
Congenital Myasthenic Syndromes or Inherited Disorders of Neuromuscular Transmission: Recent Discoveries and Open Questions
Abstract
Congenital myasthenic syndromes (CMS) form a heterogeneous group of rare diseases characterized by fatigable muscle weakness. They are genetically-inherited and caused by defective synaptic transmission at the cholinergic neuromuscular junction (NMJ). The number of genes known to cause CMS when mutated is currently 30, and the relationship between fatigable muscle weakness and defective functions is quite well-understood for many of them. However, some of the most recent discoveries in individuals with CMS challenge our knowledge of the NMJ, where the basis of the pathology has mostly been investigated in animal models. Frontier forms between CMS and congenital myopathy, which have been genetically and clinically identified, underline the poorly understood interplay between the synaptic and extrasynaptic molecules in the neuromuscular system. In addition, precise electrophysiological and histopathological investigations of individuals with CMS suggest an important role of NMJ plasticity in the response to CMS pathogenesis. While efficient drug-based treatments are already available to improve neuromuscular transmission for most forms of CMS, others, as well as neurological and muscular comorbidities, remain resistant. Taken together, the available pathological data point to physiological issues which remain to be understood in order to achieve precision medicine with efficient therapeutics for all individuals suffering from CMS.
Congenital myasthenic syndromes (CMS) form a heterogeneous group of rare diseases that are, by definition, genetically-inherited and primarily caused by defective neurotransmission at the peripheral cholinergic synapse known as the neuromuscular junction (NMJ). Clinical diagnosis of CMS is confirmed by electromyographic investigations (EMG) with a decrement of the compound muscle action potential (CMAP) in response to repetitive nerve stimulation (RNS) at low (usually 3 Hz) frequency. However, this decrement is often not systematically recorded, mainly because of the fluctuating nature of CMS symptoms, and gene sequencing for clinical purpose is now a gold standard for ascertaining the diagnosis.
The use of reverse genetics to identify the genes which cause CMS has greatly improved our basic knowledge of the NMJ the last 20 years. The first mutation causing CMS was identified by a candidate gene approach in the CHRNE gene, which encodes the ɛ-subunit of the pentameric acetylcholine receptor (AChR) [7]. The number of known causative genes is now 30, with mutation frequencies ranging from extremely low (private gene with one single individual reported in the literature) to a high value (up to 42 % for CHRNE), and the relationship between fatigable muscle weakness and defective functions are quite well-understood for many of them [8]. This knowledge led to the use of drug-based treatments, aimed at improving neuromuscular transmission, which are frequently effective in reducing fatigable muscle weakness. Excellent reviews of the current knowledge of the clinical features, genetics, pathophysiology and drug-based treatment of CMS have recently been published elsewhere, and we recommend them to the reader [9–12].
In this review, we would like to focus on some of the most recent discoveries concerning CMS in humans which challenge our knowledge of the NMJ. First, we consider forms of CMS with unexpected comorbidities, where the basis of the pathology has mostly been investigated in animal models. We also discuss frontier forms with congenital myopathy (CM) with all the associated diagnosis complexity that these forms represent for the clinician and for the management of the disease. Next, we discuss some possible implications of the findings of electrophysiological and histopathological investigations on individuals with newly identified forms of CMS and how they may reflect the role of functional plasticity of the NMJ in the response to CMS pathogenesis. Finally, we address some of the issues remaining to be resolved in order to propose efficient treatment and achieve precision medicine for all the individuals suffering from this highly heterogeneous group of rare diseases.
CMS, COMORBIDITIES AND NEUROMUSCULAR FRONTIER FORMS
During the last four years, there has been an impressive increase in the number of identified genes known to cause rare diseases, including CMS, with the systematic use of whole exome sequencing technology in genetically-undiagnosed individuals. The genes causing CMS encode both proteins that are critical for the NMJ as well as other proteins with still unknown synaptic functions (Fig. 1, Table 1). Most of the reported mutations are recessively inherited, with the exception of those observed in SYT2 [13], SNAP25 [14] and in the genes encoding the AChR subunits resulting in the slow channel form of CMS (SCCMS) [15]. No X-linked form of CMS has been reported so far. Some recently identified causal genes are linked to forms of CMS with neurological or muscular comorbidities which are direct results of the gene defect but are not improved by the treatments commonly used to treat defective neuromuscular transmission (Table 2).
Fig.1
presynaptic CMS causal genes and related proteins recently identified as causing CMS when mutated. Schematic representation of one NMJ with the pre- (nerve terminal) and post-synaptic (muscle fiber) compartments separated by the synaptic cleft containing a specific basal lamina. The terminal Schwann cells are not represented for clarity. The causal genes (written in italics and in brackets) resulting in presynaptic forms of CMS encode proteins involved in cholinergic metabolism (CHT1, ChAT, VAChT) or synaptic vesicles docking and release. Uptake by the nerve terminal of choline resulting from hydrolysis of acetylcholine (ACh) by AChE from the synaptic cleft is assumed by CHT1. ChAT will use choline to synthesize new molecules of ACh that will be collected into synaptic vesicles by VAChT. Synaptic vesicles filled with ACh are docked to the nerve terminal membrane close to the synaptic cleft by SNARE proteins including synaptobrevin-1, synaptotagomin-2 and SNAP-25. Munc13-1 helps the maturation of synaptic vesicles by acting in their priming prior to vesicle fusion and participates in the activity-dependent refilling of readily releasable vesicle pool (RRP). The basal lamina laminin α5 protein would interact with the synaptic vesicle protein SV2A to favor the vesicular exocytosis. Agrin would have a presynaptic impact, either directly or through a retrograde signaling pathway, which remains to be better characterized.
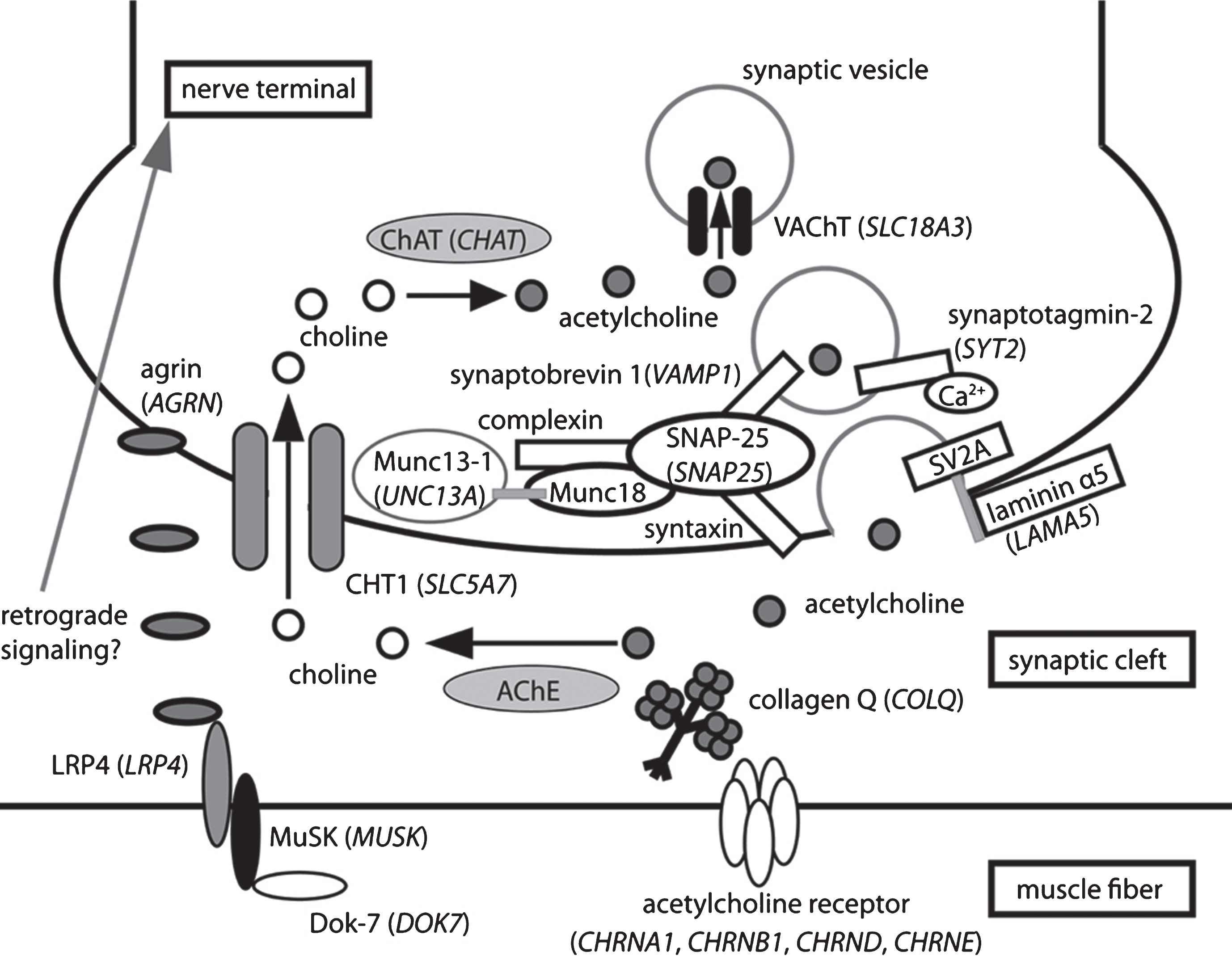
Table 1
Genes currently known to cause CMS when mutated and function primarily impaired by the mutations
| Primary impaired function | Protein (Gene symbol) |
| Presynaptic cholinergic metabolism | choline acetyl-transferase (CHAT), high-affinity choline transporter 1 (SLC5A7), vesicular acetylcholine transporter (SLC18A3) |
| Neuronal ACh release | synaptotagmin 2 (SYT2), vesicle-associated membrane protein 1 (VAMP1), synaptosomal-associated protein 25 (SNAP25), Munc13-1 (UNC13A) |
| Synaptic anchoring of AChE | collagenic tail of AChE (COLQ) |
| Muscle action potential genesis | subunits of AChR, adult isoform (CHRNE, CHRNA1, CHRND, CHRNB1), skeletal muscle voltage-dependent sodium channel (SCN4A) |
| Post-synaptic AChR aggregation | rapsyn (RAPSN), DOK7 (DOK7), agrin (AGRN), LRP4 (LRP4), MuSK (MUSK) |
| Protein glycosylation | GFPT1 (GFPT1), DPAGT1 (DPAGT1), GMPPB (GMPPB), ALG2 (ALG2), ALG14 (ALG14) |
| Presynaptic differentiation or organization | laminin β2 (LAMB2), laminin α5 (LAMA5) |
| Mitochondrial energetic metabolism | mitochondrial tricarboxylate transport protein (SLC25A1) |
| Structural proteins with unknown synaptic function | plectin (PLEC), myosin 9A (MYO9A), collagen α-1 (XIII) chain (COL13A1), prolyl endopeptidase-like (PREPL) |
Gene symbols are according to the HUGO Gene Nomenclature Committee (HGNC). Genes are classified from the most frequently mutated to the last frequently mutated for a same impaired function. NMJ = neuromuscular junction; ACh = acetylcholine; AChR = acetylcholine receptor, DOK7 = dowstream of kinase 7, LRP4 = low-density lipoprotein receptor-related protein 4, MuSK = muscle specific kinase, GFPT1 = Glutamine-fructose-6-phosphate aminotransferase 1, DPAGT1 = UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase, GMPPB = Mannose-1-phosphate guanyltransferase beta, ALG2 = alpha-1,3/1,6-mannosyltransferase, ALG14 = UDP-N-acetylglucosaminyltransferase subunit.
