
Anti-HMGCR Myopathy
Abstract
Anti-HMGCR myopathy was first recognized and characterized in patients with a history of statin exposure and immune-mediated necrotizing myopathy. After the discovery of anti-HMGCR autoantibodies, several international groups identified and characterized more patients, expanding the phenotypic spectrum of this disease to include pediatric patients and young adults without statin exposure and those with a chronic myopathy resembling limb-girdle muscular dystrophy. We provide a summary of clinical findings, pathologic features, muscle imaging, and immunogenetic risk factors of the disease. We also discuss the current treatment strategies and approaches to monitoring the therapeutic response. Lastly, we briefly summarize the current understanding of the pathophysiology of the disease and postulate a model for autoimmunity initiation and propagation in this disease.
INTRODUCTION
Autoimmune myopathies are acquired diseases of muscle resulting from the direct or indirect injury of myofibers by the immune system. Although the phenotypes of patients with autoimmune myopathies vary widely, most modern classification schemes have divided them into just three main groups based on different clinical and myopathologic features: polymyositis, dermatomyositis, and immune-mediated necrotizing myopathy [1]. However, these classification criteria rely only on a few clinical and pathologic features and fail to capture the full complexity of these diseases.
Myositis-specific autoantibodies (MSA) are defined as autoantibodies found exclusively in patients with autoimmune myopathies. Importantly, the majority of patients with autoimmune myopathy have a single MSA and each MSA is associated with a unique clinical phenotype. For example, patients with autoantibodies recognizing one of the aminoacyl tRNA synthetases (aaRS) have a syndrome that includes two or more of the following: a muscle biopsy with lymphocytic infiltrates, arthritis, interstitial lung disease, and Raynaud phenomenon. Of note, some patients with antisynthetase autoantibodies have a dermatomyositis-like rash whereas others do not. However, rather than designating those with a rash as having “dermatomyositis” and those without a rash as having “polymyositis”, most evidence suggests that these patients all have the same disease, the antisynthetase syndrome, which can be expressed differently in different individuals. As this example demonstrates, MSAs can be thought to be analogous to myopathy-related genes in muscular dystrophies, unifying a potentially divergent disease phenotype under common molecular mechanisms [2–4].
In this review, we discuss the wide range of clinical phenotypes in myositis patients with a recently identified MSA against 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR). We will use the term anti-HMGCR myopathy to refer to all phenotypes associated with this disease consistent with the most recent ENMC meeting recommendations [5]. We will also briefly discuss our current understanding of the pathomechanisms of this disease.
HISTORICAL CONTEXT
The increasing use of statin drugs as lipid lowering agents unmasked a distinct form of autoimmune myopathy associated with their use. Nearly 10 case reports of polymyositis or dermatomyositis in patients treated with statins are found in the literature from mid-1990s to early 2000s [6–14]. Subsequently, three case series published a few years apart further described 38 patients in detail with a distinct autoimmune myopathy associated with statin use [15–17]. Nearly all these patients were classified as having polymyositis or immune-mediated necrotizing myopathy (IMNM) based on their clinicopathologic features. Unlike patients with statin intolerance or statin-induced toxic myopathy who improve after discontinuation of the offending drug, the vast majority of these patients had persistent muscle weakness and creatine kinase (CK) elevation long after discontinuation of statins and only improved after immunosuppressive therapies were initiated. Thus, this distinct clinical entity, known as statin-associated autoimmune myopathy, became increasingly recognized.
Antibody discovery
Independent autoantibody discovery efforts in patients with IMNM identified a novel autoantibody using patient sera to immunoprecipitate (IP) autoantigens from radioactively labeled cell extracts. These unknown autoantibodies immunoprecipitated a pair of proteins with molecular weights of 200 and 100 kDa [18]. Unexpectedly, the majority of the patients with anti-200/100 antibody had a history of statin exposure, providing the first link with statin-associated autoimmune myopathy. Shortly thereafter, HMGCR, which has a molecular weight of about 100 kDa and also forms a 200 kDa dimer, was identified as the autoantigen targeted by this antibody [19]. The fact that HMGCR is the pharmacologic target of statins further linked statin use with this distinct disease entity.
CLINICOPATHOLOGIC SPECTRUM OF ANTI-HMGCR MYOPATHY
Anti-HMGCR myopathy was first described in older adults with a history of statin exposure. However, an expanding group of patients with variations in clinical phenotypes and ages of presentation have been reported by multiple international groups, albeit with some variations [20–25]. Nonetheless, as we will show, these wide-ranging phenotypes can be grouped as one disease, anti-HMGCR myopathy, based on shared muscle biopsy features (∼80% have a predominantly necrotizing myopathy) and the presence of anti-HMGCR autoantibodies (Fig. 1).
