
Myofibrillar Myopathies: New Perspectives from Animal Models to Potential Therapeutic Approaches
Abstract
Myofibrillar myopathies (MFMs) are muscular disorders involving proteins that play a role in the structure, maintenance processes and protein quality control mechanisms closely related to the Z-disc in the muscular fibers. MFMs share common histological characteristics including progressive disorganization of the interfibrillar network and protein aggregation. Currently no treatment is available. In this review, we describe first clinical symptoms associated with mutations of the six genes (DES, CRYAB, MYOT, ZASP, FLNC and BAG3) primary involved in MFM and defining the origin of this pathology. As mechanisms determining the aetiology of the disease remain unclear yet, several research teams have developed animal models from invertebrates to mammalians species. Thus we describe here these different models that often recapitulate human clinical symptoms. Therefore they are very useful for deeper studies to understand early molecular and progressive mechanisms determining the pathology. Finally in the last part, we emphasize on the potential therapeutic approaches for MFM that could be conducted in the future. In conclusion, this review offers a link from patients to future therapy through the use of MFMs animal models.
INTRODUCTION
Myofibrillar myopathies (MFM) are genetic muscle disorders characterized by histological abnormalities beginning in the Z-disc and causing progressive disorganization of the intermyofibrillar network, abnormal protein inclusions and vacuole formation within the sarcoplasm. The term MFM was coined in the mid-90 s [1, 2], and shortly thereafter it came to be associated with a series of genes encoding mainly proteins involved in Z-disc structure or function. The precise placement of MFM in the classification of muscle diseases remains debated: it has been classified as a congenital myopathy, but is more often considered as a dystrophy [3]. Genes implicated in MFM include DES [4], CRYAB [5], MYOT [6], ZASP/LDB3 [7], FLNC [8], BAG3 [9], and lately, two other genes FHL1 [10] and DNAJB6. In this review, we will focus on the first widely accepted six genes implicated in MFM.
MUSCLE HISTOLOGY
MFM lesions can be more or less conspicuous in muscle samples, and depend in many cases on the biopsied muscle. Care should be taken in choosing the biopsy site, with clinical examination, EMG (Electromyogram) and muscle MRI (Magnetic Resonance Imaging) used as supportive tests to choose the most appropriate biopsy location. The main histological abnormalities seen on optical microscopy include:
• amorphous granular or hyaline deposits on trichrome-stained sections;
• a modification of oxidative enzyme activities leading in some fibers to core-like or rubbed-out patterns;
• the presence of rimmed and non-rimmed vacuoles, more frequent in myotilinopathies (pathologies due to MYOT mutations) and zaspopathies (pathologies due to ZASP/LDB3 mutations);
• a large variation in fiber size;
• an abnormal and ectopic expression of multiple proteins in fibers of normal size (such expression is common in atrophic fibers).
On electron microscopy, myofibrillar disorganization starting from the Z-disc, with streaming and progressive destruction of its structure, are common, and this pattern is associated with focal accumulation of compacted and degraded myofibrillar elements and abnormal repartition of the mitochondria. The predominance of some abnormalities, such as “sandwich” formations in desminopathies (pathologies due to DES mutations) and alpha-B crystallinopathies (pathologies due to CRYAB mutations) or filamentous “bundles” in zaspopathies may aid in diagnosis [11], but are not totally specific. Histological features similar to those of MFM have been described in occasional cases of laminopathies [12] or selenopathies [13], leading to questions about the boundaries of MFM. However, as these disorders have distinct pathophysiology and presentations, they are generally not included as MFM. In addition, except for myotilin, mutations in the other MFM genes (DES, FLNC, CRYAB, ZASP, BAG3) have been reported to be linked to isolated cardiac disorders, i.e. various forms of cardiomyopathy, which show clinical variability and incomplete penetrance for those genes in MFM.
Desminopathy
First described in 1998, desminopathy cases have since been reported from many countries. Indeed, desminopathy is the most frequent MFM in France, and more than 20 affected families are fully characterized. Desminopathy is caused by mutations in DES, the gene encoding desmin [4]. As in most cases of MFM, except FHL1 (which is X-linked), the transmission is typically autosomal dominant. However, its penetrance seems to be incomplete: relatives of severely affected patients harboring a causal DES mutation may present with mild MFM lesions on muscle biopsy, but no clinical disease expression [14].
Clinically, most patients exhibit cardiac and skeletal muscle involvement. Symptom onset usually occurs before the 5th decade, with a variable initial expression. Some patients develop a distal, or less often proximal, myopathy beginning in the lower limbs, others a cardiac disease that can take the form of a conduction defect, arrhythmia or any type of cardiomyopathy (Fig. 1). This heart involvement is responsible for a high rate of complications, including high-degree conduction blocks, sustained ventricular tachycardia, cardiac insufficiency, cardio-embolic stroke or sudden death, and often leads to treatment with a pacemaker or implantable cardioverter/defibrillator, or even heart transplantation (60% and 10% of cases, respectively, in the French-Swiss experience [15]). These complications affect disease prognosis, as does the occurrence of respiratory insufficiency, which often leads to supportive ventilation. Patients rapidly develop axial weakness and, as the disease progresses, there is proximo-distal involvement of the four limbs. Dysarthria and swallowing difficulties may affect around one-third of patients [16]. Achilles and finger contractures are possible. An atypical presentation is the Stark-Kaeser phenotype [17], characterized by predominant scapulo-peroneal weakness (Fig. 1).
The diagnosis of desminopathy is facilitated by muscle MRI, which shows a rather typical pattern in the thighs, in which a fatty infiltration predominates in the semi-tendinosus, gracilis and sartorius muscles, even at late stages of the disease. In the legs, the pattern is less specific, with an initial involvement of the peroneal and soleus muscles (Fig. 1).
Genetic testing has revealed many mutations (around 70) in DES that are disease-causing; most of them are missense, and they affect almost every part of the gene. No clear genotype-phenotype correlation has been defined; however, p.R406W and p.R454W mutations are associated with a particularly severe heart involvement [18, 19].
Alpha-B crystallinopathy
Mutations in CRYAB cause alpha-B crystallinopathy [5]. Although much less common than desminopathy, this disease shares an overall common clinical presentation of muscle impairment, but with less severe heart involvement and a typically later onset [20]. A distinctive feature of alpha-B crystallinopathy is early-onset cataract, but it is not present in all families [21] (Fig. 1). Data on muscle imaging are too sparse to describe an unequivocal pattern, but it seems to differ from that of desminopathy [22].
Myotilinopathy
Myotilinopathy is caused by variations in MYOT [6]. Initially described as a cause of limb-girdle muscular dystrophy (LGMD1A) [23], myotilinopathy seems to have a more often distal presentation. It is a late-onset myopathy, usually beginning around the fifth decade, with a slow course; many patients consult a neurologist years after the initial symptoms. Axial weakness occurs late, and dysarthric speech has been reported in some patients. Myalgia, muscle hypertrophy and stiffness are common [6, 24], and a diffuse pseudo-hypertrophy resulting in a herculean phenotype was described in one family [25]. Cardiac involvement is rare in our [26] as well as in Spanish cohorts [24, 27]. Respiratory insufficiency may affect up to 23% of patients [27].