Table 2
Clinical features of forms of CMS with causative genes recently reported
| Gene symbol | GMPPB | COL13A1 | MYO9A | LAMA5 | SLC5A7 | SLC18A3 | VAMP1 | SYT2 | SNAP25 | UNC13A |
| Protein | GMPPB | collagen α-1 (XIII) chain | myosin 9A | laminin α5 | CHT1 | VAChT | VAMP1 | synaptotagmin-2 | SNAP25 | Munc13-1 |
| Reported families | 10 | 2 | 2 | 1 | 5 | 3 | 3 | 2 | 1 | 1 |
| Inheritance | AR | AR | AR | AR | AR | AR | AR | AD | de novo | AR |
| Reported cases | 14 | 3 | 3 | 1 | 5 | 4 | 5 | 11 | 1 | 1 |
| Ptosis | yes (3) | yes (3) | yes (3) | yes | yes (5) | yes (2) | no (5) | slight (2) | yes | yes |
| Apnea | N.D. | N.D. | yes (1) | no | yes (5) | yes (1), no (1), ventilation (2) | no (5) | N.D. | no | N.D. |
| Cognitive delay | mild (1) | LD (1) | yes (1), LD (1) | no | yes (3) | LD (2), yes (1), | N.D. | mild language delay (1) | yes | yes |
| Response to AChEI | yes (12/14)a | no (2/2) | yes (3/3) | mild | yes (4/5) | yes (2/2) | yes (5/5) | no (2/2) | no | no |
| Other medications | DAP and/or salbutamol | DAP+salbutamol | DAP (1), DAP+FLX (1)b | DAP | N.D. | DAP+salbutamol (1) | N.D. | DAP (2) | DAP | DAP |
| Other specific features | raised CK level (10/10) | micrognathia (3) | N.D. | facial tics, mild volume loss of brain | N.D. | JC (3), retrognathia (2), CF (1) | JC (2), joint laxity (1) | foot deformities | JC | cortical hyperexcitability |
aThe denominator of the fraction indicates the number of patients who received examination or medication. bCrisis after this combination was reported. AR = autosomal recessive; AD = autosomal dominant; N.D. = no data, LD = learning difficulties; AChEI = acetylcholinesterase inhibitor; DAP = 3,4-Diaminopyridine; FLX = fluoxetine; JC = joint contractures; CF = mild reduction of left ventricular cardiac function.
Neurological comorbidities
Two years ago, only one form of presynaptic CMS had been identified, linked to loss-of-function mutations in the gene encoding the presynaptic choline acetyltransferase enzyme ChAT (Fig. 1). The number of presynaptic forms of CMS is now 7, with an extremely low frequency of occurrence in all of them which, with the exception of CHAT and SLC5A7 related forms (Table 1), have only been reported in isolated individuals. SLC5A7 encodes the hemicholinium sensitive-high-affinity choline transporter 1 (CHT1), expressed by all cholinergic neurons, which imports choline, generated by hydrolysis of ACh by acetylcholinesterase (AChE), from the synaptic cleft into the nerve terminal (Fig. 1). One dominant-negative frameshift mutation in this gene has been reported to cause hereditary motor neuropathy [16]. On the other hand, loss-of-function mutations (missense and nonsense) in SLC5A7 cause CMS with episodic apnea (EA) during early infancy (CMS-EA), as do loss-of-function mutations in CHAT. A third gene recently shown to cause CMS-EA is SLC18A3, which encodes the vesicular ACh transporter (VAChT) required for uptake of newly synthesized ACh into presynaptic vesicles [18, 19].
These three presynaptic proteins, critical for cholinergic metabolism, are expressed in all cholinergic neurons. It is well known that nicotinic cholinergic neurotransmission plays an important role in “higher-level” functions such as behavior and cognition [20]. Reflecting this role, five of the eight infants with CMS-EA due to SLC5A7 or SLC18A3 mutations suffer from severe cognitive impairment (Table 2). Whether this comorbidity results from episodes of brain hypoxia during EA, or is a direct consequence of defective central cholinergic neurons, has to be further investigated. Sudden apneic episodes are one of the most severe clinical features of CMS. They have sometimes been reported in CMS as a result of mutations in genes encoding subunits of AChR (fast channel syndrome), rapsyn and the skeletal muscle voltage-gated sodium channel NaV1.4. However, the systematic association of EA with loss-of-function in genes encoding neuronal proteins critical for presynaptic cholinergic homeostasis (uptake, local synthesis and vesicular upload of the neurotransmitter, Table 2) suggests that these episodes result from the critical role of cholinergic transmission in respiratory functions during development [21, 22].
A further presynaptic group of CMS, due to mutations in genes encoding members of the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex, have also emerged from high-throughput sequencing of human samples. This complex drives the docking or fusion of the synaptic vesicles (SV) with the presynaptic motoneuronal membrane (Fig. 1). Dominant mutations in SYT2 and SNAP25B genes, encoding the synaptotagmin 2 and the SNAP25B proteins of the vesicle-associated (v-SNARE) and presynaptic membrane-associated (t-SNAREs) complex respectively, were shown to cause CMS without (SYT2) or with (SNAP25B) neurological comorbidities in individuals with isolated forms of CMS (cortical hyperexcitability, cerebellar ataxia, and intellectual disability, Table 2) [14, 23].
The absence of signs suggesting defective high-level functions in individuals with SYT2 dominant mutations may be due to the functional redundancy of synaptotagmin paralogs and to the restricted expression of the B isoform of synaptotagmin 2 encoded by this gene. However, they presented with a Charcot-Marie Tooth-like clinical pattern in two unrelated families [13, 23]. In one family, affected members developed foot deformities in childhood including pes cavus and hammer toes. In the second family, congenital hip dislocation was reported in addition to foot deformities. Neurophysiological study showed a Lambert-Eaton Myasthenic Syndrome-like pattern with low-amplitude of CMAP and marked increment after brief exercise (see below). In addition, a slight reinnervation pattern on needle electromyography of distal muscles, consistent with motor neuropathy was recorded in one family [13]. This CMS-like form benefited both at the clinical and neurophysiological level from treatment with 3,4-diaminopyridine (DAP), a chemical inhibitor of presynaptic K+ channels.
The clinical association between CMS and neurological disorders due to defective docking or exocytosis of SV has been further emphasized by the recent identification of loss-of-function mutations in the genes encoding VAMP1 and Munc13-1 (Fig. 1, Table 2). VAMP1 is a member of the synaptobrevin family (v-SNARE). Four individuals with CMS from two unrelated families, were shown to possess loss-of-function mutations in VAMP1 [4]. All presented severe impairment of developmental milestones. Munc13-1 is a partner of syntaxin 1B – a t-SNARE component – and is critical for the assembly of the SNARE complex, which promotes priming of the SV in preparation for release in response to Ca2+ entry [1]. Loss-of-function of UNC13A has been reported in one patient with CMS, microcephaly and cortical hyperexcitability that could be related to the expression of Munc13-1 in glutamatergic neurons [24]. Interestingly, one amino-acid substitution exerting a gain-of-function effect in Munc13-1 has subsequently been reported in one isolated individual with dyskinesia, developmental delay and autism [25]. The impact of this gain of function mutation on neuromuscular transmission was not investigated.