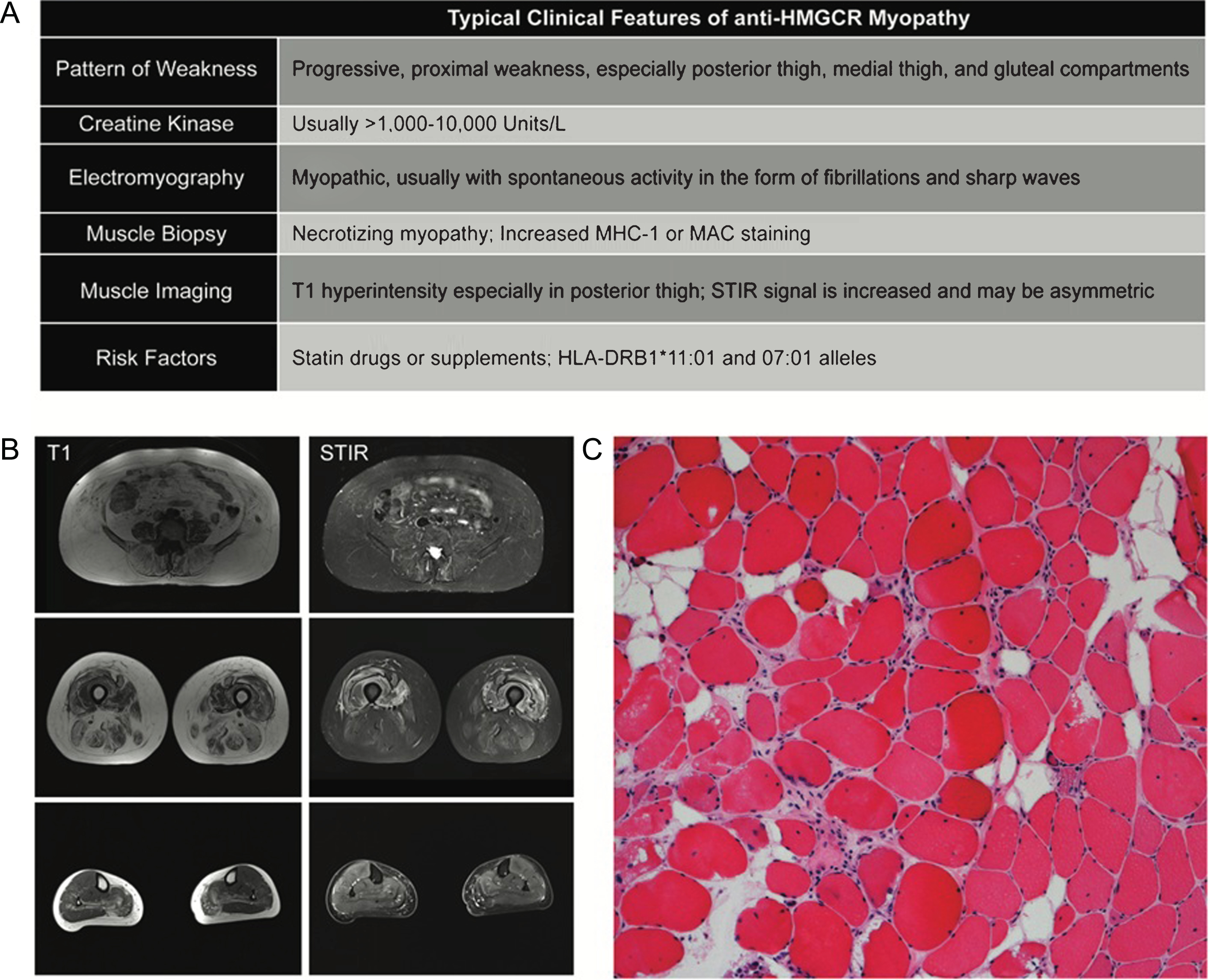
Fig.1
Typical Clinicopathologic features of anti-HMGCR Myopathy. (A) Summary of clinical findings on exam and ancillary testing. (B) muscle MRI of the lower extremities. Note selective involvement of paraspinal, gluteals in the hip and posterior and medial compartments of the thigh. Short tau inversion recovery (STIR) signal increase can be patchy and asymmetric. (C) A typical hematoxylin and eosin stain of a patient with anti-HMGCR myopathy showing myofiber atrophy, degeneration, and regeneration without prominent lymphocytic inflammation.
Classic adult-onset anti-HMGCR myopathy
The majority of patients with anti-HMGCR myopathy have an adult-onset disease characterized by subacute, progressive, proximal weakness, and highly elevated CK levels, usually in the ∼1,000–20,000 IU/L range. Similar to other autoimmune diseases, a slight female predominance has been reported [18, 20, 21, 23]. Most patients have a history of exposure to statins, at rates much higher than other autoimmune myopathies, and their symptoms fail to resolve after discontinuation of the offending drug. Fatigue and myalgia are reported in 20–60% of the patients. Dysphagia is reported in a sizable minority (16–30%) of patients [17, 18, 20] and others have highlighted truncal weakness as a unique clinical feature [22].
In general, anti-HMGCR myopathy predominantly effects skeletal muscle. Non-specific systemic and extramuscular symptoms (e.g. rash, arthritis, Raynaud’s phenomenon) are uncommon. Severe restrictive lung disease and cardiomyopathy are not typical features, although atrial tachyarrhythmias have been noted in a few patients with other comorbidities [20].
Children and young adults with anti-HMGCR myopathy
A sizable minority or, in some cohorts, the majority of anti-HMGCR myopathy patients do not have a history of exposure to statin drugs. These statin-unexposed adult patients are generally younger but their overall clinical presentation is similar to the classic statin-associated phenotype. Most present with a subacute-onset myopathy and biopsy findings consistent with an IMNM. Perhaps counterintuitively, long-term follow up of younger adults with anti-HMGCR myopathy has demonstrated that they generally tend to have more severe disease and a worse prognosis in response to immunotherapies compared to the older, statin-exposed group [26]. More recently, several pediatric patients with anti-HMGCR myopathy have been identified [27, 28]. When treated with immunotherapies, most pediatric patients show a favorable response, though long-term follow up studies are not available in this group of children.
