
Functional Characterization of C-terminal Ryanodine Receptor 1 Variants Associated with Central Core Disease or Malignant Hyperthermia
Abstract
Background: Central core disease and malignant hyperthermia are human disorders of skeletal muscle resulting from aberrant Ca2+ handling. Most malignant hyperthermia and central core disease cases are associated with amino acid changes in the type 1 ryanodine receptor (RyR1), the skeletal muscle Ca2+-release channel. Malignant hyperthermia exhibits a gain-of-function phenotype, and central core disease results from loss of channel function. For a variant to be classified as pathogenic, functional studies must demonstrate a correlation with the pathophysiology of malignant hyperthermia or central core disease.
Objective: We assessed the pathogenicity of four C-terminal variants of the ryanodine receptor using functional analysis. The variants were identified in families affected by either malignant hyperthermia or central core disease.
Methods: Four variants were introduced separately into human cDNA encoding the skeletal muscle ryanodine receptor. Following transient expression in HEK-293T cells, functional studies were carried out using calcium release assays in response to an agonist. Two previously characterized variants and wild-type skeletal muscle ryanodine receptor were used as controls.
Results: The p.Met4640Ile variant associated with central core disease showed no difference in calcium release compared to wild-type. The p.Val4849Ile variant associated with malignant hyperthermia was more sensitive to agonist than wild-type but did not reach statistical significance and two variants (p.Phe4857Ser and p.Asp4918Asn) associated with central core disease were completely inactive.
Conclusions: The p.Val4849Ile variant should be considered a risk factor for malignant hyperthermia, while the p.Phe4857Ser and p.Asp4918Asn variants should be classified as pathogenic for central core disease.
INTRODUCTION
Central core disease (CCD) is a slowly progressive or non-progressive myopathy with variable penetrance and a phenotype ranging from mild to very severe weakness, even within the same family [1, 2]. In some patients there are skeletal abnormalities and respiratory muscle weakness. CCD is diagnosed by suggestive clinical features and the presence, on histological examination of skeletal muscle fibres, of a central region or ‘core’ lacking mitochondrial and oxidative enzyme activity [3].
Central core disease is typically an autosomal dominant disorder linked to variants in the skeletal muscle ryanodine receptor gene (RYR1), although recessive inheritance has been reported [4, 5]. Most CCD-associated variants have been located in the C-terminal region of RyR1 containing the transmembrane pore through which Ca2+ is gated. Although variants associated with CCD have been found throughout the gene, patients with C-terminal variants in RyR1 have generally had more severe phenotypes compared with those with variants outside the C-terminal region [6].
Individuals with CCD are considered to be susceptible to malignant hyperthermia (MH) with RyR1 defects being the common causal factor [7]. MH is an autosomal dominant pharmacogenetic disorder that predisposes patients to a reaction triggered by exposure to depolarising muscle relaxants or potent volatile anesthetics [8]. Upon exposure of an MH-susceptible person to a triggering agent, there is a sustained increase of cytoplasmic calcium within skeletal muscle fibres, causing sustained muscle contraction. This creates a hypermetabolic state, whose other features include rapid elevation of body temperature, tachycardia, hyperkalaemia, acidosis, and elevated end-tidal carbon dioxide levels.
It has been estimated that 50–70% of all cases of MH are caused by altered RyR1 function [9]. The RyR1 protein is a homotetrameric Ca2+ channel embedded in the sarcoplasmic reticulum membrane. It is responsible for releasing Ca2+ from the sarcoplasmic reticulum stores to the cytosol to initiate muscle contraction [10]. Ca2+ release is initiated by physical contact between the RyR1 tetramer and the voltage-sensing dihydropyridine receptor (DHPR) located in the sarcolemma [11]. While many other proteins contact and regulate channel activity [12], the CACNA1S gene encoding the α1s subunit of the DHPR (accounting for less than 1% of MH-susceptible cases), STAC3 [13] and CASQ1 [14] are the only other genes identified thus far with variants that have been associated with MH-susceptibility.
While MH was originally thought to be a rare disorder occurring between 1 in 10,000 and 1 in 250,000 anesthetics [15, 16], recent estimates suggest that as many as 1 in 400 individuals may carry a pathogenic variant in RYR1 [17]. Variants that cause MH susceptibility are often classed as gain-of-function mutations as they result in an accelerated release of Ca2+ from the sarcoplasmic reticulum. In contrast, those that result in CCD are generally considered loss-of-function and result in EC-uncoupling [18]. In addition there are individuals in whom both CCD and MH-susceptibility co-exist. Variants that cause both MH and CCD have been suggested to result in an uncompensated Ca2+ leak from the sarcoplasmic reticulum and have been reported as being more severe than those that result in MH only [19]. RyR1 variants in the central region of the protein have recently been found to increase calcium induced calcium release [20], while those in the N-terminal region showed either a stabilization of the closed state or an increase in calcium induced calcium release depending on their position [19]. While RyR1 variants can be associated with either MH or CCD, and some with both disorders, there is to date no well-defined molecular mechanism to explain this apparent conundrum.
We have used the human RYR1 cDNA [21] transiently expressed in HEK-293T cells together with Fura-2 Ca2+ imaging and the RyR1 agonist 4-chloro-m-cresol (4-cmc), to functionally characterize C-terminal RyR1 variants associated with either MH or CCD. HEK-293T cells do not normally express RyR1 at detectable levels or in a functional form [22] and are therefore often used to investigate directly the impact that RYR1 substitutions have on the function of RyR1 as a Ca2+ channel [23–25].
PATIENTS, MATERIALS AND METHODS
Patients
Family A (variant c.14570T>C, p.Phe4857Ser; CCD)
Patient AII:1 (Fig. 1A) came to neurological attention in her very early years. She was floppy as a baby and had difficulties with feeding in the first days of life. She also had congenital dislocation of the hips. At 9 months she had scoliosis surgery and did not walk until she was four. At 9 years she required surgery to the left hip and at 12 years, neurological and electromyographic examinations suggested a myopathy. Her condition remained non-progressive and at the age of 48 she lives relatively independently. She manages mobility with a wheelchair but can transfer independently and is able to drive. Muscle biopsy using the right tibialis anterior showed typical central core formation and myofibrillar disorganisation with Z band fragmentation and accumulation of Z band material. She has had one general anesthetic in 1973, but the agents were not recorded and another in 1985 using thiopentone and alcuronium. No adverse reactions were noted.
Fig.1
Pedigree diagrams. Probands are indicated in each pedigree by an arrow. Family A has the c.14570T>C, p.Phe4857Ser variant as a de novo RYR1 mutation. Family B carries the c.14752G>A, p.Asp4918Asn RYR1 variant. Families C and D both carry the c.14545G>A, p.Val4849 RYR1 variant with one example of discordance. Family E has the c.13920G>A, p.Met4640Ile RYR1 variant. MHN, MH negative; MHS(hc), MH susceptible to both halothane and caffeine; MHS(h), MH susceptible to halothane; MHS(c), MH susceptible to caffeine; CCD, central core disease.