The gene most recently identified as harboring mutations causing CMS with possible primary presynaptic dysfunction is LAMA5, which encodes the α-5 isoform of laminin, a basal lamina protein [26]. In vitro analyses showed that the pathogenic amino-acid substitution decreased binding of laminin α5 to the vesicular protein SV2, and impaired neural cell-adhesion and extension of cell projections. Whether this isoform of laminin is expressed in the central nervous system is unknown, and the individual with the LAMA5 loss-of-function mutation suffered from myopia and facial tics but no intellectual disability. As emphasized above, most of these presynaptic forms are observed at a very low frequency. This could be due to undiagnosed individuals suffering from forms of CMS masked by most prominent neurological signs.
Muscular comorbidities
The clinical presentations of CMS are highly variable, and one well-known differential diagnosis of CMS is congenital disorders of skeletal muscles, including the congenital myopathies (CM) and the congenital muscular dystrophies and their later onset limb-girdle muscular dystrophy (LGMD) forms. What was already a complex picture has recently become even more challenging with new clinical presentations that confound standard anatomical classifications. Atypical phenotypes associated with classical CMS genes resembling other types of neuromuscular diseases (LGMD, distal myopathies) have been observed. Conversely, significant fatigue and a positive response to AChE inhibitors have been reported in individuals with CM/CMD diagnosed at the molecular level. This striking overlap may be explained by the fact that numerous molecules involved in NMJ formation, maintenance and functioning are expressed in other regions of the muscle fiber, playing critical roles in key cell functions such as membrane trafficking, nucleus migration, or sarcolemma stability.
Some forms of CMS mimic specific types of CM and CMD/LGMD
GMPPB encodes a GDP-mannose pyrophosphorylase that catalyzes the synthesis of GDP-mannose, a sugar nucleotide essential for both N- and O-glycosylation of proteins. GMPPB mutations were first reported to cause LGMD without any clinical evidence of synaptopathy [27]. Two years later, loss-of-function mutations of GMPPB were identified in 11 individuals suffering from CMS [28]. All had elevated CK levels and muscle level of α-dystroglycan was decreased. Age at presentation was from 1.5 to 31 years. Four individuals were first diagnosed as suffering from LGMD. An episode of generalized sudden weakness and fluctuations of muscle strength was reported in one individual and fatigability during effort in another one. All individuals had CMAP decrement in response to nerve stimulation and benefited from the use of AChE inhibitors (prostigmin or pyridostigmin) as did most of the individuals with CMS. Additional individuals suffering from overlapping forms of LGMD and CMS due to loss-of-function mutations in GMPPB have been described since this first report, strengthening the inclusion of the GMPPB-linked form of CMS in the emerging group of CM with synaptopathy or CMS with myopathy [29–31]. The first member of this group is probably Fukuyama-type congenital muscular dystrophy (FCMD) since aberrant neuromuscular junctions were also observed in this secondary alpha-dystroglycanopathy due to mutations in the gene encoding fukutin [32]. The latter group may also include the limb-girdle form of CMS with tubular aggregates resulting from mutations in GFPT1, which encodes glutamine-fructose-6-phosphate transaminase 1, another enzyme critical for protein glycosylation. A recently published long-term follow-up of 11 individuals stressed a clinical evolution toward myopathic weakness that occurred concomitantly to ineffectiveness of usual CMS treatments [33].
Five individuals from three unrelated families were reported with a recessively-inherited form of CMS due to mutations in the gene encoding agrin, a basal lamina protein whose neural isoform is well known to play a key role in NMJ formation and maintenance by triggering post-synaptic AChR clustering [34]. All patients displayed CMS with distal muscle weakness and atrophy reminiscent of distal myopathy [5]. Age at onset was from birth to 15 years. MRI and neurophysiological studies were compatible with a mild myopathy restricted to distal limb muscles. However, CMAP decrement in response to RNS as well as immunostaining and ultrastructural analyses of muscle end-plate regions pointed towards neuromuscular transmission defect and a diagnosis of CMS (see below).
Some forms of CM display secondary neuromuscular transmission defects
Conversely, a series of individuals with CM and myasthenic features (fatigability, neurophysiological or histopathological evidence of NMJ remodeling) have been published in recent years (see [35] for review). Most were linked to genes causing centronuclear myopathies (MTM1, BIN1 and DNM2), which may be due to the role played by the related proteins (myotubularin 1, amphiphysin-2 and dynamin-2, respectively) in membrane trafficking and recycling which is a key cell process for post-synaptic AChRs [36–38]. Isolated individuals with myasthenic features were also reported as having mutations in the TPM2 and TPM3 genes encoding tropomyosin 2 and 3, and in RYR1 coding for the ryanodine receptor [39, 40]. The myasthenic features suggest that the neuromuscular transmission defect contributes to muscle weakness in these individuals. Accordingly, most were reported to benefit from treatment with AChE inhibitors and this should be kept in mind when clinically managing individuals with CM.
FUNCTIONAL PLASTICITY OF THE NMJ UNDERLINED BY NEUROPHYSIOLOGICAL INVESTIGATION OF CMS
Electrophysiological investigation of the NMJ
Clinical evidence of muscle fatigability resulting from impaired neuromuscular transmission is mainly obtained from EMG investigation. Decrement of the fifth CMAP, in response to 2–5 Hz RNS, by more than 10% of the initial value, is usually taken to confirm the diagnosis of CMS, in the knowledge that the amplitude of the CMAP in normal individuals changes little at these frequencies [13]. Because the CMAP reflects the summed effect of the AP of many muscle fibers, it may not detect ‘subthreshold’ impairment of transmission at individual NMJs during RNS. By contrast Single Fiber EMG (SFEMG), which measures the temporal instability, or ‘jitter’, of the activation of individual muscle fibers by the nerve, is a highly sensitive, but unspecific, method that can detect subthreshold impaired neurotransmission at even 1-2 NMJs [41].
While these approaches can provide diagnostically useful evidence of impaired neuromuscular transmission, they are generally unable to identify which steps in the complex sequence of events leading to transmission are primarily affected. Detail at this level can only be obtained by making intracellular recordings of synaptic events from NMJs in isolated motor point biopsy samples e.g. [42–44]. Unfortunately, very few centers world-wide are equipped to do this on human muscle biopsies, with the result that we remain ignorant of the exact nature and site of neurotransmission impairment in many forms of CMS.
Post-exercise CMAP increment in presynaptic CMS
In the increasing number of presynaptic forms of CMS described above, as in virtually all forms of CMS, significant decrement of the CMAP is observed during RNS at 2–5 Hz. Two important exceptions to this rule are CMS caused by mutations in CHAT, in which there is no decrement at this low frequency in most of the individuals [2, 3], and in the individual with CMS related to VAMP1, in whom increment was observed in response to RNS [4]. Seemingly paradoxically, in a number of these presynaptic forms, voluntary exercise (e.g. for 10 s) or nerve stimulation at high frequency (e.g. 20 Hz for 20 seconds or 10 Hz for 5 min) results in a transient reduction of this decrement, and may even lead to the CMAP becoming substantially bigger [4, 5]. To interpret these somewhat confusing findings, it is helpful to review some basic features of neuromuscular transmission.
Neuromuscular transmission during repetitive activity in normal conditions
Nerve impulses (nerve action potentials, or ‘nAPs’) activate muscle fibers by the process of neuromuscular transmission [12]. Each nAP opens a number of CaV2.1 channels in the nerve terminal allowing an influx of Ca2+. The resulting transient increase in Ca2+ concentration within the nerve terminal causes the release of a number (20–200 in vertebrates) of multi-molecular ‘quanta’ of the transmitter, acetylcholine (ACh) into the synaptic cleft. These quanta correspond to the amount of ACh (about 10,000 molecules) contained in each SV. This release occurs by exocytosis at specialized structures in the presynaptic membrane, the ‘active zones’ (AZs). The action of transmitter on the muscle fiber causes a transient increase of its resting membrane potential by about 35–40 mV, the so-called endplate potential (EPP). The EPP is normally great enough to cause opening of the voltage-gated sodium channels (NaV1.4) in the postsynaptic muscle membrane, thus triggering a mAP and activating contraction. In addition to this evoked release, transmitter quanta are also released spontaneously, at random, each one generating a small (about 1 mV) ‘miniature EPP’ (mEPP).
Most muscles are activated by short bursts, or longer trains, of nerve impulses at frequencies between 10–100 Hz [45]. During such activity, the amplitude of the EPP usually declines. However, because substantially more quanta are normally released than are required to trigger a mAP, the muscle is still effectively activated, i.e. transmission has a high ‘safety factor’ [46]. Most of the decline of EPP amplitude results from changes in the number of transmitter quanta released by each nAP, the so-called ‘quantal content’ (QC) of the evoked response. These changes occur over a wide range of time scales. A number of factors influence the value of the QC, and hence the amplitude of the EPP, at any moment. In Fig. 2 we show, very schematically, the likely relationships between some of the main factors, and their impact on the EPP and CMAP, in normal and pathological muscles. We emphasize that most of the changes in Ca2+ and RRP depicted represent our best estimate of what occurs rather than actual observations which, for the most part, have not been made. Collectively, these various forms of modulation of QC are often referred to as ‘short-term synaptic plasticity’ [47].
