
The New Zealand Neuromuscular Disease Patient Registry; Five Years and a Thousand Patients
Abstract
The New Zealand Neuromuscular Disease Patient Registry has been recruiting for five years. Its primary aim is to enable people with neuromuscular disease to participate in research including clinical trials. It has contributed data to large anonymised cohort studies and many feasibility studies, and has provided practical information and advice to researchers wanting to work with people with neuromuscular conditions. 1019 people have enrolled since the Registry’s launch in August 2011 with over 70 different diagnoses. Of these; 8 patients have been involved in clinical trials, 134 in other disease-specific research and 757 have contributed anonymised data to cohort studies. As a result the Registry is now effectively facilitating almost all neuromuscular research currently taking place in New Zealand.
INTRODUCTION
The New Zealand Neuromuscular Disease Patient Registry (NZNMD Registry) is a nation-wide registry for people living in New Zealand (NZ) diagnosed with any disorder supported by the Muscular Dystrophy Association of New Zealand (MDANZ). Because NZ has a relatively small population of approximately 4 250 000 as determined by 2013 NZ population census data [1] MDANZ has become the patient support organisation of choice for people diagnosed with myopathies and neuropathies as well as disorders less commonly associated with neuromuscular patient support organisations; inherited ataxias, hereditary spastic parapareses, leucodystrophies and neurocutaneous disorders. Patients diagnosed clinically with or without molecular confirmation and those who test positive predictively are included. MDANZ is the primary sponsor of the Registry and actively promotes its work.
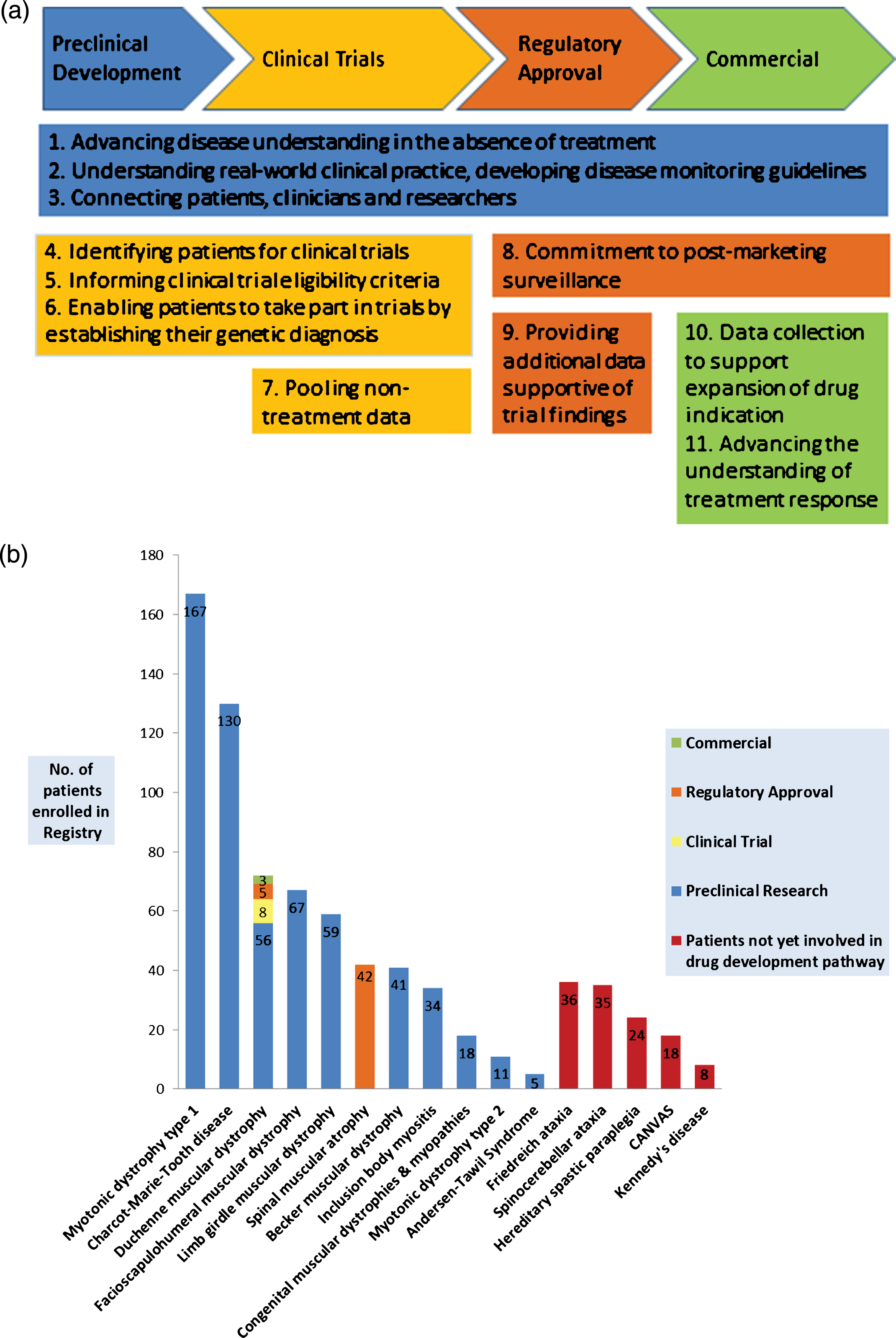
The primary aim of the Registry is to facilitate neuromuscular research by lowering the barriers faced by researchers in finding study participants, and faced by patients in finding studies to take part in [2]. The nature of rare disease research, and therefore the role of registries, evolves as effective treatments are developed. At the time of the Registry’s establishment there were very few treatments for these conditions and so the first way of achieving this aim was to contribute data to international natural history studies [3–7], mainly facilitated by the TREAT-NMD Neuromuscular Network of which the Registry is a member. More recently patients have been enrolled in preclinical research and even in clinical trials and we are considering the Registry taking up roles in post-marketing surveillance. This process closely follows the model of registry development proposed by Betsy Bogard (personal communication - see acknowledgements) (Fig. 1a).
