
Aberrant Caspase Activation in Laminin-α2-Deficient Human Myogenic Cells is Mediated by p53 and Sirtuin Activity
Abstract
Background:
Mutations in the LAMA2 gene encoding laminin-α2 cause congenital muscular dystrophy Type 1A (MDC1A), a severe recessive disease with no effective treatment. Previous studies have shown that aberrant activation of caspases and cell death through a pathway regulated by BAX and KU70 is a significant contributor to pathogenesis in laminin-α2-deficiency.
Objectives:
To identify mechanisms of pathogenesis in MDC1A.
Methods:
We used immunocytochemical and molecular studies of human myogenic cells and mouse muscles—comparing laminin-α2-deficient vs. healthy controls—to identify mechanisms that regulate pathological activation of caspase in laminin-α2-deficiency.
Results:
In cultures of myogenic cells from MDC1A donors, p53 accumulated in a subset of nuclei and aberrant caspase activation was inhibited by the p53 inhibitor pifithrin-alpha. Also, the p53 target BBC3 (PUMA) was upregulated in both MDC1A myogenic cells and Lama2–/– mouse muscles. In addition, studies with sirtuin inhibitors and SIRT1 overexpression showed that caspase activation in MDC1A myotubes was inversely related to sirtuin deacetylase activity. Caspase activation in laminin-α2-deficiency was, however, not associated with increased phosphorylation of p38 MAPK.
Conclusions:
Aberrant caspase activation in MDC1A cells was mediated both by sirtuin deacetylase activity and by p53. Interventions that inhibit aberrant caspase activation by targeting sirtuin or p53 function could potentially be useful in ameliorating MDC1A.
INTRODUCTION
Congenital muscular dystrophy Type 1A (MDC1A) is a severe, autosomal recessive disease caused by mutations in the LAMA2 gene that encodes laminin-α2 [1, 2]. Most LAMA2 mutations are in exons 1–31 (of 64) and lead to absence of laminin-α2, whereas mutations in exons 58–64 often produce partial deficiency with less severe outcomes [3–5]. Laminin-α2 links the extracellular matrix (ECM) to the sarcolemma through interactions with its two cell surface receptors, α−dystroglycan and α7-integrin. Laminin-α2 is abundant in skeletal muscle and is in other tissues, including peripheral nerves and CNS, so that, in addition to muscle pathology, laminin-α2-deficiency leads to adverse effects on peripheral nerve Schwann cells [6] and CNS oligodendrocytes [7].
Recent studies have identified molecular pathways that are disrupted by loss of laminin-α2 function and have suggested possible methods for therapeutic intervention in MDC1A. Both autophagy and proteostasis are affected in laminin-α2-deficient (Lama2–/–) mouse muscles and pathology is ameliorated by interventions designed to restore normal function of these pathways [8, 9]. We have found that aberrant induction of cell death—mediated both by caspase-3 and by the pro-death protein BAX with its cytosolic binding partner KU70—is a significant pathogenetic mechanism in laminin-α2-deficiency [10, 11]. For example, we found that inhibition of BAX-mediated cell death in Lama2–/– mice improves neuromuscular function and produces a several-fold increase in lifespan [10, 12]. This result was replicated and extended by others [13–15]. Outcomes in Lama2–/– mice can be further improved by combining cell death inhibition with expression of mini-agrin or a chimeric laminin to improve extracellular matrix (ECM)-sarcolemma linkage [14, 16] or by treatment with IGF-1 to improve myoblast survival and muscle regeneration [15]. Pathology in Lama2–/– mice also can be ameliorated by providing laminin-111 as a replacement for laminin-211 [17, 18].
To extend our knowledge of pathogenesis in MDC1A, we have now examined additional molecular pathways as possible mediators of aberrant caspase activation in laminin-α2-deficiency. Our results showed that caspase activation was not mediated by activation of p38 MAPK, a mechanism which underlies BAX-mediated pathogenesis in dystrophin–deficiency and α−sarcoglycan-deficiency [19] and may contribute to pathogenesis in emerin-deficiency [20]. On the other hand, we found that aberrant caspase activation was mediated both by p53 and by sirtuin deacetylase activity. These results raise the possibility that pathogenesis in laminin-α2-deficiency could potentially be ameliorated by interventions designed to inhibit sirtuin- or p53-mediated activation of cell death.
MATERIALS AND METHODS
Cells and culture
For this study, all of the human MDC1A myogenic cells were obtained from the Muscle Tissue Culture Collection (MTCC) at the University of Munich and the healthy control myogenic cells were obtained from the Wellstone biobank at the University of Massachusetts School of Medicine. All cells were anonymized and no personal identifying information was available to us. Each of the cultures had been produced prior to our study from muscle biopsies which had been collected under protocols approved by the appropriate institution. Protocols included informed donor consent and approval to publish results in accordance with standards of the Helsinki Declaration. Because the human cells used in our studies were obtained from cell banks with no personal identification data obtainable by us, these studies were classified as exempt from Human Studies review by the Boston University Institutional Review Board (Protocol H-33419) in accordance with policy of the U.S.A. Department of Heath and Human Services. Myogenic cells from six healthy control donors (07Ubic, 09Ubic, 14Vbic, 15Vbic, 16Ubic, 21Ubic) and from four patients with MDC1A (38/03, 50/04, 9/03, and 96/04) were as described previously [11, 21–25]. To confirm authenticity, cells were regularly assessed for genetic mutation, gender, SNP pattern [24], or lack of laminin-α2 expression [11, 25]. The human primary myogenic cells were grown on gelatin-coated dishes in high serum medium for proliferation and switched when near confluence to low serum medium for differentiation as described [11, 23]. Unless stated otherwise, all differentiated cultures were examined after four days in low serum medium.
Mice
Heterozygous Lama2dy - W/+ mice, which carry the targeted dy-W mutation in the Lama2 gene [26], were obtained from Dr. Eva Engvall and were maintained in our laboratory for >5 years by breeding with C57BL/6J mice [10–12, 27]. Mice obtained from crosses of Lama2+/– heterozygotes were genotyped at weaning by PCR [10], and quadriceps muscle tissues were obtained from the resulting wild-type, heterogyzous Lama2+/–, and homozygous Lama2–/– littermates at 4–6 weeks after birth. Approximately equal numbers of male and female mice were analyzed for each genotype. No differences between male and female mice were noted with the assays used here. Mouse studies were conducted under protocols approved by the Institutional Animal Care and Use Committee at the Boston University School of Medicine.
Antibodies
Phospho-MAPK was detected with rabbit mAb against phospho-(Thr180/Tyr182)-p38MAPK (cat. 9211, Cell Signaling Technology, Beverly MA) used at 1 : 1000 dilution. Total MAPK was detected with rabbit mAb against p38MAPK (cat. 9212, Cell Signaling Technology) used at 1 : 1000 dilution. As positive controls, cell extracts containing phospho-p38MAPK or non-phosphorylated p38MAPK (cat. 9213, Cell Signaling Technology) were used per manufacturer’s instructions for immunoblots. BBC3 (also known as PUMA) was detected with either anti-PUMAα goat pAb (cat. SC-19187, Santa Cruz Biotechnology) used at 1 : 200 dilution or with anti-PUMA pAb (cat. PA5-20007, Thermo Fisher, Rockford IL) at a concentration of 2μg/ml. GAPDH was detected with a mouse mAb (cat. 10R-G109A, Fitzgerald, Acton MA) used at 1 : 5000 dilution. Detection of p53 was with mouse mAb 1C12 (cat. 2524, Cell Signaling Technology) used at 1 : 250 dilution. Myosin heavy chain (MyHC) isoforms were detected with mouse mAb F59 [28] (Developmental Studies Hybridoma Bank, Iowa City IA) used at 1 : 10 dilution of hybridoma supernatant or with rabbit MYH3 pAb (cat# HPA021808, Sigma-Aldrich) used at 1 : 500 dilution. DDIT3 (also known as CHOP) was detected with mouse mAb L63F7 (cat. 2895, Cell Signaling Technology) used at 1 : 100 dilution.
The specificity of each primary antibody was validated using one or more techniques, including reference to previous publications with the same mAb or polyclonal antiserum; reference to manufacturers’ validation assays including knockouts; generation in our lab of expected immunofluorescence staining patterns, e.g., appearance of p53 in nuclei upon actinomycin treatment; detection in our lab of appropriate band sizes on immunoblots in the absence of non-specific bands; and detection of recombinant protein when expressed in cells that normally do not express the protein. Primary antibody binding was visualized with appropriate species-specific secondary antibodies (Thermo-Fisher, Waltham MA) conjugated to either Alexa Fluor 488 or Alexa Fluor 594 and used at 1 : 500. Nuclei were stained with bisbenzimide.
Chemicals
Staurosporine (cat. 9953, Cell Signaling Technology) was freshly prepared before use as a 1 mM stock solution of active compound in DMSO and used at final concentration of 1μM. Sirtinol (cat. 566320, EMD-Millipore, Billerica MA) was prepared as a 50 mM stock solution in DMSO and used at a final concentration of 50μM or as noted. Nutlin-3a (cat. S8059, Selleck Chemicals) was prepared as a 5 mM stock solution and used at indicated concentrations.