Fig.2
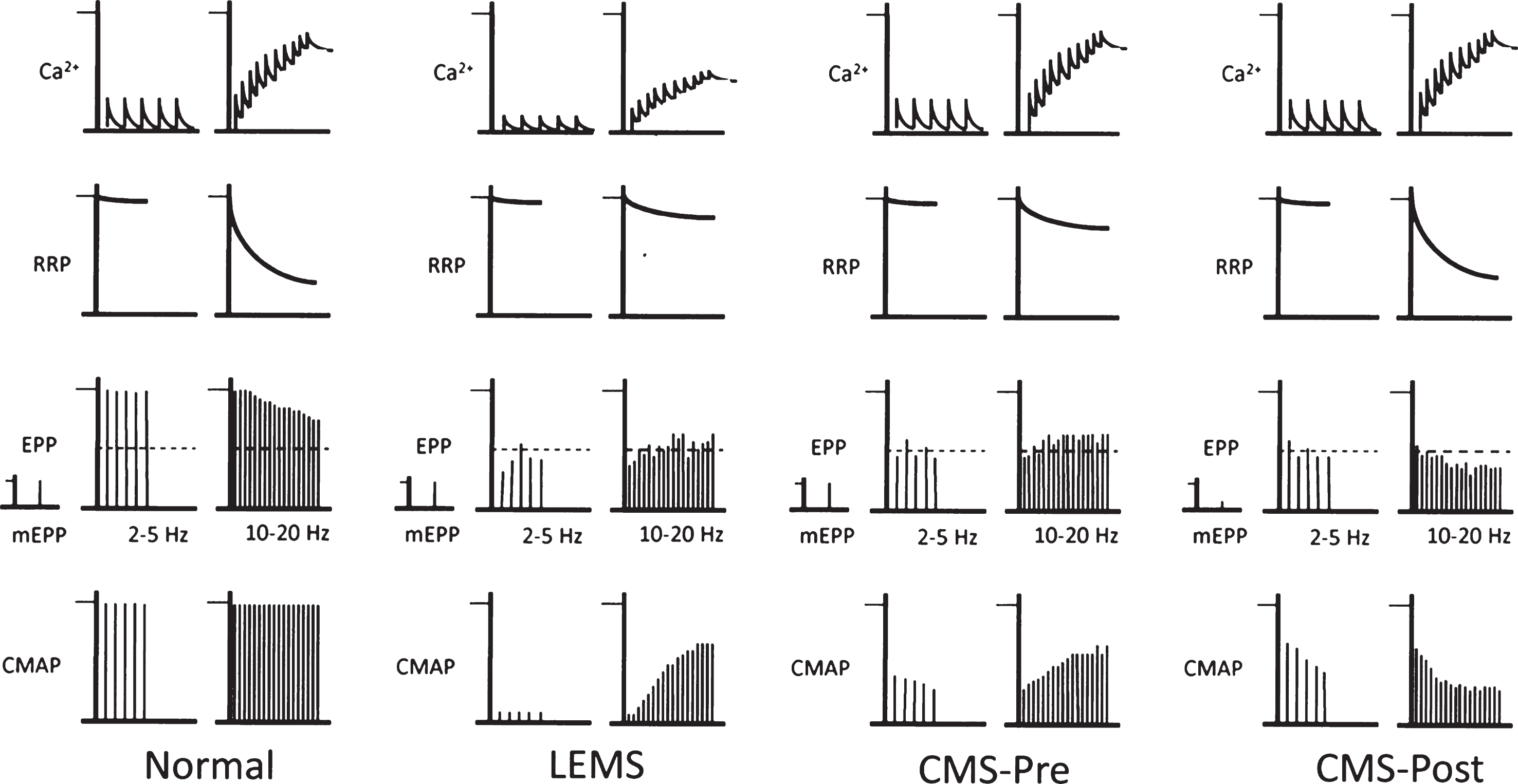
patterns of transmitter release from motor nerve terminals. This figure provides a qualitative view of how different patterns of NMJ response to repetitive activity (2–5 Hz and 10–20 Hz) may arise, and their impact on the CMAP. Top row, level of Ca2+ in the nerve terminal. Second row, size of ‘readily releasable pool’ (RRP) of transmitter quanta. Third row, mEPP followed by EPPs. Dotted horizontal line shows mAP threshold. Bottom row, CMAPs. At normal NMJs, at 2–5 Hz, each nAP causes a brief increase in Ca2+ which triggers release and generates large EPPs. These all trigger mAPs, so the CMAP shows no decrement. The RRP is almost fully replenished between the responses, although there may be a little decline. At 10–20 Hz, the individual Ca2+ transients summate, raising the level of Ca2 + . However, now there is not time for the RRP to be fully replenished between responses, so it declines significantly. The net effect is a modest decline in quantal content (QC) and EPP amplitude. However, the EPPs remain suprathreshold, so there is no decrement of the CMAP. In LEMS, the individual Ca2+ transients are much smaller than normal, so the QC at low frequency is very low and few EPPs reach threshold, so the CMAP is greatly reduced. At higher frequency, the Ca2+ transients summate, so the Ca2+ level increases. At the same time, because the QC is low, there is little decline of the RRP. As a result, the increased Ca2+ level causes a significant increase in the QC and EPP amplitude, so more EPPs reach threshold and the CMAP increases. In forms of CMS in which presynaptic function is impaired (for reasons other than a decrease in Ca2+ entry), QC is reduced at low frequency, even though mEPP amplitude, Ca2+ entry and RRP size are all normal. As a result, there is little decline in the RRP. At high frequency, the build-up of Ca2+ causes enhanced release and some EPPs reach threshold, causing some increase in the CMAP. In forms of CMS that act on the postsynaptic membrane, the amplitude of the mEPPs (‘quantal size’) is generally much smaller than normal. During low frequency activity, although the QC is normal, the EPPs are small and shows some decline. As a result, many EPPs do not reach threshold so the CMAP is small and may show some decrement as the RRP gets smaller. At high frequency, the decline in RRP results in a decline in EPP amplitude, in spite of the increase in Ca2+ level, leading to significant decrement of the CMAP.
The Ca2+ that enters following each nAP is subsequently removed by a combination of processes: binding to soluble buffers, extrusion from the terminal by Ca2+-ATPases or Na+-Ca2+ transporters, and sequestration by mitochondria and components of the ER. Each of these processes has a distinctive time course, some requiring seconds [48]. As a result, during ‘physiological’ activity at frequencies of 10 Hz and more, there is a gradual build-up of the basal level of Ca2+ in the terminal (Fig. 2, top row, Normal). Since the relationship between Ca2+ concentration and QC is very non-linear [49], the effect of the ‘residual’ Ca2+ from previous activity enhances the effect of the Ca2+ entering during subsequent nAPs. This potentiating effect would be expected to cause an increase in the amplitude of the EPP and, hence, the efficacy of transmission.
In reality, the potentiating effect of Ca2+ build up is counteracted by a decline in the number of transmitter quanta capable of being released. These quanta normally represent only about 0.1% of the SVs present in the terminal and are considered to represent a distinct ‘pool’ of ‘readily releasable’ quanta, the RRP (Fig. 2, second row) [45]. Following depletion, the RRP is only relatively slowly replenished (1–10 seconds) [45, 50]. The RRP at mammalian NMJs is currently believed to consist of the 2-3 SVs closely associated with each AZ, making a total of several hundred SVs at a typical human NMJ [43, 50]. During low frequency activation (e.g. 0.1–1 Hz), about 5–10% of the initial RRP is released by each nerve impulse. During a train of 10 impulses, the RRP would be reduced to about 60% of its initial value. However, although the resting rate of RRP replenishment is relatively slow, it speeds up during activity, helping to maintain adequate release [50].
In clinical practice, these complex interacting effects on the efficacy of transmission are typically investigated by stimulating first at 2–5 Hz and then at 10–20 Hz or by recording CMAPs at 2–5 Hz after 10 s of voluntary activation of muscles.
Neuromuscular transmission in Lambert-Eaton Myasthenic syndrome (LEMS)
An important and informative example of presynaptic transmission impairment is LEMS, an autoimmune condition in which autoantibodies directed against CaV channels, located at the AZ, cause a decline in their number [51, 52]. As a result, fewer CaV channels open in response to individual nAPs and so fewer SVs are released during stimulation at 2–5 Hz, resulting in much smaller, and often subthreshold, EPPs (Fig. 2, LEMS). As a result, the amplitude of the CMAP during low frequency stimulation is much smaller than normal. During stimulation at 10–20 Hz, as described above, the cytoplasmic Ca2+ concentration progressively increases. Because of the reduced quantal release in LEMS, the RRP initially remains much larger than at normal NMJs, so the increase in basal Ca2+ leads to a substantial increase in EPP amplitude. This, in turn, causes the activation of many more muscle fibers, and an increase in the amplitude of the CMAP.
Neuromuscular transmission in CMS due to defective synaptotagmin-2, VAMP1 and Munc13-1 SNARE proteins
By analogy with LEMS, a post-exercise increment is generally interpreted as a sign of impaired presynaptic function in CMS (Fig. 2, CMS-Pre). Such an increment has been reported in individuals with CMS related to synaptotagmin 2 [13, 23], VAMP1 [4] and Munc13-1 [24], which all play a role in evoked SV exocytosis. In these conditions, the CMAP during 2–5 Hz stimulation is reduced, though generally not as much as in LEMS. None of these proteins is known to have a direct effect on nerve-evoked Ca2+ entry. This suggests that the post-exercise increment results primarily from the action of the increase in residual Ca2+ on a RRP that is larger than normal as a result of the reduced rate of depletion associated with impaired SV release.
Neuromuscular transmission in CMS related to agrin
Particularly intriguing is the incrementing response reported in some patients with CMS caused by mutations in the gene encoding agrin [5]. Agrin induces the post-synaptic clustering of AChRs through its binding to the Lrp4-MuSK complex [34]. Patients with agrin mutations have NMJs that are abnormally fragmented and sometimes show dispersed postsynaptic gutters with evanescent borders of AChR staining, resulting in a streaky pattern of staining [5, 53, 54].