The MRI pattern of myotilinopathy includes early soleus and medial gastrocnemius involvement in the legs, and a predominant fatty infiltration of the semi-membranosus, hip adductors and biceps femoris in the thigh [27] (Fig. 1). The most frequent mutations observed in Western Europe are p.S55F and p.S60F, the former being related to a founder effect in Spain [24].
Zaspopathy
Caused by ZASP/LDB3 mutations [7], zaspopathy shares in many cases the late and distal onset of myotilinopathy, with frequent initial asymmetry of the leg weakness. However, initially proximal or proximo-distal forms have also been described, albeit less frequently. The course of the disease is slow, but at late stages (usually over 70 years of age), patients can be severely disabled. Cardiac involvement is uncommon and usually mild [5, 23], and respiratory insufficiency seems to be absent in the vast majority of cases.
MRI shows a predominant involvement of soleus and medial gastrocnemius in the legs, and of the biceps femoris and semi-membranosus in the thighs (Fig. 1). Adductor magnus, gracilis and semi-tendinosus seem to be relatively spared [24, 28]. Several mutations have been described in LDB3, p.A165V being the most frequently found in Western Europe.
Filaminopathy
FLNC mutations are implicated in filaminopathy [8]. Initially described as a proximal myopathy, filaminopathy also appears to be a cause of distal myopathy. The first description, of German families with a p.W2710X mutation, revealed a late-onset myopathy beginning around 45 years of age, with proximal weakness and wasting of the lower limbs, progressing to the upper limbs [8]. Scapular winging can be part of the phenotype. Respiratory involvement is common and may be severe. Cardiac involvement affects a little less than 50% of patients in the German families [29] (Fig. 1). A proximal and predominantly axial phenotype associated with cardiac arrhythmia has also been reported recently in a French family [30]. In Chinese patients, diarrhea and severe cardiopathy appeared as consequences of a complex p.[K899_V904del] + [5V899_C900ins] mutation that also caused proximal myopathy [31]. Distal phenotypes seem to occur earlier and affect both upper and lower limbs, especially wrist flexors and foot extensors (Fig. 1). The disease progresses toward more diffuse weakness, and heart involvement is possible, although rare.
Muscle MRI shows an involvement of the gastrocnemius and soleus muscles in the legs, and a fatty degeneration predominating in the semi-membranosus, biceps femoris, adductor magnus, vastus intermedius and medialis in the thighs (Fig. 1).
Bag3-opathy
Finally, though very rare, is a myopathy caused by mutations in BAG3 [9]. The majority of cases of BAG3-opathies affect young children, who present with severe weakness with a variable proximal, proximo-distal or distal pattern associated with nasal speech and rigid spine [9] (Fig. 1). Nerve conduction studies are important as they may show polyneuropathy, which is an important clue to the diagnosis. Creatin kinase levels (CKs) are generally higher (3–15 N) than in most MFM. The course of the disease is severe, with respiratory insufficiency or conduction defects leading to premature death in some patients (Fig. 1). Patients generally share a p.P209L mutation. Cases with later onset and milder course are also possible [unpublished case of Dr. Tanya Stojkovic].
MFM ANIMAL MODELS
No treatment is currently available for MFM diseases. To conceptualize therapeutic solutions for MFM, it may be necessary to integrate all the mechanisms that may be involved in their pathology. The pathology can be complex, however; thus, non-mammalian or murine animal models are critical to understanding the molecular mechanisms underlying MFM and to test therapeutic options.
Non-mammalian models
Among the non-mammalian model organisms, zebrafish and Drosophila have been used successfully to study the impact of pathological mutations associated with muscular diseases [32], including MFM pathologies. Indeed, Drosophila and zebrafish muscles share many structural, histological and functional similarities with human muscles [67]. Below we provide some instances of how these model systems were applied to gain insights into the physiopathological role of the six proteins associated with MFM diseases.
To date, not all of the pathological variants involved in MFM pathologies have been recapitulated in non-mammalian model systems. However, these models enable an understanding of the physiological functions of the associated genes. For instance, mutations in the heat shock protein alpha-B crystallin have not yet been studied in Drosophila or zebrafish, but the overexpression of the wild-type (WT) gene has been used as potential therapeutic approach in Drosophila models of neurodegeneration (such as Huntington alteration) to reduce aggregation of a protein with expanded polyglutamine repeats [33].
For the other genes associated with MFM, non-mammalian models have provided a better understanding of MFM molecular mechanisms. For example, post-transcriptional silencing of the Drosophila ortholog of human ZASP recapitulate some of the key aspects typical of human zaspopathies [34], despite the description of only missense mutations in patients. In particular, the affected flies display locomotor defects associated with alterations of muscle structure and ultrastructure. Recently, transgenic zebrafish have been generated by inserting fluorescent labels (citrine or mCherry) into the zebrafish desmin “a” gene [35]. The expression of the modified genes leads to desmin aggregation in skeletal and cardiac muscles, 48 h after the beginning of embryonic development. Although these zebrafish lines do not constitute “real” models of MFM mutations, desmin loss-of-function, in addition to desmin aggregation, promote skeletal and cardiac muscle defects. These defects are associated with perturbations of excitation-contraction coupling mechanisms involving calcium, sarcoplasmic reticulum and abnormal distribution of ryanodine receptors. Interestingly, desmin knockdown or pharmacological inhibition of desmin aggregates reverted these phenotypes [35].
In the same way, a FLNC nonsense mutation called Zacro (Zac), resulting in the truncation at the 15th immunoglobulin-like repeat, was established in the filamin C ortholog of the medaka teleost fish (Oryzias latipes), which presents similar advantages to the zebrafish [36]. Although myogenesis is normal in the mutant during early stages of development, its myofibrils progressively degenerate to become disaggregated at later stages, and ultimately cause myocardial rupture in the ventricle; that phenomenon can be related to MFM alteration during aging in some MFM patients.
Similarly, the invalidation of BAG3 in zebrafish using a specific morpholino is associated with myofiber disorganization, but without aggregate formation. This result is similar to those observed in a Drosophila knockout model for starvin (BAG3 ortholog) [37]. The major drawback in these studies is the loss of function of the MFM-associated genes and not the mutant expression. Indeed, MFM are mainly autosomal-dominant pathologies in which both a WT and a mutant allele are present. Moreover, injection of a mutant form of BAG3 (BAG3-P209L, p.P209L involved in human MFM) restores myofiber function in the knockout context, suggesting that the mutation does not completely abolish myofiber function [38]. Therefore, overexpression of the BAG3-P209L mutant promotes aggregate formation, leading to a reduced cellular pool of BAG3 [38].
The facility of using these model systems, when combined with new developments in CRISPR/Cas9 technologies, will enable future studies involving mutant MFM genes and large-scale screens for new potential therapeutic compounds.
Murine models
Although knockout models have been instrumental in characterizing functions of the proteins for genes implicated in MFM, such as for desmin, few murine models of MFM have been described. Desminopathy is the most studied, with 6 already published and 2 unpublished different mouse models of desmin variation (Table 1), compared to the other MFM-linked genes. For desmin and alpha-B crystallin, the majority of the studies have focused on heart phenotypes; the skeletal muscle involvement remains less documented. In contrast, animal models of filamin and myotilin alterations have predominantly assessed the skeletal muscle phenotype. Here, we will summarize common and specific alterations in the murine models of MFM.