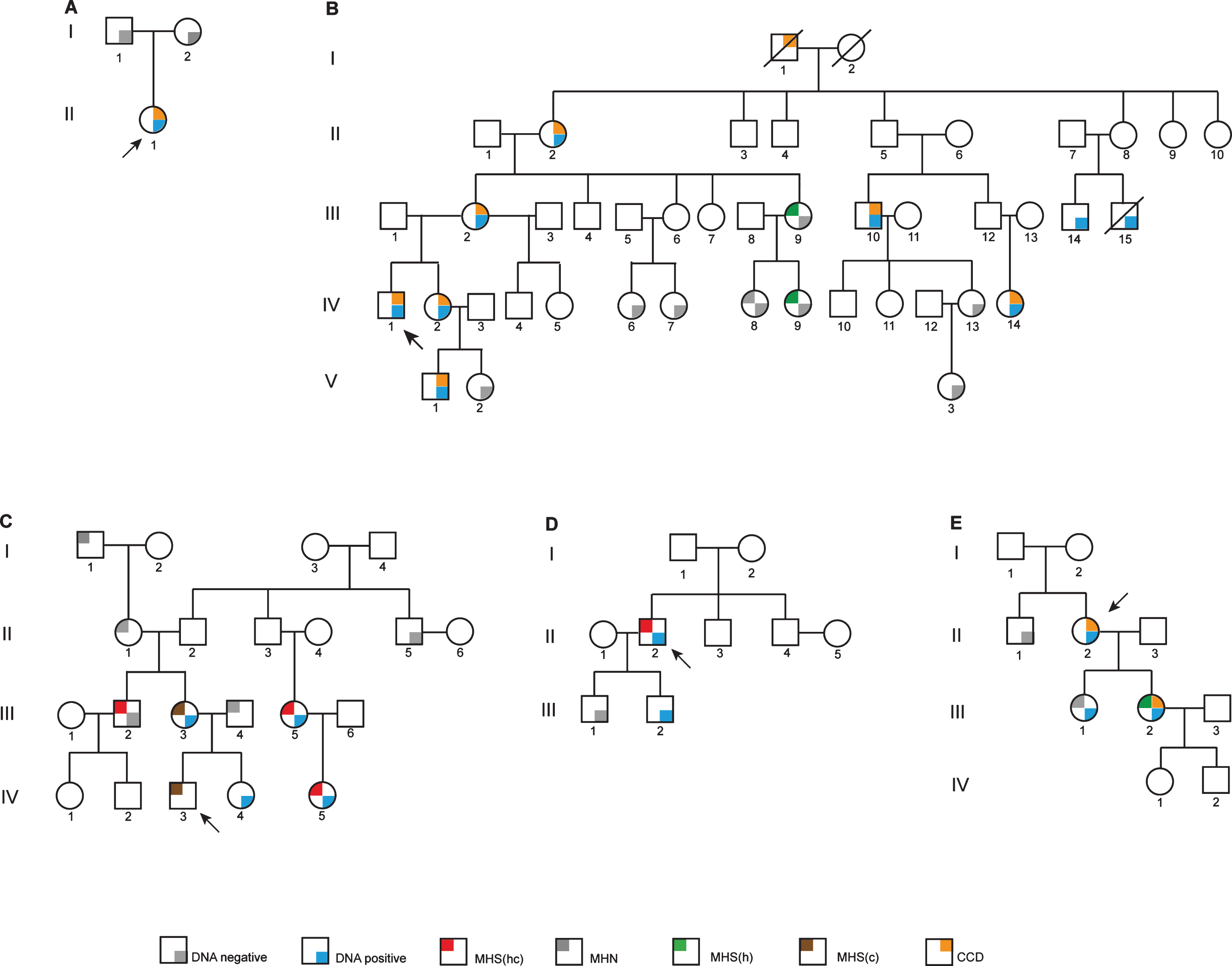
Family B (c. 14752G>A, p.Asp4918Asn; CCD)
The proband, BIV:1 (Fig. 1B) had disease onset at age 5 and when examined at age 8 had severe progressive proximal muscle weakness. At age 26, he is wheelchair-bound and has had scoliosis surgery using total intravenous anesthesia with a combination of ketamine, propofol and reminfentanil. Atracurium (0.76 mg/kg) was used for intubation. There was no evidence of MH during this procedure. He has been diagnosed with central core disease from biopsy of the right lateral thigh. Prior to diagnosis he had received two anesthetics containing volatile agents. One at age 2 used thiopentone induction and spontaneous ventilation with nitrous oxide, oxygen and isoflurane. The other at age 8 was for muscle biopsy using gas induction with nitrous oxide, oxygen and sevoflurane and spontaneous ventilation. Both were brief procedures with no signs of MH. The proband’s sister BIV:2, has proximal muscle weakness, exhibits Gower’s sign, difficulty burying eyelashes and slight facial weakness. She has had three general anesthetics before the family diagnosis was known, two with suxamethonium and all three with volatile agents and had no recorded reaction. The mother of the proband BIII:2, has subtle facial weakness, wasting of the thenar eminence, mildly reduced power in shoulder girdle, positive Gower’s sign and reduced ankle reflexes. She has had several minor procedures under general anesthesia, all with non-triggering agents and no abnormalities noted. Individual BII:2 has mild muscle weakness associated with CCD. Individual BI:1 was reported to have been “weak” as a child and the family thought that CCD was inherited from him as his wife BI:2 was asymptomatic into her nineties. Two sisters, cousins of the proband, BIV:6 and BIV:7 have no signs of CCD or muscle weakness. Individuals BIV:13 and BV:2 appear to be asymptomatic for CCD, while BV:1 is symptomatic of CCD and has had one non-triggering general anesthetic with no abnormalities noted.
Family C (variant c.14545G>A, p. Val4849Ile; MH)
Few clinical details are available for this family (Fig. 1C). The proband CIV:3 reportedly had an MH reaction in the Royal Children’s Hospital, Melbourne during anesthesia for squint surgery in 1997. The episode was characterized by a rapid rise in CO2 levels (to 98 mm Hg), muscle rigidity and a rapid rise in temperature associated with tachycardia. Blood gases revealed a mixed metabolic and respiratory acidosis. There was a rapid response to dantrolene. His Clinical Grading Score was at least 58 indicating almost certain MH. He was confirmed as MH-susceptible by in vitro contracture test in Australia in 1997. Patient CIII:5 was tested positive for MH by in vitro contracture testing in New Zealand. Her daughter CIV:5 also tested MH-susceptible. Individual CIII:2 tested MH-susceptible by in vitro contracture test in Australia in 2001. There is no evidence of CCD in this family.
Family D (variant c.14545G>A, p. Val4849Ile; MH)
Patient DII:2 (Fig. 1D) is a Caucasian male who presented for wisdom teeth extraction at age 17 in 1991, with no significant past history. During an anesthetic using both halothane and suxamethonium he developed tachycardia of 120 bpm as well as supraventricular and ventricular ectopic beats which settled when the volatile anesthetic was changed to isoflurane. Because of increasing end-tidal CO2 the procedure was abandoned. The patient did not have muscle rigidity and dantrolene was not administered. There is no evidence of CCD in this family.
Family E (variant c.13920G>A, p.Met4640Ile; CCD)
The proband EII:2 (Fig. 1E) is currently aged 73. She presented with symptoms and signs consistent with congenital myopathy at birth. She had two general anesthetics prior to 2005, but details were not available. Muscle histology indicated changes consistent with CCD in one daughter (EIII:2). Her other daughter (EIII:1) is asymptomatic. Prior to her biopsy for the IVCT she had two non-triggering anesthetics using total intravenous anesthesia. Since then she has had one anesthetic with sevoflurane for a laparoscopy lasting 1 hour. There was no evidence of MH.
