in myasthenia gravis: Effects of therapy. J Neurol. (2011) ;258: (12):2284–5. |
[62] | Kang SY , Kang CH , Lee KH .B-cell-activating factor is elevated in serum of patients with myasthenia gravis. Muscle Nerve. (2016) ;54: (6):1030–33. |
[63] | Cufi P , Dragin N , Ruhlmann N , Weiss JM , Fadel E , Serraf A , et al. Central role of interferon-beta in thymic events leading to myasthenia gravis. J Autoimmun. (2014) ;52: :44–52. |
[64] | Avidan N , Le Panse R , Harbo HF , Bernasconi P , Poulas K , Ginzburg E , et al. VAV1 and BAFF, via NFkB pathway, are genetic risk factors for myasthenia gravis. Annals of Clinical and Translational Neurology. (2014) ;1: (5):329–339. |
[65] | Stohl W .Inhibition of B cell activating factor (BAFF) in the management of systemic lupus erythematosus (SLE). Expert Review of Clinical Immunology. (2017) ;13: (6):623–33. |
[66] | Lenert A , Niewold TB , Lenert P .Spotlight on blisibimod and its potential in the treatment of systemic lupus erythematosus: Evidence to date. Drug Design, Development and Therapy. (2017) ;11: :747–57. |
[67] | Méraouna A , Cizeron-Clairac G , Le Panse R , Bismuth J , Truffault F , Talaksen C , et al. The chemokine CXCL13 is a key molecule in autoimmune Myasthenia Gravis. Blood. (2006) ;108: (2):432–40. |
[68] | Luo C , Li Y , Liu W , Feng H , Wang H , Huang X , et al. Expansion of circulating counterparts of follicular helper T cells in patients with myasthenia gravis. J Neuroimmunol. (2013) ;256: (1-2):55–61. |
[69] | Klimatcheva E , Pandina T , Reilly C , Torno S , Bussler H , Scrivens M , et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. (2015) ;16: (6):1–17. |
[70] | Schaffert H , Pelz A , Saxena A , Losen M , Meisel A , Thiel A , et al. IL-17-producing CD4(+) T cells contribute to the loss of B-cell tolerance in experimental autoimmune myasthenia gravis. Eur J Immunol. (2015) :45: (5):1339–47. |
[71] | Aguilo-Seara G , Xie Y , Sheehan J , Kusner LL , Kaminski HJ .Ablation of IL-17 expression moderates experimental autoimmune myasthenia gravis disease severity. Cytokine. (2017) ;96: :279–85. |
[72] | Burkett PR , Kuchroo VK .IL-17 blockade in psoriasis. Cell. (2016) ;167: (7):1669. |
[73] | Glatt S , Helmer E , Haier B , Strimenopoulou F , Price G , Vajjah P , et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. (2017) ;83: (5):991–1001. |
[74] | Nirula A , Nilsen J , Klekotka P , Kricorian G , Erondu N , Towne JE , et al. Effect of IL-17 receptor A blockade with brodalumab in inflammatory diseases. Rheumatology (Oxford). (2016) ;55: (suppl 2):ii43-ii55. |
[75] | Pestka S , Krause CD , Walter MR .Interferons, interferon-like cytokines, and their receptors. Immunol Rev. (2004) ;202: :8–32. |
[76] | Meager A , Wadhwa M , Dilger P , Bird C , Thorpe R , Newsom-Davis J , et al. Anti-cytokine autoantibodies in autoimmunity: Preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clin Exp Immunol. (2003) ;132: (1):128–36. |
[77] | Kalunian KC , Merrill JT , Maciuca R , McBride JM , Townsend MJ , Wei X , et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. (2016) ;75: (1):196–202. |
[78] | O’Shea JJ , Kontzias A , Yamaoka K , Tanaka Y , Laurence A . Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. (2013) :72: Suppl 2:ii111–5. |
[79] | Hunter CA , Jones SA .IL-6 as a keystone cytokine in health and disease. Nat Immunol. (2015) ;16: (5):448–57. |
[80] | Matusevicius D , Navikas V , Palasik W , Pirskanen R , Fredrikson S , Link H .Tumor necrosis factor-alpha, lymphotoxin, interleukin (IL)-6, IL-10, IL-12 and perforin mRNA expression in mononuclear cells in response to acetylcholine receptor is augmented in myasthenia gravis. J Neuroimmunol. (1996) ;71: (1-2):191–8. |
[81] | Cohen-Kaminsky S , Devergne O , Delattre RM , Klingel-Schmitt I , Emilie D , Galanaud P , et al. Interleukin-6 overproduction by cultured thymic epithelial cells from patients with myasthenia gravis is potentially involved in thymic hyperplasia. Eur Cytokine Netw. (1993) ;4: (2):121–32. |