Skeletal muscle weakness
Protein aggregates and myofibrillar disorganization in muscular tissue, leading to skeletal muscle weakness, are hallmarks of MFM pathology. Several of the skeletal muscle alterations in patients with MFM are found in mouse models. Indeed, AAV-mediated overexpression of desmin mutants (p.R406W) and (p.E413K) in the tibialis anterior of 10-week-old mice causes protein aggregation (as well as p.D399Y mutant shown in Fig. 2A), decreases in contractile function and morphological changes as observed in patients with MFM [39] (Table 1). However, some specific differences should be noted; for instance, the p.R406W desmin mutant induces muscle regeneration, in contrast to the p.E413K mutant. Such differences help to explain the heterogeneous phenotype observed in patients. Although the AAV-mediated overexpression affects only one muscle, it does offer the possibility of screening for several mutants in parallel to decipher the potential heterogeneous pathological mechanisms among desmin mutants as well as between MFM types.
A low-level overexpression of the desmin p.L345P mutant (hemagglutinin-tagged) driven by the mouse desmin promoter also induces a specific muscle phenotype in transgenic mice [40]. In isolated p.L345P soleus muscles, contractile function and recovery from fatigue are impaired, and motor function is reduced. However, this model exhibits no protein aggregates in cardiac or skeletal muscles (Table 1).
In contrast, a recently described model [7] bears a desmin p.R349P missense knock-in (KI). This mutation corresponds to the most frequently occurring human desmin missense mutation, p.R350P. Thus, unlike the transgenic model, the knock-in model does not have overexpression of desmin, and therefore more closely resembles the patho-physiological conditions found in patients. The p.R349P-KI mice present skeletal muscle weakness associated with age-dependent desmin aggregation [41]; we have found similar features in our unpublished p.R405W-KI mice (Fig. 2B). Moreover, a specific modification of p.R349P desmin turnover is associated with its potential post-translational modification. Together, these mouse models of desminopathy suggest that each desmin mutant represents some unique pathology, leading to the heterogeneous phenotypes reported for desminopathy (Table 1).
The same pattern holds for all of the MFM-associated proteins. Indeed, a recently described filamin-C knock-in mouse model, harbouring the p.W2710X mutation to mimic the human variation, also develops muscle weakness and myofibrillar instability with formation of filamin-C–positive lesions streaming between Z-discs [42] (Table 1). For myotilin, transgenic mice expressing mutant p.T57I under the control of the human skeletal muscle actin promoter also develop progressive myofibrillar pathology, including myofibrillar aggregation close to the Z-discs, as well as myofibrillar vacuolization [43] (Table 1). However, the effect differs from the muscle phenotype observed in patients. Indeed, whereas the extensor digitorum longus significantly loses maximum specific isometric force, the soleus and diaphragm muscles are not affected. These results indicate an aggregate-dependent contractile dysfunction that does not appear to occur with the desmin R349P mutation, delineating some common and unique molecular mechanisms.
Alteration of heart functions
Cardiac function is commonly altered in MFM. Mouse models recapitulate well the range of cardiac phenotypes. Indeed, the first transgenic mouse model, “D7-des”, bears a 7–amino acid deletion (p.R173 to E179) in the desmin sequence, placed under the control of the alpha-myosin heavy chain promoter, for heart-specific expression [44]. The D7-des mouse heart shows aberrant intrasarcoplasmic and electron-dense granular filamentous aggregates that are desmin-positive and characteristic of human desmin-related cardiomyopathy (Table 1). The desmin filament network is significantly disrupted, and myofibril alignment is compromised. Further, the ability of the heart to respond to β-agonist stimulation is significantly blunted.
Desmin p.L345P mice present echocardiographic changes including left ventricular wall hypertrophy and a decreased left ventricular chamber dimension [40] (Table 1). Moreover, the missense desmin mutation p.I451M, targeted to the heart in transgenic mice, results in an absence of desmin Z-disc localization (though desmin can associate with the intercalated discs) and characteristics typical of dilated cardiomyopathy [45] (Table 1). Finally, the desmin p.R349P-KI mice also exhibit dilated cardiomyopathy, cardiac arrhythmias and conduction defects (Table 1). Similarly, the well-studied transgenic mice with overexpression of the alpha-B crystallin p.R120G mutant in cardiomyocytes, cardiomyopathy is accompanied by aberrant alpha-B crystallin and desmin aggregation [46] (Table 1). In contrast, the overexpression of WT alpha-B crystallin does not induce cardiomyopathy, reinforcing the idea that individual mutants have distinct pathology. In addition, a transgenic model with cardiac-restricted expression of a ZASP p.S196L missense mutation presents with cardiac conduction defects and atrioventricular block at three months old, whereas older mice (10 months) develop hemodynamic dysfunctions consistent with dilated cardiomyopathy [47] (Table 1).
Ca2 + changes and cardiac features
In addition to histological features of the MFM pathologies, murine models have permitted investigation into the molecular mechanisms involved in MFM. For instance, in the transgenic desmin p.L345P model [40], the most striking ultrastructural changes are mitochondrial swelling and vacuolization, with calcium levels significantly increased in skeletal and cardiac myocytes during and after contractions (Table 1). Interestingly, L-type calcium and sodium currents are also altered in isolated p.S196L murine cardiomyocytes, which may explain conduction alterations observed in the transgenic model bearing the ZASP p.S196L missense mutation [47]. Similar studies on the CRYAB p.R120G transgenic mouse also show endoplasmic reticulum stress and impairment in calcium regulation, leading to cardiac dysfunction, arrhythmias and shortened lifespan [48] (Table 1). These results suggest that investigations into calcium and mitochondria should be undertaken for every model to determine the importance of such mechanisms in the pathogenesis of MFM.
Proteasome dysfunction and autophagy pathway alterations
The ubiquitin-proteasome system (UPS) has been implicated in MFM pathologies. Indeed, the UPS is impaired in the D7-des transgenic hearts [49]. Moreover, in the case of proteasomal impairment, BAG3 is involved in the switch toward autophagy after localization of ubiquitinated proteins into cytoplasmic puncta co-labeled with different canonical autophagy markers [50]. Thus, a mutation in BAG3, as in BAG3opathy, could affect this switch, even in the absence of a BAG3-MFM model. In a CRYAB-p.R120G transgenic model [51], which exhibits high levels of cytotoxic amyloid oligomers in heart and mitochondrial dysfunction [52], proteasomal impairment [49] and an increased autophagic flux are observed, which may correspond to an adaptive response of the protein quality control (PQC) system to destroy and recycle misfolded proteins [53] (Table 1).
The accumulation and aggregation of abnormal proteins suggest a common mechanism that may underlie the development of all MFM. These features should be systematically investigated in different models of the disease.
