
Long-Term Pulmonary Function in Duchenne Muscular Dystrophy: Comparison of Eteplirsen-Treated Patients to Natural History
Abstract
Background:
Duchenne muscular dystrophy (DMD) is a rare, degenerative, X-linked genetic disease that results in progressive muscle loss and premature death, most commonly from respiratory or cardiac failure. DMD is primarily caused by whole exon deletions, resulting in a shift of the dystrophin mRNA reading frame that prevents production of functional dystrophin protein. Eteplirsen, a phosphorodiamidate morpholino oligomer (PMO), is designed to skip exon 51, restore the reading frame, and induce production of internally shortened dystrophin in patients with mutations amenable to such treatment.
Objective:
Describe lung function assessed throughout eteplirsen studies 201/202.
Methods:
Studies 201/202 included 12 patients treated with eteplirsen over 5 years. Pulmonary function tests included forced vital capacity (FVC), maximum expiratory pressure (MEP), and maximum inspiratory pressure (MIP). With no long-term placebo control, FVC results were compared with data from the United Dystrophinopathy Project (UDP). MIP and MEP were compared to published natural history.
Results:
Age-adjusted mixed-model repeated-measures analysis showed decreases of 2.3% and 2.6% annually for FVC% p and MEP% p, and an annual increase of 0.6% for MIP% p for the eteplirsen-treated cohort. Data from the UDP demonstrated a 4.1% decline in FVC% p. The published natural history reports annual declines of at least 2.7% and 3.8% for MEP% p and MIP% p, respectively, in patients with DMD.
Conclusions:
With eteplirsen treatment, deterioration of respiratory muscle function based on FVC% p was half of that seen in the UDP; MEP% p and MIP% p compared favorably with natural history.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked recessive neuromuscular disease that affects approximately one out of 3500 to 5000 male newborns [1–4]. It is caused by mutations in the dystrophin gene, which result in an abnormal dystrophin protein, altered myocyte integrity, muscle wasting and relentlessly progressive weakness. Affected boys show initial signs in the first years of life and eventually lose the ability to walk during childhood [5]. DMD patients ultimately succumb to respiratory and cardiac failure in their late teens or early 20s [6, 7]. The DMD locus has a high spontaneous mutation rate, which is a reflection of the large gene size; approximately one third of sporadic cases of DMD are due to de novo mutations. Large rearrangements, including deletions or duplications of one or more exons, are the most frequently seen gene defect, and point mutations account for 13% to 15% of cases [8]. The majority of these mutations alter the mRNA reading frame and prevent expression of functional dystrophin protein. Eteplirsen is a phosphorodiamidate morpholino oligomer based RNA analog targeting patients amenable to exon 51 skipping [9]. It induces skipping of exon 51 to restore the mRNA reading frame with the goal of allowing for the production of an internally deleted but functional dystrophin protein. This strategy is supported by the finding that internally deleted dystrophin proteins occur in patients with Becker muscular dystrophy who present with a less severe phenotype [10]. Eteplirsen has been shown to increase dystrophin-positive fibers in skeletal muscle [11–13] and to restore the dystrophin-associated glycoprotein complex at the sarcolemma [11].
In DMD patients, pulmonary function becomes progressively impaired as the dystrophic process affects respiratory muscles, including the diaphragm, and leads to significant morbidity and mortality. This can be measured by assessing a number of different parameters of lung function, including the total amount of air that can be moved through the lungs after a maximal inspiration and then exhalation (forced vital capacity [FVC]) and the maximal sustained pressure generated during inspiration (maximum inspiratory pressure [MIP]) and expiration (maximum expiratory pressure [MEP]). FVC measures output of both inspiratory and expiratory muscles, is an excellent measure of respiratory function reserve, and is widely used in DMD to assess respiratory function [14–20]. MEP and MIP provide a more specific assessment of expiratory and inspiratory muscle function [14–16, 19]. In children, FVC, MIP, and MEP normally increase with growth; to adjust for this, evaluations over time are done using percent predicted (% p) relative to expected measurements in healthy controls rather than using absolute values.
Study 201 comprised a 24-week, randomized, placebo-controlled period followed by a 4-week open-label period, and study 202 was a 212-week, open-label safety extension study. These consecutive studies assessed the safety and efficacy of eteplirsen. The primary end point was the 6-minute walk test (6MWT), and data on this and other clinical and biomarker end points were reported separately [12, 13]. Lung function parameters were collected as exploratory assessments throughout the study. We hypothesized that FVC% p, MIP% p, and MEP% p would remain stable or decline less than the annual decline observed in observational patient-level data (FVC) and in natural history studies of patients with DMD reported in the literature (MIP% p, MEP% p, FVC) [14–17, 19].
MATERIALS AND METHODS
Eteplirsen-treated cohort
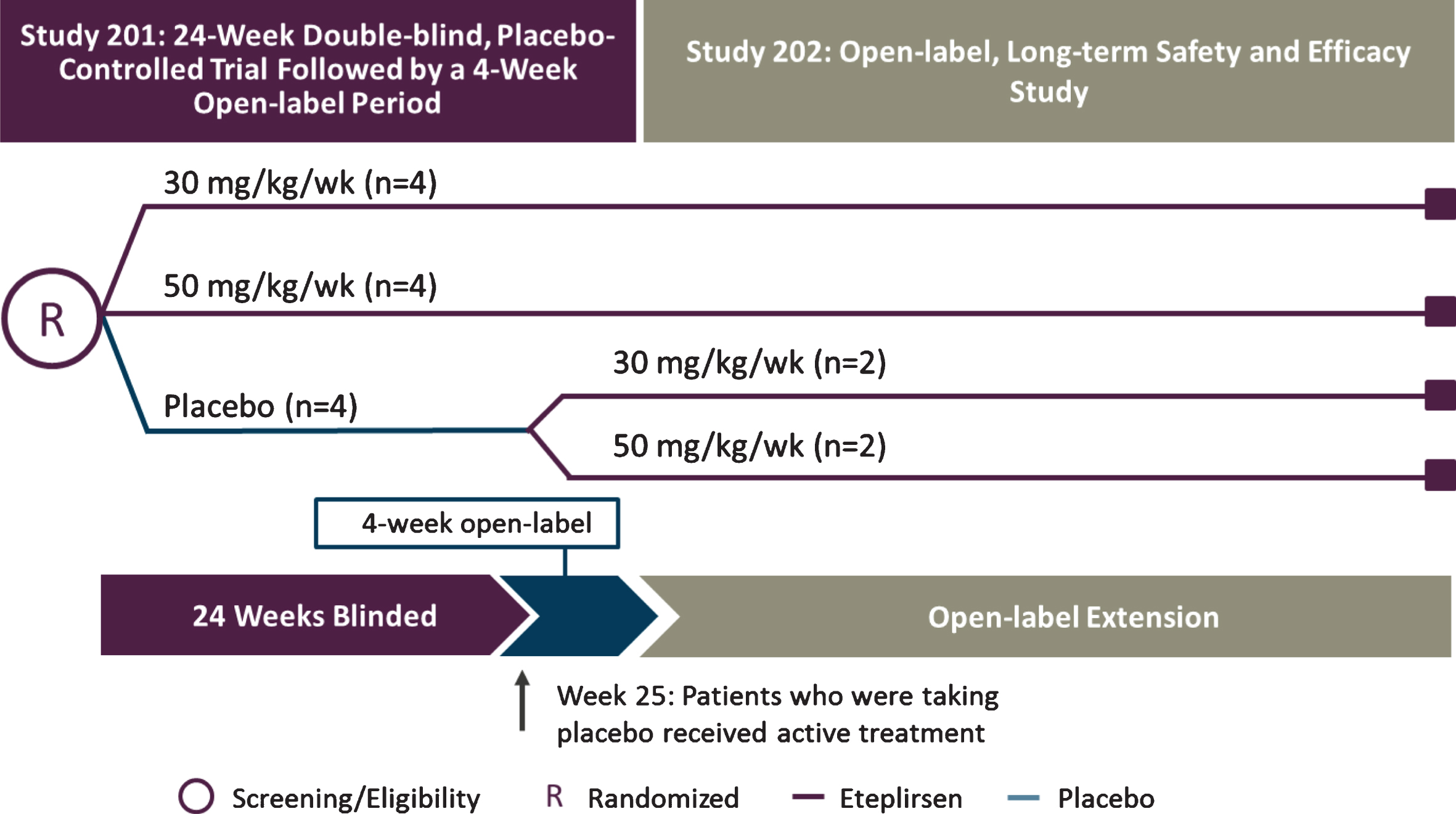
The eligibility criteria for eteplirsen studies 201 and 202 (NCT01396239 and NCT01540409) included age of 7 to 13 years old, dystrophin mutations amenable to exon 51 skipping, and stable corticosteroid therapy for≥24 weeks prior to entry, with maintenance of steroid therapy throughout the study. Patients were randomly assigned to 3 cohorts (n = 4 each) in a double-blind fashion, receiving weekly intravenous (IV) eteplirsen 30 mg/kg or 50 mg/kg or placebo for 24 weeks during study 201 (Fig. 1) [13]. Patients who had received eteplirsen continued at the same dose after transitioning to study 202, whereas placebo-treated patients were randomized 1:1 to 30 mg/kg or 50 mg/kg eteplirsen. Eteplirsen studies 201 and 202 were approved by their respective institutional review boards. Informed assent was obtained from each patient and written informed consent was obtained from a parent or legal guardian for each patient prior to enrollment. Both studies were conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines.