Regulatory authority approvals
This study has been approved by the Central Regional Ethics Committee, Ministry of Health, Wellington, New Zealand (MWH/03/04/018) and the Massey University Genetic Technology Committee (GMO 05/MU/01 and GMO 00/MU/60) acting as an Institutional Biological Safety Committee for the Environmental Protection Authority, Wellington, New Zealand.
In vitro contracture tests
Muscle biopsies were obtained and in vitro contracture testing was performed at the Palmerston North Hospital (or the Royal Melbourne Hospital for some members of family C) using the standard diagnostic procedure for MH-susceptibility according to the European Malignant Hyperthermia Group protocol and local laboratory control data [26–28]. Briefly, a diagnosis of MH-susceptible (MHShc) is made if the responses to both halothane and caffeine are above the threshold of 0.4 g (in Palmerston North) or 0.2 g (in Melbourne) using 2% halothane and 0.2 g using 2 mM caffeine. An MH-negative (MHN) diagnosis is made if the response is below threshold for both agents. If the response is above threshold for only one of the agents the diagnosis is MHSh or MHSc for halothane and caffeine, respectively [29]. A variation on the European Malignant Hyperthermia Group protocol was used for family C (Fig. 1C) in Australia, where most individuals (CI:1, CII:1, CII:5, CIII:3, CIII:4, CIV:3 and CIV:4) reside. A saturated solution of halothane bubbled with carbogen was used for the halothane component of the test before the European Malignant Hyperthermia Group protocol was instituted in the year 2000, hence the concentration of halothane is unknown.
Variant detection
Variants in RYR1 were identified by DNA sequence analysis of either genomic DNA as described previously [30] or cDNA by reverse-transcription PCR followed by Sanger sequencing as previously described [31]. The complete coding region of RYR1 was screened for each proband with the exception of the patient with the c.14570T>C, p.Phe4857Ser variant. In this case, the following exons were sequenced, which cover all of the current causative variants in RYR1: 2–9, 11–17, 39–48, 95–98 and 100–106. In some cases RYR1 and CACNA1S were screened using next generation sequencing as previously described [30]. Screening for the absence of a novel variant in DNA samples from MHN individuals was carried out using high resolution amplicon melting analysis (HRM) [32]. SsoFast EvaGreen™ DNA polymerase (BioRad laboratories) was used for HRM according to the manufacturer’s instructions. Primer sequences for HRM assays will be provided on request. Classification of genetic variants identified in patients is according to GenBank accession numbers NM_000540.2 and NP_000531.2 for cDNA and protein, respectively.
Site-directed mutagenesis
Plasmids representing the C-terminal coding region of RYR1 were constructed by PCR-amplification using a protocol based on the QuikChange™ method (Stratagene). Complementary primer pairs were obtained from Integrated DNA Technologies (Supplemental Table 1). Amplification conditions were one cycle at 98°C for 30 seconds, then 18 cycles of 95°C for two minutes; 57°C for 30 seconds; 72°C for six minutes, followed by one cycle at 72°C for five minutes. The PCR products were digested with restriction endonuclease DpnI (New England BioLabs) according to the manufacturer’s instructions to remove all methylated template DNA, creating a homogeneous mutant population.
Cloning RYR1 cDNA
The full-length RYR1 cDNA, containing a specified variant was generated in two sequential cloning steps by standard methods. The non-mutated full-length RYR1 cDNA was as previously described [21]. Constructs were confirmed by Sanger DNA sequencing using a capillary ABI3730 Genetic Analyzer with BigDye™ Terminator Version 3.1 (Applied Biosystems) chemistry at the Massey Genome Service, Palmerston North,New Zealand.
Transient expression
HEK-293T cells, maintained in Dulbecco’s modified Eagle’s medium using standard mammalian cell culture procedures, were transfected with the pcDNA3.1+ vector containing RYR1 cDNA for transient expression or empty pcDNA3.1+ vector using FuGENE HD (Promega). For Ca2+ release assays cells were transfected at 80% confluence in 96 well plates with 300 ng of plasmid DNA to 1 μL of FuGENE HD per well. For western blotting cells were transfected at 80% confluence in T25 flasks with 6 μg of plasmid and 24 μL of FuGENE HD per flask. Ca2+ release assays or western blots were carried out 72 hours after transfection.
Western blotting
Total protein was extracted from transiently-expressing HEK-293T cells with cell lysis buffer (0.1 M Tris-HCl pH 7.8, 0.5% Triton X-100) containing cOmplete Mini EDTA-free protease inhibitor (Roche). Concentration of protein extracts was determined using the Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories) according to the manufacturers’ instructions. Protein samples (∼250 μg) were resolved by sodium dodecylsulphate-polyacrylamide gel electrophoresis (7.5% polyacrylamide) and transferred to a polyvinylidene fluoride membrane at 70 mA for 20 hours at 4°C. The membrane was blocked using 5% skim milk in TBST before addition of the primary antibodies; 34C (Sigma) for detection of RyR1 or anti-α-tubulin (Sigma) as a loading control, followed by horseradish peroxidase-conjugated anti-mouse secondary antibody (Jackson laboratories). Proteins were visualized using an X-ray developer after brief exposure with chemiluminescence blotting substrate (Roche) according to the manufacturer’s instructions. Successful expression of RyR1 protein was assessed by visual inspection of the film image, comparing the RyR1 band with the matching tubulin band.
Ca2+ release assays
HEK-293T cells were transfected with plasmid DNA in a UV-transparent 96-well plate and incubated in balanced salt solution (140 mM NaCl, 2.8 mM KCl, 2 mM MgCl2, 10 mM glucose and 10 mM HEPES buffer pH 7.3) containing 2 mM CaCl2, 2 μM Fura-2 AM and 0.01% pluronic F-127 per well for one hour at 37°C in the dark. Cells were washed in Ca2+-free balanced salt solution. Activation of the RyR1 using 4-cmc as an agonist was measured by the change in the fluorescence emission ratio at 510 nm when excited by wavelengths 340 nm and 380 nm using an Olympus IX81 fluorescence microscope. Each well was assayed by establishing a fluorescence ratio baseline (indicative of basal cytosolic [Ca2+]) using Ca2+-free balanced salt solution before adding Ca2+-free balanced salt solution containing 4-cmc. The final concentrations of 4-cmc used were 0, 200, 300, 400, 600, 800 and 1000 μM.
Statistical analysis
To account for any differences in cell density the amount of Ca2+ released at each concentration of 4-cmc was normalized for each assay by expressing Ca2+ release as a percentage of the total Ca2+ released with 1000 μM 4-cmc. A minimum of eight replicates was used for each RyR1 variant including five replicates for each concentration of 4-cmc. The data were pooled and the mean±standard error of the mean (SEM) was calculated for each value of 4-cmc used. A sigmoidal curve was fitted for each dataset using the Sigmoidal Logistic function, type 3 within OriginLab Origin 8 software and the half maximal effective concentration (EC50) values were calculated from individual sigmoidal curves. Statistical significance of the difference between the EC50 values for each of the variants and the normal (wild-type control) variant was calculated in the form of p-values using an unpaired Student’s t-test using OriginLab Origin 8 software. An alpha level of 0.0017 was used for all statistical tests according to the Bonferroni correction for 99% significance.
RESULTS
Muscle testing and DNA analysis
Results of the in vitro contracture test for susceptibility to MH, CCD and RYR1 variant status are summarized in Table 1.