Immunocytology
As in our previous studies [23], cultures of differentiated cells were washed twice with PBS and then fixed either with 2% paraformaldehyde for 10 min or with ice-cold 100% methanol as found to be appropriate for the primary antibody in preliminary validation studies. Fixed cultures were washed three times with PBS. Paraformaldehyde-fixed cultures were additionally permeabilized with 0.5% Triton X-100 for 10 min at room temperature. All fixed cultures were blocked at room temperature for 60 min in 4% horse serum, 4% goat serum (Thermo Fisher), and 4% bovine serum albumin (EMD Millipore, Billerica, MA) in PBS plus 0.1% Triton X-100. Fixed and blocked cultures were incubated overnight at 4°C with primary antibody diluted as noted in blocking solution. The following day, cells were rinsed three times with PBS and incubated for 1 h with the appropriate secondary antibody diluted 1 : 500 in blocking solution. For double immunostaining, cultures were subsequently incubated with the second primary antibody, washed, and incubated with the second secondary antibody as above for the primary staining.
Microscopy
Immunostaining was analyzed by manually scanning the entire culture area followed by digital imaging and manual quantitation. Images were acquired using a Nikon E800 microscope with Spot camera and software version 5.1 (Diagnostic Instruments Inc., Sterling Heights, Michigan).
Immunoblotting
Mouse quadriceps muscles or cell cultures were homogenized and lysates were analyzed by SDS-PAGE and immunoblotting as in our previous work [10–12, 25, 27]. The concentration of protein in muscle and cell lysates was determined by Bradford dye-binding assay (Thermo-Fisher), and equal amounts of protein were analyzed in each lane of each individual gel. Immunoblotted proteins were detected with an appropriate primary antibody and an Alexa-680-conjugated secondary antibody with appropriate species specificity. Immunoblots were quantified using the quantification tool in the Odyssey 2.0 software that accompanies the LI-COR Odyssey infrared system (LI-COR Biosciences, Lincoln NE).
Enzyme and viability assays
DEVDase activity (i.e., Caspase 3 and 7) was measured using the Caspase-Glo 3/7 enzymatic assay kit (cat. G8090, Promega, Madison WI). The Cell-titer Fluor assay kit (cat. G6080, Promega) was used to measure relative cell numbers. Total cellular deacetylase activity was measured with the Sirt Glo assay kit (cat. G6450, Promega). All enzyme assays were carried out according to manufacturer’s instructions. The proprietary recombinant luciferase used in the Promega assay kits is not inhibited by pifithrin-alpha, as determined both by the manufacturer [29] and our own studies (not shown). Enzyme assay results were normalized for cell number by dividing the enzyme activity by the cell titer.
Adenoviral vectors
Adenovirus vectors to express SIRT1 or, as a control, LacZ, were produced as described [30] and used at a multiplicity of infection = 10 for 72 h.
Statistical tests
GraphPad Prism 7 (GraphPad Software, LaJolla CA) was used to carry out appropriate T-tests or one way ANOVA as noted in figure legends. Equal variance was not assumed for two sample T-tests. For ANOVA, Bonferroni post-tests were used with α= 0.01. All sample sizes (n) used for statistical tests and for figures were biological replicates, i.e., measurements from independent samples.
RESULTS
Laminin-α2-deficiency was not associated with increased phosphorylation of p38 MAPK
Recent work showed that phosphorylation of p38 MAPK was increased and mechanistically linked to BAX activation in both dystrophin-deficient (mdx) and delta-sarcoglycan-deficient (Sgcd–/–) mouse muscles [19]. In our previous studies, we showed that aberrant activation of BAX and caspase-3 contributes to pathology in laminin-α2-deficient mice and also occurs in differentiated cultures of myogenic cells obtained from MDC1A patients [10, 11, 27]. Thus, to determine if the phospho-MAPK activation mechanism also occurred in laminin-α2-deficiency, we examined MAPK phosphorylation in human MDC1A myogenic cells and in Lama2–/– mouse muscles.
We first determined if p38 MAPK phosphorylation was increased in differentiated cultures of MDC1A vs. healthy control myogenic cells. By immunoblotting, however, the differentiated cultures of MDC1A and healthy myogenic cells had similar ratios of phosphorylated to total p38 MAPK in (Fig. 1A, C). In addition, total p38 MAPK appeared to be unchanged between MDC1A and control cell cultures (Fig. 1A). Each of the cultures was comprised of >90% desmin-positive myogenic cells and showed similar extents of myotube formation (∼50% of nuclei in myotubes). For this work, we used the same phospho-p38 MAPK and total p38 MAPK mAbs as used by Wissing et al. [19]. Thus, phosphorylation of p38 MAPK did not appear to differ between differentiated cultures of laminin-α2-deficient (MDC1A) and healthy control myogenic cells.
Fig.1
Increased phosphorylation of p38 MAPK is not associated with laminin-alpha-2-deficiency. A. Immunoblots of lysates of 4d differentiated human myogenic cell lysates with antibodies specific for phospho-p38 MAPK (P-p38 MAPK) and total p38 MAPK showed that differentiated cultures of laminin-α2-deficient (MDC1A) and healthy control myogenic cells had similar levels and ratios of P-p38 MAPK to total p38 MAPK. One representative immunoblot is shown, see panel C for quantitation. 70μg of protein per lane. B. Similar immunoblots of mouse muscle lysates showed that phosphorylation of p38 MAPK tended to be decreased in quadriceps muscles from Lama2–/– mice compared to muscles from wild-type mice. One representative immunoblot is shown, see panel C for quantitation. 30μg of protein per lane. Loading control = Ponceau S stain. Lanes 4 and 5 included purified phospho-MAPK and MAPK proteins to serve as positive controls for antibody specificity and thus showed little or no staining in the loading controls. C. Quantitative densitometry of immunoblots showed that the P-p38 MAPK/Total p38 MAPK ratio was decreased at P = 0.02 in Lama2–/– compared to wild-type mouse muscles, whereas the ratio was similar in MDC1A compared to healthy control myogenic cells. Identities of individual control and MDC1A donor cells are as indicated. All = average of results from cell cultures of all healthy control (light gray) or all MDC1A (dark gray) donors. Error bars = SE. P values from unpaired T-test of all control vs. all laminin-α2-deficient samples with n as indicated.
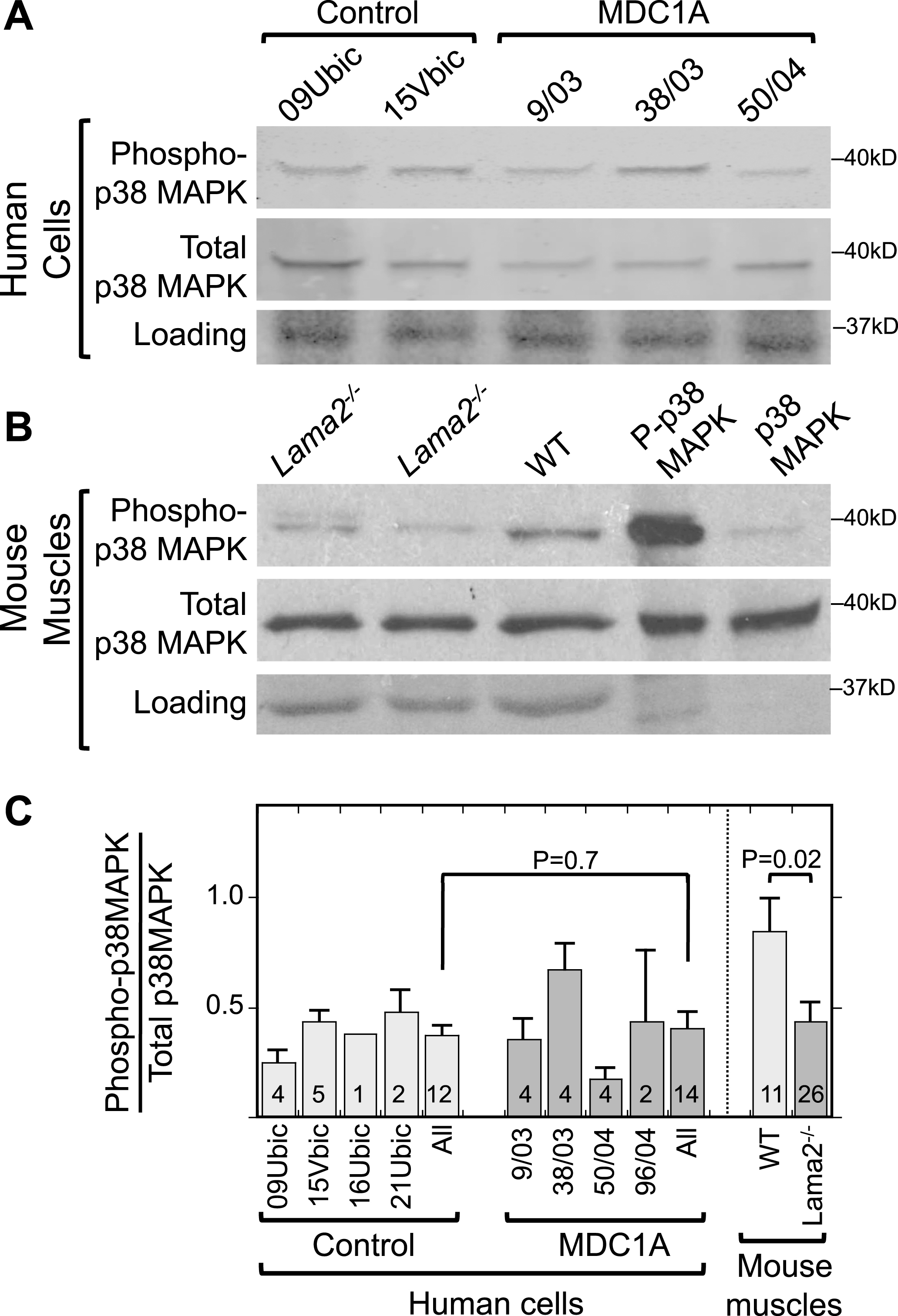
We next examined p38 MAPK phosphorylation in Lama2–/– vs. wild-type mouse muscles, because intact muscles have cell-type interactions (e.g., with nerves, fibroblasts, and inflammatory cells) that are not recapitulated in myogenic cell cultures. We found that, on average, Lama2–/– mouse quadriceps muscles tended to have less phosphorylation of p38 MAPK than wild-type muscles obtained from age-matched littermates (Fig. 1B, C). Total p38 MAPK appeared to be unchanged between Lama2–/– and wild-type mouse muscles (Fig. 1B). We examined Lama2–/– muscles at three and four weeks after birth when pathology was obvious. The difference in the average ratio of phospho-p38 MAPK to total p38 MAPK in Lama2–/– (n = 26) vs. wild-type (n = 11) muscles reached P = 0.02 by t-test (Fig. 1C).