Fig.1
Study design for eteplirsen studies 201/202. Twelve patients with DMD were randomly assigned to 1 of 3 cohorts receiving weekly infusions in a 24-week, double-blind, placebo-controlled study: eteplirsen 30 mg/kg or 50 mg/kg (purple lines), or placebo (blue line). At week 25, eteplirsen-treated patients continued the same weekly dose as open-label, while placebo patients were randomized to open-label treatment with eteplirsen 30 mg/kg or 50 mg/kg weekly IV (study 202). Pulmonary function tests were assessed in compliance with American Thoracic Society guidelines at least every 24 weeks. The 5-year data represent time on drug, beginning at week 0 for patients in treatment arms and at week 24 for those in the placebo arm of study 201. DMD, Duchenne muscular dystrophy; IV, intravenous.
All functional assessments were done at Nationwide Children’s Hospital, (Columbus, OH). A MicroLab ML3500 spirometer with a pediatric mouthpiece and adapter was used for all testing. Pulmonary function tests (PFTs) were performed at each functional assessment visit by experienced physical therapists who were trained in performing spirometry in compliance with American Thoracic Society/European Respiratory Society (ATS/ERS) guidelines and included MIP, MEP, and FVC [21]. At least 3 successful trials for each test were completed, but testing continued until the patient’s maximal effort was recorded. In order to establish normal values, the height or a surrogate for height was obtained for each patient. Standing height was measured for the 10 patients who maintained ambulation during the study. For the 2 patients who lost ambulation during the study, standing height was measured from baseline until week 24, after which loss of ambulation occurred. After loss of ambulation, height was estimated from ulnar length measurements because measurement of height can be complicated by inability to stand, scoliosis, and joint contractures [22].
Natural history cohort
Since pulmonary function declines relatively slowly over time in patients with DMD and the placebo-controlled period in study 201 was limited to 24 weeks, patient-level data from 34 patients who participated in the United Dystrophinopathy Project (UDP), who were in the age range of eteplirsen-treated patients (7–15.5 years old) and who underwent pulmonary function testing, were used as a comparator [17]. The UDP seeks to understand how variations in the dystrophin gene affect the clinical symptoms of DMD. The UDP was chosen based on availability of data and comparability with the eteplirsen group with regard to patient baseline characteristics and the use of similar treatment guidelines in the care of the patients [7]. Characteristics of this cohort have been reported previously [17]. All patients with a DMD phenotype and confirmed dystrophin mutation were eligible for participation in the UDP, in which DMD was clinically defined as symptom onset by the age of 5 years, having a positive Gowers’ sign, having an abnormal gait, having a markedly elevated creatine kinase level, and losing ambulation by the age of 12 years [17]. As part of the UDP, prospective spirometry data were collected in the Neuromuscular Clinic at The Children’s Hospital of Philadelphia (CHOP; Philadelphia, PA) from 2005–2010. DMD care guidelines [6, 7] were generally followed. Standing height was measured when the patient was ambulatory. If the patient could no longer stand, the larger of sitting arm span or recumbent segmental length (head to hip, hip to knee, and knee to foot) was used to calculate a surrogate measure of height.
Like the eteplirsen cohorts, standardized pulmonary function testing was performed in the UDP by trained expert evaluators according to ATS/ERS guidelines [21, 23]. Assessments included FVC and FVC% predicted. MIP and MEP were not performed. A minimum of three trials with maximal effort were attempted by each patient, and the therapist performing the testing made the initial determination if the effort was acceptable. PFTs were performed during regular Neuromuscular Clinic visits, which usually occur quarterly or semiannually. The data from the UDP utilized for this comparison included patient number, visit number, age at visit, height at visit, weight at visit, FVC, and FVC% p.
Conversion of PFT values to percent predicted
Since pulmonary function is impacted by body growth and age, FVC was normalized to height and age and expressed as % predicted (FVC% p, MIP% p and MEP% p). Normal values were defined based on height according to the predicted normal values that were established by Polgar et al. [24] and Hankinson et al. (NHANES III) [23]. The most widely used correction for predicting MEP and MIP was employed in this study and adjusts by age (MEP) or weight (MIP) [25].
Statistical analysis
For the patients who were originally randomized to eteplirsen, baseline was considered the randomization visit and follow-up was 240 weeks. For patients who were originally randomized to placebo (n = 4), baseline was considered the study visit prior to transition to eteplirsen (week 24 visit). Therefore, summary statistics and analyses for all patients (n = 12) were based on 216 weeks of eteplirsen treatment. Consistent with the prior analysis of this study, the data for both dose groups were pooled for this report [12, 13]. Mean age was calculated based on the number of whole years for each subject (i.e., ages of 7, 8, 9, etc.).
The FVC% p, MEP% p, and MIP% p were plotted over time both by weeks on treatment and by age. Annual change was calculated as change from baseline at 216 weeks of treatment divided by 4.5 years (a year is considered 48 weeks).