Table 1
MH/CCD status
| Family ID | RyR1 variant | 2% halothane (g) | 2 mM caffeine (g) | MH Status | CCD status | |
| histology | symptoms | |||||
| AII:1 | p.Phe4857Ser | n.d. | n.d. | n.d. | CCD | CCD |
| BI:1 | n.d. | n.d. | n.d. | n.d. | n.d. | CCD |
| BII:2 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BIII:2 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BIII:9*** | wt | 0.7 | 0.2 | MHSh | n.d. | none |
| BIII:10 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BIII:14 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | n.d. |
| BIII:15 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | n.d. |
| BIV:1 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BIV:2 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BIV:6 | wt | n.d. | n.d. | n.d. | n.d. | none |
| BIV:7 | wt | n.d. | n.d. | n.d. | n.d. | none |
| BIV:8 | wt | 0 | 0 | MHN | n.d. | none |
| BIV:9*** | wt | 0.6 | 0.2 | MHSh | n.d. | none |
| BIV:13 | wt | n.d. | n.d. | n.d. | n.d. | none |
| BIV:14 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BV:1 | p.Asp4918Asn | n.d. | n.d. | n.d. | n.d. | CCD |
| BV:2 | wt | n.d. | n.d. | n.d. | n.d. | none |
| BV:3 | wt | n.d. | n.d. | n.d. | n.d. | none |
| CI:1 | n.d. | 0** | 0 | MHN | n.d. | none |
| CII:1 | n.d. | 0.28** | 0.23* | MHN | n.d. | none |
| CII:5 | wt | n.d. | n.d. | n.d. | n.d. | none |
| CIII:2*** | wt | 0.65 | 0.3 | MHShc | n.d. | none |
| CIII:3 | p.Val4849Ile | 0.55** | 1.8 | MHSc | n.d. | none |
| CIII:4 | n.d. | n.d. | 0 | MHN | n.d. | none |
| CIII:5 | p.Val4849Ile | 3.8 | 1.9 | MHShc | n.d. | none |
| CIV:3 | n.d. | 1.35** | 1.7 | MHSc | n.d. | none |
| CIV:4 | p.Val4949Ile | n.d. | n.d. | n.d. | n.d. | none |
| CIV:5 | p.Val4849Ile | 5.4 | 3.4 | MHShc | n.d. | none |
| DII:2 | p.Val4849Ile | 5.5 | 3.5 | MHShc | n.d. | none |
| DIII:1 | wt | n.d. | n.d. | n.d. | n.d. | none |
| DIII:2 | p.Val4849Ile | n.d. | n.d. | n.d. | n.d. | none |
| EII:1 | wt | n.d. | n.d. | n.d. | n.d. | none |
| EII:2 | p.Met4640Ile | n.d. | n.d. | n.d. | n.d. | CCD |
| EIII:1*** | p.Met4640Ile | 0.1 | 0 | MHN | n.d. | none |
| EIII:2 | p.Met4640Ile | 1.1 | 0.2 | MHSh | CCD | CCD |
*Test not carried out according to EMHG protocol. **Halothane concentration unknown (test prior to EMHG protocol). ***Discordant. n.d: not done. wt: wild-type. All variants show the amino acid change according to GenBank accession NP_000531.2.
The proband AII:1 (Fig. 1A) with the c.14570T>C, p.Phe4857Ser RYR1 variant was confirmed histologically to have CCD. The variant was absent in both parents and from a panel of 150 MHN samples screened using HRM analysis as well as from the ExAc database (http://exac.broadinstitute.org).
The proband BIV:1 (Fig. 1B) was diagnosed with severe CCD by clinical examination but has not been tested for MH susceptibility. He carries the c. 14752G>A, p.Asp4918Asn RYR1 variant. Only three in vitro contracture tests have been carried out in family B. Individuals BIII:9 and BIV:9 were both MHSh with low contractures for halothane. Individual BIV:8 was MHN and lacks the familial variant. None of these individuals show any evidence of CCD. All individuals diagnosed with CCD also carry the c. 14752G>A, p.Asp4918Asn RYR1 variant. BIII:14 and BIII:15 both carry the familial variant, but their CCD status is unknown.
Individuals CIII:5 and CIV:5 (Fig. 1C), show strong contractures for both halothane and caffeine. This is also the case for proband DII:2 (Fig. 1D). These strong contractures show concordance with the c.14545G>A, p.Val4849Ile RYR1 variant. On the other hand, the individuals (CIII:2, CIII:3 and the proband CIV:3) tested in Australia exhibit rather weaker contractures and discordance was observed between genotype and phenotype in one individual (CIII:2). In addition, the complete coding regions of the RYR1 and CACNA1S genes were completely sequenced in CIII:5 (Fig. 1C) and no other variants were identified. For the proband DII:2, the complete coding regions of fifty genes with potential involvement in calcium homeostasis in skeletal muscle, including RYR1 and CACNA1S, were sequenced and no other rare variants wereidentified.
The proband EII:2 (Fig. 1E) has been diagnosed clinically with CCD and carries the c.13920G>A, p.Met4640Ile RYR1 variant but has not been tested for MH susceptibility. Only one family member has been diagnosed MHSh by IVCT in pedigree E (EIII:2, Fig. 1E) and she also has a phenotype consistent with CCD. While this coincides with the presence of the familial RYR1 variant, her sister, EIII:1 who also carries the variant has been diagnosed MHN by in vitro contracture test and appears asymptomatic for CCD. The coding regions of the RYR1 and CACNA1S genes have been completely sequenced in the proband (Fig. 1, EII:2) and no other variants wereidentified.