Taken together, our results from mouse muscles and human cell cultures suggested that activation of p38 MAPK (as measured by increased phosphorylation) did not underlie the aberrant activation of BAX and cell death in laminin-α2-deficiency. Because our results did not support a role for activation of p38 MAPK in pathogenesis of laminin-α2-deficiency, we decided to leave the cellular origin and functional consequences of decreased p38 MAPK activity in mouse muscles for future studies. We turned, therefore, to examining alternative possible pathways by which aberrant caspase activation might be regulated in laminin-α2-deficiency.
Mediation of caspase activation by p53 in MDC1A myogenic cells
Because p53 can be an effector of BAX-mediated cell death [31], we next examined whether inhibition of p53 function could limit the aberrant activation of caspase activity in differentiated MDC1A cultures. Low level caspase (DEVDase) activity is an intrinsic component of the differentiation program in all myogenic cells [32], but MDC1A myotubes show a caspase activation pattern that is qualitatively and quantitatively different from that in control myotubes. In particular, we consistently find that a small fraction (∼2–5%) of MDC1A myotubes show strong immunostaining for activated caspase-3 [11, 25], whereas such strong staining is very rare in healthy myotubes. As a result, the amount of activated caspase-3 is several-fold higher in MDC1A myotube cultures than in control cultures (ranging from ∼2.5X to ∼5X in different experiments), and we use this aberrant activation of caspase (DEVDase) as a marker for pathogenesis.
Thus, to determine if p53 functioned in MDC1A pathogenesis, we measured caspase activity (DEVDase) in parallel differentiated cultures of MDC1A and healthy control myogenic cells that were either left untreated or treated with different concentrations of pifithrin-alpha, which is an inhibitor of p53 function [33]. As before, untreated MDC1A cultures had a several-fold higher caspase activity than untreated healthy control cultures. When treated with 10μM or 25μM pifithrin-alpha, however, the caspase activity in MDC1A cultures was decreased by about two-thirds, in some cases to near the level found in untreated healthy control cultures (Fig. 2A). We next examined whether pifithrin-alpha would inhibit staurosporine-induced cell death, because we had shown that staurosporine, which is a non-selective protein kinase inhibitor [34], induces BAX-mediated cell death in myogenic cells [10–12, 35]. Treatment with pifithrin-alpha also lowered the amount of staurosporine-induced caspase activation (Fig. 2B) and ameliorated staurosporine-induced morphological abnormalities such as “blebbing” of nuclei and disorganization of myosin filaments (Fig. 2C, D). Thus, p53 function appeared to be required in differentiated MDC1A cultures for spontaneous and staurosporine-induced caspase activation, both of which we have previously shown to be BAX-dependent [11].
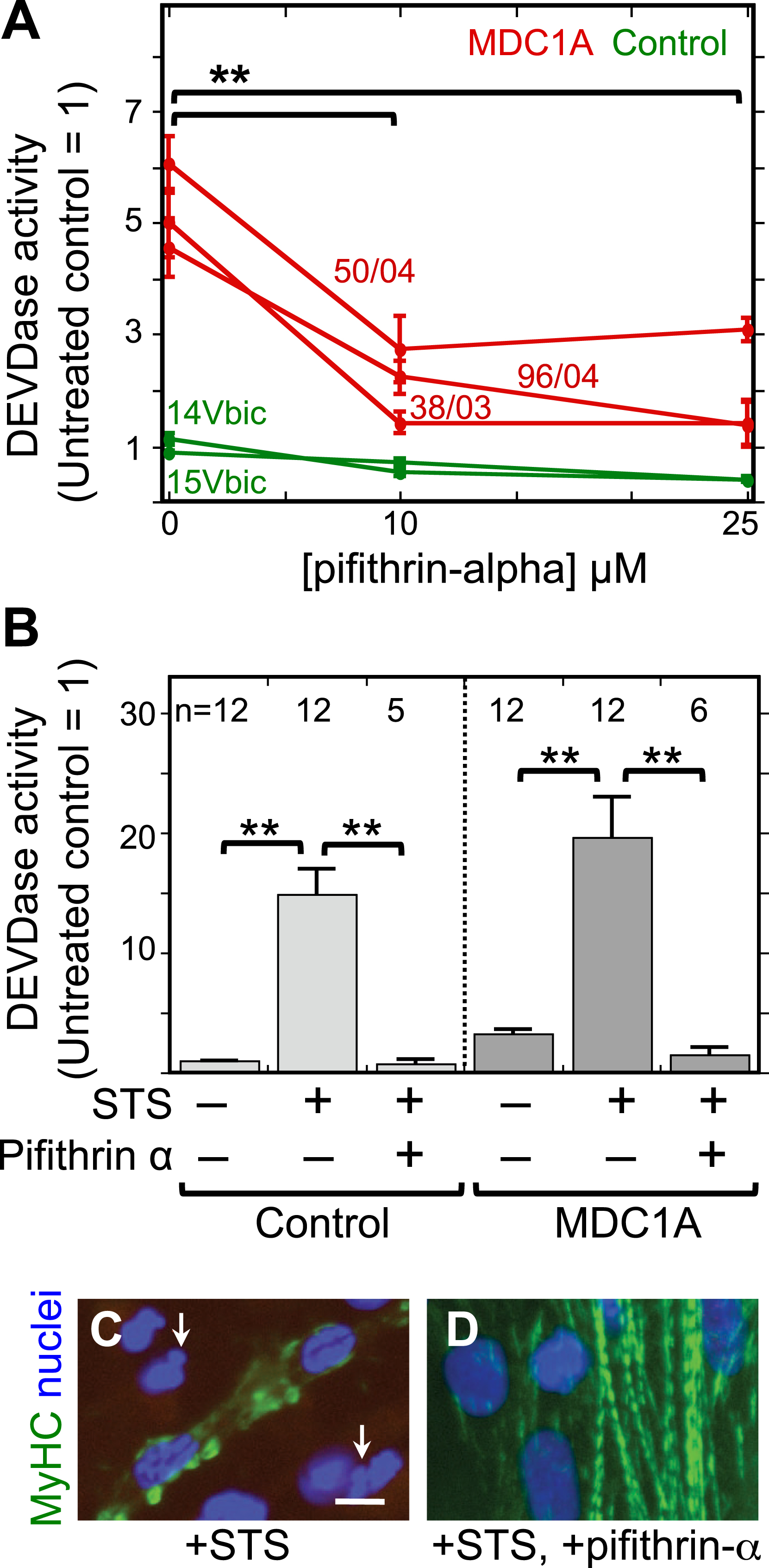
Fig.2
The p53 inhibitor pifithrin-alpha decreased both spontaneous and staurosporine-induced caspase activity in MDC1A myogenic cells. A. Cultures of MDC1A (red lines) and healthy control (green lines) myogenic cells were incubated with pifithrin-alpha at the indicated concentrations and assayed for caspase 3/7 (DEVDase) enzymatic activity after four days in differentiation medium. Caspase values were normalized so that the average of the untreated healthy controls = 1. Error bars = SE; **P < 0.01 by ANOVA to compare values at 0, 10μM, and 25μM; n = 3 for cells of each individual donor. B. As indicated, cells were either left untreated or treated with 25μM pifithrin-alpha as in panel A either without staurosporine (STS) or with staurosporine at 1μM for the final 4.5 h prior to harvest. Pifithrin-alpha treatment reduced both spontaneous and staurosporine-induced caspase activity in MDC1A cultures. Error bars = SE; **P < 0.01 by ANOVA; n as indicated. Healthy control cells included 15Vbic, 16Ubic, and 21Ubic. MDC1A cells included 38/03, 50/04, and 96/04. C, D. Treatment with staurosporine (+STS) at 1μM for 4.5 h hours generated morphological abnormalities of nuclei (blue) including blebbing and fragmentation (e.g., arrows in panel C) and disrupted the striated organization of myosin heavy chain (MyHC, green) in MDC1A (50/04) cultures (panel C). However, these staurosporine-induced changes were largely eliminated when the p53 inhibitor pifithrin-alpha (+pifithrin-α) at 25μM was included in combination with staurosporine (panel D). Bar in panel C = 20μm.