In all these patients with agrin mutations described so far, there was clear decrement of the CMAP during 2–5 Hz stimulation, suggesting low amplitude EPPs resulting in a critically reduced safety factor [5, 53, 54]. This is consistent with the pattern expected for reduced AChRs, as seen classically in myasthenia gravis and in inherited AChR deficiencies (Fig. 2, CMS-Post). However, by contrast with typical ’postsynaptic’ forms of CMS, a period of exercise resulted in an increment in CMAP amplitude in each of 5 patients investigated in one study [5]. This suggests some form of presynaptic abnormality, reminiscent of the CMS-Pre forms described above.
In searching for an explanation of this intriguing result, it is of interest that agrin’s binding partner, Lrp4, also serves as a ‘retrograde’ signal, which regulates the properties and behavior of the nerve terminal during synaptogenesis [55, 56] and may mediate forms of structural plasticity (‘synaptic homeostasis’) in the adult [57, 58]. One possibility is that impaired agrin function somehow interferes with that retrograde signaling, leading to altered presynaptic maturation. However, no CMAP potentiation was observed in the few patients with CMS related to Lrp4, despite a severe impairment of neurotransmission with reduced quantal content and EPP amplitude [10].
Post-exercise increment was also reported in the one individual identified with loss-of-function mutations in LAMA5, which impair interaction of the basal lamina laminin α5 protein with SV2. SV2 is a vesicular protein critical for SV fusion with the presynaptic membrane in response to elevated cytoplasmic Ca2+. A dramatic reduction of number of quanta released was recorded from NMJs in muscle biopsy samples of this individual [26]. As with agrin, the details of the pathway leading to impaired neuromuscular transmission with post-exercise increment when laminin α5 is missing remain to be documented.
It is noteworthy that there is great variation in the kinetics of the incremental response seen in these various individuals. In LEMS, the decline of CMAP potentiation occurs on a time scale of 10–15 seconds [59, 60]. This decline is significantly slower (2–60 min) in CMS related to synaptotagmin 2 [13] and agrin [5]. This suggests that quite different mechanisms are involved in the neurotransmission potentiation observed in these presynaptic forms of CMS. More detailed clinical neurophysiological studies by EMG in vivo and on isolated biopsy samples in vitro would help to clarify this situation.
HISTOPATHOLOGICAL INVESTIGATION OF HUMAN MUSCLE BIOPSY SAMPLES IN CMS
Overview
The impairment of neuromuscular transmission that gives rise to the defining features of CMS is generally accompanied by structural changes in the cellular components of the NMJ. Because these are often more easily investigated than the functional defects themselves, they form an important component of many recent studies of CMS. Immunohistological as well as ultrastructural analyses help to confirm the diagnosis of CMS versus CM and, in some situations, may point clinicians to a specific form of CMS.
The mature NMJ is formed by the presynaptic nerve terminal of one α motor neuron closely apposed to the postsynaptic gutters of the skeletal muscle fiber membrane and ensheathed by several non-myelinating terminal Schwann cells. Most current knowledge of the NMJ in CMS comes from the investigation of mouse models in which key synaptic components have been deleted or inactivated. However, the human NMJ is quite distinct from the rodent NMJ in at least two aspects. The first concerns its development. The first steps of synaptic maturation, including the loss of polyaxonal innervation and the elaboration of the mature form of the NMJ, occur postnatally in mice but take place in utero in humans [61]. The second aspect concerns the architecture of the adult NMJ. The human NMJ is smaller in diameter (20μm vs 30μm in mice) and is composed of smaller and more distinct post-synaptic elements with an aspect called “cat’s paw” compared to the nice “pretzel-like form” observed in rodents [43]. In spite of these differences, morphological investigations of human samples show many similarities with observations made in mice, which is critical for developing precision therapeutics using this species as a preclinical model.
The classical Koelle and Friedenwald histochemical method was one of the first methods used to visualize the motor endplate by revealing the synaptic cholinesterase activity [62]. The use of immuno-histo-fluorescent techniques to label the innervated zone of myofibers teased from skeletal muscle biopsies can give a global structural view of several NMJs by simultaneously staining the terminal axons (anti-neurofilaments), the Schwann cells (anti-cytoplasmic S100 glial protein), and the post-synaptic AChRs (fluorescent α−bungarotoxin conjugates). Ultrastructural analyses complete this investigation by exploring the fine structure of the presynaptic, synaptic and postsynaptic elements in a smaller number of synapses.
Application to CMS
Morphological investigation of the NMJ sometimes suggest a particular form of CMS, and also give essential cues about the functional plasticity of this giant cholinergic synapse in pathological situations. For example, the form of CMS due to loss-of-function mutations in the gene encoding the ColQ collagenic tail of the synaptic form of AChE was specifically recognized by the lack of AChE staining, despite the presence of nerve terminals and well defined synaptic gutters at the NMJ, well before the identification of the causative gene [63]. Whatever the primary functional impact of the defective protein on the NMJ may be most, if not all, forms of CMS are eventually associated with a global dysfunctional organization due to the complex and dynamic molecular dialogs occurring between its three cell components [64].
Presynaptic CMS-EA
The global architecture of the NMJ has been investigated by immunostaining of teased muscle fibers for only a few patients with a presynaptic form of CMS. Consistent with the critical role of ACh for NMJ formation and maintenance, CMS-EA due to loss-of-function of CHT1 is associated with several axons innervating a single NMJ, suggesting persisting polyaxonal innervation, and a poorly-differentiated post-synaptic apparatus and generally defective maintenance of the NMJ, suggestive of repeated denervation-reinnervation events, as illustrated in Fig. 3 [17]. Interestingly, NMJs in this form of CMS display an increased level of synaptic butyryl cholinesterase (BChE), whose synaptic functions have not been investigated in detail. This form of cholinesterase is bound to perisynaptic Schwann cells and is believed to finely tune synaptic transmission by regulating the binding of ACh to AChRs located on these glial cells [65]. How a decrease of CHT1 level results in increased expression of BChE remains to be investigated, as does the specificity of this inverse correlation, since the synaptic level of BChE has never been investigated in muscle samples from individuals with ChAT or VAChT-related forms of CMS-EA.
Fig.3
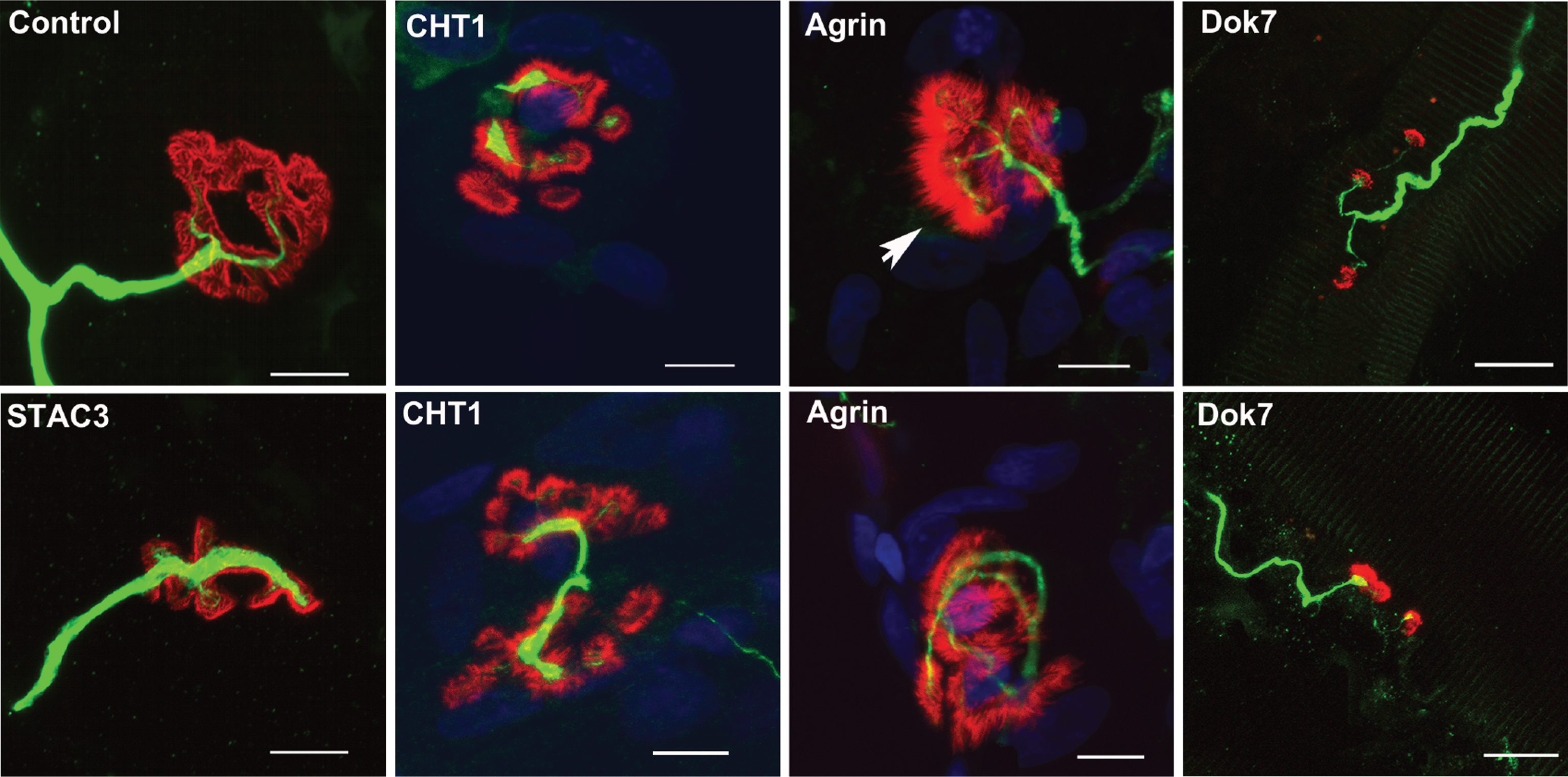
morphological changes occurring at the NMJ in some pre- and post-synaptic forms of CMS. Representative whole-mount pictures of neuromuscular biopsies of individuals with CMS due to mutations in genes encoding presynaptic, synaptic, and postsynaptic proteins. Dilacerated muscle fibers were used for immunostaining of motor axons using an anti-neurofilament (NF 165 kDa, in green) and for fluorescent staining of postsynaptic AChRs using tetra-rhodamine-conjugated α-bungarotoxin (in red). In one adult control sample, the terminal axonal branches typically end as a fork innervating a well-defined nAChR structure. Morphological analysis of NMJs from one young adult suffering from presynaptic CMS related to CHT1 shows a disorganized post-synaptic element with fragmented synaptic gutters partially innervated by thin terminal axons. An unusual spike-like area pattern of nAChR staining (arrow) contacted by thin terminal axonal branch was observed in individual with synaptic CMS related to agrin. Strong decrease of axonal branching with innervation “en passant” contacting small isolated synaptic cups was observed in one adult suffering from post-synaptic CMS related to DOK7. One muscle sample from one adolescent suffering from congenital myopathy due to an homozygous missense mutation in the STAC3 gene, encoding a component of the excitation-contraction coupling machinery (6), is shown for comparison with congenital myopathy without evidence for secondary synaptopathy. In this myopathic condition, the NMJs look nearly normal, with terminal axonal branches contacting well-defined synaptic gutters. Scale bar = 10μm (applies to all pictures).