Confirmation of RyR1 expression
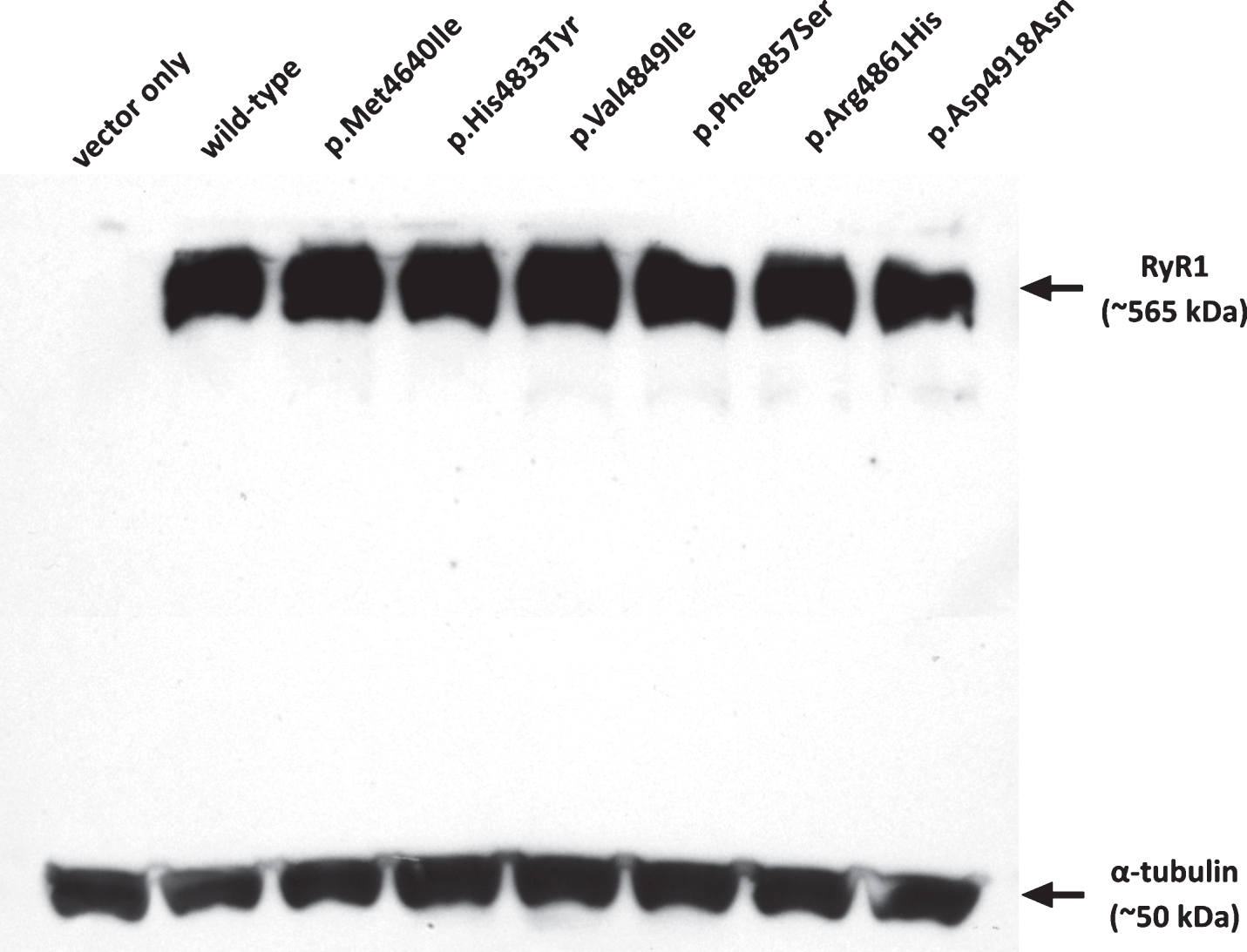
Visualization of the western blot for RYR1 expression relative to α-tubulin confirmed robust production of RyR1 from all constructs and was confirmed as negative in the vector-only control (Fig. 2). Immunofluorescence was carried out as previously described [24, 33] and confirmed that recombinant RyR1 was targeted to the endoplasmic reticulum (data not shown).
Fig.2
Expression of RyR1 protein in HEK-293T cells. Western blot showing RyR1 protein expression for each of the constructs in comparison with α-tubulin as a loading control. Approximately 250 μg of total protein extract was loaded in each lane. Variants are indicated by their amino acid change.
Analysis of RyR1 activity
Ca2+-release in response to the agonist 4-cmc in HEK-293T cells transiently expressing human wild-type or mutated RYR1 cDNA was used as an EMHG-approved method for functional analysis [29]. The MH-associated c.14497C>T, p.His4833Tyr RyR1 mutation was chosen as a control for gain-of-function as we have previously demonstrated hypersensitivity in different experimental systems [21, 33, 34]. The CCD-associated c.14581C>T, p.Arg4861His RyR1 mutation has been previously reported to result in a hyposensitive channel in patient-derived B-lymphoblastoid cells [35]. Both of these channels had decreased EC50 values (290.2±11.5 and 322.4±19.7 μM for p.His4833Tyr and p.Arg4861His, respectively) to 4-cmc compared with wild-type (468.8±17.2 μM) at a confidence limit of >99% (Fig. 3 and Supplemental Table 2). This was expected for the MH-causative c.14497C>T, p.His4833Tyr variant but not for the c.14581C>T, p.Arg4861His CCD-associatedvariant.
Fig.3
Calcium release in HEK-293T cells expressing RyR1 channel variants. HEK-293T cells transfected with wild-type or variant RYR1 cDNAs were exposed to incremental doses of the RyR1-specific agonist 4-cmc in the presence of the Ca2+-indicator Fura-2. Increases in intracellular calcium upon introduction of the agonist were normalised to the calcium released in response to 1000 μM 4-cmc. Each dataset represents a minimum of eight replicates for each RyR1 variant including five replicates for each concentration of 4-cmc, and is expressed±SEM and fitted with a sigmoidal curve. Variants are indicated by their amino acid change - p.His4833Tyr and p.Arg4861His represent positive controls. Statistically significant difference from the control “normal” channel sample is denoted by *(p < 0.0017, ≥99% confidence).
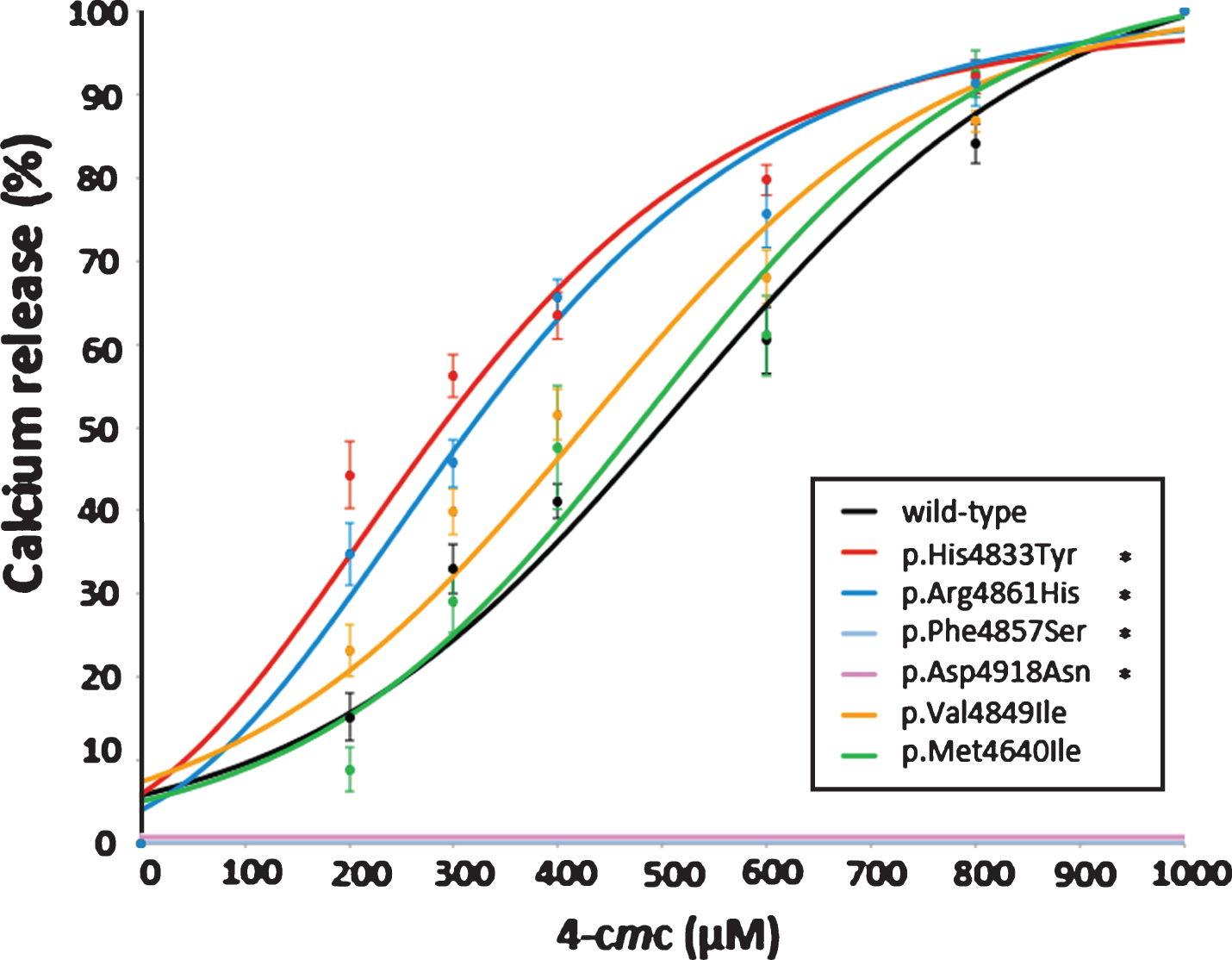
The two CCD-associated variants c.14570T>C, p.Phe4857Ser (Family A) and c.14752G>A, p.Asp4918Asn (Family B) both showed a complete lack of measurable change in intracellular Ca2+ in response to all concentrations of 4-cmc. The MH-associated c.14545G>A, p. Val4849Ile variant (Families C and D) showed a marginally increased response to 4-cmc while the CCD-associated c.13920G>A, p.Met4640Ile variant (Family E) showed no significant difference in Ca2+ release when compared with the wild-type RyR1 (Fig. 3). EC50 values are presented in Supplemental Table 2. There was no significant difference in basal cytosolic Ca2+ levels, as measured by resting fluorescence, between the wild-type and any of the variant constructs.