Furthermore, we found by quantitative immunoblotting that the p53-regulated BBC3 protein (also known as PUMA) was on average more abundant in differentiated MDC1A myogenic cell cultures than in parallel healthy control cultures (Fig. 3). Similarly, Lama2–/– quadriceps muscle samples also had higher levels of BBC3 than did age- and gender-matched quadriceps muscles from wild-type mice (Fig. 3). Because BBC3 upregulation requires active p53 [36], this result suggested that p53 is, on average, more active in laminin-α2-deficient human myogenic cells and mouse muscles than in healthy controls. Consistent with this possibility, we found that a subset of MDC1A nuclei showed p53 immunostaining that was plainly above background (Fig. 4A, B), whereas nuclei of healthy control cells seldom showed such p53 immunostaining. For example, in two quantitative studies of differentiated cultures, we found that nuclei with p53 immunostaining that was obviously above background amounted to 9.8% (25 of 255) of the nuclei in 38/03 MDC1A cells and 11.9% (147 of 1,234) of the nuclei in 50/04 MDC1A cells, whereas only 0.4% (3 of 864) and 0.9% (9 of 949) of the nuclei were p53-positive in two independent cultures of 15Vbic healthy control cells.
Fig.3
Laminin-α2-deficient human myogenic cells and mouse muscles show increased levels of the p53-regulated BBC3 protein. A. Expression of BBC3 (PUMA) was increased both in differentiated MDC1A compared to healthy control myogenic cell cultures and in Lama2–/– compared to wild-type control mouse quadriceps muscles. Results are presented as the BBC3 to GAPDH ratio determined from densitometry of immunoblots. Individual cell donors are indicated. All = average of results from cell cultures of all healthy control (light gray) or all MDC1A (dark gray) donors as indicated. Error bars = SE; P values from T-test of all control vs. all laminin-α2-deficient samples with n as indicated. B. Representative full-length immunoblots demonstrating specificity of BBC3 antibody for human cell culture (left) and mouse muscle (right) samples as indicated. All cell samples were analyzed on one immunoblot and all mouse samples were also analyzed on one immunoblot, but lanes were re-arranged for presentation as shown by the dotted lines. The separate lower immunoblots show the GAPDH loading control band used for densitometry. The BBC3/GAPDH ratio determined by densitometry is shown at the top of each lane. MW = molecular weight standards.
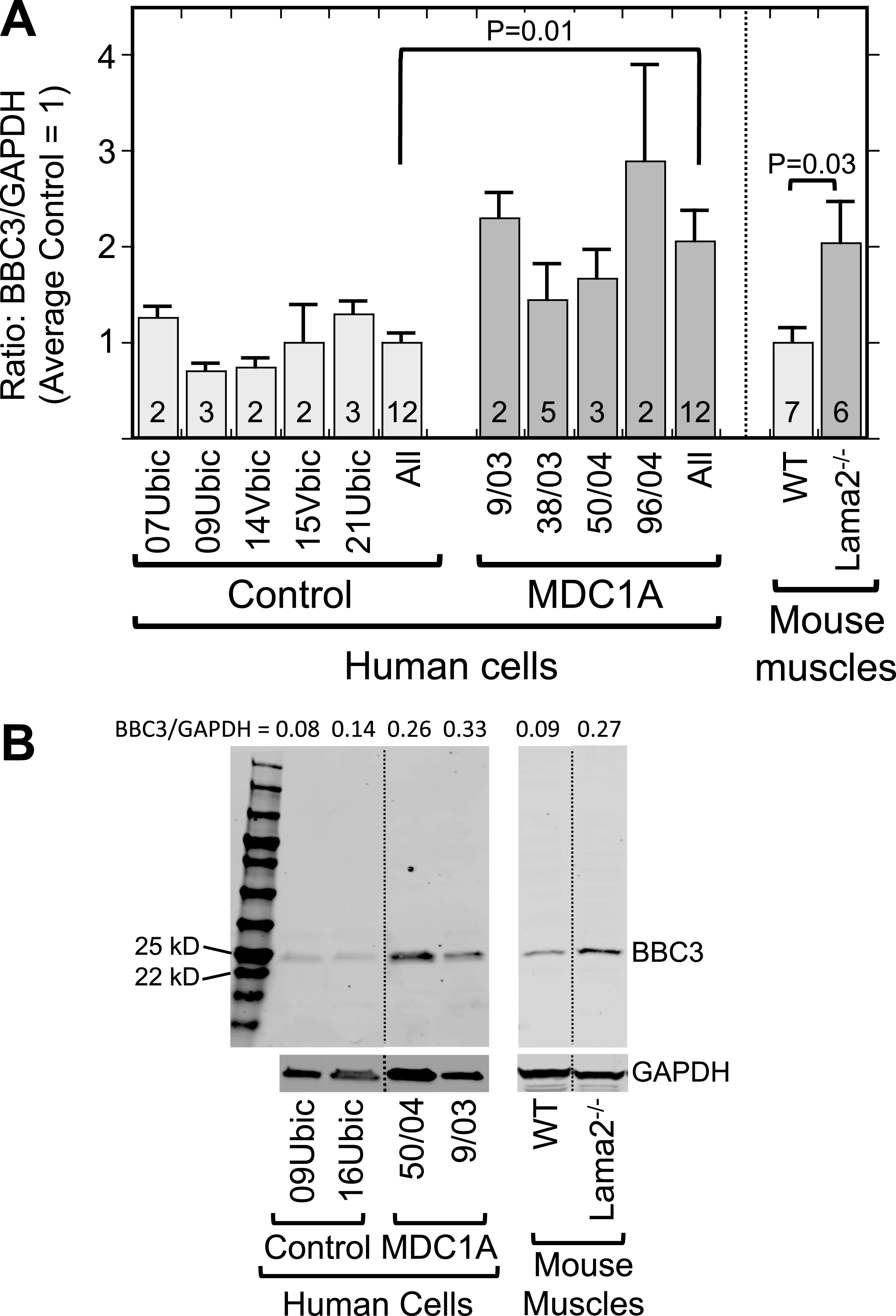
Fig.4
MDC1A myogenic cells show atypical accumulation of p53 and sensitivity to HDM2 inhibition. A, A’. Lower power view of a 4d differentiated culture of 50/04 human myogenic cells. Four nuclei, indicated by arrows, showed nuclear accumulation of p53 (red), whereas many surrounding nuclei (blue) did not show such immunostaining for p53. Immunostaining for myosin heavy chain (MyHC, green) identified differentiated myotubes and showed that the four p53-positive nuclei in this image were within a myotube. Typically, p53 was found in about 10% of MDC1A nuclei, but in <0.5% of healthy control nuclei; see text for quantitation. Bar in panel A = 20μM. B, B’. A higher power view of a 4d differentiated culture of 38/03 cells. Arrows indicate p53-positive nuclei; asterisk indicates a p53-negative nucleus. Bar in panel B = 20μM. C. Treatment with nutlin-3a, which prevents HDM2 from inhibiting p53, generated a larger increase in DEVDase activity in differentiated cultures of MDC1A myogenic cells (red lines, donors 96/04 and 38/03 as indicated) than in healthy control cultures (green lines, donors 15Vbic and 21Ubic). Single asterisks indicate that the control and MDC1A average values differed with P < 0.01 at 5μM and 10μM nutlin-3a. **P < 0.01 to indicate that DEVDase activity was higher in MDC1A cultures at nutlin-3a concentrations of 5μM and 10μM vs. MDC1A cultures that were untreated or treated with 1μM nutlin-3a. Statistical test by ANOVA. Error bars = SE; n = 3 for each individual donor.