Other dysfunctions
The expression of mutant forms of BAG3 (p.R128W and p.L462P) in rat neonatal cardiomyocytes (RNC) induces an increase in sensitivity to apoptosis due to serum depletion [54] (Table 1). BAG-3 binds the anti-apoptotic protein BCL2 [55], enhancing the anti-apoptotic effect of BCL2 in cells of various tumors [56–58]. This function has yet to be confirmed in vivo because of the lack of a specific animal model of BAG3-related MFM. However, the CRYAB R120G transgenic mice exhibit alterations in apoptosis markers, such as increased cytoplasmic cytochrome c, down-regulated BCL-2, up-regulated BAX and activated caspase-3 in mutant myocytes, all of which indicate an activation of apoptotic cell death [52] (Table 1).
In addition, CRYAB p.R120G knock-in mice exhibit cataract features that are age- and mutant gene dosage-dependent [59]. This recapitulates early-onset cataract as seen in the human alpha-B crystallinopathy (Table 1).
THERAPEUTIC APPROACHES
The production of an abnormal protein can results in loss or gain of functions. For instance, the loss of sarcomeric structure due to a defect in one of the structural proteins (desmin, filamin C, myotilin, ZASP) or a maintenance protein (CRYAB, BAG3) will not only lead to structural defects, but also to altered mitochondrial positioning and function, which may then generate oxidative stress [60, 61]. Aberrant proteins are detected by the PQC, which mobilizes heat-shock proteins (HSPs), the main chaperone molecules in cells, to refold any misfolded proteins. When correct refolding is not possible, HSPs, with the help of co-chaperones, target abnormal proteins to the UPS and/or autophagy degradative systems [62]. When these protective systems are overloaded, mutant proteins accumulate; further, they may adopt a beta-amyloid conformation that is toxic for cells, leading to apoptosis or necrosis [63, 64]. Therapeutic options that have been explored for MFM have targeted these mechanisms: i.e., stimulating the PQC system (through HSPs) or the degradative system, inhibiting aggregate formation particularly to avoid accumulation of beta-amyloid toxic structures, rescuing mitochondria or preventing oxidative stress, or, as a last resort, inhibiting apoptosis (an overview is presented in Fig. 3).
Using HSP as therapeutic compounds
In the PQC system, the modulation of HSP expression is an excellent target for some pathologies. Indeed, the induction of the HSP system improves skeletal function in aged mice [65], but also presents an anti-aggregative effect in neurodegenerative pathologies such as Alzheimer’s disease (for review see 23). These findings provide some hope with these regard to MFM pathologies, in which inducing chaperone activity should be explored (Fig. 3). Importantly, alpha-B crystallin is the most abundant HSP in the heart. When CRYAB p.R120G transgenic mice are crossed with D7-des mice with desmin-related cardiomyopathy, double-mutant mice exhibit dramatically more abundant aggregation of desmin than the D7-des line, show a significantly stronger cardiac hypertrophic response, and die from congestive heart failure before 7 weeks [67]. Thus, normal alpha-B crystalline function is protective against some effects of mutant desmin, a hypothesis that has been confirmed with stably transfected cells, but not yet in animals [67]. HSP27 (HSPB1), another molecular chaperone, may also provide protection against toxicity mediated by mis-folded proteins [68]. Directly delivering HSP27 (HSPB1) to muscles using viral vectors may therefore be a promising treatment approach. Finally, a transgenic line with cardiac-specific HSP22 (HSPB8) expression also shows a reduced progression of cardiomyopathy compared with CRYAB p.R120G mice [69].
Another way to induce HSPs is to administer pharmacological inducers of HSP response. For example, geranylgeranylacetone, an inhibitor of protein binding to HSP90 that induces the HSP response, reduces amyloid oligomer levels in the CRYAB p.R120G transgenic model; this treatment also reduces heart size and fibrosis and improves cardiac function and survival compared to untreated mice [69]. The chemical chaperone compounds 4-phenyl-butyrate (4-PBA) and trimethylamine N-oxide (TMAO) have been used to treat primary myotubes and mouse models deficient in plectin, which present abnormal aggregation of wild-type desmin. Indeed, 4-PBA, but not TMAO, treatment induces a significant reduction in desmin aggregation [70]. Moreover, muscular function improves with 4-PBA. These data suggest an interesting potential therapeutic role for chaperone molecules in MFM pathologies. However, we note that care should be taken in the case of alpha-B crystallinopathies, to avoid inducing the endogenous mutated (p.R120G) gene using glucocorticoids, for instance [71].
Redox and mitochondria
Production of aggregates in muscle fibers may generate indirect consequences such as oxido-reductive stress. This stress is due in part to the accumulation of aggregated proteins with a slow turnover rate, which are more prone to oxidation [72]. Additionally, oxidative damage may be generated by mitochondria that are damaged through mislocalization in response to defects in Z-line structure, and/or that accumulate because mitophagy is less effective due to the saturation of the autophagic apparatus by aggregated proteins. Therefore, antioxidant treatment appears promising for counterbalancing the effects of accumulated abnormal proteins and damaged mitochondria (Fig. 3). In the CRYAB p.R120G mouse model, re-establishing a redox equilibrium is essential for rescuing mice from cardiomyopathy [73, 74]. Further, inhibiting xanthine oxidase in this model with oxypurinol for 1 to 3 months restores mitochondrial function [75]. However, cardiac contractile function and compliance do not improve because of mechanical deficits in passive cytoskeletal stiffness [75]. Importantly, CRYAB p.R120G mice show an increased activation of apoptotic cell death [76]. Drugs acting on mitochondrial permeability, such as nicorandil, a mitochondrial ATP-sensitive potassium channel opener, reduce mitochondrial impairment and apoptotic death in CRYAB p.R120G transgenic mice [76]. Mitochondria are essential in the pathway leading to apoptosis [77] and thus represent an important therapeutic target. The restoration of mitochondrial function and inhibition of apoptosis was tested a decade ago: mice over-expressing the anti-apoptotic protein BCL-2 were crossed with desmin-null mice [78], and a mortality significantly declined. A similar outcome occurred when CRYAB p.R120G mice were crossed with a transgenic line specifically over-expressing BCL-2 in the heart [79].
Proteasome and autophagy
Aberrant protein aggregation with accumulation of ubiquitinated proteins is a common process in many neurodegenerative diseases [80], as well as in MFM [81], and leads to proteasomal malfunction. The use of CRYAB p.R120G mice as a model of MFM confirmed that aberrant protein aggregation impairs UPS proteolytic function in the heart [82]. However, no pharmacological inducer of the UPS is known, which precludes this therapeutic option. In contrast, macroautophagy (here referred to as “autophagy”) constitutes a rescue system that is stimulated when UPS function is impaired (reviewed in 83). In CRYAB p.R120G mice, there is more than 2-fold increase in cardiomyocyte autophagic activity [84]. Increased level of basal autophagy in these mice decreases cardiac hypertrophy and intracellular aggregates and prolongs survival [85]. In addition, large aggregates called “aggresomes,” which are actively transported by the microtubule-dependent retrograde transport to the perinuclear area, are more efficiently degraded by autophagy [86]. It is possible to further stimulate autophagy with fasting [87, 88] or with treatment with AMPK [89], cyclosporine A [90], or rapamycin [91], among many compounds described in the literature [92] (Fig. 3). Although some of these drugs have been successfully tested in animal models of muscular dystrophy [89, 90], they have not yet been tried in MFM models.