Chronic anti-HMGCR myopathy with a LGMD-like phenotype
A careful assessment of published studies of anti-HMGCR cohorts identifies a very small subset of pediatric and adult anti-HMGCR patients with a limb-girdle muscular dystrophy (LGMD) phenotype. These patients generally have had a very chronic course of disease, at times with asymptomatic elevation of CK several years before developing muscle weakness [20, 23, 28–30]. Some were diagnosed with LGMD for many years before eventually being diagnosed with anti-HMGCR myopathy. Only a minority of these patients have undergone extensive genetic testing for known muscular dystrophy genes [26]. But so far, none have been found to have pathogenic mutations in LGMD genes. Conversely, large patient cohorts with LGMD have not been comprehensively evaluated for presence of anti-HMGCR antibodies, though it is conceivable that a non-negligible portion of these patients have anti-HMGCR myopathy. Identifying such patients would be extremely important due to the therapeutic implications.
Muscle biopsy
Muscle biopsy findings in anti-HMGCR myopathy are typical of a pauci-immune necrotizing myopathy, showing myofiber degeneration and necrosis with a variable density of regenerating fibers (Fig. 1C). Quantification of myofiber atrophy has also highlighted this as a dramatic feature of the muscle pathology [31]. In patients with chronic disease, fiber size variability, internally placed nuclei, and increased endomysial fibrosis can be seen. Perivascular chronic inflammation can occasionally be seen but lymphocytic invasion of healthy myofibers, as seen in polymyositis, is rarely observed. Major histocompatibility complex 1 (MHC-1) may show increased sarcolemmal staining in non-necrotic fibers and membrane attack complex immunostaining (e.g. C5b-9) may stain the sarcolemma with variable intensity as well as endothelial cells occasionally [24, 28, 32]. A few studies have reported abnormalities in a perifascicular or perimysial distribution [16, 23]. However, these findings vary and are not universally present.
Muscle imaging
Muscle imaging generally shows a heterogeneous pattern of muscle involvement with a predilection for proximal muscles. The three most affected muscle groups in the proximal lower extremities include the posterior thigh, gluteal, and medial thigh compartments [22, 26, 28]. Distal muscles in the lower leg compartment are less studied. Short tau inversion recovery (STIR) signal is usually increased more diffusely and may be asymmetric (Fig. 1B). T1 hyperintensity reflects connective tissue and fatty replacement in the muscle; this is not likely to be reversed by immunosuppressive therapy. The portion of muscle weakness that is reflected by STIR signal increase without T1 change highlights the muscle at risk that can potentially benefit from treatment.
Immunogenetic and environmental risk factors
As in other autoimmune diseases, an interaction between the host immune system and an environmental trigger has been postulated to underlie anti-HMGCR myopathy. As discussed above, statin drugs are a known environmental risk factor for anti-HMGCR myopathy. Other environmental exposures to statins (e.g. mushrooms, a natural source of statins) could possibly trigger the disease in the susceptible host.
The immunogenetic background of the patient can also play a role in their predisposition to develop anti-HMGCR myopathy. The presence of HLA-DRB1*11 : 01 allele is a well-established immunogenetic risk factor for developing this disease. Originally identified in nearly 70% of patients, it has now been validated in multiple cohorts with different racial and ethnic origins as an immunogenetic risk factor [33–35]. A second allele, HLA-DRB1*07 : 01 has been reported in pediatric patients with the disease [27, 29]. Although enriched in patients with anti-HMGCR myopathy, these susceptibility loci are present in the general population at a prevalence of 7–15%. Thus, assaying for these immunogenetic risk factors would not be a practical means of identifying which patients are susceptible to statin-associated autoimmune myopathy. However, they potentially provide insight into the immunopathogenesis of the disease, an area of active investigation.
Association with cancer
Several recent studies of patients with anti-HMGCR myopathy have identified a slightly increased risk of cancer [36–40]. This apparent risk remains lower than other inflammatory myopathies (e.g. dermatomyositis) or antibody-negative IMNMs, but is higher than the general population. Commonly reported cancers include adenocarcinoma of the GI tract, breast, uterus or ovaries. A few cases with lymphoma, melanoma, and thyroid cancer are also reported in the literature. It is unclear whether heightened screening tests are warranted in anti-HMGCR myopathy patients but at the very least, age-appropriate cancer screening guidelines should be meticulously followed.
ANTI-HMGCR AUTOANTIBODY TESTING
Anti-HMGCR autoantibodies were first discovered using an IP assay. Subsequently, several assays for antibody detection have been developed by different laboratories including several enzyme-linked immunosorbent assays (ELISA), immunofluorescence using rat liver as antigen, and addressable laser bead immunoassay (ALBIA) [19, 20, 41, 42]. Some, but not all, of these assays have been validated carefully using the IP assay as a gold standard with reported specificity of ∼94–100% and sensitivity of ∼95–99% [21, 43, 44].
From a clinical standpoint, validated ELISA-positive sera are extremely good indicators of the disease in patients with high pretest probability (i.e. those with muscle weakness, elevated CK, or IMNM on biopsy) [45]. However, some caution in the interpretation should be exercised, especially when the assays are used for research purposes in large cohorts of patients without muscle weakness or CK elevation, reducing the positive predictive value of the test [46]. This may partly explain the detection of anti-HMGCR autoantibodies in a few patients without any signs of muscle weakness or CK elevation, or in those with clearly defined alternative clinical diagnoses. Thus, a positive anti-HMGCR test in association with a new disease entity or novel phenotype should always be confirmed with more specific and sensitive assays (e.g. IP).