A developing aim of the Registry is to obtain molecular confirmation of diagnoses. The Registry facilitates this in two ways; either by highlighting a clinical genetic test to the patient’s clinician or, for patients who remain without molecular confirmation of their diagnosis despite clinical genetic testing, by identifying appropriate research studies concerned with finding the genetic basis of their disorder.
Fig.1
The changing roles of a rare disease patient registry. (a) Ten roles performed by registries along the pathway of treatment development – adapted with permission from Betsy Bogard (see acknowledgements*). (b) The NZ NMD Registry’s current role in the pathway of treatment development for its respective patient populations.
METHODS
To achieve these aims the NZNMD Registry operates as a longitudinal opt-in registry for both children and adults living with neuromuscular disease in NZ. Led by a principal investigator, who is a consultant neurologist with the Auckland District Health Board, and managed by a genetic counsellor who is the Registry Curator, demographic, pre-specified, disease-specific clinical and genetic information is collected, curated and regularly updated. More recently patient self-reported data has been incorporated for some disorders. Data are stored with the same degree of security as clinical data within the hospital firewall. Some data for specific disorders; Duchenne muscular dystrophy (DMD), spinal muscular atrophy (SMA), myotonic dystrophy, facioscapulohumeral muscular dystrophy (FSHD), and Charcot-Marie-Tooth disease (CMT) are housed on secure platforms provided by overseas collaborators [2, 7]. The NZNMD Registry is an ongoing research project. The Registry is physically located within the neurology department at ADHB, the largest neurology centre in NZ. The Registry has an oversight committee made up of individuals living with neuromuscular disorders, clinical scientists, adult and paediatric neurologists, geneticists and representation from the sponsor, MDANZ.
The NZNMD Registry has a simple referral process. Referrals can come from any source including self-referral, doctor or patient support organisations such as MDANZ, and require, at a minimum, the NZ national health unique patient identifier (NHI). The MDANZ has tasked its fieldworkers with informing and consenting patients to the Registry. Clinicians are not involved in collecting or entering data though their assistance may be required for obtaining genetic tests or clinical records to support the patient’s diagnosis.