Antiaggregation
Inhibiting protein aggregation seems to be a “simple” way of reducing the burden in muscle cells. Doxycycline administered via drinking water to CRYAB p.R120G transgenic mice results in reduced CRYAB aggregates, attenuated cardiac hypertrophy, and delayed premature death [93]. Another way to reduce pre-amyloid toxic oligomer accumulation is long-term voluntary exercise. CRYAB p.R120G mice housed in cages with running wheels exhibit a significant reduction in beta-amyloid oligomers with a concomitant increase in lifespan [94]. However, acute exercise induces lesions in soleus and diaphragm muscles in filamin C p.W2710X knock-in mice [42]. Moreover, two recent cellular studies on myoblasts seem to show that antioxidant or pro-autophagic compounds could reduce aggregation, linking antioxidant activity and/or the PQC system with aggregation modulation [95, 96]. Such compounds (NAC, trolox, pp242) will be future candidates to test efficacy in existing animal models (Fig. 3).
CONCLUSION
Interestingly, one of the older animal models, the CRYAB p.R120G mice, has contributed a large amount of data involving protein aggregation, beta-amyloid toxicity, calcium and mitochondrial defects leading to oxido-reduction stress, alteration of the machinery controlling protein turnover and apoptosis. These findings have likely uncovered essential mechanisms forming the basis of generation of MFM diseases (Fig. 3). Thus, laboratories with existing animal models or planning to develop new ones should be encouraged to investigate these mechanisms. Advancing our understanding of MFM will promote new therapeutic solutions for these pathologies.
Conflict of interest statement
We declare that no conflict of interest exists.
ACKNOWLEDGMENTS
A.L., S.B.P., F.D., E.C. and P.V. are supported by Université Paris Diderot, CNRS and ANR (3BSV5-0017-DESMECA) and AFM grant n°15454 and 18358.
Animal experiments were conducted with respect to animal health and well-being in accordance with the ethics committee (authorization number CEB-16-2016). We thank the “plateforme d’Hébergement et d’expérimentation animale Buffon”.
Confocal images were acquired at the “plateau d’imagerie” of the BFA unit. AAV have been obtained from “plateforme de production de vecteurs viraux, INSERM UMR 1089” (thanks to F.Baltier and P.Moullier).
REFERENCES
[1] | Nakano S , Engel AG , Waclawik AJ , Emslie-Smith AM , Busis NA . Myofibrillar myopathy with abnormal foci of desmin positivity. 1. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol. (1996) ;55: :549–62. |
[2] | De Bleecker JL , Engel AG , Ertl BB . Myofibrillar Myopathy with Abnormal Foci of Desmin Positivity. II. Immunocytochemical Analysis Reveals Accumulation of Multiple Other Proteins. J Neuropathol Amp Exp Neurol. (1996) ;55: (5):563–77. |
[3] | Selcen D , Engel AG . Chapter 11 – Myofibrillar myopathies In: Griggs Robert C. and Amato Anthony A., editor. Handbook of Clinical Neurology [Internet]. Elsevier; (2011) . pp. 143–54. Available from: http://www.sciencedirect.com/science/article/pii/B9780080450315000116 |
[4] | Goldfarb LG , Park K-Y , Cervenakova L , Gorokhova S , Lee H-S , Vasconcelos O , et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. (1998) ;19: (4):402–3. |
[5] | Vicart P , Caron A , Guicheney P , Li Z , Prevost M-C , Faure A , et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. (1998) ;20: (1):92–5. |
[6] | Selcen D . Mutations in myotilin cause myofibrillar myopathy. Neurology. (2004) ;63: (2):405. |
[7] | Selcen D , Engel AG . Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. (2005) ;57: (2):269–76. |
[8] | Vorgerd M , van der Ven PFM , Bruchertseifer V , Löwe T , Kley RA , Schröder R , et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. (2005) ;77: (2):297–304. |
[9] | Selcen D , Muntoni F , Burton BK , Pegoraro E , Sewry C , Bite AV , et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. (2009) ;65: (1):83–9. |
[10] | Selcen D , Bromberg MB , Chin SS , Engel AG . Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology. (2011) ;77: (22):1951–9. |
[11] | Zheng J , Chen S , Chen Y , Zhu M , Hong DH . A novel mutation in the PDZ-like motif of ZASP causes distal ZASP-related myofibrillar myopathy. Neuropathology. (2016) ;37: (1):45–51. |
[12] | D’Amico A , Benedetti S , Petrini S , Sambuughin N , Boldrini R , Menditto I , et al. Major myofibrillar changes in early onset myopathy due to de novo heterozygous missense mutation in lamin A/C gene. Neuromuscul Disord. (2005) ;15: (12):847–50. |
[13] | Ferreiro A , Ceuterick-de Groote C , Marks JJ , Goemans N , Schreiber G , Hanefeld F , et al. Desmin-related myopathy with mallory body–like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol. (2004) ;55: (5):676–86. |
[14] | Dalakas MC , Dagvadorj A , Goudeau B , Park K-Y , Takeda K , Simon-Casteras M , et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. (2003) ;13: (3):252–8. |
[15] | Wahbi K , Béhin A , Charron P , Dunand M , Richard P , Meune C , et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul Disord. (2012) ;22: (3):211–8. |
[16] | Olivé M , Armstrong J , Miralles F , Pou A , Fardeau M , Gonzalez L , et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord NMD. (2007) ;17: (6):443–50. |
[17] | Walter MC , Reilich P , Huebner A , Fischer D , Schröder R , Vorgerd M , et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain. (2007) ;130: (6):1485. |
[18] | Dagvadorj A , Olivé M , Urtizberea J-A , Halle M , Shatunov A , Bönnemann C , et al. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J Neurol. (2004) ;251: (2):143–9. |
[19] | Olivé M , Goldfarb L , Moreno D , Laforet E , Dagvadorj A , Sambuughin N , et al. Desmin-related myopathy: Clinical, electrophysiological, radiological, neuropathological and genetic studies. J Neurol Sci. (2004) ;219: (1-2):125–37. |
[20] | Selcen D , Engel AG . Myofibrillar myopathy caused by novel dominant negative αB-crystallin mutations. Ann Neurol. (2003) ;54: (6):804–10. |
[21] | Sacconi S , Féasson L , Antoine JC , Pécheux C , Bernard R , Cobo AM , et al. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Disord. (2012) ;22: (1):66–72. |
[22] | Reilich P , Schoser B , Schramm N , Krause S , Schessl J , Kress W , et al. The G154S mutation of the alpha-B crystallin gene (CRYAB) causes late-onset distal myopathy. Neuromuscul Disord. (2010) ;20: (4):255–9. |
[23] | Hauser MA , Horrigan SK , Salmikangas P , Torian UM , Viles KD , Dancel R , et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet. (2000) ;9: (14):2141–7. |
[24] | Olivé M , Odgerel Z , Martínez A , Poza JJ , Bragado FG , Zabalza RJ , et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul Disord. (2011) ;21: (8):533–42. |
[25] | Gamez J , Armstrong J , Shatunov A , Selva-O’Callaghan A , Dominguez-Oronoz R , Ortega A , et al. Generalized muscle pseudo-hypertrophy and stiffness associated with the myotilin Ser55Phe mutation: A novel myotilinopathy phenotype? J Neurol Sci. (2009) ;277: (1-2):167–71. |
[26] | Pénisson-Besnier I , Talvinen K , Dumez C , Vihola A , Dubas F , Fardeau M , et al. Myotilinopathy in a family with late onset myopathy. Neuromuscul Disord. (2006) ;16: (7):427–31. |