Disease specificity of anti-HMGCR autoantibody
Anti-HMGCR autoantibodies are highly specific and are not found in patients with other related diseases. For example, a few studies of patients with statin intolerance or self-limited statin myopathy have failed to identify anti-HMGCR positive patients in these cohorts [44, 47, 48]. In addition, patients with genetically proven muscular dystrophy do not have anti-HMGCR autoantibodies [49].
On the other hand, the co-occurrence of anti-HMGCR antibodies with other MSAs (e.g. SRP [50] and aaRS [22]), inclusion body myositis [21, 23] and other autoimmune diseases (e.g. scleroderma [42], myasthenia gravis [21]) has been reported. Some of these cases may be due to lower specificity of the test used for autoantibody detection in these studies (e.g., ELISA). However, some of these cases are genuine co-occurrence of two diseases in the same patient with clinical features of both disorders [22]. Nonetheless, compared to other MSAs, anti-HMGCR is one of the most specific autoantibodies and, in the right clinical setting, is almost always diagnostic and clinically valuable.
TREATMENT OF ANTI-HMGCR MYOPATHY
Immunosuppressive regimens
Discontinuation of the offending statin drug and avoidance of the class is the first, and perhaps the most important, step in treatment of patients with anti-HMGCR myopathy. Rarely, patients may slowly improve in strength without any further interventions and some may only have persistently elevated CK without weakness. However, the majority of patients eventually require immunosuppressive therapy due to progressive weakness.
No published controlled studies have compared different therapeutic regimens in anti-HMGCR myopathy. Based on observational studies, most patients require more than one immunosuppressive agent. Typically used drugs include daily steroids, methotrexate, azathioprine, mycophenolate mofetil, or rituximab. The most effective regimens especially in patients with refractory disease usually include intravenous immunoglobulins (IVIg) and some patients have been successfully treated with IVIg monotherapy [51]. In patients who have plateaued in recovery, a decrease in dosing should be attempted, with close monitoring.
Therapy maintenance, escalation, and tapering can also vary and these issues have not been fully investigated. Controlled studies are necessary to fully assess dosing and efficacy of IVIg and/or other immunosuppressive regimens in this disease both as initiation therapy and maintenance.
Therapeutic monitoring
Therapeutic monitoring of patients with anti-HMGCR myopathy may include several measurements to guide decision making about therapy initiation, escalation, or termination. Most importantly, these should include patient-reported functional changes and clinical examination including semi-quantitative muscle strength testing (or when available quantitative dynamometry). The most sensitive measures usually include timed tests and can be extremely helpful in monitoring improvement. In addition, since myofiber necrosis leads to elevation of the serum CK, changes in serum CK levels can be used as a surrogate biomarker of disease activity and nearly always decrease substantially in parallel with improved strength. It is worth noting that some patients may have markedly improved or even normal muscle strength with persistently elevated CK levels [26]. Whether such patients warrant more aggressive therapy to decrease the CK levels and prevent chronic muscle damage remains unclear.
Anti-HMGCR antibody levels are reported to correlate with disease severity [20, 52]. However, they rarely, if ever, normalize even in patients that appear to have recovered fully [22]. Thus, anti-HMGCR antibody titers may not be useful for therapeutic monitoring.
Muscle imaging with MRI can be helpful in two ways. First, STIR signal hyperintensity generally reflects areas of active disease. Consequently, response to treatment can be inferred when these areas of hyperintensity diminish and eventually resolve. Of note, we have observed that STIR signal reduction may sometimes lag behind clinical improvement. Second, MRI can identify areas of permanent fatty replacement, which are visualized as T1 signal hyperintense areas within muscles. In persistently weak patients with significant fatty replacement (seen on T1) and minimal active disease (seen on STIR), escalation of immunosuppressive therapy is unlikely to result in further clinical improvements.
PATHOPHYSIOLOGY AND MECHANISM OF DISEASE
The etiology and mechanisms of disease in anti-HMGCR myopathy are areas of active investigation. However, several studies in recent years have provided clues regarding the immunopathogenesis of anti-HMGCR myopathy. Based on the available data, an emerging model of disease can be postulated, enabling predictions about pathomechanisms of anti-HMGCR myopathy that can subsequently be tested in laboratory and clinical studies and revised as needed. Given the wide spectrum of anti-HMGCR myopathy phenotypes, we propose a general framework of interaction between host immunogenetic background with environmental triggers, with multiple paths ultimately converging on sustained autoimmune muscle injury and a sabotaged or inadequate attempt at regeneration (Fig. 2). In this section, we use HMGCR to refer to the protein/autoantigen and anti-HMGCR to the autoantibodies that target the HMGCR protein.