DISCUSSION
The objective of this study was to identify genetic variants in RYR1 in families with either CCD or MH and then determine their functional effects in order to assess pathogenicity. This is particularly important for MH-susceptible patients as DNA testing can be used only if a variant has been accepted by the EMHG as functionally causative [29]. As patients with CCD are also considered susceptible to MH, it is of obvious benefit to characterize variants that segregate with CCD as well as those that segregate with MH. There is always the possibility of a muscle disorder with a variant in RYR1, therefore such patients would be given non-triggering anesthesia so as to avoid any potential adverse outcomes [36].
Four C-terminal RyR1 variants were identified in five families with either CCD (c.13920G>A, p.Met4640Ile; c.14570T>C, p.Phe4857Ser and c.14752G>A, p.Asp4918Asn) or MH (c.14545G>A, p.Val4849Ile). While three of these variants have been identified previously, none to date have been formerly included in the EMHG list of causative mutations. Each variant was introduced into the full-length human RYR1 cDNA and transiently expressed in HEK-293T cells. Ca2+-release assays were then carried out to assess the functional significance of each variant compared to controls. None of the introduced variants caused a change in basal Ca2+ concentration therefore there is no evidence for any variant rendering the channel leaky in our experimental system. Our results demonstrating a hypersensitive channel for the c.14581C>T, p.Arg4861His variant are at odds with a previous report [35] where only one concentration of 4-cmc was used rather than a dose-response curve. It is possible that the genetic background in the patient-derived B-lymphocytes could have had an effect on results. It is note-worthy that we have seen a New Zealand family carrying the c.14581C>T, p.Arg4861His RyR1 variant who are symptomatic of CCD but whose IVCT results indicate MH susceptibility consistent with a gain-of-function. Other RYR1 variants have also been shown to be associated with both CCD and MH phenotypes [37].
c.14570T>C, p. Phe4857Ser (Family A)
The c.14570T>C, p.Phe4857Ser RYR1 variant associated with CCD resulted in a non-functional RyR1 channel. This is a loss-of-function phenotype consistent with CCD. Both parents (confirmed by paternity testing), who were asymptomatic, were shown not to carry the variant, confirming that it has occurred de novo in the proband (Fig. 1A). This variant was also absent from 150 MHN DNA samples, and from the ExAc database of >60,000 human exomes (http://exac.broadinstitute.org), suggesting that it is a rare pathogenic variant. The p.Phe4857Ser residue, which is highly conserved (Supplemental Figure 1) is predicted to be at one end of the S5 transmembrane helix in a relatively hydrophobic region of the protein [38]. Replacing a bulky hydrophobic phenylalanine residue with a small polar serine residue could feasibly alter the tertiary structure of the channel. A number of other CCD-linked variants reside near this position [38]. Taken together, these observations suggest that the c.14570T>C, p.Phe4857Ser variant is a loss-of-function mutation consistent with the CCD phenotype.
c.14752G>A, p.Asp4918Asn (Family B)
The c. 14752G>A, p.Asp4918Asn variant segregates with CCD in this reasonably large New Zealand pedigree (Fig. 1B) and has been identified previously as associated with CCD [23]. Here we have shown its complete lack of activity in our assay system. This is consistent with two previous studies [23, 39], which show impaired RyR1 function with this variant. The c. 14752G>A, p.Asp4918Asn RYR1 variant does not occur in the ExAc database. The p.Asp4918 amino acid is located within the S6 transmembrane helix, which forms the major part of the ion-conducting pathway through the channel [38]. The change from the negatively charged carboxylate-containing aspartic acid residue to the polar, carboxamide-containing asparagine is likely to affect ion conduction. This is consistent with EC-uncoupling, stabilization of the closed state of the channel and inhibition of opening [18, 40]. Each of these lines of evidence as well as segregation with CCD in more than one family, suggest the c. 14752G>A, p.Asp4918Asn RYR1 variant is likely to cause CCD and should be classed as pathogenic.
c.14545G>A,p.Val4849Ile (Families C and D)
The c.14545G>A, p.Val4849Ile variant is associated with strong contracture responses in the in vitro contracture test and while the two families that carry this variant are too small to confirm segregation, the variant has been identified in the heterozygous state in other families with MH [41] and with CCD in the homozygous state [42]. The variant is heterozygous in both families in this study, with no other rare variant in RYR1 or CACNA1S, and appears to segregate with MH-susceptibility (Table 1) without evidence of CCD in any of the family members. Our functional studies show that RyR1 bearing this variant releases Ca2+ at lower concentrations of agonist (411.6±18.5 μM 4-cmc) compared to the wild-type RyR1 (468.8±17.2 μM 4-cmc) although this difference is not statistically significant when a post hoc analysis is applied. Previous functional studies of this variant in several different systems have provided inconsistent results [43, 44], while our results are in agreement with a more recent study in HEK-293 cells that used caffeine as the agonist [45]. This variant has a minor allele frequency of 8.254×10–6, occurring only once, in the ExAc database. The substitution of a valine to the larger isoleucine residue at this site in the middle of the S5 transmembrane helix could impair the hypothesized interaction between the S5 and S6 helices that are proposed to make up the Ca2+ pore of RyR1 [38]. These collective observations suggest this variant may result in a hypersensitive channel in the HEK-293T cell system, indicating a gain-of-function mutation consistent with MH pathophysiology. The existence of other variants in as yet unknown genes cannot be ruled out, especially since some discordance was noted in family C, and the p.Val4849Ile variant may be a functional modifier, rather than a pathogenic variant per se. Additional more definitive functional analysis will be required, as well as whole exome sequencing of family C and D before pathogenicity of this variant in these two families can be confirmed.