Patient-level data from the UDP were used in the analysis of FVC [17] and were compared to patient-level data for eteplirsen-treated patients from studies 201/202. FVC% p versus age (in years) was plotted for eteplirsen-treated patients from studies 201/202 and separately for the natural history cohort patients from the UDP.
The UDP study included a broader age range than data from Studies 201/202, including patients through 24 years of age, when pulmonary function is expected to have declined substantially and a majority of patients would be expected to be on assisted ventilation. Therefore, the natural history cohort that is analyzed and presented here was restricted to only include untreated patients whose age would have qualified them for studies 201/202 (N = 34). Using Proc Glimmix in SAS, the slope and corresponding 95% confidence interval of the best-fit lines for each group were estimated from a mixed-effects model, with age as a fixed effect and patient as a random effect to account for each value not being independent.
Statistical analysis was performed using SAS version 9.2 or 9.4 (SAS Institute Inc; Cary, North Carolina).
RESULTS
Baseline demographic and disease characteristics for the eteplirsen-treated and natural history cohorts are provided in Table 1. Eteplirsen-treated patients were, on average, 9 years of age, 123.9 cm tall, and weighed 33.0 kg, with a body mass index (BMI) of 21.13 kg/m2. Mean FVC% p and MIP% p were within the normal range at 97.7% and 91.7%, respectively, and MEP% p was 80.7%. All patients were taking concomitant steroid treatment throughout the study. At baseline, natural history patients were on average 10 years of age, 131.2 cm tall, weighed 35.0 kg, and had a BMI of 19.7 kg/m2. At baseline, 68% of natural history patients were treated with corticosteroids, and mean FVC% p was 96.8%.
Table 1
Demographics and baseline disease characteristics of the eteplirsen-treated and natural history cohorts
| Parametera | Eteplirsen-Treated Cohort | Natural History Cohort | |
| Baseline (N = 12) | Week 216 (N = 12) | Baseline (N = 34) | |
| Age, years | 9.0 (1.13) | 13.1 (1.08) | 10.1 (2.22) |
| Height, cm | 123.9 (8.37) | 137.0 (13.82) | 131.2 (15.37) |
| Weight, kg | 33.0 (7.29) | 47.8 (16.07) | 35.0 (14.15) |
| Body mass index, kg/m2 | 21.1 (3.52) | 25.0 (5.04) | 19.7 (4.36) |
| Ambulatory, n (%) | 12 (100) | 10 (83) | 27 (79) |
| Steroid-treated, n (%) | 12 (100) | 12 (100) | 23 (68) |
| FVC, l | 1.66 (0.310) | 1.88 (0.405) | 1.80 (0.463) |
| FVC% p | 97.7 (14.00) | 85.3 (15.95) | 96.8 (23.30) |
| MEP, cm H2O | 70.2 (30.45) | 75.9 (22.76) | NA |
| MEP% p | 80.7 (34.21) | 69.2 (20.23) | NA |
| MIP, cm H2O | 63.5 (17.09) | 69.3 (17.32) | NA |
| MIP% p | 91.7 (21.35) | 87.1 (23.79) | NA |
aData are presented as mean (SD) unless otherwise indicated. % p, percent predicted; FVC, forced vital capacity; MEP, maximum expiratory pressure; MIP, maximum inspiratory pressure; NA, not available; SD, standard deviation.
Absolute FVC and FVC% p were plotted by time on treatment and by age. In eteplirsen-treated patients, FVC absolute volume continued to increase in nearly all patients (Fig. 2). Mean FVC% p decreased from a mean 97.7% to 85.3% over 216 weeks, a decrease of 2.8% per study year (Fig. 3).
Fig.2
(A) FVC in liters by weeks on treatment, and (B) FVC in liters versus age (rounded to nearest 0.5 year for mean line). Only assessments performed every 24 weeks are represented graphically, although additional time points were assessed during the first 96 weeks. In Figures 2 through 7, the thick black line represents the mean while individual patients are represented by the same colored line throughout. FVC, forced vital capacity.
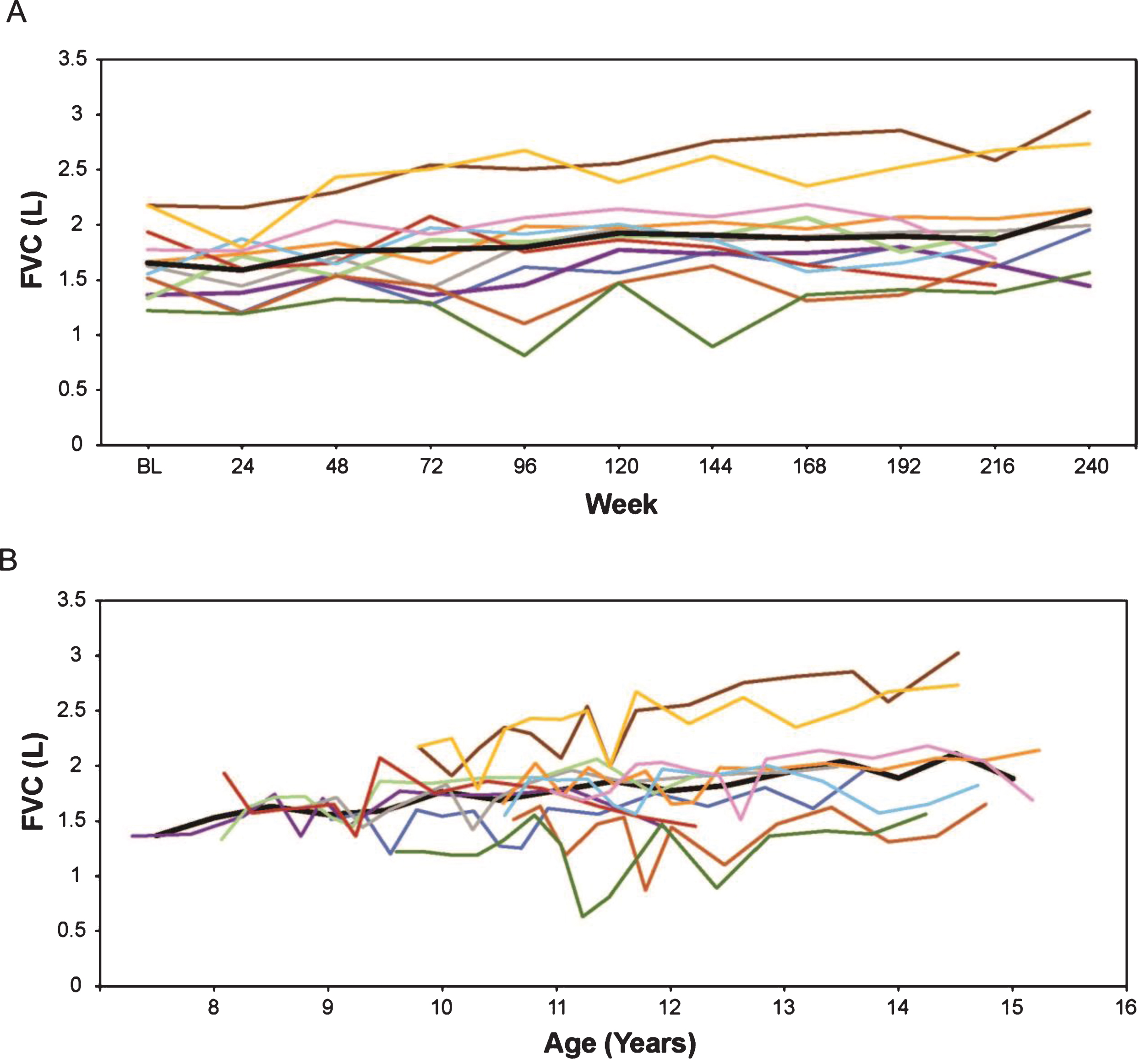
Fig.3
(A) FVC% p by weeks on treatment, and (B) FVC% p versus age (rounded to nearest 0.5 year for mean line). Only assessments performed every 24 weeks are represented graphically although additional time points were assessed during the first 96 weeks. FVC% p, percent predicted forced vital capacity.