Synaptic form of CMS due to agrin dysfunction
As indicated above, some synaptic forms of CMS linked to mutations in the gene encoding agrin display distinct features at the clinical as well as neurophysiological level. However, this is not related to specific structural changes of the NMJ. Morphological investigations of muscle biopsies from one individual with a clinically unspecific form of CMS due to AGRN mutations showed profound structural changes including denervated and remodeled endplates with a decrease of postsynaptic AChRs, as expected from the critical role of this synaptic protein for their aggregation (Fig. 3) [53]. Ultrastructural analysis confirmed denervation processes and stressed major presynaptic abnormalities in this individual. Similar structural changes were observed in the agrin-related form of CMS with distal muscle atrophy and post-exercise increment [5]. In addition, a less common pattern of postsynaptic gutters with evanescent borders of AChR staining, resulting in a postsynaptic streaky staining pattern, was observed (Fig. 3). This streaky pattern may result from the presence of secondary synaptic folds which are not covered by a nerve terminal or perisynaptic Schwann cells, as observed by electron microscopy, pointing towards a presynaptic defect.
Post-synaptic CMS with reduced AChR density
The most frequent form of CMS is due to mutations in the CHRNE gene, which result primarily in reduced post-synaptic nAChR density as seen in human muscle biopsies [66–68]. Reduced density of postsynaptic nAChR has also been demonstrated in muscle biopsies from individuals with CMS due to mutations affecting other subunits of AChRs [69–71]. Interestingly, in post-synaptic CMS due to mutations in the gene encoding DOK7, the synaptic morphology displays specific and major changes in the presynaptic compartment with thin nodal axonal sprouts contacting small separated synaptic cups characteristics of innervation “en passant” (Fig. 3) [72, 73]. This further underlines the ability of the presynaptic plasticity of this giant peripheral synapse to compensate for defective neurotransmission, plasticity that remains to be further understood at the molecular level since its understanding may open the door to innovative therapeutics against defective neuromuscular transmission whatever its etiology.
TOWARD PRECISION MEDICINE FOR ALL INDIVIDUALS SUFFERING FROM CMS
In the near future, the use of high-throughput sequencing technologies for diagnostic purposes will be further developed worldwide with national programs of genomic medicine [74]. Whole-exome or genome sequencing has been found to be particularly pertinent for CMS, where some years ago, no causative genes had been recognized for about 50% of individuals with a clinically-suspected CMS [8]. Even though mutations causing CMS have now been identified in 30 genes, a huge number of individuals suffering from isolated forms of fatigable muscle weakness still await clinical diagnosis and effective treatment in clinical referral centers worldwide, and the undoubtful impact of this clinical uncertainty on their quality of life deserves further investigation. Careful neurophysiological investigation of neuromuscular transmission in several nerve-muscle pairs during distinct periods of life, as well as histopathological investigations of muscle biopsy samples and, wherever possible, electrophysiological studies of motor point biopsy samples, are mandatory to ascertain the nature of the impairment of neuromuscular transmission in these individuals so that accurate differential diagnoses, excluding alternatives such as CM or functional somatic syndromes, can be made.
Particular challenges
Many of the most recently identified mutations causing CMS are ‘private’, known to occur in only 1 or 2 people. In these situations there are major challenges, both technical and experimental, arising from the high number of variants existing for each of us and from the mandatory demonstration of pathogenicity of the candidate variant when the causative gene to be searched for has no known physiological relationship with synapses [75]. Moreover, not only are most of the remaining individuals with undiagnosed fatigable muscle weakness sporadic cases, but this complex trait may have a polygenic origin with association with susceptibility variants, whose identification remains challenging in the field of rare diseases. This challenging situation is a feature of all forms of orphan diseases, and international programs of data sharing, such as matchmaker exchange and RD-connect platforms, have now emerged to favor emergence of pertinent bioinformatics tools, and to facilitate transfer of clinical genomic information and biological samples between research teams to resolve this critical issue [76, 77].
Therapeutics
Identifying the pathogenic mechanism of each individual suffering from CMS is also critical to guide therapy since it is well know that drugs that benefit one form of CMS can be harmful in another form [10, 11]. From the earliest recognition of CMS as a distinct, if heterogeneous, group of conditions, the main therapeutic tools have been the use of anticholinesterases [78]. The second main tool has been the drug 3,4-DAP, which prolongs the nAP by blocking the neuronal K+ channels whose opening terminates the nAP [79]. By using these drugs in tandem, it is often possible to use lower doses, resulting in fewer side effects. However, a number of conditions have been identified in which anticholinesterase drugs are not only ineffective, but may actually exacerbate the symptoms, e.g. inherited AChE deficiency and ‘slow-channel’ CMS, in which the prolonged AChR-gated channel openings lead to harmful entry of Ca2+ into the postsynaptic region and secondary degeneration of the post-synaptic element [10].
A further new therapeutic dimension, based on the initially empirical use of sympathomimetics, has emerged in recent years from efforts to treat patients whose symptoms are refractory to cholinesterase inhibitors. Treatment with a range of β2-adrenergic agonists (ephedrine, albutamol, salbutamol, ...) has proven to be effective in autoimmune myasthenia as well as in several forms of CMS [10, 11, 35, 80–83]. These drugs, as well as adrenaline itself, are known to have a variety of short-term (minutes) effects on neuromuscular transmission in vitro, some apparently presynaptic and others postsynaptic [84–86]. However, these effects occur only at concentrations that are an order of magnitude or so greater than the concentrations attainable in humans [85, 86]. Furthermore, most of the therapeutic benefits that have been reported for sympathomimetics in patients with CMS require days, weeks or even months to be fully achieved, suggesting that these drugs enhance neuromuscular transmission by inducing long-term structural changes. The mechanism(s) underlying their effects, or even whether their main action is on the central or peripheral neurons, the skeletal muscle, the Schwann cells, or even the vasculature, is not known.
The success of sympathomimetics on impaired neuromuscular transmission has prompted reinvestigation of the role of sympathetic innervation of skeletal muscle. A recent study confirms the association of unmyelinated sympathetic neurons and NMJs in mouse leg muscles [87]. It further reports that stimulation of the appropriate sympathetic nerve activates postsynaptic β2-adrenoceptors and elevates postsynaptic cAMP production at NMJs. Interestingly, chemical sympathectomy was found to cause a reduction in NMJ size, AChR density and CMAP amplitude, and these deleterious effects were largely reversed by systemic treatment with clenbuterol for 10 days [87]. These findings seem to demonstrate significant effects of sympathetic innervation on the maintenance of NMJs. It will be important to learn more about how these effects are mediated and how they relate to the clinically beneficial effects of sympathomimetics on neuromuscular transmission.
CONCLUSION
The recent development of sequencing technology has greatly improved our knowledge of the etiology of CMS. Despite the increasing power of these molecular genetic investigations, physiological studies are ultimately the only way to define the underlying basis of impaired neuromuscular transmission in the context of a highly plastic synapse. While EMG studies in a clinical context can indicate the presence of impaired neuromuscular transmission, they cannot define the particular steps in the synaptic transmission process which are defective following a developmental or structural defect. Detailed studies of pathologic NMJs using histological tools and intracellular recording in motor point biopsies are the only way such defects can be defined. Whatever the practical difficulties, such studies remain essential if the pathogenic mechanisms that give rise to the various forms of CMS are ever to be understood and targetable pathways for promoting novel efficient therapies to be identified for muscle weakness resulting from impaired neuromuscular transmission.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank all the families agreeing to participate to research works and the members of national networks on CMS for fruitful discussions. This work is supported by INSERM, CNRS, Sorbonne universités UPMC-Paris 06, Association Française contre les Myopathies (AFM) –Téléthon (research grants AFM16573 and AFM20030), and Investissements d’avenir (ANR-10-IAIHU-06) to SN, and the Medical Research Council as part of the MRC Centre for Neuromuscular Diseases (reference G1002274, grant ID 98482), the Wellcome Trust Pathfinder Award, and by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 305444 (RD-Connect) and no. 2012-305121 (NeurOmics) to HL.