c.13920G>A,p.Met4640Ile (Family E)
The c.13920G>A, p.Met4640Ile variant, while appearing to associate with CCD, does not completely segregate with the disorder or with a positive in vitro contracture test for MH-susceptibility, although the family is very small (Fig. 1E). While Ca2+ release in response to 4-cmc in our functional model was indistinguishable from a wild-type control, the p.Met4640 residue is conserved across RyR1 isoforms and species [23, 46], and it does not occur in the ExAc database. The cryo-electron microscopy model for RyR1 structure [38] places the p.Met4640 residue at the edge of the S2 transmembrane helix, which faces the lumen of the sarcoplasmic reticulum. In this position, the substitution to an isoleucine residue is unlikely to have a functional effect on the channel, unless this region of the RyR1 is involved in interaction with a regulatory protein, which would not be expressed in our HEK-293T cell system. Two other amino acid substitutions at the same residue (p.Met4640Val and p.Met4640Arg) have been identified and even though these occurred in individuals with CCD [23, 46, 47], modeling of the p.Met4640Val RyR1 variant in dyspedic mouse myotubes showed no effect on excitation-contraction coupling or bi-directional coupling compared with the wild-type [46]. It remains possible that the p.Met4640Ile RyR1 variant contributes to the CCD phenotype in the presence of other genetic factors. On the other hand, CCD in this family could be caused by a variant in a gene other than RYR1. There is no current evidence to support the p.Met4640Ile variant causing the MH phenotype.
The HEK-293T cell system has the advantage that the effect of variants can be compared in an identical genetic background. The model is not perfect however, as the RYR1 cDNA is expressed in a homozygous manner, while affected patients in this study are heterozygous for their familial variants. The RyR1 tetramer can be formed in vivo containing zero to four mutant channels as, in general, both alleles are expressed. Further, only every second RyR1 tetramer, in a characteristic array, contacts the dihydropyridine receptor to allow excitation-contraction coupling [48]. There is no way of predicting the arrangement of normal or mutant tetramers in vivo but the possibility that some may contain dysfunctional channels is consistent with either the CCD or MH phenotype.
CONCLUSIONS
Two neuromuscular disorders caused by changes in skeletal muscle Ca2+ homeostasis, CCD and MH, have been associated with dysfunctional RyR1 channels. We report functional analysis on a novel de novo RYR1 variant, c.14570T>C, p.Phe4857Ser and a previously reported RYR1 variant c.14752G>A, p.Asp4918Asn, associated with CCD which both show complete lack of channel opening in response to agonists. As both variants are expressed heterozygously in patients it is feasible that a RyR1 tetramer containing one or more defective monomers would be hypoactive and lead to the CCDphenotype.
The c.14545G>A, p.Val4849Ile RYR1 variant segregating with MH-susceptibility in two NZ families exhibits a mildly hypersensitive phenotype in our system. Given the in vivo phenotypic data as well as our functional analysis of the variant, it is possible that the RyR1 tetramer with one or more defective monomers may show hyperactivity in response to agonists. Alternatively, this variant may be a functional modifier.
Finally, we have shown that the c.13920G>A, p.Met4640Ile RYR1 variant, potentially associated with CCD in one of our families, appears to behave as a normal channel when expressed in HEK-293T cells. This, together with in silico analysis and previous modeling of a different variant at this site, makes it likely that CCD is caused by a variant in another gene in this family. Alternatively, a phenotypic effect may depend on protein-protein interactions in skeletal muscle that do not occur in HEK-293T cells.
Based on these findings we suggest that the c.14545G>A, p.Val4849Ile RYR1 variant should be considered a risk factor for MH, and the c.14752G>A, p.Asp4918Asn and c.14570T>C, p.Phe4857Ser loss-of-function mutations should be considered causative of CCD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank Mrs Lili Rhodes and Dr Natisha Magan (Institute of Fundamental Sciences, Massey University, Palmerston North, New Zealand) for their expertise in solving cloning problems and Professor Phil Hopkins and Dr Dorota Miller (UK MH Unit, St James University Hospital, Leeds, UK) for some of the DNA sequence analysis. This work was funded in part by the Neuromuscular Foundation Research Trust of the Muscular Dystrophy Association of New Zealand Inc. Salary for AHS was provided by grant 15/11 from the Australian and New Zealand College of Anaesthetists.
REFERENCES
[1] | Lynch PL , Tong J , Lehane M , Mallet A , Giblin L , Heffron JJA , et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci USA. (1999) ;96: :4164–9. |
[2] | Monnier N , Romero NB , Lerale J , Nivoche Y , Qi D , MacLennan DH , et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. (2000) ;9: (18):2599–608. |
[3] | Jungbluth H . Central core disease. Orphanet Journal of Rare Diseases. (2007) ;2: :25. |
[4] | Duarte ST , Oliveira J , Santos R , Pereira P , Barroso C , Conceicao I , et al. Dominant and recessive RYR1 mutations in adults with core lesions and mild muscle symptoms. Muscle & Nerve. (2011) ;44: (1):102–8. |
[5] | Kossugue PM , Paim JF , Navarro MM , Silva HC , Pavanello RC , Gurgel-Giannetti J , et al. Central core disease due to recessive mutations in RYR1 gene: Is it more common than described? Muscle & Nerve. (2007) ;35: (5):670–4. |
[6] | Wu S , Ibarra MC , Malicdan MC , Murayama K , Ichihara Y , Kikuchi H , et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain: A Journal of Neurology. (2006) ;129: (Pt 6):1470–80. |
[7] | McCarthy TV , Quane KA , Lynch PJ . Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. (2000) ;15: (5):410–7. |
[8] | Rosenberg H , Pollock N , Schiemann A , Bulger T , Stowell K . Malignant hyperthermia: A review. Orphanet Journal of Rare Diseases. (2015) ;10: :93. |
[9] | Sambuughin N , Holley H , Muldoon S , Brandom BW , de Bantel AM , Tobin JR , et al. Screening of the entire ryanodine receptor type 1 coding region for sequence variants associated with malignant hyperthermia susceptibility in the north american population. Anesthesiology. (2005) ;102: (3):515–21. |
[10] | Phillips MS , Fujii J , Khanna VK , DeLeon S , Yokobata K , de Jong PJ , et al. The structural organization of the human skeletal muscle ryanodine receptor (RYR1) gene. Genomics. (1996) ;34: (1):24–41. |
[11] | Melzer W , Dietze B . Malignant hyperthermia and excitation-contraction coupling. Acta Physiol Scand. (2001) ;171: (3):367–78. PubMed PMID: 11412150. |
[12] | Hwang JH , Zorzato F , Clarke NF , Treves S . Mapping domains and mutations on the skeletal muscle ryanodine receptor channel. Trends Mol Med. (2012) ;18: (11):644–57. |
[13] | Horstick EJ , Linsley JW , Dowling JJ , Hauser MA , McDonald KK , Ashley-Koch A , et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nature Communications. (2013) ;4: :1952. |
[14] | Lewis KM , Ronish LA , Rios E , Kang C . Characterization of two human skeletal calsequestrin mutants implicated in malignant hyperthermia and vacuolar aggregate myopathy. J Biol Chem. (2015) ;290: (48):28665–74. |