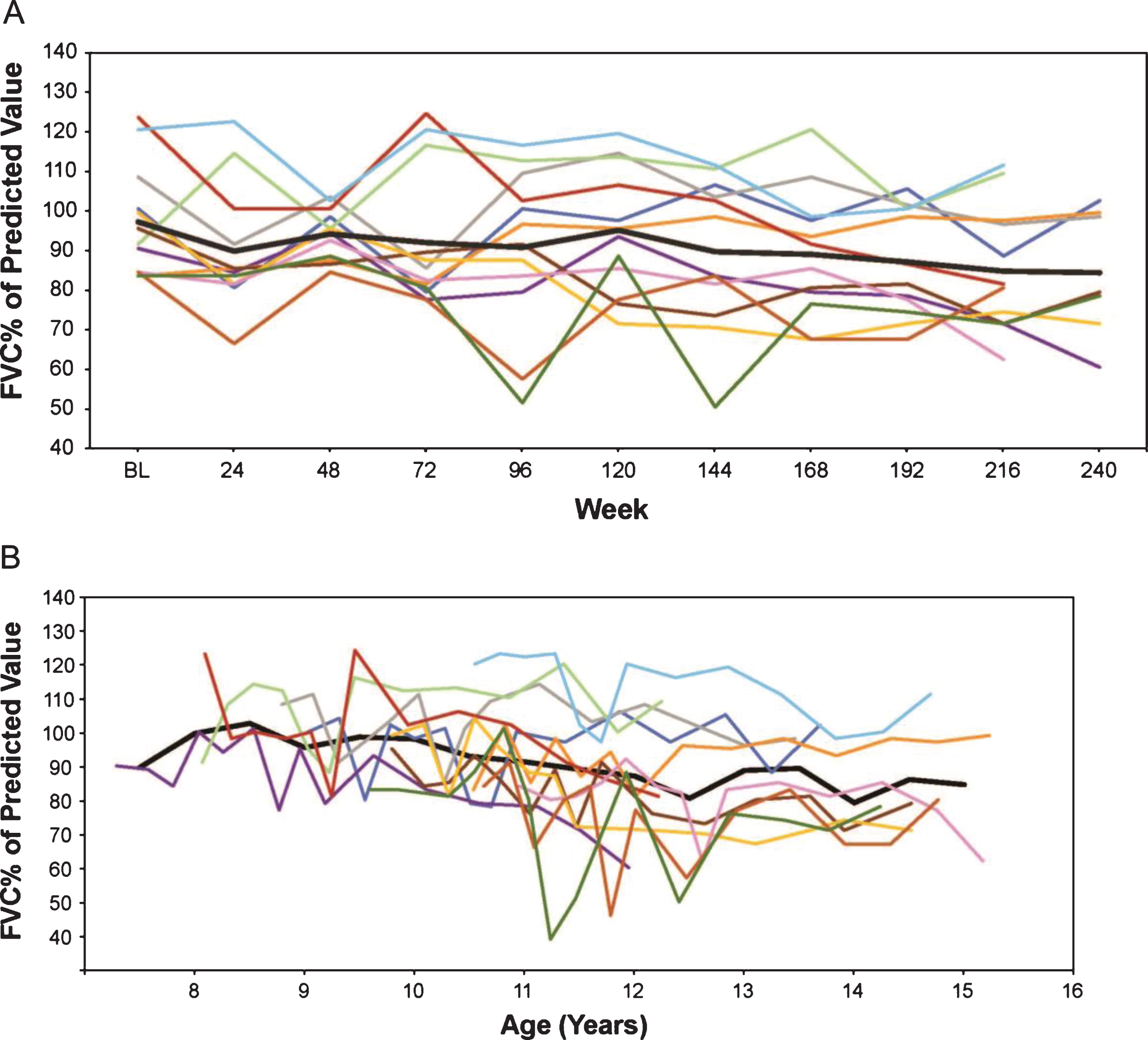
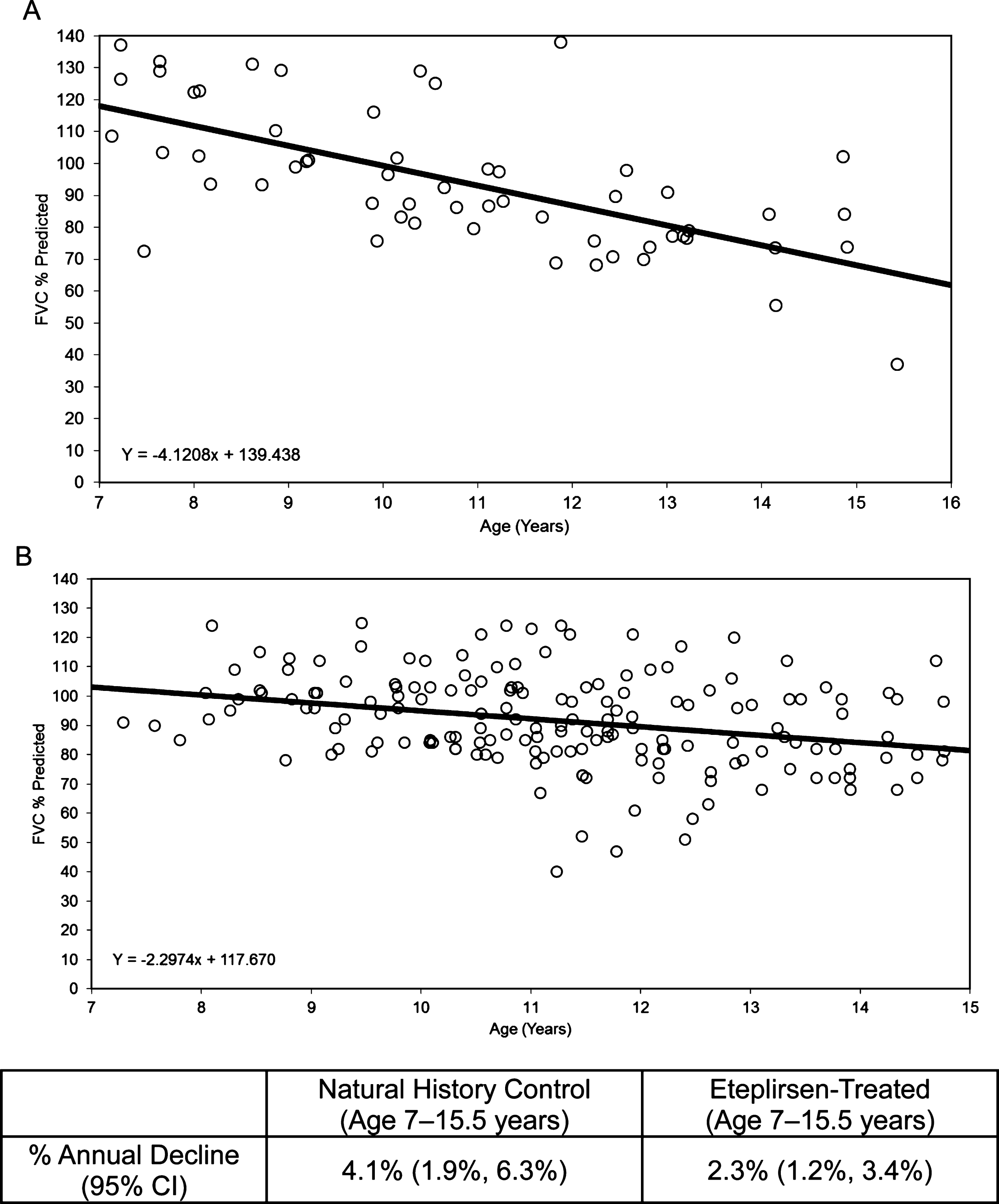
Using an age-adjusted mixed-effects analysis, the natural history cohort from UDP was compared with the eteplirsen-treated cohort, revealing a difference in the rate of decline of FVC% p. The slope of the natural history cohort was –4.1, or a loss of 4.1% per year (Fig. 4A), while the slope of the eteplirsen-treated cohort was –2.3, or a loss of 2.3% per year (Fig. 4B).