REFERENCES
[1] | Wang S , Choi UB , Gong J , Yang X , Li Y , Wang AL , et al. Conformational change of syntaxin linker region induced by Munc13s initiates SNARE complex formation in synaptic exocytosis. The EMBO Journal. (2017) ;36: (6):816–29. |
[2] | Engel AG , Shen XM , Selcen D , Sine SM . What have we learned from the congenital myasthenic syndromes. J Mol Neurosci. (2010) ;40: (1-2):143–53. |
[3] | Arredondo J , Lara M , Gospe SM Jr , Mazia CG , Vaccarezza M , Garcia-Erro M , et al. Choline Acetyltransferase Mutations Causing Congenital Myasthenic Syndrome: Molecular Findings and Genotype-Phenotype Correlations. Hum Mutat. (2015) ;36: (9):881–93. |
[4] | Salpietro V , Lin W , Delle Vedove A , Storbeck M , Liu Y , Efthymiou S , et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann Neurol. (2017) ;81: (4):597–603. |
[5] | Nicole S , Chaouch A , Torbergsen T , Bauche S , de Bruyckere E , Fontenille MJ , et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. (2014) ;137: (Pt 9):2429–43. |
[6] | Horstick EJ , Linsley JW , Dowling JJ , Hauser MA , McDonald KK , Ashley-Koch A , et al. Stac3 is a component of theexcitation-contraction coupling machinery and mutated in NativeAmerican myopathy. Nat Commun. (2013) ;4: :1952. |
[7] | Ohno K , Hutchinson DO , Milone M , Brengman JM , Bouzat C , Sine SM , et al. Congenital myasthenic syndrome caused by prolongedacetylcholine receptor channel openings due to a mutation in theM2 domain of the epsilon subunit. Proc Natl Acad Sci USA. (1995) ;92: (3):758–62. |
[8] | Abicht A , Dusl M , Gallenmuller C , Guergueltcheva V , Schara U , Della Marina A , et al. Congenital myasthenic syndromes:Achievements and limitations of phenotype-guided gene-after-genesequencing in diagnostic practice: A study of 680 patients. Hum Mutat. (2012) ;33: (10):1474–84. |
[9] | Beeson D . Congenital myasthenic syndromes: Recent advances. Current opinion in neurology. (2016) ;29: (5):565–71. |
[10] | Engel AG , Shen XM , Selcen D , Sine SM . Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. The Lancet Neurology. (2015) ;14: (5):461. |
[11] | McMacken G , Abicht A , Evangelista T , Spendiff S , Lochmuller H . The Increasing Genetic and Phenotypical Diversity of Congenital Myasthenic Syndromes. Neuropediatrics. (2017) ;48: (4):294–308. |
[12] | Slater CR . Reliability of neuromuscular transmission and how it is maintained. Handb Clin Neurol. (2008) ;91: :27–101. |
[13] | Whittaker RG , Herrmann DN , Bansagi B , Hasan BA , Lofra RM , Logigian EL , et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology. (2015) ;85: (22):1964–71. |
[14] | Shen XM , Selcen D , Brengman J , Engel AG . Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. (2014) ;83: (24):2247–55. |
[15] | Chaouch A , Muller JS , Guergueltcheva V , Dusl M , Schara U , Rakocevic-Stojanovic V , et al. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J Neurol. (2012) ;259: (3):474–81. |
[16] | Barwick KE , Wright J , Al-Turki S , McEntagart MM , Nair A , Chioza B , et al. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am J Hum Genet. (2012) ;91: (6):1103–7. |
[17] | Bauche S , O’Regan S , Azuma Y , Laffargue F , McMacken G , Sternberg D , et al. Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea. Am J Hum Genet. (2016) ;99: (3):753–61. |
[18] | Aran A , Segel R , Kaneshige K , Gulsuner S , Renbaum P , Oliphant S , et al. Vesicular acetylcholine transporter defect underlies devastating congenital myasthenia syndrome. Neurology. (2017) ;88: (11):1021–8. |
[19] | O’Grady GL , Verschuuren C , Yuen M , Webster R , Menezes M , Fock JM , et al. Variants in SLC18A3, vesicular acetylcholinetransporter, cause congenital myasthenic syndrome. Neurology. (2016) ;87: (14):1442–8. |
[20] | Changeux J , Corringer P , Maskos U . The Nicotinic Acetylcholine Receptor: From Molecular Biology to Cognition. Neuropharmacology. (2015) ;96: (B):135–6. |
[21] | Haji A , Takeda R , Okazaki M . Neuropharmacology of control of respiratory rhythm and pattern in mature mammals. Pharmacol Ther. (2000) ;86: (3):277–304. |
[22] | Shao XM , Feldman JL . Cholinergic neurotransmission in the preBotzinger Complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience. (2005) ;130: (4):1069–81. |
[23] | Herrmann DN , Horvath R , Sowden JE , Gonzalez M , Sanchez-Mejias A , Guan Z , et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet. (2014) ;95: (3):332–9. |
[24] | Engel AG , Selcen D , Shen XM , Milone M , Harper CM . Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet. (2016) ;2: (5):e105. |
[25] | Lipstein N , Verhoeven-Duif NM , Michelassi FE , Calloway N , van Hasselt PM , Pienkowska K , et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. The Journal of Clinical Investigation. (2017) ;127: (3):1005–18. |
[26] | Maselli RA , Arredondo J , Vazquez J , Chong JX , Bamshad MJ , Nickerson DA , et al. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. American Journal of Medical Genetics Part A. (2017) ;173: (8):2240–5. |
[27] | Carss KJ , Stevens E , Foley AR , Cirak S , Riemersma M , Torelli S , et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am J Hum Genet. (2013) ;93: (1):29–41. |
[28] | Belaya K , Rodriguez Cruz PM , Liu WW , Maxwell S , McGowan S , Farrugia ME , et al. Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain. (2015) ;138: (Pt 9):2493–504. |
[29] | Luo S , Cai S , Maxwell S , Yue D , Zhu W , Qiao K , et al. Novel mutations in the C-terminal region of GMPPB causing limb-girdle muscular dystrophy overlapping with congenital myasthenic syndrome. Neuromuscul Disord. (2017) ;27: (6):557–64. |
[30] | Montagnese F , Klupp E , Karampinos DC , Biskup S , Glaser D , Kirschke JS , et al. Two patients with GMPPB mutation: The overlapping phenotypes of limb-girdle myasthenic syndrome and limb-girdle muscular dystrophy dystroglycanopathy. Muscle Nerve. (2017) ;56: (2):334–40. |
[31] | Rodriguez Cruz PM , Belaya K , Basiri K , Sedghi M , Farrugia ME , Holton JL , et al. Clinical features of the myasthenic syndrome arising from mutations in GMPPB. J Neurol Neurosurg Psychiatry. (2016) ;87: (8):802–9. |
[32] | Taniguchi M , Kurahashi H , Noguchi S , Fukudome T , Okinaga T , Tsukahara T , et al. Aberrant neuromuscular junctions and delayedterminal muscle fiber maturation inalpha-dystroglycanopathies. Hum Mol Genet. (2006) ;15: (8):1279–89. |
[33] | Bauche S , Vellieux G , Sternberg D , Fontenille MJ , De Bruyckere E , Davoine CS , et al. Mutations in GFPT1-related congenital myasthenic syndromes are associated with synaptic morphological defects and underlie a tubular aggregate myopathy with synaptopathy. J Neurol. (2017) ;264: (8):1791–803. |
[34] | Wu H , Xiong WC , Mei L . To build a synapse: Signaling pathways in neuromuscular junction assembly. Development. (2010) ;137: (7):1017–33. |
[35] | Rodriguez Cruz PM , Palace J , Beeson D . Congenital myasthenic syndromes and the neuromuscular junction. Current Opinion in Neurology. (2014) ;27: (5):566–75. |
[36] | Claeys KG , Maisonobe T , Bohm J , Laporte J , Hezode M , Romero NB , et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. (2010) ;74: (6):519–21. |
[37] | Gibbs EM , Clarke NF , Rose K , Oates EC , Webster R , Feldman EL , et al. Neuromuscular junction abnormalities in DNM2-related centronuclear myopathy. J Mol Med (Berl). (2013) ;91: (6):727–37. |
[38] | Liewluck T , Shen XM , Milone M , Engel AG . Endplate structure andparameters of neuromuscular transmission in sporadic centronuclearmyopathy associated with myasthenia. Neuromuscul Disord. (2011) ;21: (6):387–95. |
[39] | Munot P , Lashley D , Jungbluth H , Feng L , Pitt M , Robb SA , et al. Congenital fibre type disproportion associated with mutations in the tropomyosin 3 (TPM3) gene mimicking congenital myasthenia. Neuromuscul Disord. (2010) ;20: (12):796–800. |
[40] | Illingworth MA , Main M , Pitt M , Feng L , Sewry CA , Gunny R , et al. RYR1-related congenital myopathy with fatigable weakness, responding to pyridostigimine. Neuromuscul Disord. (2014) ;24: (8):707–12. |
[41] | Stalberg E , Trontelj JV . The study of normal and abnormal neuromuscular transmission with single fibre electromyography. J Neurosci Methods. (1997) ;74: (2):145–54. |