[27] | Olivé M , Goldfarb LG , Shatunov A , Fischer D , Ferrer I . Myotilinopathy: Refining the clinical and myopathological phenotype. Brain. (2005) ;128: (10):2315–26. |
[28] | Fischer D , Kley RA , Strach K , Meyer C , Sommer T , Eger K , et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology. (2008) ;71: (10):758–65. |
[29] | Kley RA , Hellenbroich Y , van der Ven PFM , Fürst DO , Huebner A , Bruchertseifer V , et al. Clinical and morphological phenotype of the filamin myopathy: A study of 31 German patients. Brain. (2007) ;130: (12):3250–64. |
[30] | Avila-Smirnow D , Gueneau L , Batonnet-Pichon L , Delort F , Bécane H.-M , Claeys K, et al. Cardiac arrhythmia and late-onset muscle weakness caused by a myofibrillar myopathy with unusual histopathological features due to a novel missense mutation in FLNC. Rev Neurol (Paris). (2016) ;172: (10):594–606. |
[31] | Luan X , Hong D , Zhang W , Wang Z , Yuan Y . A novel heterozygous deletion–insertion mutation (2695–2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscul Disord. (2010) ;20: (6):390–6. |
[32] | Plantié E , Migocka-Patrzałek M , Daczewska M , Jagla K . Model Organisms in the Fight against Muscular Dystrophy: Lessons from Drosophila and Zebrafish. Molecules. (2015) ;20: (4):6237–53. |
[33] | Tue NT , Shimaji K , Tanaka N , Yamaguchi M . Effect of αB-Crystallin on Protein Aggregation in Drosophila. J Biomed Biotechnol. (2012) ;2012: :252049. |
[34] | Benna C , Peron S , Rizzo G , Faulkner G , Megighian A , Perini G , et al. Post-transcriptional silencing of the Drosophila homolog of human ZASP: A molecular and functional analysis. Cell Tissue Res. (2009) ;337: (3):463–76. |
[35] | Ramspacher C , Steed E , Boselli F , Ferreira R , Faggianelli N , Roth S , et al. Developmental Alterations in Heart Biomechanics and Skeletal Muscle Function in Desmin Mutants Suggest an Early Pathological Root for Desminopathies. Cell Rep. (2015) ;11: (10):1564–76. |
[36] | Fujita M , Mitsuhashi H , Isogai S , Nakata T , Kawakami A , Nonaka I , et al. Filamin C plays an essential role in the maintenance of the structural integrity of cardiac and skeletal muscles, revealed by the medaka mutant zacro. Dev Biol. (2012) ;361: (1):79–89. |
[37] | Arndt V , Dick N , Tawo R , Dreiseidler M , Wenzel D , Hesse M , et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. (2010) ;20: (2):143–8. |
[38] | Ruparelia AA , Oorschot V , Vaz R , Ramm G , Bryson-Richardson RJ . Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol (Berl). (2014) ;128: (6):821–33. |
[39] | Joanne P , Chourbagi O , Hourde C , Ferry A , Butler-Browne G , Vicart P , et al. Viral-mediated expression of desmin mutants to create mouse models of myofibrillar myopathy. Skelet Muscle. (2013) ;3: (1):4. |
[40] | Kostareva A , Sjöberg G , Bruton J , Zhang S-J , Balogh J , Gudkova A , et al. Mice expressing L345P mutant desmin exhibit morphological and functional changes of skeletal and cardiac mitochondria. J Muscle Res Cell Motil. (2008) ;29: (1):25–36. |
[41] | Clemen C , Stöckigt F , Strucksberg K-H , Chevessier F , Winter L , Schütz J , et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol (Berl). (2015) ;129: (2):297–315. |
[42] | Chevessier F , Schuld J , Orfanos Z , Plank A-C , Wolf L , Maerkens A , et al. Myofibrillar instability exacerbated by acute exercise in filaminopathy. Hum Mol Genet. (2015) ;24: (25):7207–20. |
[43] | Garvey SM , Miller SE , Claflin DR , Faulkner JA , Hauser MA . Transgenic mice expressing the myotilin T57I mutation unite the pathology associated with LGMD1A and MFM. Hum Mol Genet. (2006) ;15: (15):2348–62. |
[44] | Wang X , Osinska H , Dorn GW , Nieman M , Lorenz JN , Gerdes AM , et al. Mouse model of desmin-related cardiomyopathy. Circulation. (2001) ;103: (19):2402–7. |
[45] | Mavroidis M , Panagopoulou P , Kostavasili I , Weisleder N , Capetanaki Y . A missense mutation in desmin tail domain linked to human dilated cardiomyopathy promotes cleavage of the head domain and abolishes its Z-disc localization. FASEB J. (2008) ;22: (9):3318–27. |
[46] | Wang X , Osinska H , Klevitsky R , Gerdes AM , Nieman M , Lorenz J , et al. Expression of R120G–αB-Crystallin Causes Aberrant Desmin and αB-Crystallin Aggregation and Cardiomyopathy in Mice. Circ Res. (2001) ;89: (1):84–91. |
[47] | Li Z , Ai T , Samani K , Xi Y , Tzeng H-P , Xie M , et al. A ZASP Missense Mutation, S196L, Leads to Cytoskeletal and Electrical Abnormalities in a Mouse Model of Cardiomyopathy. Circ Arrhythm Electrophysiol. (2010) ;3: (6):646–56. |
[48] | Jiao Q , Sanbe A , Zhang X , Liu J-P , Minamisawa S . αB-Crystallin R120G variant causes cardiac arrhythmias and alterations in the expression of Ca2+-handling proteins and endoplasmic reticulum stress in mice. Clin Exp Pharmacol Physiol. (2014) ;41: (8):589–99. |
[49] | Liu J , Chen Q , Huang W , Horak KM , Zheng H , Mestril R , et al. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. (2006) ;20: (2):362–4. |
[50] | Minoia M , Boncoraglio A , Vinet J , Morelli FF , Brunsting JF , Poletti A , et al. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-to-autophagy switch. Autophagy. (2014) ;10: (9):1603–21. |
[51] | Sanbe A , Osinska H , Saffitz JE , Glabe CG , Kayed R , Maloyan A , et al. Desmin-related cardiomyopathy in transgenic mice: A cardiac amyloidosis. Proc Natl Acad Sci U S A. (2004) ;101: (27):10132–6. |
[52] | Maloyan A , Sanbe A , Osinska H , Westfall M , Robinson D , Imahashi K , et al. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-b-crystallin desmin related cardiomyopathy. Circulation. (2005) ;112: (22):3451–61. |
[53] | Zheng Q , Su H , Ranek MJ , Wang X . Autophagy and p62 in Cardiac Proteinopathy. Circ Res. (2011) ;109: (3):296–308. |
[54] | Arimura T , Ishikawa T , Nunoda S , Kawai S , Kimura A . Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat. (2011) ;32: (12):1481–91. |
[55] | Lee J-H , Takahashi T , Yasuhara N , Inazawa J , Kamada S , Tsujimoto Y . Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene. (1999) ;18: (46):6183–90. |
[56] | Festa M , Del Valle L , Khalili K , Franco R , Scognamiglio G , Graziano V , et al. BAG3 protein is overexpressed in human glioblastoma and is a potential target for therapy. Am J Pathol. (2011) ;178: (6):2504–12. |
[57] | Franco R , Scognamiglio G , Salerno V , Sebastiani A , Cennamo G , Ascierto PA , et al. Expression of the anti-apoptotic protein BAG3 in human melanomas. J Invest Dermatol. (2012) ;132: (1):252–4. |
[58] | Rosati A , Bersani S , Tavano F , Dalla Pozza E , De Marco M , Palmieri M , et al. Expression of the antiapoptotic protein BAG3 Is a feature of pancreatic adenocarcinoma and its overexpression is associated with poorer survival. Am J Pathol. (2012) ;181: (5):1524–9. |
[59] | Andley UP , Hamilton PD , Ravi N , Weihl CC . A knock-in mouse model for the R120G mutation of αB-crystallin recapitulates human hereditary myopathy and cataracts. PLoS One. (2011) ;6: (3):e17671. |
[60] | Diokmetzidou A , Soumaka E , Kloukina I , Tsikitis M , Makridakis M , Varela A , et al. Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J Cell Sci. (2016) ;129: (20):3705–20. |