Formal written consent is obtained from all participants before joining the Registry using a short information sheet and consent form, in keeping with the low risks associated with being on the Registry. The Registry receives ethics approval annually.
Researchers, from both within NZ and internationally, contact the Registry Curator to request access to the Registry’s services. Enquiries to the Registry take different forms. They may be from industry conducting feasibility studies for clinical trials, or from clinicians or academics seeking data for service planning or for natural history studies. In these cases, the relevant population is identified from the database and anonymised data is disseminated in aggregate form to researchers. For studies where researchers are using the Registry to recruit participants, those who are likely to be eligible are identified by the curator and information about the study is disseminated through a variety of means including email, telephone, post and advertisement.
Academic research is facilitated free of charge, except for the recovery of direct costs associated with the dissemination of information, not including the time of the Registry Curator. Industry-led research is facilitated along with a request for costs to be met including a contribution towards overheads and the time of the Registry Curator.
The NZNMD Registry collects ethnicity data and, in this way, it is able to support and facilitate research involving people with neuromuscular disease from minority ethnic groups. Māori are the indigenous people of NZ and their rights to self-determination are confirmed in the Treaty of Waitangi. Māori have specific values regarding (but not limited to) collection, processing and disposal of DNA. The Registry has policies in place to act in a culturally responsive manner in these regards.
Participants’ information and confidentiality is highly respected and protected. Their time is also valued and requests for participation in studies for which the participant are ineligible are avoided. Researchers contacting the registry are provided with rapid response times for information requests, which are approved through the oversight committee.
RESULTS
The NZNMD Registry has enrolled 1019 people with neuromuscular disorders over the past five years with over 70 different diagnoses, which is estimated to represent around one quarter of all people living in NZ with a neuromuscular disorder [8]. The average age of patients was 44 years with 150 under the age of 16. The commonest four diagnoses were myotonic dystrophy type 1 (168), CMT 136, dystrophinopathies (122), FHSD (70) which together comprised 49% of the enrolment to date. For disorders such as DMD the proportion of the population enrolled is close to 100% and for others, such as myotonic dystrophy, where the interest from researchers has been lower, the proportion of the population enrolled is around 50% (unpublished data, Alice Theadom on behalf of the MD Prev, New Zealand National Prevalence of Genetic Muscle Disease, study group). 58% of people enrolled in the Registry have a molecular diagnosis, where applicable. 34 registry participants have died. The oldest participants (3) are 92 years old and the youngest less than one year old; 452 (44%) are female (Table 1).
Table 1
Characteristics of NZ NMD registry patients
| Neuromuscular Disorder | Total no. | % Molecular | % Molecular | Mean Age | % Female |
| of patients | Diagnosis* | Diagnosis* | |||
| Sept. 2013 [13] | Mar. 2017 | ||||
| Myopathies | |||||
| Myotonic dystrophy type 1 | 168 | 56 | 80 | 45.0 (1–84) | 55 |
| Myotonic dystrophy type 2 | 11 | 14 | 100 | 59.7 (35–71) | 55 |
| Duchenne muscular dystrophy | 73 | 73 | 93 | 14.7 (3–39) | 0 |
| Becker muscular dystrophy | 42 | 68 | 74 | 37.1 (6–71) | 0 |
| Manifesting carrier of dystrophinopathy | 7 | not specified | 86 | 43.7 (16–79) | 100 |
| Facioscapulohumeral muscular dystrophy | 70 | 50 | 66 | 49.6 (19–81) | 40 |
| Limb girdle muscular dystrophy | 58 | 10 | 28 | 48.9 (10–79) | 50 |
| Emery-Dreifuss muscular dystrophy | 8 | not specified | 63 | 33.0 (17–49) | 25 |
| Ion channel disorders (myotonia congenita, | 17 | not specified | 82 | 35.6 (3–81) | 18 |
| periodic paralysis, ATS) | |||||