[42] | Plomp JJ , Morsch M , Phillips WD , Verschuuren JJ . Electrophysiological analysis of neuromuscular synaptic function in myasthenia gravis patients and animal models. Experimental Neurology. (2015) ;270: :41–54. |
[43] | Slater CR , Lyons PR , Walls TJ , Fawcett PR , Young C . Structure and function of neuromuscular junctions in the vastus lateralis of man. A motor point biopsy study of two groups of patients. Brain. (1992) ;115: (Pt 2):451–78. |
[44] | Engel AG . Congenital myasthenic syndromes. Handb Clin Neurol. (2008) ;91: :285–331. |
[45] | Slater CR . The functional organization of motor nerve terminals. Progress in Neurobiology. (2015) ;134: :55–103. |
[46] | Wood SJ , Slater CR . Safety factor at the neuromuscular junction. Progress in Neurobiology. (2001) ;64: (4):393–429. |
[47] | Zucker RS , Regehr WG . Short-term synaptic plasticity. Annu Rev Physiol. (2002) ;64: :355–405. |
[48] | Magleby KL . Facilitation, augmentation, and potentiation of transmitter release. Progress in Brain Research. (1979) ;49: :175–82. |
[49] | Dodge FA , Rahamimoff R . Co-operative action a calcium ions in transmitter release at the neuromuscular junction. Journal of Physiology. (1967) ;193: (2):419–32. |
[50] | Ruiz R , Cano R , Casanas JJ , Gaffield MA , Betz WJ , Tabares L . Active zones and the readily releasable pool of synaptic vesicles at the neuromuscular junction of the mouse. Journal of Neuroscience. (2011) ;31: (6):2000–8. |
[51] | Tarr TB , Wipf P , Meriney SD . Synaptic Pathophysiology and Treatment of Lambert-Eaton Myasthenic Syndrome. Molecular Neurobiology. (2015) ;52: (1):456–63. |
[52] | Engel AG , Nagel A , Fukuoka T , Fukunaga H , Osame M , Lang B , et al. Motor nerve terminal calcium channels in Lambert-Eaton myasthenic syndrome. Morphologic evidence for depletion and that the depletion is mediated by autoantibodies. Annals of the New York Academy of Sciences. (1989) ;560: :278–90. |
[53] | Huze C , Bauche S , Richard P , Chevessier F , Goillot E , Gaudon K , et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. (2009) ;85: (2):155–67. |
[54] | Maselli RA , Fernandez JM , Arredondo J , Navarro C , Ngo M , Beeson D , et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Human Genetics. (2012) ;131: (7):1123–35. |
[55] | Yumoto N , Kim N , Burden SJ . Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. (2012) ;489: (7416):438–42. |
[56] | Wu H , Lu Y , Shen C , Patel N , Gan L , Xiong WC , et al. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. (2012) ;75: (1):94–107. |
[57] | Plomp JJ . Trans-synaptic homeostasis at the myasthenic neuromuscular junction. Front Biosci (Landmark Ed). (2017) ;22: :1033–51. |
[58] | Takamori M . Synaptic Homeostasis and Its Immunological Disturbance in Neuromuscular Junction Disorders. Int J Mol Sci. (2017) :18: (4). |
[59] | Maddison P , Newsom-Davis J , Mills KR . Decay of postexerciseaugmentation in the Lambert-Eaton myasthenic syndrome: Effect ofcooling. Neurology. (1998) ;50: (4):1083–7. |
[60] | Maddison P , Newsom-Davis J , Mills KR . Effect of3,4-diaminopyridine on the time course of decay of compound muscleaction potential augmentation in the Lambert-Eaton myasthenicsyndrome. Muscle Nerve. (1998) ;21: (9):1196–8. |
[61] | Hesselmans LF , Jennekens FG , Van den Oord CJ , Veldman H , Vincent A . Development of innervation of skeletal muscle fibers in man: Relation to acetylcholine receptors. Anat Rec. (1993) ;236: (3):553–62. |
[62] | Couteaux R , Taxi J . Recherches histochimiques sur la distributiondes activités cholinestérasiques au niveau de la synapsemyoneurale. Arch Anat Micr Morphol Ex. (1952) ;41: :352–92. |
[63] | Engel AG , Lambert EH , Gomez MR . A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol. (1977) ;1: (4):315–30. |
[64] | Darabid H , Perez-Gonzalez AP , Robitaille R . Neuromuscularsynaptogenesis: Coordinating partners with multiple functions. NatRev Neurosci. (2014) ;15: (11):703–18. |
[65] | Petrov KA , Girard E , Nikitashina AD , Colasante C , Bernard V , Nurullin L , et al. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by alpha7 nicotinic receptors and butyrylcholinesterase. J Neurosci. (2014) ;34: (36):11870–83. |
[66] | Milone M , Wang HL , Ohno K , Prince R , Fukudome T , Shen XM , et al. Mode switching kinetics produced by a naturally occurring mutation in the cytoplasmic loop of the human acetylcholine receptor epsilon subunit. Neuron. (1998) ;20: (3):575–88. |
[67] | Ohno K , Quiram PA , Milone M , Wang HL , Harper MC , Pruitt JN 2nd , et al. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: Identification and functional characterization of six new mutations. Hum Mol Genet. (1997) ;6: (5):753–66. |
[68] | Shen XM , Ohno K , Sine SM , Engel AG . Subunit-specific contributionto agonist binding and channel gating revealed by inheritedmutation in muscle acetylcholine receptor M3-M4 linker. Brain. (2005) ;128: (Pt 2):345–55. |
[69] | Quiram PA , Ohno K , Milone M , Patterson MC , Pruitt NJ , Brengman JM , et al. Mutation causing congenital myasthenia reveals acetylcholine receptor beta/delta subunit interaction essential for assembly. The Journal of Clinical Investigation. (1999) ;104: (10):1403–10. |
[70] | Shen XM , Ohno K , Fukudome T , Tsujino A , Brengman JM , De Vivo DC , et al. Congenital myasthenic syndrome caused by low-expressor fast-channel AChR delta subunit mutation. Neurology. (2002) ;59: (12):1881–8. |
[71] | Shen XM , Fukuda T , Ohno K , Sine SM , Engel AG . Congenital myasthenia-related AChR delta subunit mutation interferes with intersubunit communication essential for channel gating. The Journal of Clinical Investigation. (2008) ;118: (5):1867–76. |
[72] | Ben Ammar A , Petit F , Alexandri N , Gaudon K , Bauche S , Rouche A , et al. Phenotype genotype analysis in 15 patients presenting a congenital myasthenic syndrome due to mutations in DOK7. J Neurol. (2010) ;257: (5):754–66. |
[73] | Slater CR , Fawcett PR , Walls TJ , Lyons PR , Bailey SJ , Beeson D , et al. Pre- and post-synaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. (2006) ;129: (Pt 8):2061–76. |
[74] | Boycott KM , Rath A , Chong JX , Hartley T , Alkuraya FS , Baynam G , et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am J Hum Genet. (2017) ;100: (5):695–705. |
[75] | Marx V . The DNA of a nation. Nature. (2015) ;524: (7566):503–5. |
[76] | Philippakis AA , Azzariti DR , Beltran S , Brookes AJ , Brownstein CA , Brudno M , et al. The Matchmaker Exchange: A platform for raredisease gene discovery. Hum Mutat. (2015) ;36: (10):915–21. |
[77] | Thompson R , Johnston L , Taruscio D , Monaco L , Beroud C , Gut IG , et al. RD-Connect: An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. J Gen Intern Med. (2014) ;29: (Suppl 3):S780–7. |
[78] | Schara U, Della Marina A, Abicht A. Congenital myasthenic syndromes: Current diagnosis and therapeutic approaches. Neuropediatrics. (2012) ;43: (4):184–93. |
[79] | Kirsch GE , Narahashi T . 3, 4-diaminopyridine. A potent new potassium channel blocker. Biophys J. (1978) ;22: (3):507–12. |
[80] | Engel AG . The therapy of congenital myasthenic syndromes. Neurotherapeutics. (2007) ;4: (2):252–7. |
[81] | Lashley D , Palace J , Jayawant S , Robb S , Beeson D . Ephedrinetreatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology. (2010) ;74: (19):1517–23. |
[82] | Burke G , Hiscock A , Klein A , Niks EH , Main M , Manzur AY , et al. Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul Disord. (2013) ;23: (2):170–5. |
[83] | Lorenzoni PJ , Scola RH , Kay CS , Filla L , Miranda AP , Pinheiro JM , et al. Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J Neurol Sci. (2013) ;331: (1-2):155–7. |
[84] | Krnjevic K , Miledi R . Adrenaline and failure of neuromuscular transmission. Nature. (1957) ;180: (4590):814–5. |
[85] | Sieb JP , Engel AG . Ephedrine: Effects on neuromuscular transmission. Brain Research. (1993) ;623: (1):167–71. |
[86] | Milone M , Engel AG . Block of the endplate acetylcholine receptor channel by the sympathomimetic agents ephedrine, pseudoephedrine, and albuterol. Brain Research. (1996) ;740: (1-2):346–52. |
[87] | Khan MM , Lustrino D , Silveira WA , Wild F , Straka T , Issop Y , et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc Natl Acad Sci U S A. (2016) ;113: (3):746–50. |