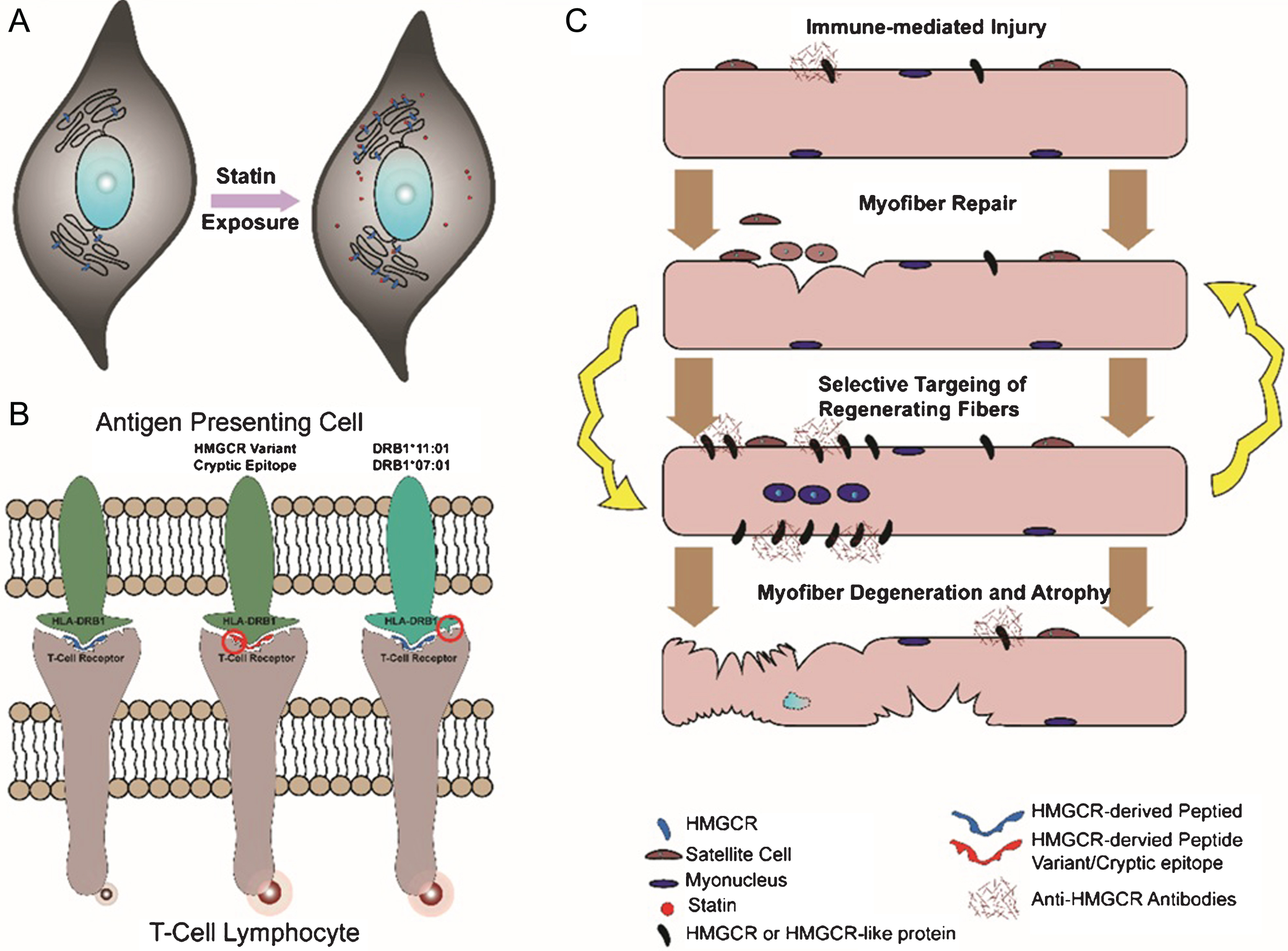
Fig.2
Schematic of postulated pathomechanisms of anti-HMGCR myopathy. (A) Statin exposure results in overexpression of HMGCR in multiple tissues and cell types. (B) Processing of HMGCR by antigen-presenting cells under different scenarios. Variants in HMGCR or cryptic epitopes unmasked by drugs (e.g. statins) and/or polymorphisms in the HLA molecule may increase T-cell receptor activation and initiate autoimmunity. (C) Immune-mediated injury to the muscle is directed against HMGCR or other antigens recognized by anti-HMGCR antibody. Attempts at myofiber repair results in increased HMGCR (or its mimic) expression and sustained autoimmunity with ultimate atrophy and degeneration of myofibers.
Initiation of autoimmunity
Association of anti-HMGCR antibodies with statin intake and/or specific class II HLA alleles (i.e. HLA-DRB1*11 : 01 and 07 : 01) implicates the processing of HMGCR by the host immune system as an important step in pathogenesis. Statin exposure and immunogenetic alleles are risk factors present in all ethnic and racial backgrounds, marking a common pathogenic pathway in the disease.
One possible mechanistic explanation for autoimmunity initiation suggests that certain epitopes (natural or cryptic) of HMGCR, when processed by antigen presenting cells, can potentially trigger an immune response (Fig. 2B). This is likely a function of several factors:
1. HMGCR expression levels. It is now well established that initiation of statin drugs increases HMGCR expression in muscle and other tissues [53–55]. Elevated HMGCR protein levels could result in aberrant processing by antigen presenting cells and the subsequent production of cryptic epitopes that might play a role in provoking an autoimmune response. (Fig. 2A).
2. Immunogenicity of HMGCR-derived peptides. In addition to absolute HMGCR quantity, other factors could confer increased immunogenicity to HMGCR-derived peptides. For example, statin binding to HMGCR could cause conformational changes of the protein and alter its cleavage by antigen presenting cells, resulting in the generation of cryptic epitopes. Somatic (e.g. in case of cancer associated anti-HMGCR myopathy) or genomic sequence variants in HMGCR could potentiate its immunogenicity, as noted for other autoimmune diseases such as scleroderma [56]. Finally, statins may exert immunomodulatory effects on the immune system and alter autoantigen processing.