Fig.4
FVC% p: Natural history controls compared with eteplirsen-treated patients. (A) Natural history controls (ages 7–15.5 years), with an annual decline of 4.1%, and (B) eteplirsen-treated patients (30 or 50 mg/kg/wk IV) from studies 201/202, all available on-treatment data plotted versus age, with an annual decline of 2.3%. FVC% p, percent predicted forced vital capacity.
In eteplirsen-treated patients, MEP% p (calculated using the Wilson et al. formula [25]) decreased from a mean 80.7% to 69.2% over 216 weeks, an annual decrease of 2.6%. In an age-adjusted mixed-effects analysis of MEP% p, an annual decrease of 2.6% was seen. Similar trajectories were seen across all patients (Fig. 5).
Fig.5
MEP% p in eteplirsen-treated patients (A) by weeks on treatment, and (B) by average age in years (rounded to the nearest 0.5 years for mean line). Only assessments performed every 24 weeks are represented graphically, although additional time points were assessed during the first 96 weeks. MEP% p, percent predicted maximum expiratory pressure.
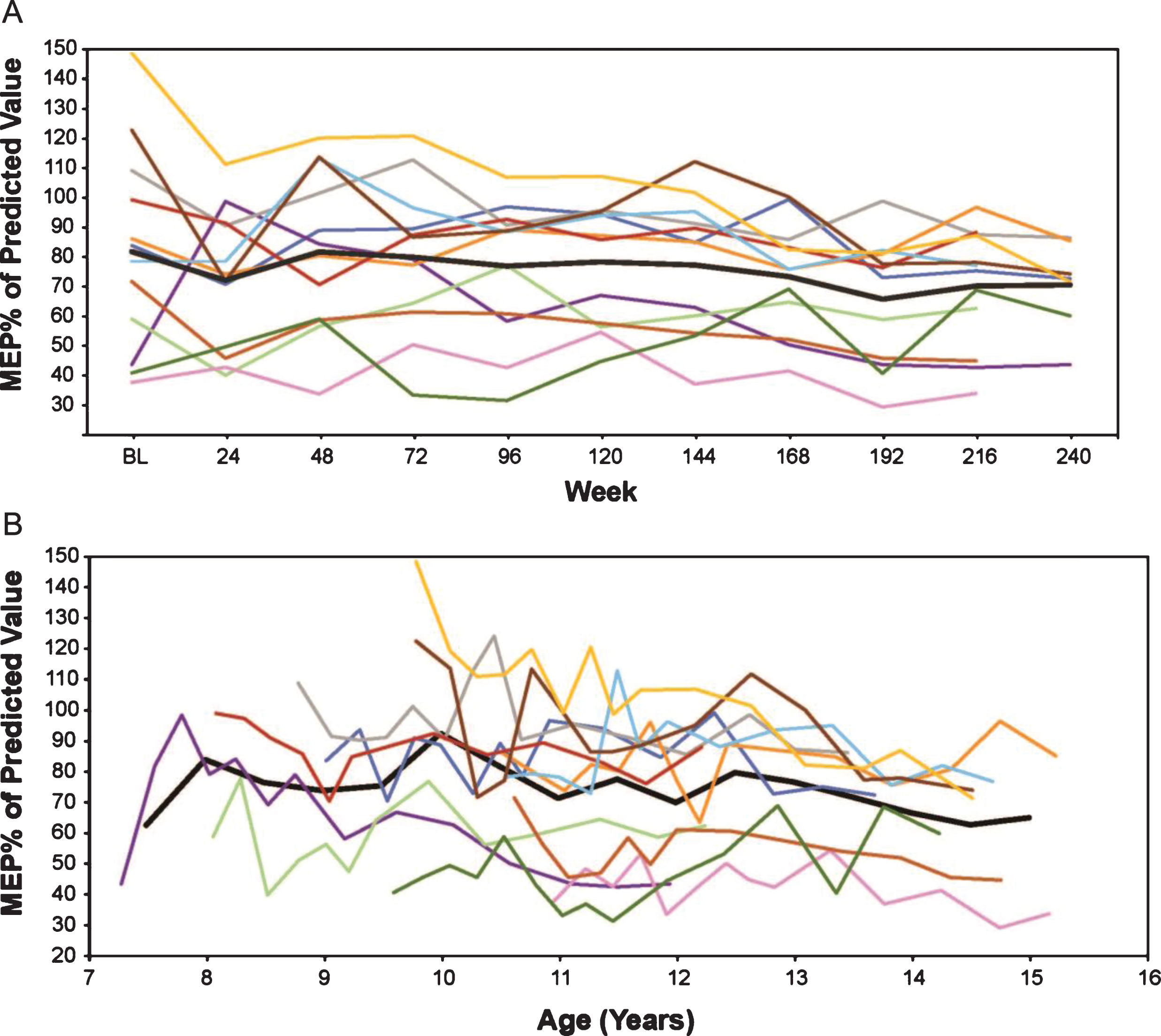
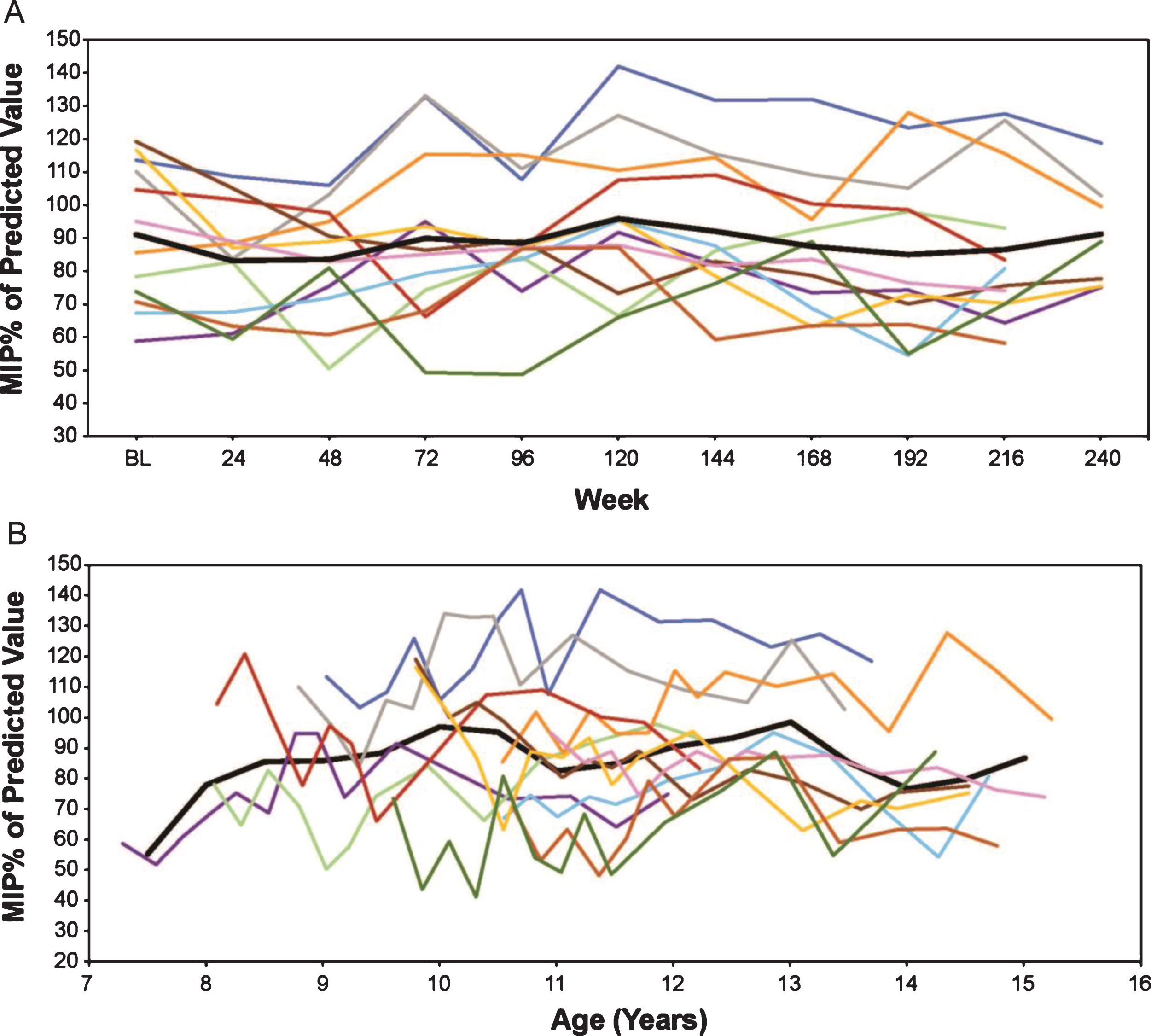
Eteplirsen-treated patients show a very modest decline on MIP% p over 5 years, with a numeric decrease from a mean 91.7% to 87.1% over 216 weeks, an annual decrease of 1.0%. In an age-adjusted mixed-effects analysis of MIP% p, the eteplirsen-treated patients show a stable performance with annual increase of 0.6%. Similar trajectories were seen across all patients (Fig. 6).
Fig.6
MIP% p in eteplirsen-treated patients (A) by weeks on treatment, and (B) by average age in years (rounded to the nearest 0.5 years for mean line). Only assessments performed every 24 weeks are represented graphically, although additional time points were assessed during the first 96 weeks. % pMIP, percent predicted maximum inspiratory pressure.
DISCUSSION
Loss of pulmonary function is a key contributor to morbidity and mortality in patients with DMD [26, 27]. Loss of pulmonary function is secondary to a decline in respiratory muscle strength. Early reports suggest that FVC increases and plateaus around the ages of 13 to 14 years [14] before it decreases due to loss of respiratory muscle strength and restrictive respiratory disease. However, more recent natural history publications show that FVC% p decreases almost as soon as pulmonary function can be reliably tested in children [15–17]. Progressive pulmonary failure requires the use of noninvasive and invasive ventilation and is a key milestone in the progression of DMD, significantly impacting quality of life. By increasing dystrophin production, eteplirsen may be preserving respiratory muscle, slowing the decline of pulmonary function and ultimately having a positive impact on morbidity and mortality.
Three spirometry measures, FVC, MIP and MEP, were used to characterize changes in pulmonary function in patients with DMD who were treated with eteplirsen for up to 5 years. In contrast to what would be expected in untreated DMD patients, eteplirsen-treated patients demonstrated slowing of the rate of decline in FVC% p, MEP% p and MIP% p throughout the study.