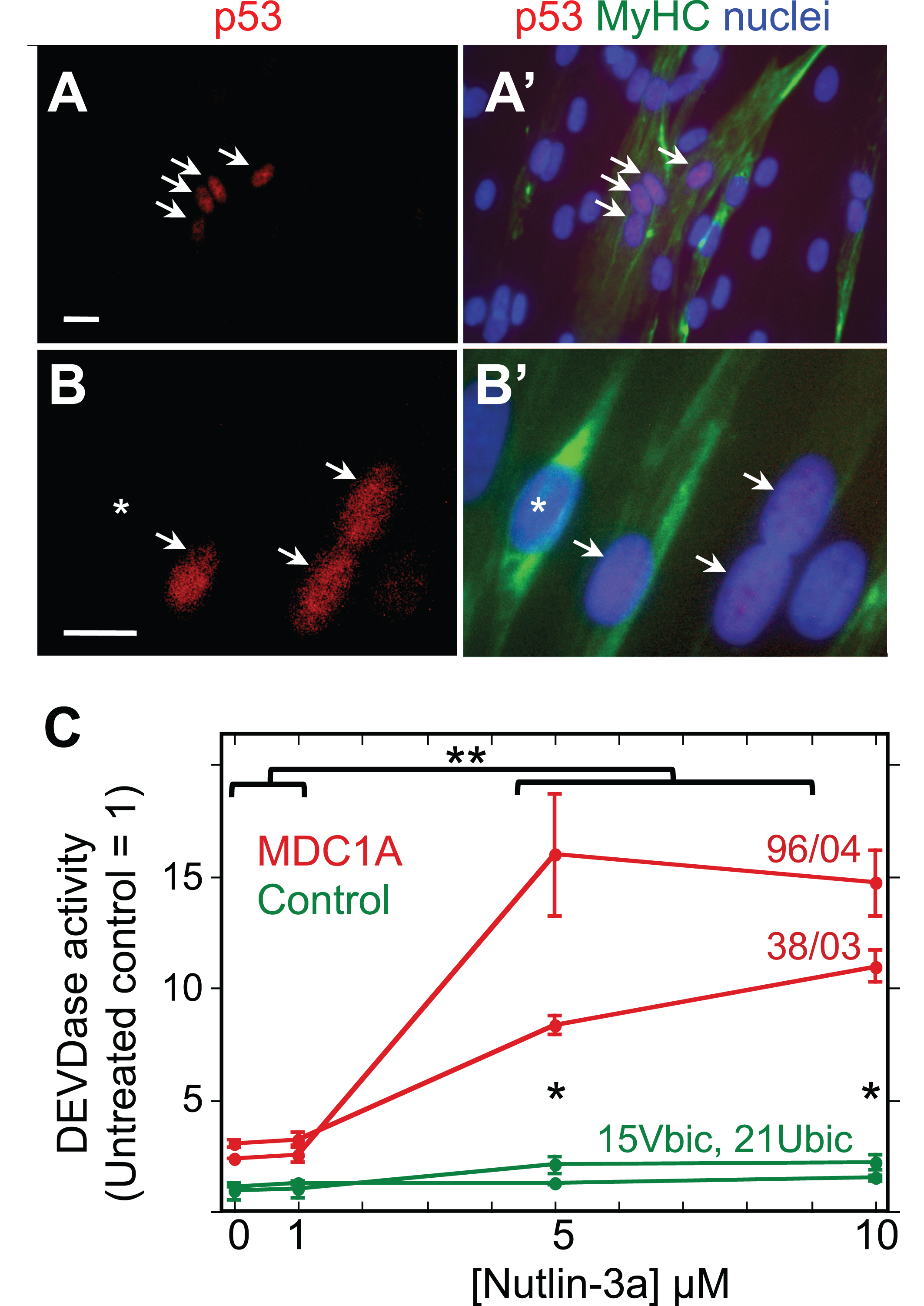
Furthermore, we found that caspase activity in four day differentiated cultures of MDC1A cells was increased 3-5X by a 24 h treatment with 5μM or 10μM nutlin-3a, a drug that prevents HDM2 from binding to and inhibiting p53 (Fig. 4B) [37]. Differentiated cultures of healthy control cells also showed increased caspase activity upon treatment with Nutlin-3a, but both the ∼2X increase and final caspase levels were less in the differentiated control cultures than in MDC1A cultures. Much as seen previously in cultures of mouse C2C12 myogenic cells [38], we also found that nutlin-3a inhibited myotube formation when it was added to proliferating MDC1A or control myoblasts at the time of switching to differentiation medium (not shown). Because nutlin-3a prevents HDM2 from binding to and inhibiting p53, these findings suggest that MDC1A cells, as well as healthy control cells, have functional HDM2 that at least partially restrains p53-mediated caspase activation.
Mediation of caspase activation by sirtuin deacetylase activity in MDC1A myogenic cells
We next carried out experiments to determine how altering sirtuin deacetylase activity affected caspase activation in MDC1A myogenic cells in comparison to healthy control cells. Previous studies had shown that sirtuins could regulate cell death pathways, including p53 and KU70 acetylation, in some non-skeletal muscle cell types [39–42], First, we found that treatment with sirtinol, when used at a concentration that inhibits both SIRT1 and SIRT2, caused a 3-4X increase in caspase activity (DEVDase) in differentiated MDC1A cultures but had little or no effect in parallel cultures of healthy control cells (Fig. 5A). Sirtinol-induced caspase activation was prevented by treatment with 25μM pifithrin-alpha (Fig. 5B) indicating that it was p53-dependent. In addition, sirtinol treatment was accompanied by accumulation of DDIT3 (also known as CHOP) in almost all nuclei of both MDC1A (Fig. 5C) and healthy control (not shown) human myogenic cells. We examined DDIT3 because it is a marker for ER stress, and sustained ER stress can activate caspase-3 and cell death. Staurosporine treatment was not accompanied by nuclear accumulation of DDIT3 (Fig. 5D).
Fig.5
Sirtinol treatment increased caspase activity in MDC1A cells in a p53-dependent process and also induced nuclear accumulation of the ER stress marker DDIT3 (CHOP). A. Treatment with sirtinol at 5μM for 24 h caused a 3–4X increase in caspase 3/7 (DEVDase) activity in differentiated cultures of 38/03 and 50/04 MDC1A myogenic cells, but had little effect on caspase activity in cultures of 09Ubic and 15Vbic healthy control myogenic cells. **P < 0.01 by ANOVA with n as indicated. B. As in panel A, treatment with sirtinol at 5μM for 24 h was accompanied by a 3–4X increase of caspase (DEVDase) activity. This sirtinol-induced caspase activation was prevented when 25μM pifithrin-alpha was added at the same time as sirtinol, indicating that the sirtinol-induced caspase activation was p53 dependent. For healthy controls, 15Vbic cells were used. For MDC1A, 50/04 cells were used. Error bars = SE. **P < 0.01 by ANOVA with n = 3. C-E. Differentiated MDC1A 50/04 cultures were treated with sirtinol (5μM for 24 h) or staurosporine (1μM for 4.5 h), i.e., conditions we had previously shown to increase caspase (DEVDase) activity. Under these conditions, DDIT3 (also known as CHOP; red), a marker for ER stress, accumulated in the nuclei (blue) of cells treated with sirtinol but not in the nuclei of untreated or staurosporine-treated cells. Though not shown, CHOP also accumulated in the nuclei of sirtinol-treated healthy control cells 15Vbic. Bar in panel C = 20μm.
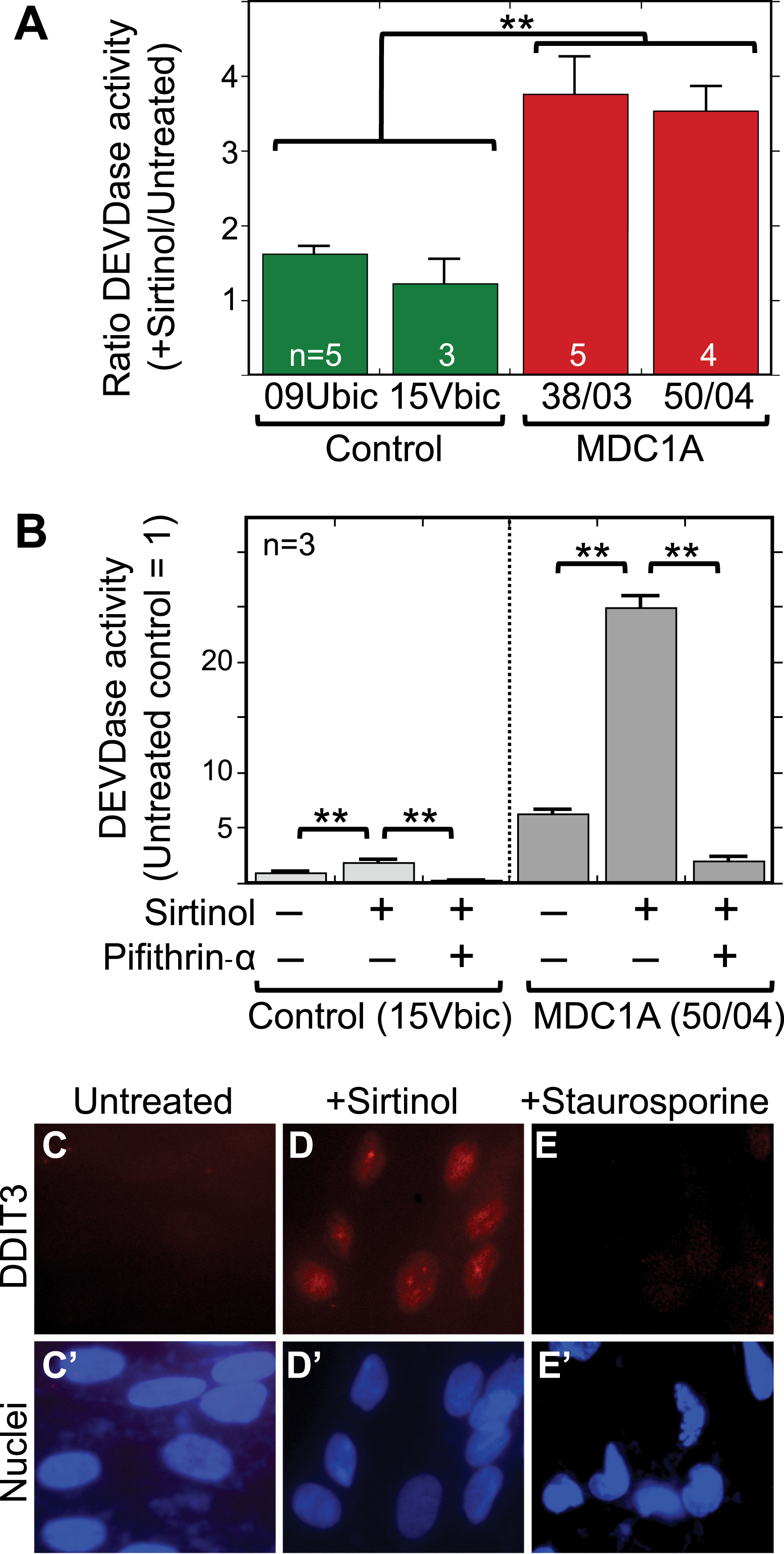
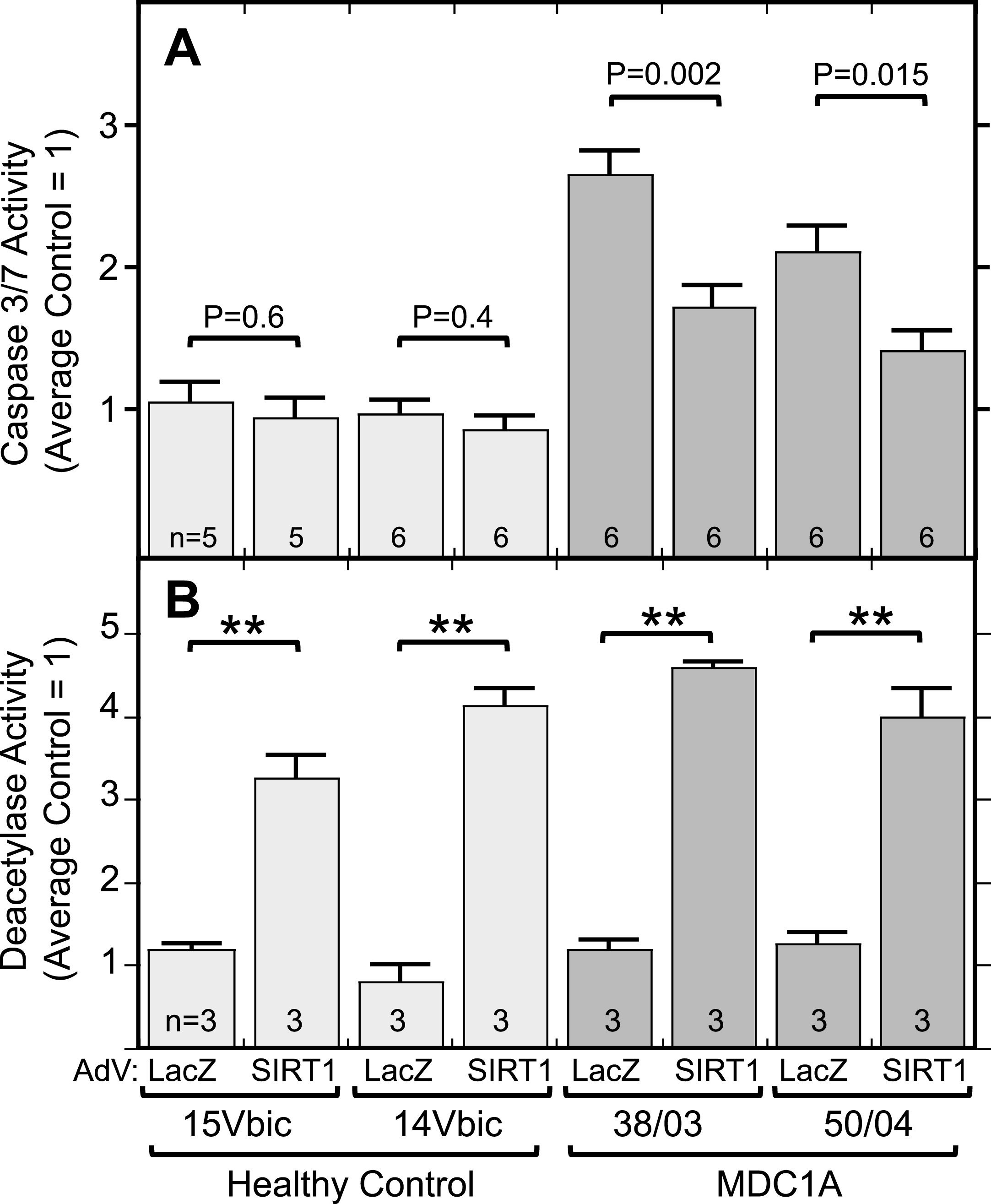
Because inhibition of SIRT1 led to increased caspase activation, we next asked if the opposite also occurred, that is, if activation of SIRT1 would decrease caspase activation in MDC1A cells. We found that adenoviral-mediated overexpression of SIRT1 did lead to a significant decrease in DEVDase enzymatic activity in differentiated cultures of MDC1A, but not healthy control, cells (Fig. 6A). As a control, infection with an adenovirus-LacZ vector did not affect caspase or sirtuin activity in MDC1A or healthy control cells. Total deacetylase activity was increased 3-4X in the SIRT1 adenovirus-treated cultures (Fig. 6B). Thus, increased expression of SIRT1 was sufficient to moderate the spontaneous, aberrant increase of caspase activity that was seen in MDC1A myogenic cells compared to healthy controls.