3. Immunogenic HLA alleles. Processed peptides on the surface of antigen presenting cells are always presented in the context of HLA molecules. It is possible that certain peptides derived from HMGCR protein are significantly more immunogenic when presented in the context of risk factor HLA alleles (i.e. HLA-DRB1*11 : 01 and 07 : 01). Study of HMGCR derived peptides and their immunogenicity in different HLA contexts can shed light on this aspect of the disease.
Obviously these three factors need not exist in isolation. Risk factors such as statin exposure, immunogenetic background, cancer, and/or sequence variants in HMGCR may merely decrease the threshold to develop autoimmunity. Variability in initiation of autoimmunity may explain the highly variable age of onset and rate of progression for the disease.
Immune-mediated injury
The mechanisms of immune-mediated injury and specific targeting of muscle is another important, yet poorly understood, aspect of disease pathogenesis in anti-HMGCR myopathy.
Anti-HMGCR antibodies may be directly pathogenic. HMGCR is a membrane associated protein that is predominantly localized to the endoplasmic reticulum, and to a lesser degree may be present in peroxisomes based on in vitro studies in different cell types [57, 58]. While its subcellular localization in normal myofibers is likely to be similar, some studies have suggested that HMGCR expression is highly increased in regenerating myofibers, and perhaps even on the cell surface, serving as the target for antibody-mediated injury (Fig. 2C). Consistently, a recent study showed anti-HMGCR antibodies cause myofiber atrophy, albeit not necrosis, in vitro [31]. However, passive transfer experiments would be necessary to further assess their direct pathogenicity.
If anti-HMGCR antibodies bind HMGCR protein – or some other target – on the myofiber surface, they may facilitate immune mediated injury by complement fixation. A recent study of 19 anti-HMGCR muscle biopsies has shown complement, including both C5b-9 and C1q, on the sarcolemma of necrotic fibers, suggestive of antibody mediated complement activation [59]. These findings support a direct pathogenic role for anti-HMGCR antibodies, targeting the sarcolemma and inducing complement activation and subsequent myofiber injury. Alternatively, anti-HMGCR antibodies might be merely a byproduct of a T-cell mediated cytotoxic attack on myofibers. Elucidating the mechanisms of muscle injury remains a priority as this could identify appropriate therapeutic targets. For example, if muscle damage is a result of complement mediated cytotoxicity, then complement inhibitors could be an effective therapy for anti-HMGCR myopathy.
CONCLUSIONS
In summary, we have presented a general overview of the clinical and pathological aspects of anti-HMGCR myopathy as a specific disease entity. Though rare, with appropriate clinical suspicion and judicious use of autoantibody testing, anti-HMGCR myopathy can be diagnosed effectively and in most cases treated successfully. This is of special importance in anti-HMGCR myopathy patients who carry incorrect, untreatable diagnoses such as LGMD. From a clinical standpoint, controlled studies of different immunosuppressive regimens are lacking and if conducted carefully would have immediate clinical consequences in treatment of patients with anti-HMGCR myopathy. Studies of the pathomechanism of the disease are in their early stages but can also inform future clinical studies and provide a better understanding of the disease process. They can also potentially pave the way to identify novel, and disease-specific therapeutic targets for patients with anti-HMGCR myopathy while expanding our knowledge of how the immune system interacts with muscle in disease states.
CONFLICT OF INTEREST
The authors do not have any conflicts to report.
ACKNOWLEDGMENTS AND SOURCES OF FUNDING
The funding for this study is provided through the intramural research program of the National Institutes of Health.
REFERENCES
[1] | Hoogendijk JE , Amato AA , Lecky BR , Choy EH , Lundberg IE , Rose MR , et al. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. (2004) ;14: (5):337–45. |
[2] | Stenzel W , Preusse C , Allenbach Y , Pehl D , Junckerstorff R , Heppner FL , et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology. (2015) ;84: (13):1346–54. |
[3] | De Bleecker JL , De Paepe B , Aronica E , de Visser M , EMMBS Group, Amato A , et al. 205th ENMC International Workshop: Pathology diagnosis of idiopathic inflammatory myopathies part II 28-30 March 2014, Naarden, The Netherlands. Neuromuscul Disord. (2015) ;25: (3):268–72. |
[4] | Uruha A , Suzuki S , Suzuki N , Nishino I . Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain. (2016) ;139: (Pt 9):e50. |
[5] | Allenbach Y , Mammen AL , Stenzel W , Benveniste O , Immune-Mediated Necrotizing Myopathies Working G. 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord. (2017) . |
[6] | Takagi A , Shiio Y . Pravastatin-associated polymyositis, a case report. Rinsho Shinkeigaku. (2004) ;44: (1):25–7. |