[61] | Winter L , Wittig I , Peeva V , Eggers B , Heidler J , Chevessier F , et al. Mutant desmin substantially perturbs mitochondrial morphology, function and maintenance in skeletal muscle tissue. Acta Neuropathol (Berl). (2016) ;132: :453–73. |
[62] | Chen B , Retzlaff M , Roos T , Frydman J . Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. (2011) ;3: (8):a004374. |
[63] | Ben-Gedalya T , Cohen E . Quality control compartments coming of age. Traffic. (2012) ;13: (5):635–42. |
[64] | Dunlop RA , Brunk UT , Rodgers KJ . Oxidized proteins: Mechanisms of removal and consequences of accumulation. IUBMB Life. (2009) ;61: (5):522–7. |
[65] | Silverstein MG , Ordanes D , Wylie AT , Files DC , Milligan C , Presley TD , et al. Inducing muscle heat shock protein 70 improves insulin sensitivity and muscular performance in aged mice. J Gerontol A Biol Sci Med Sci. (2015) ;70: (7):800–8. |
[66] | Blair LJ , Sabbagh JJ , Dickey CA . Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin Ther Targets. (2014) ;18: (10):1219–32. |
[67] | Wang X , Klevitsky R , Huang W , Glasford J , Li F , Robbins J . αB-Crystallin modulates protein aggregation of abnormal desmin. Circ Res. (2003) ;93: (10):998–1005. |
[68] | Arrigo A-P , Simon S , Gibert B , Kretz-Remy C , Nivon M , Czekalla A , et al. Hsp27 (HspB1) and αB-crystallin (HspB5) as therapeutic targets. FEBS Lett. (2007) ;581: (19):3665–74. |
[69] | Sanbe A , Daicho T , Mizutani R , Endo T , Miyauchi N , Yamauchi J , et al. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS One. (2009) ;4: (4):e5351. |
[70] | Winter L , Staszewska I , Mihailovska E , Fischer I , Goldmann WH , Schröder R , et al. Chemical chaperone ameliorates pathological protein aggregation in plectin-deficient muscle. J Clin Invest. (2014) ;124: (3):1144–57. |
[71] | Nédellec P , Edling Y , Perret E , Fardeau M , Vicart P . Glucocorticoid treatment induces expression of small heat shock proteins in human satellite cell populations: Consequences for a desmin-related myopathy involving the R120G alpha B-crystallin mutation. Neuromuscul Disord. (2002) ;12: (5):457–65. |
[72] | Janué A , Olivé M , Ferrer I . Oxidative stress in desminopathies and myotilinopathies: A link between oxidative damage and abnormal protein aggregation. Brain Pathol. (2007) ;17: (4):377–88. |
[73] | Rajasekaran NS , Connell P , Christians ES , Yan L-J , Taylor RP , Orosz A , et al. Human alphaB-crystallin mutation causes Oxido-reductive Stress and protein aggregation cardiomyopathy in R120GCryAB Mice. Cell. (2007) ;130: (3):427–39. |
[74] | Kannan S , Muthusamy VR , Whitehead KJ , Wang L , Gomes AV , Litwin SE , et al. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc Res. (2013) ;100: (1):63–73. |
[75] | Maloyan A , Osinska H , Lammerding J , Lee RT , Cingolani OH , Kass DA , et al. Biochemical and mechanical dysfunction in a mouse model of desmin-related myopathy. Circ Res. (2009) ;104: (8):1021–8. |
[76] | Sanbe A , Marunouchi T , Yamauchi J , Tanonaka K , Nishigori H , Tanoue A . Cardioprotective effect of nicorandil, a mitochondrial ATP-sensitive potassium channel opener, prolongs survival in HSPB5 R120G transgenic mice. PLoS One. (2011) ;6: (4):e18922. |
[77] | Bhola PD , Letai A . Mitochondria—judges and executioners of cell death sentences. Mol Cell. (2016) ;61: (5):695–704. |
[78] | Weisleder N , Taffet GE , Capetanaki Y . Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc Natl Acad Sci U S A. (2004) ;101: (3):769–74. |
[79] | Maloyan A , Sayegh J , Osinska H , Chua BH , Robbins J . Manipulation of death pathways in desmin related cardiomyopathy. Circ Res. (2010) ;106: (9):1524–32. |
[80] | Yerbury JJ , Ooi L , Dillin A , Saunders DN , Hatters DM , Beart PM , et al. Walking the tightrope: Proteostasis and neurodegenerative disease. J Neurochem. (2016) ;137: (4):489–505. |
[81] | Ferrer I , Olivé M . Molecular pathology of myofibrillar myopathies. Expert Rev Mol Med. (2008) ;10: :e25. |
[82] | Chen Q , Liu J-B , Horak KM , Zheng H , Kumarapeli ARK , Li J , et al. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. (2005) ;97: (10):1018–26. |
[83] | Ravikumar B , Sarkar S , Davies JE , Futter M , Garcia-Arencibia M , Green-Thompson ZW , et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. (2010) ;90: (4):1383–435. |
[84] | Tannous P , Zhu H , Johnstone JL , Shelton JM , Rajasekaran NS , Benjamin IJ , et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. (2008) ;105: (28):9745–50. |
[85] | Bhuiyan MS , Pattison JS , Osinska H , James J , Gulick J , McLendon PM , et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. (2013) ;123: (12):5284–97. |
[86] | Hyttinen JMT , Amadio M , Viiri J , Pascale A , Salminen A , Kaarniranta K . Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev. (2014) ;18: :16–28. |
[87] | Michalsen A , Li C . Fasting therapy for treating and preventing disease – current state of evidence. Forsch Komplementärmedizin Res Complement Med. (2013) ;20: (6):444–53. |
[88] | Riehle C , Abel ED . Insulin regulation of myocardial autophagy. Circ J. (2014) ;78: (11):2569–76. |
[89] | Pauly M , Daussin F , Burelle Y , Li T , Godin R , Fauconnier J , et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol. (2012) ;181: (2):583–92. |
[90] | Grumati P , Coletto L , Sandri M , Bonaldo P . Autophagy induction rescues muscular dystrophy. Autophagy. (2011) ;7: (4):426–8. |
[91] | Eghtesad S , Jhunjhunwala S , Little SR , Clemens PR . Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol Med. (2011) ;17: (9-10):917–24. |
[92] | Rubinsztein DC , Codogno P , Levine B . Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. (2012) ;11: (9):709–30. |
[93] | Zheng H , Tang M , Zheng Q , Kumarapeli ARK , Horak KM , Tian Z , et al. Doxycycline attenuates protein aggregation in cardiomyocytes and improves survival of a mouse model of cardiac proteinopathy. J Am Coll Cardiol. (2010) ;56: (17):1418–26. |
[94] | Maloyan A , Gulick J , Glabe CG , Kayed R , Robbins J . Exercise reverses preamyloid oligomer and prolongs survival in αB-crystallin-based desmin-related cardiomyopathy. Proc Natl Acad Sci. (2007) ;104: (14):5995–6000. |
[95] | Segard B-D , Delort F , Bailleux V , Simon S , Leccia E , Gausseres B , et al. N-acetyl-L-cysteine prevents stress-induced desmin aggregation in cellular models of desminopathy. PLoS One. (2013) ;8: (10):e76361. |
[96] | Cabet E , Batonnet-Pichon S , Delort F , Gausserès B , Vicart P , Lilienbaum A . Antioxidant treatment and induction of autophagy cooperate to reduce desmin aggregation in a cellular model of desminopathy. Wiche G, editor. PLoS One. (2015) ;10: (9):e0137009. |
Figures and Tables
Fig.1
Clinical overview of MFM alterations. Each color represent the features found in one pathology. Legend color is detailed in the box localized in up right corner. The text in italic correspond to specific pattern visualized in MRI studies. Alpha-B-crystallinopathies shares common features with desmin. So only specific pattern of alpha-B-crystallinopathies are indicated.