Fig.6
Overexpression of SIRT1 lowered caspase activity in MDC1A cultures. A. Differentiated MDC1A cultures (38/03 and 50/04) in which SIRT1 was expressed from an adenovirus (AdV) vector had lower levels of active caspase (DEVDase) than parallel cultures in which ß-galactoside (LacZ) was overexpressed. SIRT1 overexpression had little effect on healthy control cultures (14Vbic and 15Vbic). Adenovirus vectors were added for 72 h at multiplicity of infection of 10. Caspase activity was normalized so that the average of LacZ-expressing healthy control cultures = 1.0. B. Total cellular deacetylase activity was increased ∼3-4X by adenoviral-mediated expression of SIRT1 as in panel A. Error bars = SE. **P < 0.001 or as indicated by unpaired T-test of LacZ vs. SIRT1 with cells and n as indicated.
DISCUSSION
In this study, we examined plausible mechanisms to identify those that could mediate aberrant activation of cell death pathways in laminin-α2-deficiency. Our results suggest that sirtuin deacetylase and p53 activity, but not activation of p38 MAPK, can mediate caspase activation in laminin-α2-deficiency. These results raise the possibility that interventions targeted at p53 or sirtuins could have positive effects in laminin-α2-deficiency.
A recent comprehensive study showed that p38 MAPK is more highly phosphorylated in muscles of dystrophin-deficient (mdx) and γ-sarcoglycan-deficient mice than in muscles of wild-type mice, and that this increased p38 MAPK activation is an important contributor to pathogenesis in these mouse disease models [19]. In contrast, we found that homozygous, laminin-α2-deficient dyW/dyW mouse muscles had, on average, decreased (not increased) phosphorylation of p38 MAPK compared to wild-type mouse muscles. The cellular and molecular mechanisms underlying decreased p38 MAPK phosphorylation in Lama2–/– muscles remains to be investigated. However, because p38 MAPK phosphorylation was similar in our cultures of laminin-α2-deficient and healthy control cells, it is likely that mechanisms extrinsic to muscle cells (e.g., inflammation, fibrosis) contributed to altered p38 MAPK phosphorylation in dyW/dyW muscle tissues that we examined. A previous study reported that muscles of 18 week old wild-type and dy2J/dy2J mice had similar levels of p38 MAPK phosphorylation [43]. Compared to the dyW/dyW mice used in our study, which lack almost all functional laminin-α2 and have a rapid onset of disease, dy2J/dy2J mice retain a more functional laminin-α2 and develop pathology more slowly. Though p38 MAPK activity is an important regulator of normal myogenesis [44] and is increased in mouse models of both dystrophin-deficiency and γ-sarcoglycan-deficiency [19], our results and those of Elbaz et al. [43] show that pathogenesis in laminin-α2-deficiency (e.g., as measured by caspase activation) is not associated with increased activity of p38 MAPK.
Though p38 MAPK did not appear to be linked to caspase activation in laminin-α2-deficiency, our studies did support a role for p53 in aberrant activation of caspase-3 in MDC1A myotubes. In particular, we found that treatment of MDC1A myotube cultures with an inhibitor of p53, pifithrin-alpha, reduced the aberrantly increased caspase activity in untreated, differentiated cultures of MDC1A myogenic cells, in some cases to near the level found in untreated cultures of healthy controls. In addition, we found that ∼10% of the nuclei in MDC1A cultures, but <0.5% of the nuclei in controls, showed immunostaining for p53. Translocation to and accumulation of p53 in the nucleus is expected during p53-mediated cell death. Furthermore, the p53-regulated BBC3 (also called PUMA) protein was more abundant in laminin-α2-deficient human cells and mouse muscles than in healthy controls. BBC3 transcription is p53-dependent and thus the amount of BBC3 protein serves as a proxy for the amount of p53 transcriptional activation. Finally, treatment with nutlin-3a, which prevents HDM2-mediated inhibition of p53 function, produced more caspase activation in MDC1A than healthy control cells.