| Congenital muscular dystrophies &myopathies | 32 | not specified | 39 | 26.0 (3–76) | 53 |
| Mitochondrial myopathies | 17 | not specified | 53 | 51.7 (31–M) | 53 |
| Inclusion body myositis | 34 | not applicable | not applicable | 57.3 (43–92) | 56 |
| Neuropathies | |||||
| Charcot-Marie-Tooth disease | 136 | 31 | 40 | 51.4 (6–89) | 54 |
| Hereditary Sensory &Autonomic Neuropathy | 3 | not specified | 100 | 41.0 (21–65) | 67 |
| Anterior Horn cell Disease | |||||
| Spinal muscular atrophy | 42 | 73 | 88 | 23.1 (4–81) | 48 |
| Kennedy’s disease | 8 | not specified | 100 | 54.9 (29–71) | 0 |
| Ataxias | |||||
| Spinocerebellar ataxia | 50 | 83 | 65 | 62.8 (37–83) | 35 |
| Friedreich ataxia | 36 | 50 | 64 | 39.9 (16–67) | 47 |
| Cerebellar ataxia, neuropathy &vestibular | 19 | not applicable | 0 | 71.2 (53–92) | 67 |
| areflexia syndrome (CANVAS) | |||||
| Hereditary spastic paraplegia | |||||
| Hereditary spastic paraplegia | 25 | not specified | 36 | 38.0 (8–89) | 40 |
| Neurocutaneous disorders | |||||
| Neurofibromatosis type 1 | 16 | not specified | 50 | 30.8 (3–73) | 50 |
| Tuberous Sclerosis | 4 | not specified | 75 | 34 (20–44) | 75 |
| Other neuromuscular disorders | |||||
| Other neuromuscular disorders e.g | 34 | not applicable | not applicable | 63 (15–85) | 70 |
| myasthenia gravis, polymyositis | |||||
| Other genetic neuromuscular disorders | 109 | not specified | 9 | 40.7 (4–71) | 31 |
| e.g Pompe disease | |||||
| Overall | 1019 | 51% | 58% | 44.0 (1 to 92) | 44 |
Diagnosis, demographics and diagnosis rates compared with our earlier publication in 2013 [13]. “not specified” means that these diagnoses were not separated out in the graphs in our previous publication.
Referral sources of patients to the registry include: MDANZ (181 self-referrals and 453 consented through fieldworkers); the Principal Investigator, who runs neurogenetic clinics (207); other neurologists (80); Genetic Health Service of New Zealand (47); paediatric neurologists (27); prevalence studies (18); GPs (2); National Metabolic Service (1); and General Paediatricians (3).
The Registry has contributed over 300 records containing de-identified patient data to global databases for dystrophinopathies (135) [3, 4, 5], spinal muscular atrophy (SMA) (42) [6] and Charcot-Marie-Tooth disease (CMT) (129) [7]. It also acts as a conduit for recruitment for patients to self-report and/or clinicians to report to global registries for ultra-rare neuromuscular disorders, for example, GNE myopathy (2) [9, 10], myotubular myopathy (2) [11], and FKRP gene disorders (3) [12] (Fig. 1b).
The Registry has contributed data to 23 feasibility studies of which 17 were coordinated through the TREAT NMD Neuromuscular Network. It has also disseminated information and assisted in recruitment for nine disease-specific studies [13–16] including the landmark ocular coherence tomography study that first discovered the presence of epiretinal membranes as a feature of myotonic dystrophy [13] and has had an active role in patient recruitment for 2 clinical trials for treating DMD with a further 2 planned. It has played a vital role in large prevalence studies of genetic muscle disorders and CMT.
The Registry has assisted in recruiting 28 patients with limb girdle muscular dystrophy (LGMD) [17] and 12 patients with CMT, all without molecular diagnoses, into whole exome sequencing studies that aim to find the genetic cause of their condition. Molecular diagnoses have been obtained in 19 patients with significant changes in treatment and management indicated in 6, for example the diagnosis in one case of congenital myasthenic syndrome treatable with salbutamol [18]. Vitally important genetic counselling implications for family members have been identified in several patients.
DISCUSSION
We have demonstrated that an overarching registry serving all neuromuscular diseases managed by a single project team is effective; this is in contrast to countries such as the UK, Spain and Germany where disease-specific registries are commonly deployed but similar to Canada where the Canadian Neuromuscular Disease Registry (CNDR) covers a range of disorders [19]. Population size, health-system structure, clinician interest and make-up of patient support organisations are all factors that can influence registry design in this regard.