[15] | Halliday NJ . Malignant hyperthermia. J Craniofac Surg. (2003) ;14: (5):800–2. |
[16] | Ording H . Incidence of malignant hyperthermia in Denmark. Anesth Analg. (1985) ;64: (7):700–4. |
[17] | Gonsalves SG , Ng D , Johnston JJ , Teer JK , Stenson PD , Cooper DN , et al. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology. (2013) ;119: (5):1043–53. |
[18] | Avila G , O’Connell KM , Dirksen RT . The pore region of the skeletal muscle ryanodine receptor is a primary locus for excitation-contraction uncoupling in central core disease. J Gen Physiol. (2003) ;121: (4):277–86. |
[19] | Murayama T , Kurebayashi N , Yamazawa T , Oyamada H , Suzuki J , Kanemaru K , et al. Divergent activity profiles of type 1 ryanodinereceptor channels carrying malignant hyperthermia and central coredisease mutations in the amino-terminal region. PLoS One. (2015) ;10: (6):e0130606. |
[20] | Murayama T , Kurebayashi N , Ogawa H , Yamazawa T , Oyamada H , Suzuki J , et al. Genotype-phenotype correlations of malignant hyperthermia and central core disease mutations in the central region of the RYR1 channel. Hum Mutat. (2016) ;37: (11):1231–41. |
[21] | Sato K , Pollock N , Stowell KM . Functional studies of RYR1 mutations in the skeletal muscle ryanodine receptor using human RYR1 complementary DNA. Anesthesiology. (2010) ;112: (6):1350–4. |
[22] | Tong J , Du GG , Chen SR , MacLennan DH . HEK-293 cells possess a carbachol- and thapsigargin-sensitive intracellular Ca2+ store that is responsive to stop-flow medium changes and insensitive to caffeine and ryanodine. Biochem J. (1999) ;343: (Pt 1):39–44. |
[23] | Kraeva N , Zvaritch E , Rossi AE , Goonasekera SA , Zaid H , Frodis W , et al. Novel excitation-contraction uncoupled RYR1 mutations in patients with central core disease. Neuromuscul Disord. (2013) ;23: (2):120–32. |
[24] | Roesl C , Sato K , Schiemann AH , Pollock N , Stowell KM . Functional characterisation of the R2452W ryanodine receptor variant associated with malignant hyperthermia susceptibility. Cell Calcium. (2014) ;56: (3):1950201. |
[25] | Zhou H , Yamaguchi N , Xu L , Wang Y , Sewry C , Jungbluth H , et al. Characterization of recessive RYR1 mutations in core myopathies. Hum Mol Genet. (2006) ;15: (18):2791–803. |
[26] | Ording H . Diagnosis of susceptibility to malignant hyperthermia in man. Br J Anaesth. (1988) ;60: (3):287–302. |
[27] | Ording H , Brancadoro V , Cozzolino S , Ellis FR , Glauber V , Gonano EF , et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: Results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group: Acta Anaesthesiologica Scandinavica. (1997) ;41: (8):955–66. |
[28] | Pollock N , Langton EE , Stowell KM , Bulger TF . Safety of exposure of malignant hyperthermia non-susceptible patients and their relatives to anaesthetic triggering agents. Anaesthesia and Intensive Care. (2011) ;39: (5):887–94. |
[29] | Hopkins PM , Ruffert H , Snoeck MM , Girard T , Glahn KP , Ellis FR , et al. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth. (2015) ;115: (4):531–9. |
[30] | Stephens J , Schiemann AH , Roesl C , Miller D , Massey S , Pollock N , et al. Functional analysis of RYR1 variants linked to malignant hyperthermia. Temperature. (2016) ;3: (2):1–12. |
[31] | Gillies R , Bjorksten A , Du Sart D , Hockey B . Analysis of the entire ryanodine receptor type 1 and alpha 1 subunit of the dihydropyridine receptor (CACNA1S) coding regions for variants associated with malignant hyperthermia in Australian families. Anaesthesia and Intensive Care. (2015) ;43: (2):157–66. |
[32] | Grievink H , Stowell KM . Identification of ryanodine receptor 1 single-nucleotide polymorphisms by high-resolution melting using the LightCycler 480 System. Analytical Biochemistry. (2008) ;374: (2):396–404. |
[33] | Sato K , Roesl C , Pollock N , Stowell KM . Skeletal muscle ryanodine receptor mutations associated with malignant hyperthermia showed enhanced intensity and sensitivity to triggering drugs when expressed in human embryonic kidney cells. Anesthesiology. (2013) ;119: (1):111–8. |
[34] | Anderson AA , Brown RL , Polster B , Pollock N , Stowell KM . Identification and biochemical characterization of a novel ryanodine receptor gene mutation associated with malignant hyperthermia. Anesthesiology. (2008) ;108: (2):208–15. |
[35] | Tilgen N , Zorzato F , Halliger-Keller B , Muntoni F , Sewry C , Palmucci LM , et al. Identification of four novel mutations in the C-terminal membrane spanning domain of the ryanodine receptor Association with central core disease and alteration of calcium homeostasis. Hum Mol Genet. (2001) ;10: (25):2879–87. |
[36] | Racca F , Mongini T , Wolfler A , Vianello A , Cutrera R , Del Sorbo L , et al. Recommendations for anesthesia and perioperative management of patients with neuromuscular disorders. Minerva anestesiologica. (2013) ;79: (4):419–33. |
[37] | Robinson RL , Brooks C , Brown SL , Ellis FR , Halsall PJ , Quinnell RJ , et al. RYR1 mutations causing central core disease are associated with more severe malignant hyperthermia in vitro contracture test phenotypes. Hum Mutat. (2002) ;20: (2):88–97. |
[38] | Yan Z , Bai XC , Yan C , Wu J , Li Z , Xie T , et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. (2015) :517: (7532):50–5. |
[39] | Gao L , Balshaw D , Xu L , Tripathy A , Xin C , Meissner G . Evidence for a role of the lumenal M3-M4 loop in skeletal muscle Ca2+ release channel (ryanodine receptor) activity and conductance. Biophys J. (2000) ;79: (2):828–40. |
[40] | Avila G , Dirksen RT . Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J Gen Physiol. (2001) ;118: (3):277–90. |
[41] | Robinson R , Carpenter D , Shaw MA , Halsall J , Hopkins P . Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat. (2006) ;27: (10):977–89. |
[42] | Jungbluth H , Muller CR , Halliger-Keller B , Brockington M , Brown SC , Feng L , et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology. (2002) ;59: (2):284–7. |
[43] | Ducreux S , Zorzato F , Ferreiro A , Jungbluth H , Muntoni F , Monnier N , et al. Functional properties of ryanodine receptors carrying three amino acid substitutions identified in patients affected by multi-minicore disease and central core disease, expressed in immortalized lymphocytes. Biochem J. (2006) ;395: (2):259–66. |
[44] | Monnier N , Kozak-Ribbens G , Krivosic-Horber R , Nivoche Y , Qi D , Kraev N , et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat. (2005) ;26: (5):413–25. |
[45] | Merritt A , Booms P , Shaw M-A , Miller D , Daly C , Bilmen J , et al. Assessing the pathogenicity of RYR1 variants in malignant hyperthermia. British Journal of Anaesthesia. 2017 accepted. |
[46] | Dulhunty AF , Groom L , Goonasekera S , Mirza S , Hanna AD , Gallant EM , et al. Proceedings of the australian physiological society. (2012) ;43: :186P. |
[47] | Zhao Y , Hu J , Zhao Z , Shen H , Bing Q , Li N . Next generation sequencing reveals ryanodine receptor 1 mutations in a Chinese central core disease cohort. Muscle & Nerve. (2016) ;54: (3):432–8. |
[48] | Paolini C , Protasi F , Franzini-Armstrong C . The relative position of RyR feet and DHPR tetrads in skeletal muscle. J Mol Biol. (2004) ;342: (1):145–53. |