In MDC1A myogenic cells, our results, when taken together, suggest that (i) p53 is partially activated, though still inhibited by interaction with HDM2; and (ii) p53 is required for aberrant caspase activation. Previous work has shown that regulated p53 function is likely required for normal function of skeletal muscles. For example, though myogenic cell proliferation and regeneration after injury occur normally in p53-null mice [45], other studies have found that p53-null mouse myogenic cells in culture show moderately lower fusion [34, 46], as well as decreased mitochondrial synthesis and autophagy [47]. Further studies are needed to identify the molecular mechanisms that link laminin-α2-deficiency to p53-mediated caspase activation and to determine if a therapeutic strategy based on appropriate manipulation of the p53 pathway might be possible [48]. In addition, HDM2 has been reported to bind to and regulate KU70, as well as p53, in the cytoplasm [49]. Thus, it will likely be informative to investigate how the interaction of HDM2 with KU70 may be altered in laminin-α2-deficiency.
Finally, our studies suggest that caspase activation in laminin-α2-deficient myogenic cells is inversely related to SIRT1 deacetylase activity. In particular, we found that overexpression of SIRT1 in differentiated MDC1A myogenic cell cultures led to decreased caspase activity, whereas inhibition of sirtuin activity (with sirtinol) led to an increase in caspase activity. The seven members of the sirtuin family play significant roles in a very large number of cellular functions, including regulation of homeostasis in skeletal muscles [50–55]. Our finding that sirtinol had a negative effect on MDC1A myogenic cells, as measured by increased caspase activation, is consistent with studies showing that sirtinol can induce apoptosis in multiple cell types [56–58]. Our results are also consistent with previous work showing that sirtinol-induced apoptosis is p53-dependent [56] and that SIRT1 inhibition can increase KU70 acetylation [38, 59]. However, it remains to be determined if the decreased caspase activation caused by SIRT1 overexpression is dependent on p53. On the other hand, studies of Drosophila and nematode models of neural and muscle diseases have found positive effects upon treatment with sirtinol [60, 61]. The underlying molecular mechanisms through which inhibition of sirtuin(s), particularly by sirtinol, leads to either a positive or negative effect in a particular cell type or organism remain to be determined. Additional work is also needed to identify the roles in MDC1A of the additional members of the sirtuin family, as well as non-sirtuin deacetylases and acetyltransferases. Cell death and bioenergetic pathways are dysregulated in MDC1A [62] and are likely targets of sirtuin regulation.
In this study, our goal was to carry out initial studies of multiple pathways to identify mechanisms that could be linked to MDC1A pathogenesis and would thus be candidates for further study. As noted in the Introduction, we and other groups are examining potential therapeutic approaches for MDC1A based on techniques designed to restore normal muscle functions, such as ECM attachment, myofiber regeneration, autophagy, proteastasis, and/or intracellular signaling to prevent activation of cell death pathways [8–18]. Despite this progress, much remains unknown about pathogenetic mechanisms in laminin-α2-deficiency. Our results did not support a role in MDC1A pathogenesis for overactivity of p38 MAPK, but our study did identify p53 and sirtuin deacetylase activity as mediators of aberrant caspase activation in MDC1A myogenic cells. Our study, though limited in scope, suggests that additional work is warranted to identify the detailed molecular mechanisms by which p53 and sirtuin deacetylases regulate activation of muscle cell death pathways in laminin-α2-deficiency. Additional studies are also needed to determine if interventions designed to restore normal function to the p53 and sirtuin pathways might be useful in ameliorating pathology in MDC1A.
COMPETING INTERESTS
The authors have no conflicts of interest to report.
AUTHORS’ CONTRIBUTIONS
S.Y., M.L.B., B.Y., and J.B.M. planned, performed, and analyzed experiments. D.I. and M.B. produced adenoviruses and provided facilities and technical advice for the adenovirus studies. J.B.M. wrote the manuscript and all authors participated in editing the manuscript.
ACKNOWLEDGMENTS
We thank anonymous biopsy donors for their generosity; Dr. Sachiko Homma (Boston University School of Medicine) for much helpful advice; Dr. Mahasweta Girgenrath for intial study of p38 MAPK; Chinaemere Igwebuike with support from the Summer Training as Research Scholars Program at Boston University for help with initial p53 studies; Eric Schmidt with support from the Undergraduate Research Opportunities Program and Michaela Welch with support from the HHMI Summer Research Program in Neuroscience at Boston University for help with initial studies of acetylation regulators; and Dr. Lydia Sorokin (University of Münster) for a laminin-α2 mAb.
This work was supported by grants from the NIH (R01AR060328 to J.B.M., and R01AR062587 to Peter L. Jones with a subcontract to J.B.M.); the Muscular Dystrophy Association (#216422 to J.B.M.); the Association Française contre les Myopathies (#18248 to J.B.M.); the Undergraduate Research Opportunity Program at Boston University (to B.Y.); and funding for imaging from the Boston University Clinical and Translational Science Institute which is supported by the National Center for Advancing Translational Sciences at the NIH (1UL1TR001430). The Muscle Tissue Culture Collection (MTCC) at the University of Munich is part of the German network on muscular dystrophies (MD-NET, service structure S1, 01GM0601) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany). MTCC is a partner of Eurobiobank (www.eurobiobank.org) and TREAT-NMD (www.treat-nmd.eu). Funders had no role in design or implementation of the study. Please contact the corresponding author for data or material requests.
REFERENCES
[1] | Gawlik KI , Durbeej M . Skeletal muscle laminin and MDC1A: Pathogenesis and treatment strategies. Skelet Muscle. (2011) ;1: :9. |
[2] | Durbeej M . Laminin-α2 chain-deficient congenital muscular dystrophy: Pathophysiology and development of treatment. Curr Top Membr. (2015) ;76: :31–60. |
[3] | Guicheney P , Vignier N , Helbling-Leclerc A , Nissinen M , Zhang X , Cruaud C , et al. Genetics of laminin α2 chain (or merosin) deficient congenital muscular dystrophy: From identification of mutations to prenatal diagnosis. Neuromusc Dis. (1997) ;7: :180–6. |
[4] | Miyagoe-Suzuki Y , Nakagawa M , Takeda I . Merosin and congenital muscular dystrophy. Microscopy Research and Technique. (2000) ;48: :181–91. |
[5] | Tezak Z , Prandini P , Boscaro M , Marin A , Devaney J , Marino M , et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat. (2003) ;21: :103–11. |
[6] | Homma S , Beermann ML , Miller JB . Peripheral nerve pathology, including aberrant Schwann cell differentiation, is ameliorated by doxycycline in a laminin-α2-deficient mouse model of congenital muscular dystrophy. Hum Mol Genet. (2011) ;20: :2662–72. |
[7] | Relucio J , Menezes MJ , Miyagoe-Suzuki Y , Takeda S , Colognato H . Laminin regulates postnatal oligodendrocyte production by promoting oligodendrocyte progenitor survival in the subventricular zone. Glia. (2012) ;60: :1451–67. |
[8] | Carmignac V , Svensson M , Körner Z , Elowsson L , Matsumura C , Gawlik KI , Allamand V , Durbeej M . Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. (2011) ;20: :4891–902. |
[9] | Körner Z , Fontes-Oliveira CC , Holmberg J , Carmignac V , Durbeej M . Bortezomib partially improves laminin α2 chain-deficient muscular dystrophy. Am J Pathol. (2014) ;184: :1518–28. |
[10] | Girgenrath M , Dominov JA , Kostek CA , Miller JB . Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Investigation. (2004) ;114: :1635–39. |
[11] | Vishnudas VK , Miller JB . Ku70 regulates Bax-mediated pathogenesis in laminin-alpha2-deficient human muscle cells and mouse models of congenital muscular dystrophy. Hum Mol Genet. (2009) ;18: :4467–77. |
[12] | Dominov JA , Kravetz AJ , Ardelt M , Kostek CA , Beermann ML , Miller JB . Muscle-specific BCL2 expression ameliorates muscle disease in laminin-alpha2-deficient, but not dystrophin-deficient, mice. Hum Mol Genet. (2005) ;14: :1029–40. |
[13] | Erb M , Meinen S , Barzaghi P , Sumanovski LT , Courdier-Früh, Rüegg MA , Meier T . Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J Pharmacol Exp Ther. (2009) ;331: :787–95. |
[14] | Meinen S , Lin S , Thurnherr R , Erb M , Meier T , Rüegg MA . Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice. EMBO Mol Med. (2011) ;3: :465–79. |
[15] | Yamauchi J , Kumar A , Duarte L , Mehuron T , Girgenrath M . Triggering regeneration and tackling apoptosis: A combinatorial approach to treating congenital muscular dystrophy type 1 A. Hum Mol Genet. (2013) ;22: :4306–17. |