We observed almost half the expected decline in FVC% p per year in eteplirsen-treated patients (–2.3%) compared with the natural history cohort selected to reflect a similar age range (–4.1%). A global index of respiratory function, FVC measures the maximal amount of air that can be moved through the lungs while breathing with maximal effort, which reflects both inspiratory and expiratory muscle function. The observed difference from natural history suggests that eteplirsen helps preserve respiratory muscle function. The observed difference is particularly meaningful since the natural history cohort was selected based on the use of the same robust methodology to assess pulmonary function and had similar standard of care for DMD treatment [17]. The 4.1% per year rate of decline in FVC% p in the natural history cohort is consistent with that observed in other published reports in DMD patients (Table 2) reporting a linear decline of approximately 5% or more per year. A recent report by Khirani et al. [16] using prospectively collected data demonstrated a similar 4.9% decline in FVC% p. Findings in older publications have generally provided a higher estimated annual decrease in FVC% p than those described by Mayer et al. and Khirani et al. [14, 16–19] and, therefore, also support a positive impact of eteplirsen treatment.
Table 2
Literature-reported annual decline in FVC% p, MEP% p, and MIP% p
| Publication | Age Range, y | Patients, N | Patients With Valid PFTs, n | Study Design | Annual Decline |
| Mayer 2015 [17] | 5–24 | 60 | 44 | Prospective, observational | FVC% p: 5% |
| Buyse 2015 [32] | 10–18 | 64 | 33 | Randomized, double-blind, placebo-controlled | FVC% p: 8.95% a |
| Khirani 2014 [16] | 6–19 | 48 | 48b,c | Retrospective chart review | FVC% p: 4.9% |
| MEP% p: 3.6% | |||||
| MIP % p: 3.9% | |||||
| Henricson 2013 [15] | 7–18d | 340 | 195d | Prospective, observational | FVC% p: ≈5.0% |
| MEP% p: ≈3.1% | |||||
| MIP % p: ≈3.8% | |||||
| Hahn 1997 [14] | 7–25 | 52 | 51e | Cross-sectional analyses | FVC% p: 7.9% f |
| MEP% p: 2.7% | |||||
| MIP % p: Stability to 14 years, then decline | |||||
| McDonald 1995 [18] | 5–20+ | 160 | 39e | Prospective, observational | FVC% p (7–10 years): 0.3%; n = 8 |
| FVC% p (10–20 years): 8.5%; n = 26 | |||||
| FVC% p (>20 years): 6.2%; n = 5 | |||||
| Miller 1988 [19] | ≈5–23 | 147 | 68e | Retrospective chart review | FVC% p: ≈6% |
aAnnual decline in FVC% p was observed in the placebo arm of the study. bA total of 23 patients (48%) had successive FVC% p assessments. cA total of 24 patients (50%) had successive MEP% p and MIP% p assessments. dFor patients with available PFTs from ages 7–18 years. eNumber of patients with PFT available. fAfter FVC plateau (FVC% p,11–12 year age group). FVC% p, percent predicted forced vital capacity; MEP% p, percent predicted maximum expiratory pressure; MIP% p, percent predicted maximum inspiratory pressure; PFT, pulmonary function test.