A registry’s role is dynamic and should be responsive to the changing needs of its stakeholders; patients, researchers and clinicians as illustrated in the Bogard model of registry development (Fig. 1a). In five years the NZNMD Registry has evolved from carrying out roles important during preclinical drug development, such as Advancing disease understanding in the absence of treatment and Connecting patients with researchers, to performing vital work in the clinical trial arena by Identifying patients for clinical trials, and Informing study eligibility criteria. For DMD and SMA we are now considering the role of Post-marketing surveillance. The role of Providing additional data supportive of trial findings has been performed for a new SMA treatment and, as new drugs for both DMD and SMA enter the commercial market, we anticipate the later roles of Data collection to support expansion of drug indication and Advancing the understanding of treatment response becoming important to the Registry (See Fig. 1b).
Only 58% of people enrolled in the Registry have a molecular diagnosis (see Table 1) which is a 7% improvement compared with three years ago [20]. However, as lack of molecular diagnosis limits access to treatment, genetic counselling and research, a goal of the Registry is to gain sufficient funding so that it can facilitate access to genetic testing for patients who are not able to access testing through the usual pathways. This would be similar to other registries including Duchenne Connect [21], the Jain Foundation Dysferlin Registry [22] and the Myotubular Myopathy Registry [11].
The Registry has changed the face of neuromuscular research in NZ. The two most important factors in achieving this are the integral involvement of the patient support organisation and the minimal dependence upon clinicians, which can only occur with dedicated staff. Prior to the NZNMD Registry a person with a neuromuscular condition living in NZ could only register in an overseas registry and were very unlikely to be offered the opportunity to participate in research of any sort. Now hundreds of New Zealanders have been involved in research and the vast majority of neuromuscular research in NZ is now facilitated by the Registry.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The New Zealand Neuromuscular Disease Registry is fully funded by the research trust of MDANZ, Neuromuscular Research New Zealand.
*Betsy Bogard is the Director of Global Research Development of the International Fibrodysplasia Ossificans Progressiva Association (IFOPA). She devised her Registry model while employed by Fulcrum Therapeutics. She has kindly given permission for this to be used in this paper but there is no other relationship – real or implied, between either Fulcrum Therapeutics or IFOPA and the New Zealand Neuromuscular Disease Registry.
REFERENCES
[1] | 2013 New Zealand Census. [Internet] 2017 [Accessed 6 March 2017]. Available from: http://www.stats.govt.nz/Census/-census.aspx |
[2] | Rodrigues M , Hammond-Tooke G , Kidd A , Love D , Patel R , Dawkins H , Bellgard M , Roxburgh R . The New Zealand neuromuscular disease registry. J Clin Neurosci. (2012) ;19: (12):1749–50. doi: 10.1016/j.jocn.2012.04.008 |
[3] | Bladen, et al. The TREAT-NMD Duchenne muscular dystrophy registries: conception, design, and utilization by industry and academia. Hum Mutat. (2013) ;34: (11):1449–57. doi: 10.1002/humu.22390 |
[4] | Bladen C , Koeks, et al. Clinical outcomes in Duchenne Muscular Dystrophy: a study of 5345 patients from the TREAT-NMD DMD global database (Submitted to Orphanet Journal of Rare Diseases February 2017). |
[5] | Bladen C , Salgado D , et al. Analysis of more than 7000 Duchenne Muscular Dystrophy mutations. Hum Mutat. (2015) ;36: (4):395–402. doi: 10.1002/humu.22758 |