[16] | McKee KK , Crosson SC , Meinen S , Reinhard JR , Rüegg MA , Yurchenco PD . Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J Clin Invest. (2017) ;127: :1075–89. |
[17] | Gawlik K , Miyagoe-Suzuki Y , Ekblom P , Takeda S , Durbeej M . Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum Mol Genet. (2004) ;13: :1775–84. |
[18] | Rooney JE , Knapp JR , Hodges BL , Wuebbles RD , Burkin DJ . Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am J Pathol. (2012) ;180: :1593–602. |
[19] | Wissing ER , Boyer JG , Kwong JQ , Sargent MA , Karch J , McNally EM , Otsu K , Molkentin JD . P38α MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum Mol Genet. (2014) ;23: :5452–63. |
[20] | Koch AJ , Holaska JM . Loss of emerin alters myogenic signaling and miRNA expression in mouse myogenic progenitors. PLoS One. (2012) ;7: (5):e37262. |
[21] | Homma S , Chen JCJ , Rahimov F , Beermann ML , Hanger K , Bibat GM , et al. A unique library of myogenic cells from facioscapulohumeral muscular dystrophy subjects and unaffected relatives: Family, disease, & cell function. Eur J Hum Genet. (2012) ;20: :404–10. |
[22] | Homma S , Beermann ML , Boyce FM , Miller JB . Expression of FSHD-related DUX4-FL alters proteostasis and induces TDP-43 aggregation. Ann Clin Transl Neurol. (2015) ;2: :151–66. |
[23] | Homma S , Beermann ML , Yu B , Boyce FM , Miller JB . Nuclear bodies reorganize during myogenesis and are differentially disrupted by expression of FSHD-associated DUX4. Skelet Muscle. (2016) ;6: :42. |
[24] | Jones TI , Chen JC , Rahimov F , Homma S , Arashiro P , Beermann ML , et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: Evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. (2012) ;21: :4419–30. |
[25] | Yoon S , Stadler G , Beermann ML , Schmidt EV , Windelborn JA , Schneiderat P , Wright WE , Miller JB . Immortalized myogenic cells from congenital muscular dystrophy type1A patients recapitulate aberrant caspase activation in pathogenesis: A new tool for MDC1A research. Skelet Muscle. (2013) ;3: :28. |
[26] | Kuang W , Xu H , Vachon PH , Liu L , Loechel F , Wewer UM , Engvall E . Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest. (1998) ;102: :844–52. |
[27] | Girgenrath M , Beermann ML , Vishnudas VK , Homma S , Miller JB . Pathology is alleviated by doxycycline in a laminin-alpha2-deficient mouse model of congenital muscular dystrophy. Ann Neurol. (2008) ;65: :47–56. |
[28] | Miller JB , Crow MT , Stockdale FE . Slow and fast myosin heavy chain content defines three types of myotubes in early muscle cell cultures. J Cell Biol. (1985) ;101: :1643–50. |
[29] | Hook B , Schagat T . Comparison of luciferases as ATP sensors in the presence of inhibitors: CellTiter-Glo® Assay shows less compound interference than the Perkin Elmer ATPlite™ 1step assay. Promega Corporation Web site. http://www.promega.com/resources/pubhub/celltiter-glo-assay-shows-less-compound-interference-than-other-luminescent-assays/Updated 2011. Accessed November 20, (2017) . |
[30] | Shao D , Fry JL , Han J , Hou X , Pimentel DR , Matsui R , Cohen RA , Bachschmid MM . A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J Biol Chem. (2014) ;289: :7293–306. |
[31] | Uo T , Veenstra TD , Morrison RS . Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. (2009) ;29: :2824–32. |
[32] | Bell RA , Al-Khalaf M , Megeney LA . The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet Muscle. (2016) ;6: :16. |
[33] | Komarov PG , Komarova EA , Kondratov RV , Christov-Tselkov K , Coon JS , Chernov MV , Gudkov AV . A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. (1999) ;285: :1733–7. |
[34] | Karaman MW , Herrgard S , Treiber DK , Gallant P , Atteridge CE , Campbell BT , et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. (2008) ;26: :127–32. |
[35] | Nowak JA , Malowitz J , Girgenrath M , Kostek CA , Kravetz AJ , Dominov JA , Miller JB . Immortalization of mouse myogenic cells can occur without loss of p16INK4a, p19ARF, or p53 and is accelerated by inactivation of Bax. BMC Cell Biol. (2004) ;5: :1. |
[36] | Fischer M . Census and evaluation of p53 target genes. Oncogene. (2017) ;36: :3943–56. |
[37] | Shangary S , Wang S . Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. (2009) ;49: :223–41. |
[38] | Walsh EM , Niu M , Bergholz J , Xiao ZX . Nutlin-3 down-regulates retinoblastoma protein expression and inhibits muscle cell differentiation. Biochem Biophys Res Commun. (2015) ;461: :293–9. |
[39] | Cohen HY , Miller C , Bitterman KJ , Wall NR , Hekking B , Kessler B , Howitz KT , Gorospe M , de Cabo R , Sinclair DA . Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. (2004) ;305: :390–2. |
[40] | Sundaresan NR , Samant SA , Pillai VB , Rajamohan SB , Gupta MP . SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. (2008) ;28: :6384–401. |
[41] | Chaudhary N , Nakka KK , Chavali PL , Bhat J , Chatterjee S , Chattopadhyay S . SMAR1 coordinates HDAC6-induced deacetylation of Ku70 and dictates cell fate upon irradiation. Cell Death Dis. (2014) ;5: :e1447. |
[42] | Hada M , Kwok RP . Regulation of ku70-bax complex in cells. J Cell Death. (2014) ;7: :11–3. |
[43] | Elbaz M , Yanay N , Aga-Mizrachi S , Brunschwig Z , Kassis I , Ettinger K , Barak V , Nevo Y . Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in the dy(2J)/dy(2J) mouse. Ann Neurol. (2012) ;71: :699–708. |
[44] | Segalés J , Islam AB , Kumar R , Liu QC , Sousa-Victor P , Dilworth FJ , et al. Chromatin-wide and transcriptome profiling integration uncovers p38α MAPK as a global regulator of skeletal muscle differentiation. Skelet Muscle. (2016) ;6: :9. |
[45] | White JD , Rachel C , Vermeulen R , Davies M , Grounds MD . The role of p53 in vivo during skeletal muscle post-natal development and regeneration: Studies in p53 knockout mice. Int J Dev Biol. (2002) ;46: :577–82. |
[46] | Porrello A , Cerone MA , Coen S , Gurtner A , Fontemaggi G , Cimino L , Piaggio G , Sacchi A , Soddu S . p53 regulates myogenesis by triggering the differentiation activity of pRb. J Cell Biol. (2000) ;151: :1295–304. |
[47] | Fortini P , Ferretti C , Iorio E , Cagnin M , Garribba L , Pietraforte D , et al. The fine tuning of metabolism, autophagy and differentiation during in vitro myogenesis. Cell Death Dis. (2016) ;7: :e2168. |
[48] | Gudkov AV , Komarova EA . Dangerous habits of a security guard: The two faces of p53 as a drug target. Hum Mol Genet. (2007) ;16 Spec No 1: :R67–72. |
[49] | Gama V , Gomez JA , Mayo LD , Jackson MW , Danielpour D , Song K , Haas AL , Laughlin MJ , Matsuyama S . Hdm2 is a ubiquitin ligase of Ku70-Akt promotes cell survival by inhibiting Hdm2-dependent Ku70 destabilization. Cell Death Differ. (2009) ;16: :758–69. |
[50] | Vinciguerra M , Fulco M , Ladurner A , Sartorelli V , Rosenthal N . SirT1 in muscle physiology and disease: Lessons from mouse models. Dis Model Mech. (2010) ;3: :298–303. |
[51] | Gurd BJ . Deacetylation of PGC-1α by SIRT Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol Nutr Metab. (2011) ;36: :589–97. |
[52] | Ryall JG . The role of sirtuins in the regulation of metabolic homeostasis in skeletal muscle. Curr Opin Clin Nutr Metab Care. (2012) ;15: :561–6. |
[53] | Tonkin J , Villarroya F , Puri PL , Vinciguerra M . SIRT1 signaling as potential modulator of skeletal muscle diseases. Curr Opin Pharmacol. (2012) ;12: :372–6. |
[54] | Villalba JM , Alcaín FJ . Sirtuin activators and inhibitors. Biofactors. (2012) ;38: :349–59. |
[55] | Kupis W , Pałyga J , Tomal E , Niewiadomska E . The role of sirtuins in cellular homeostasis. J Physiol Biochem. (2016) ;72: :371–80. |
[56] | Peck B , Chen CY , Ho KK , Di Fruscia P , Myatt SS , Coombes RC , Fuchter MJ , Hsiao CD , Lam EW . SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. (2010) ;9: :844–55. |
[57] | Kumari S , Chaurasia SN , Nayak MK , Mallick RL , Dash D . Sirtuin inhibition induces apoptosis-like changes in platelets and thrombocytopenia. J Biol Chem. (2015) ;290: :12290–9. |
[58] | Paradis FH , Hales BF . The effects of class-specific histone deacetylase inhibitors on the development of limbs during organogenesis. Toxicol Sci. (2015) ;148: :220–8. |
[59] | Kim MJ , Hong KS , Kim HB , Lee SH , Bae JH , Kim DW , et al. Ku70 acetylation and modulation of c-Myc/ATF4/CHOP signaling axis by SIRT1 inhibition lead to sensitization of HepG2 cells to TRAIL through induction of DR5 and down-regulation of c-FLIP. Int J Biochem Cell Biol. (2013) ;45: :711–23. |
[60] | Catoire H , Pasco MY , Abu-Baker A , Holbert S , Tourette C , Brais B , Rouleau GA , Parker JA , Néri C . Sirtuin inhibition protects from the polyalanine muscular dystrophy protein PABPN1. Hum Mol Genet. (2008) ;17: :2108–17. |
[61] | Pallos J , Bodai L , Lukacsovich T , Purcell JM , Steffan JS , Thompson LM , Marsh JL . Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet. (2008) ;17: :3767–75. |
[62] | Fontes-Oliveira CC , Steinz M , Schneiderat P , Mulder H , Durbeej M . Bioenergetic Impairment in Congenital Muscular Dystrophy Type 1A and Leigh Syndrome Muscle Cells. Sci Rep. (2017) ;7: :45272. |