Changes in MEP and MIP in eteplirsen-treated patients also compare favorably to the published literature. MEP is a global index of expiratory muscle strength, while MIP is a global index of inspiratory muscle strength, reflecting diaphragm function in particular. The annual decline in MEP% p for eteplirsen-treated patients of 2.6% is comparable to or slightly lower than the decline of 2.7% to 3.6% observed in published reports of DMD patients (Table 2). The annual increase in MIP% p for eteplirsen-treated patients of 0.6% per year compares favorably with the annual declines observed in published reports of DMD patients of 3.8% to 3.9% per year. Taken together, these data suggest a positive impact of eteplirsen on preservation of respiratory muscle function.
Pulmonary function can be reliably monitored with FVC, MIP and MEP. FVC is the most commonly used and well understood measure of pulmonary function in neuromuscular disease, as evidenced by the natural history reports summarized in Table 2. FVC% p has been shown to be a reliable measure in school-aged patients, as shown by low within-subject coefficients of variation for successive assessments [28]. In addition, since most studies that define the natural history of pulmonary function in patients with DMD include measurement of FVC, FVC provides a more available comparator for patients treated with eteplirsen [14, 16–18, 23]. Measurement of MEP and MIP are more challenging than FVC, which may be reflected in the lower number of studies using these measures. Both are indicators of respiratory muscle strength, but are subject to variability given that patients with muscle weakness, especially very young patients, may have a progressively harder time performing the test correctly as muscle weakness progresses. The variability can be further increased by muscle fatigue, which is typically more prominent in patients with muscle diseases. Inadequate measurement methodology and insufficient cooperation from patients may underestimate the measurements. However, the cited studies used well trained assessors, making it unlikely there was systematic ascertainment bias.
In contrast to studies in the published literature, patients in the eteplirsen study were all treated with corticosteroids. Recent natural history publications have shed light on the impact of corticosteroid use on pulmonary function in patients with DMD, showing that corticosteroids delay the onset but not the rate of decline [17, 29]. Using the same data we used for the natural history cohort, Mayer et al. showed that corticosteroid treatment does not affect the slope of deterioration of FVC% p [17]. In that study, patients aged 5 to 24 years experienced a linear decline of approximately 5% per year that was not impacted by corticosteroid use or ambulatory status. A separate analysis of the CINRG database showed that corticosteroid use delays the onset of pulmonary function decline by 2 to 3 years, but that patients experienced the same rate of decline after approximately age 10, regardless of steroid use [29]. Findings from these analyses support the validity of the comparison of eteplirsen-treated patients to natural history studies that include patients with mixed corticosteroid use.
Pulmonary function correlates with general disease progression in DMD with natural history studies revealing correlations between pulmonary function and loss of ambulation [30], as well as upper limb function [29]. In addition to being indicative of general disease progression, declining pulmonary function has several specific clinical impacts. As detailed in the recent review by Mayer et al., declining FVC% p results in increased rates of hospitalization for respiratory events, increased risk of postoperative respiratory insufficiency, need for assisted cough techniques, nocturnal ventilation, and, finally, full-time ventilation [29]. A retrospective analysis of longitudinal spirometry data by Phillips et al. showed that absolute FVC correlates strongly with mortality; the 5-year survival for patients who dropped below 1 L FVC was 8% [31]. An estimate of delay in time to reach FVC <1 L for the eteplirsen-treated cohort was not possible because the majority of patients were in the phase of disease with increasing FVC (Fig. 2), but reducing the need for pulmonary support by slowing pulmonary decline can logically be predicted to improve the quality and potentially length of life for patients with DMD.
Pulmonary function data from DMD patients who received eteplirsen in studies 201/202 compare favorably to natural history data published in the scientific literature. Using these comparator data, the deterioration of respiratory muscle function with eteplirsen treatment as measured by FVC% p was half of that seen in natural history. MEP% p and MIP% p also declined more slowly in eteplirsen-treated patients compared to natural history [14, 16]. This demonstrates the potential of eteplirsen in preserving respiratory function in patients with DMD.
CONFLICTS OF INTEREST
This work was supported by Sarepta Therapeutics, Inc. Editorial support was provided by Peloton Advantage (Parsippany, NJ) and was funded by Sarepta Therapeutics. T. B. Kinane, O. H. Mayer, J. R. Mendell, and L. P. Lowes serve as remunerated consultants to Sarepta Therapeutics. S. L. Moody performed statistical analysis for this project, which was funded by Sarepta Therapeutics. P. W. Duda was an employee of Sarepta Therapeutics at the time of this study and during manuscript preparation. No author received an honorarium for participation in this project. Employees of Sarepta Therapeutics contributed to the study design, data analysis, and critical review of the manuscript. The decision to submit resided solely with the authors.
ACKNOWLEDGMENTS
This study was sponsored by Sarepta Therapeutics, Inc. Editorial support was provided by Peloton Advantage, LLC (Parsippany, NJ) and was funded by Sarepta Therapeutics.