[6] | Bladen C , et al. Mapping the differences in care for 5,000 Spinal Muscular Atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. Neurol. (2014) ;261: (1):152–63. doi: 10.1007/s00415-013-7154-1. |
[7] | Fridman V , et al. On behalf of the Inherited Neuropathies Consortium. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. (2015) ;86: (8):873–8. doi: 10.1136/jnnp-2014-308826 |
[8] | Deenen JCW , Horlings CGC , Verschurren JGM , Verbeek ALM , van Engelen BGM . The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. Journal of Neuromuscular Disorders. (2015) ;2: (1)73–85. |
[9] | Pogoryelova O , The International GNE Registry [Internet] 13 Dec 2016 [Accessed 6 March 2017] Available from: http://www.treat-nmd.eu/gne/patient-registries/international-registry/ |
[10] | Ultragenyx, The GNEM Disease Monitoring Program [Internet] 2017 [Accessed 6 March 2017] Available from: https://www.gnem-dmcom/ |
[11] | Bullivant J . The Myotubular and Centronuclear Myopathy Patient Registry [Internet] 2017 [Accessed 6 March 2017] Available from http://mtmcnmregistry.org/ |
[12] | Robertson A . The Global FKRP Registry [Internet] 2016 [Accessed 6 March 2017] Available from: https://www.fkrp-registry.org/ |
[13] | Kersten HM , Roxburgh RH , Child N , Polkinghorne PJ , Frampton C , Danesh-Meyer HV . Epiretinal membrane: a treatable cause of visual disability in myotonic dystrophy type 1. J Neurol. (2014) ;261: (1):37–44. doi: 10.1007/s00415-013-7141-6. Epub 2013 Oct 17. |
[14] | Kersten HM , Roxburgh RH , Danesh-Meyer HV . Ophthalmic manifestations of inherited neurodegenerative disorders. Nat Rev Neurol. (2014) ;10: (6):349–62. doi: 10.1038/nrneurol.2014.79. Epub 2014 May 20. |
[15] | Kersten HM , Danesh-Meyer HV , Roxburgh RH . Ophthalmic findings in myotonic dystrophy type a case series. J Neurol. (2016) ;263: (12):2552–2554. Epub 2016 Nov 22. |
[16] | Teoh LJ , Geelhoed EA , Bayley K , Leonard H , Laing NG . Health care utilization and costs for children and adults with duchenne muscular dystrophy. Muscle Nerve. (2016) ;53: (6):877–84. doi: 10.1002/mus.24965. Epub 2016 May 9. |
[17] | Ghaoui R , Cooper ST , Lek M , Jones K , Corbett A , Reddel SW , Needham M , Liang C , Waddell LB , Nicholson G , O’Grady G , Kaur S , Ong R , Davis M , Sue CM , Laing NG , North KN , MacArthur DG , Clarke NF . Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: Outcomes and lessons learned. JAMA Neurol. (2015) ;72: (12):1424–32. doi: 10.1001/jamaneurol.2015.2274. PMID: 26436962. |
[18] | Mulroy E , Ghaoui R , Hutchinson D , Rodrigues M , Lek M , MacArthur DG , Cooper ST , Clarke NF , Roxburgh R . A “Limb Girdle Muscular Dystrophy” responsive to asthma therapy. Practical Neurology In Press, (2017) . |
[19] | Korngut L , Campbell C , Johnston M , Benstead T , Genge A , Mackenzie A , McCormick A , Biggar D , Bourque P , Briemberg H , O’Connell C , Dojeiji S , Dooley J , Grant I , Hogan G , Johnston W , Kalra S , Katzberg HD , Mah JK , McAdam L , McMillan HJ , Melanson M , Selby K , Shoesmith C , Smith G , Venance SL , Wee J , CNDR Investigator Network. The CNDR: collaborating to translate new therapies for Canadians. Can J Neurol Sci. (2013) ;40: (5)698–704. |
[20] | Rodrigues M , Kidd A , Love D , Roxburgh R . New Zealand Neuromuscular Disease Registry; A review of genetic data. J Clin Neuroscience. (2015) ;22: (2):434–6. doi: 10.1016/j.jocn.2014.06.096. Epub 2014 Oct 19. |
[21] | Wang RT , Nelson SF . What can Duchenne Connect teach us about treating Duchenne muscular dystrophy? Curr Opin Neurol. (2015) ;28: (5):535–41. doi: 10.1097/WCO.0000000000000245. Review. PMID: 26356412. |
[22] | The Jain Foundation, The International Dysferlin Registry [Internet] 2016 [Accessed 6 March 2017] Available from: http://dysferlinregistry.org/ |