[7] | Riesco-Eizaguirre G , Arpa-Gutierrez FJ , Gutierrez M , Toribio E . Severe polymyositis with simvastatin use. Rev Neurol. (2003) ;37: (10):934–6. |
[8] | Giordano N , Senesi M , Mattii G , Battisti E , Villanova M , Gennari C . Polymyositis associated with simvastatin. Lancet. (1997) ;349: (9065):1600–1. |
[9] | Khattak FH , Morris IM , Branford WA . Simvastatin-associated dermatomyositis. Br J Rheumatol. (1994) ;33: (2):199. |
[10] | Vasconcelos OM , Campbell WW . Dermatomyositis-like syndrome and HMG-CoA reductase inhibitor (statin) intake. Muscle Nerve. (2004) ;30: (6):803–7. |
[11] | Noel B , Cerottini JP , Panizzon RG . Atorvastatin-induced dermatomyositis. Am J Med. (2001) ;110: (8):670–1. |
[12] | Hill C , Zeitz C , Kirkham B . Dermatomyositis with lung involvement in a patient treated with simvastatin. Aust N Z J Med. (1995) ;25: (6):745–6. |
[13] | Rodriguez-Garcia JL , Serrano Commino M . Lovastatin-associated dermatomyositis. Postgrad Med J. (1996) ;72: (853):694. |
[14] | Zuech P , Pauwels C , Duthoit C , Mery L , Somogyi A , Louboutin A , et al. Pravastatin-induced dermatomyositis. Rev Med Interne. (2005) ;26: (11):897–902. |
[15] | Fauchais AL , Iba Ba J , Maurage P , Kyndt X , Bataille D , Hachulla E , et al. Polymyositis induced or associated with lipid-lowering drugs: Five cases. Rev Med Interne. (2004) ;25: (4):294–8. |
[16] | Needham M , Fabian V , Knezevic W , Panegyres P , Zilko P , Mastaglia FL . Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. (2007) ;17: (2):194–200. |
[17] | Grable-Esposito P , Katzberg HD , Greenberg SA , Srinivasan J , Katz J , Amato AA . Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. (2010) ;41: (2):185–90. |
[18] | Christopher-Stine L , Casciola-Rosen LA , Hong G , Chung T , Corse AM , Mammen AL . A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. (2010) ;62: (9):2757–66. |
[19] | Mammen AL , Chung T , Christopher-Stine L , Rosen P , Rosen A , Doering KR , et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. (2011) ;63: (3):713–21. |
[20] | Allenbach Y , Drouot L , Rigolet A , Charuel JL , Jouen F , Romero NB , et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: Inconstant exposure to statin. Medicine (Baltimore). (2014) ;93: (3):150–7. |
[21] | Watanabe Y , Suzuki S , Nishimura H , Murata KY , Kurashige T , Ikawa M , et al. Statins and myotoxic effects associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies: An observational study in Japan. Medicine (Baltimore). (2015) ;94: (4):e416. |
[22] | Waters MJ , Limaye V . Clinico-serologic features of statin-induced necrotising autoimmune myopathy in a single-centre cohort. Clin Rheumatol. (2017) . |
[23] | Alshehri A , Choksi R , Bucelli R , Pestronk A . Myopathy with anti-HMGCR antibodies: Perimysium and myofiber pathology. Neurol Neuroimmunol Neuroinflamm. (2015) ;2: (4):e124. |
[24] | Ashton C , Junckerstorff R , Bundell C , Hollingsworth P , Needham M . Treatment and outcomes in necrotising autoimmune myopathy: An Australian perspective. Neuromuscul Disord. (2016) ;26: (11):734–40. |
[25] | Kennedy N , Keating P , O’Donnell J . HMGCR-associated myositis: A New Zealand case series and estimate of incidence. Intern Med J. (2016) ;46: (5):622–5. |
[26] | Tiniakou E , Pinal-Fernandez I , Lloyd TE , Albayda J , Paik J , Werner JL , et al. More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Rheumatology (Oxford). (2017) ;56: (5):787–94. |
[27] | Kishi T , Rider LG , Pak K , Barillas-Arias L , Henrickson M , McCarthy PL , et al. Association of Anti-3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Autoantibodies With DRB1*01 and Severe Myositis in Juvenile Myositis Patients. Arthritis Care Res (Hoboken). (2017) ;69: (7):1088–94. |
[28] | Liang WC , Uruha A , Suzuki S , Murakami N , Takeshita E , Chen WZ , et al. Pediatric necrotizing myopathy associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies. Rheumatology (Oxford). (2017) ;56: (2):287–93. |
[29] | Mohassel P , Foley AR , Donkervoort S , Fequiere PR , Pak K , Bonnemann CG , et al. Anti-3-hydroxy-3-methylglutaryl-coenzyme a reductase necrotizing myopathy masquerading as a muscular dystrophy in a child. Muscle Nerve. (2017) . |
[30] | Tard C , Tiffreau V , Jaillette E , Jouen F , Nelson I , Bonne G , et al. Anti-HMGCR antibody-related necrotizing autoimmune myopathy mimicking muscular dystrophy. Neuropediatrics. (2017) . |
[31] | Arouche-Delaperche L , Allenbach Y , Amelin D , Preusse C , Mouly V , Mauhin W , et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol. (2017) ;81: (4):538–48. |
[32] | Chung T , Christopher-Stine L , Paik JJ , Corse A , Mammen AL . The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle Nerve. (2015) ;52: (2):189–95. |
[33] | Mammen AL , Gaudet D , Brisson D , Christopher-Stine L , Lloyd TE , Leffell MS , et al. Increased frequency of DRB1*01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (Hoboken). (2012) ;64: (8):1233–7. |
[34] | Limaye V , Bundell C , Hollingsworth P , Rojana-Udomsart A , Mastaglia F , Blumbergs P , et al. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve. (2015) ;52: (2):196–203. |