REFERENCES
[1] | Emery AE . Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. (1991) ;1: (1):19–29. |
[2] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , Kneile K , Dunn DM , Duval B , Aoyagi A , Hamil C , Mahmoud M , Roush K , Bird L , Rankin C , Lilly H , Street N , Chandrasekar R , Weiss RB . Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: (3):304–13. |
[3] | Duchenne Muscular Dystrophy Danbury, CT: National Organization for Rare Disorders; 2016 [Available from: https://rarediseases.org/rare-diseases/duchenne-muscular-dystrophy/. |
[4] | Kinali M , Arechavala-Gomeza V , Feng L , Cirak S , Hunt D , Adkin C , Guglieri M , Ashton E , Abbs S , Nihoyannopoulos P , Garralda ME , Rutherford M , McCulley C , Popplewell L , Graham IR , Dickson G , Wood MJ , Wells DJ , Wilton SD , Kole R , Straub V , Bushby K , Sewry C , Morgan JE , Muntoni F . Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. (2009) ;8: (10):918–28. |
[5] | Connolly AM , Florence JM , Cradock MM , Malkus EC , Schierbecker JR , Siener CA , Wulf CO , Anand P , Golumbek PT , Zaidman CM , Philip Miller J , Lowes LP , Alfano LN , Viollet-Callendret L , Flanigan KM , Mendell JR , McDonald CM , Goude E , Johnson L , Nicorici A , Karachunski PI , Day JW , Dalton JC , Farber JM , Buser KK , Darras BT , Kang PB , Riley SO , Shriber E , Parad R , Bushby K , Eagle M , MDA DMD. Clinical Research Network. Motor and cognitive assessment of infants and young boys with Duchenne Muscular Dystrophy: Results from the Muscular Dystrophy Association DMD Clinical Research Network. Neuromuscul Disord. (2013) ;23: (7):529–39. |
[6] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , Kaul A , Kinnett K , McDonald C , Pandya S , Poysky J , Shapiro F , Tomezsko J , Constantin C , D.M.D. Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: (1):77–93. |
[7] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , Kaul A , Kinnett K , McDonald C , Pandya S , Poysky J , Shapiro F , Tomezsko J , Constantin C , D.M.D. Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. (2010) ;9: (2):177–89. |
[8] | Mendell JR , Buzin CH , Feng J , Yan J , Serrano C , Sangani DS , Wall C , Prior TW , Sommer SS . Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology. (2001) ;57: (4):645–50. |
[9] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , den Dunnen JT . Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: (3):293–9. |
[10] | Kole R , Krieg AM . Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. (2015) ;87: :104–7. |
[11] | Cirak S , Feng L , Anthony K , Arechavala-Gomeza V , Torelli S , Sewry C , Morgan JE , Muntoni F . Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. (2012) ;20: (2):462–7. |
[12] | Mendell JR , Goemans N , Lowes LP , Alfano LN , Berry K , Shao J , Kaye EM , Mercuri E . Longitudinal effect of eteplirsen vs. historical control on ambulation in DMD. Ann Neurol. (2016) ;79: (2):257–71. |
[13] | Mendell JR , Rodino-Klapac LR , Sahenk Z , Roush K , Bird L , Lowes LP , Alfano L , Gomez AM , Lewis S , Kota J , Malik V , Shontz K , Walker CM , Flanigan KM , Corridore M , Kean JR , Allen HD , Shilling C , Melia KR , Sazani P , Saoud JB , Kaye EM , Eteplirsen Study G. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. |
[14] | Hahn A , Bach JR , Delaubier A , Renardel-Irani A , Guillou C , Rideau Y . Clinical implications of maximal respiratory pressure determinations for individuals with Duchenne muscular dystrophy. Arch Phys Med Rehabil. (1997) ;78: (1):1–6. |
[15] | Henricson EK , Abresch RT , Cnaan A , Hu F , Duong T , Arrieta A , Han J , Escolar DM , Florence JM , Clemens PR , Hoffman EP , McDonald CM , CINRG Investigators. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. (2013) ;48: (1):55–67. |
[16] | Khirani S , Ramirez A , Aubertin G , Boule M , Chemouny C , Forin V , Fauroux B . Respiratory muscle decline in Duchenne muscular dystrophy. Pediatr Pulmonol. (2014) ;49: (5):473–81. |
[17] | Mayer OH , Finkel RS , Rummey C , Benton MJ , Glanzman AM , Flickinger J , Lindstrom BM , Meier T . Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr Pulmonol. (2015) ;50: (5):487–94. |
[18] | McDonald CM , Johnson ER , Abresch RT , Carter GT , Fowler WM Jr , Kilmer DD . Profiles of neuromuscular diseases. Limb-girdle syndromes. Am J Phys Med Rehabil. (1995) ;74: (5 Suppl):S117–30. |
[19] | Miller F , Moseley CF , Koreska J , Levison H . Pulmonary function and scoliosis in Duchenne dystrophy. J Pediatr Ortho. (1988) ;8: (2):133–7. |
[20] | Finder J , Mayer OH , Sheehan D , Sawnani H , Abresch RT , Benditt J , Birnkrant DJ , Duong T , Henricson E , Kinnett K , McDonald CM , Connolly AM . Pulmonary endpoints in Duchenne muscular dystrophy. A workshop summary. Am J Respir Crit Care Med. (2017) ;196: (4):512–9. |
[21] | Miller MR , Hankinson J , Brusasco V , Burgos F , Casaburi R , Coates A , Crapo R , Enright P , van der Grinten CP , Gustafsson P , Jensen R , Johnson DC , MacIntyre N , McKay R , Navajas D , Pedersen OF , Pellegrino R , Viegi G , Wanger J , Force AET. Standardisation of spirometry. Eur Respir J. (2005) ;26: (2):319–38. |
[22] | Gauld LM , Kappers J , Carlin JB , Robertson CF . Height prediction from ulna length. Dev Med Child Neurol. (2004) ;46: (7):475–80. |
[23] | Hankinson JL , Odencrantz JR , Fedan KB . Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. (1999) ;159: (1):179–87. |
[24] | Polgar G , Varuni P . Pulmonary Function Testing in Children: Techniques and Standards. Philadelphia, PA: Saunders; (1971) . p. 273. |
[25] | Wilson SH , Cooke NT , Edwards RH , Spiro SG . Predicted normal values for maximal respiratory pressures in caucasian adults and children. Thorax. (1984) ;39: (7):535–8. |
[26] | LoMauro A , D’Angelo MG , Aliverti A . Assessment and management of respiratory function in patients with Duchenne muscular dystrophy: Current and emerging options. Ther Clin Risk Manag. (2015) ;11: :1475–88. |
[27] | Roberto R , Fritz A , Hagar Y , Boice B , Skalsky A , Hwang H , Beckett L , McDonald C , Gupta M . The natural history of cardiac and pulmonary function decline in patients with duchenne muscular dystrophy. Spine. (2011) ;36: (15):E1009–17. |
[28] | Meier T , Rummey C , Leinonen M , Spagnolo P , Mayer OH , Buyse GM . Characterization of pulmonary function in 10-18 year old patients with Duchenne muscular dystrophy. Neuromuscul Disord. (2017) ;27: (4):307–14. |
[29] | Mayer OH , Henricson EK , McDonald CM , Buyse GM . Advances in pulmonary care in Duchenne muscular dystrophy. US Neurology. (2017) ;13: (1):35–41. |
[30] | Humbertclaude V , Hamroun D , Bezzou K , Berard C , Boespflug-Tanguy O , Bommelaer C , Campana-Salort E , Cances C , Chabrol B , Commare MC , Cuisset JM , de Lattre C , Desnuelle C , Echenne B , Halbert C , Jonquet O , Labarre-Vila A , N’Guyen-Morel MA , Pages M , Pepin JL , Petitjean T , Pouget J , Ollagnon-Roman E , Richelme C , Rivier F , Sacconi S , Tiffreau V , Vuillerot C , Picot MC , Claustres M , Beroud C , Tuffery-Giraud S . Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. Eur J Paediatr Neurol. (2012) ;16: (2):149–60. |
[31] | Phillips MF , Quinlivan RC , Edwards RH , Calverley PM . Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med. (2001) ;164: (12):2191–4. |
[32] | Buyse GM , Voit T , Schara U , Straathof CS , D’Angelo MG , Bernert G , Cuisset JM , Finkel RS , Goemans N , McDonald CM , Rummey C , Meier T . Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet. (2015) ;385: (9979):1748–57. |