[82] | Maurer M , Bougoin S , Feferman T , Frenkian M , Bismuth J , Mouly V , et al. IL-6 and Akt are involved in muscular pathogenesis in myasthenia gravis. Acta Neuropathologica Communications. (2015) ;3: :1–14. |
[83] | Deng C , Goluszko E , Tuzun E , Yang H , Christadoss P .Resistance to experimental autoimmune myasthenia gravis in IL-6-deficient mice is associated with reduced germinal center formation and C3 production. J Immunol. (2002) ;169: (2):1077–83. |
[84] | Aricha R , Mizrachi K , Fuchs S , Souroujon MC .Blocking of IL-6 suppresses experimental autoimmune myasthenia gravis. J Autoimmun. (2011) ;36: (2):135–41. |
[85] | Takeuchi T , Thorne C , Karpouzas G , Sheng S , Xu W , Rao R , et al. Sirukumab for rheumatoid arthritis: The phase III SIRROUND-D study. Ann Rheum Dis. (2017) ;76: (12):2001–2008. |
[86] | Sheppard M , Laskou F , Stapleton PP , Hadavi S , Dasgupta B .Tocilizumab (Actemra). Human Vaccines & Immunotherapeutics. (2017) ;13: (9):1972–88. |
[87] | Eskandari SK , Seelen MAJ , Lin G , Azzi JR . The immuno-proleasome: An old player with a novel and emerging role in alloimmunity. American Journal of Transplantation: Official Journal of the American Society of Transplantation and the American Society of Transplant Surgeons. (2017) ;17: (12):3033–3039. |
[88] | Neubert K , Meister S , Moser K , Weisel F , Maseda D , Amann K , et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. (2008) ;14: (7):748–55. |
[89] | Gomez AM , Vrolix K , Martinez-Martinez P , Molenaar PC , Phernambucq M , van der Esch E , et al. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J Immunol. (2011) ;186: (4):2503–13. |
[90] | Gomez AM , Willcox N , Vrolix K , Hummel J , Nogales-Gadea G , Saxena A , et al. Proteasome inhibition with bortezomib depletes plasma cells and specific autoantibody production in primary thymic cell cultures from early-onset myasthenia gravis patients. J Immunol. (2014) ;193: (3):1055–63. |
[91] | Liu RT , Zhang P , Yang CL , Pang Y , Zhang M , Zhang N , et al. ONX-a selective inhibitor of immunoproteasome, ameliorates experimental autoimmune myasthenia gravis by modulating humoral response. J Neuroimmunol. (2017) ;311: :71–78. |
[92] | Drachman DB .Myasthenia gravis. N Engl J Med. (1994) ;330: (25):1797–810. |
[93] | Leite MI , Jones M , Strobel P , Marx A , Gold R , Niks E , et al .Myasthenia gravis thymus: Complement vulnerability of epithelial and myoid cells, complement attack on them, and correlations with autoantibody status. Am J Pathol. (2007) ;171: (3):893–905. |
[94] | Lennon VA , Seybold ME , Lindstrom JM , Cochrane C , Ulevitch R .Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J Exp Med. (1978) ;147: (4):973–83. |
[95] | Biesecker G , Gomez CM .Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J Immunol. (1989) ;142: (8):2654–9. |
[96] | Soltys J , Kusner LL , Young A , Richmonds C , Hatala D , Gong B , et al. Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann Neurol. (2009) ;65: (1):67–75. |
[97] | Tuzun E , Scott BG , Goluszko E , Higgs S , Christadoss P .Genetic evidence for involvement of classical complement pathway in induction of experimental autoimmune myasthenia gravis. J Immunol. (2003) ;171: (7):3847–54. |
[98] | Romi F , Kristoffersen EK , Aarli JA , Gilhus NE .The role of complement in myasthenia gravis: Serological evidence of complement consumption in vivo . J Neuroimmunol. (2005) ;158: (1-2):191–4. |
[99] | Tuzun E , Huda R , Christadoss P .Complement and cytokine based therapeutic strategies in myasthenia gravis. J Autoimmun. (2011) ;37: (2):136–43. |
[100] | Howard JF Jr , Barohn RJ , Cutter GR , Freimer M , Juel VC , Mozaffar T , et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. (2013) ;48: (1):76–84. |
[101] | Howard JF Jr , Utsugisawa K , Benatar M , Murai H , Barohn RJ , Illa I , et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): A phase 3, randomised, double-blind, placebo-controlled, multicentre study. The Lancet Neurology. (2017) ;16: (12):979–86. |
[102] | McNamara LA , Topaz N , Wang X , Hariri S , Fox L , MacNeil JR . High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. Am J Transplant. (2017) ;17: (9):2481–4. |