[35] | Ohnuki Y , Suzuki S , Shiina T , Uruha A , Watanabe Y , Suzuki S , et al. HLA-DRB1 alleles in immune-mediated necrotizing myopathy. Neurology. (2016) ;87: (18):1954–5. |
[36] | Kadoya M , Hida A , Hashimoto Maeda M , Taira K , Ikenaga C , Uchio N , et al. Cancer association as a risk factor for anti-HMGCR antibody-positive myopathy. Neurol Neuroimmunol Neuroinflamm. (2016) ;3: (6):e290. |
[37] | Allenbach Y , Keraen J , Bouvier AM , Jooste V , Champtiaux N , Hervier B , et al. High risk of cancer in autoimmune necrotizing myopathies: Usefulness of myositis specific antibody. Brain. (2016) ;139: (Pt 8):2131–5. |
[38] | Mizuma A , Kouchi M , Netsu S , Yutani S , Kitao R , Suzuki S , et al. Paraneoplastic anti-3-hydroxy-3-methylglutary-coenzyme A reductase antibody-positive immune-mediated necrotizing myopathy in a patient with uterine cancer. Intern Med. (2017) ;56: (14):1915–8. |
[39] | Yamaguchi T , Matsunaga A , Ikawa M , Shirafuji N , Nishino I , Hamano T . A case of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibody-positive paraneoplastic necrotizing myopathy associated with advanced gastric cancer that responded to intravenous immunoglobulin therapy. Rinsho Shinkeigaku. (2017) ;57: (3):118–23. |
[40] | Tsujikawa K , Hara K , Muro Y , Nakanishi H , Niwa Y , Koike M , et al. HMGCR antibody-associated myopathy as a paraneoplastic manifestation of esophageal carcinoma. Neurology. (2016) ;87: (8):841–3. |
[41] | Alvarado-Cardenas M , Marin-Sanchez A , Martinez MA , Martinez-Martinez L , Pinal-Fernandez I , Labrador-Horrillo M , et al. Statin-associated autoimmune myopathy: A distinct new IFL pattern can increase the rate of HMGCR antibody detection by clinical laboratories. Autoimmun Rev. (2016) ;15: (12):1161–6. |
[42] | Hudson M , Luck Y , Stephenson M , Choi MY , Wang M , Baron M , et al. Anti-HMGCR antibodies in systemic sclerosis. Medicine (Baltimore). (2016) ;95: (44):e5280. |
[43] | Jaskowski TD , La’ulu SL , Mahler M , Tebo AE . Detection of autoantibodies to 3-hydroxy-3-methylglutaryl-coenzyme a reductase by ELISA in a reference laboratory setting. Clin Chim Acta. (2017) ;472: :30–4. |
[44] | Mammen AL , Pak K , Williams EK , Brisson D , Coresh J , Selvin E , et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken). (2012) ;64: (2):269–72. |
[45] | Klein M , Mann H , Plestilova L , Zamecnik J , Betteridge Z , McHugh N , et al. Increasing incidence of immune-mediated necrotizing myopathy: Single-centre experience. Rheumatology (Oxford). (2015) ;54: (11):2010–4. |
[46] | Droney L , Gillis D , Wong R . New immunoassays for anti-HMG-CoA reductase antibodies may lead to incorrect diagnosis in inflammatory myositis. Pathology. (2017) ;49: (6):638–9. |
[47] | Floyd JS , Brody JA , Tiniakou E , Psaty BM , Mammen A . Absence of anti-HMG-CoA reductase autoantibodies in severe self-limited statin-related myopathy. Muscle Nerve. (2016) ;54: (1):142–4. |
[48] | Keating P , Young J , George P , Florkowski C , Spellerberg M , Kennedy N . Anti-HMGCR autoantibodies in self-limiting statin-induced myopathy. Int J Rheum Dis. (2017) . |
[49] | Mammen AL , Casciola-Rosen L , Christopher-Stine L , Lloyd TE , Wagner KR . Myositis-specific autoantibodies are specific for myositis compared to genetic muscle disease. Neurol Neuroimmunol Neuroinflamm. (2015) ;2: (6):e172. |
[50] | Suzuki S , Nishikawa A , Kuwana M , Nishimura H , Watanabe Y , Nakahara J , et al. Inflammatory myopathy with anti-signal recognition particle antibodies: Case series of 100 patients. Orphanet J Rare Dis. (2015) ;10: :61. |
[51] | Mammen AL , Tiniakou E . Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. (2015) ;373: (17):1680–2. |
[52] | Werner JL , Christopher-Stine L , Ghazarian SR , Pak KS , Kus JE , Daya NR , et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. (2012) ;64: (12):4087–93. |
[53] | Brown MS , Faust JR , Goldstein JL , Kaneko I , Endo A . Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem. (1978) ;253: (4):1121–8. |
[54] | Nakanishi M , Goldstein JL , Brown MS . Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J Biol Chem. (1988) ;263: (18):8929–37. |
[55] | Morikawa S , Murakami T , Yamazaki H , Izumi A , Saito Y , Hamakubo T , et al. Analysis of the global RNA expression profiles of skeletal muscle cells treated with statins. J Atheroscler Thromb. (2005) ;12: (3):121–31. |
[56] | Joseph CG , Darrah E , Shah AA , Skora AD , Casciola-Rosen LA , Wigley FM , et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science. (2014) ;343: (6167):152–7. |
[57] | Kovacs WJ , Tape KN , Shackelford JE , Duan X , Kasumov T , Kelleher JK , et al. Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes. Histochem Cell Biol. (2007) ;127: (3):273–90. |
[58] | Luskey KL , Stevens B . Human 3-hydroxy-3-methylglutaryl coenzyme A reductase. Conserved domains responsible for catalytic activity and sterol-regulated degradation. J Biol Chem. (1985) ;260: (18):10271–7. |
[59] | Allenbach Y , Arouche-Delaperche L , Preusse C , Radbruch H , Butler-Browne G , Champtiaux N , et al. Necrosis in anti-SRP+ and anti-HMGCR+ myopathies: Role of auto-antibodies and complement. Neurology. (2018) ; In press. |