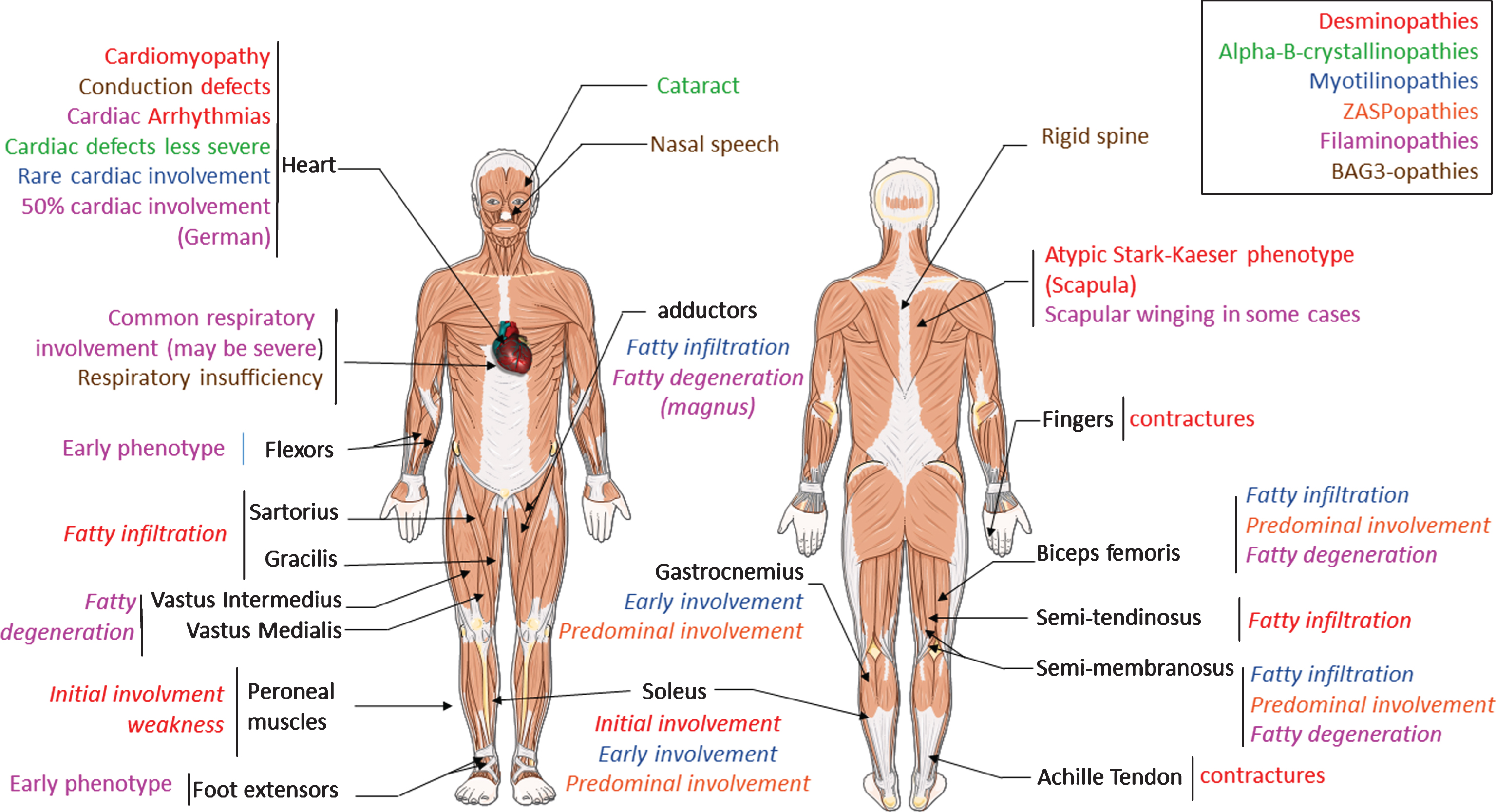
Fig.2
Altered desmin subcellular distribution in skeletal muscle tissue. A- 11-week-old mice (C57bl6J) were injected in tibialis anterior (TA) with associated-adeno-virus (AAV) encoding Myc-tagged wild-type (WT) or D399Y desmin. One month later, TA were extracted and frozen, and Myc immunostaining on transverse or longitudinal cryosections was performed to detect exogenous desmin. D399Y shows a perinuclear accumulation related to human desmin aggregation observed in patients. Scale bar = 20 μm. B- DesKI-R405W mice (homologous to R406W mutation in human, C57bl6N) were analyzed at 3 months old. Briefly, TA were extracted and frozen and desmin immunostaining was performed on transverse or longitudinal cryosections to detect endogenous desmin in homozygous (Hom), heterozygous (Het) or wild-type (WT) mice. A typical desminopathy staining pattern with predominantly subsarcolemmal and also sarcoplasmic desmin-positive protein aggregates can be observed in homozygous tissue. Moreover, a regular cross-striated desmin pattern is present in WT mice, and mainly preserved in HET mice, whereas Hom mice show an altered striation and fiber shapes. Scale bar = 20 μm.
Fig.3
Scheme of dysregulated functions in myofibrillar myopathies and potential therapeutic treatments.
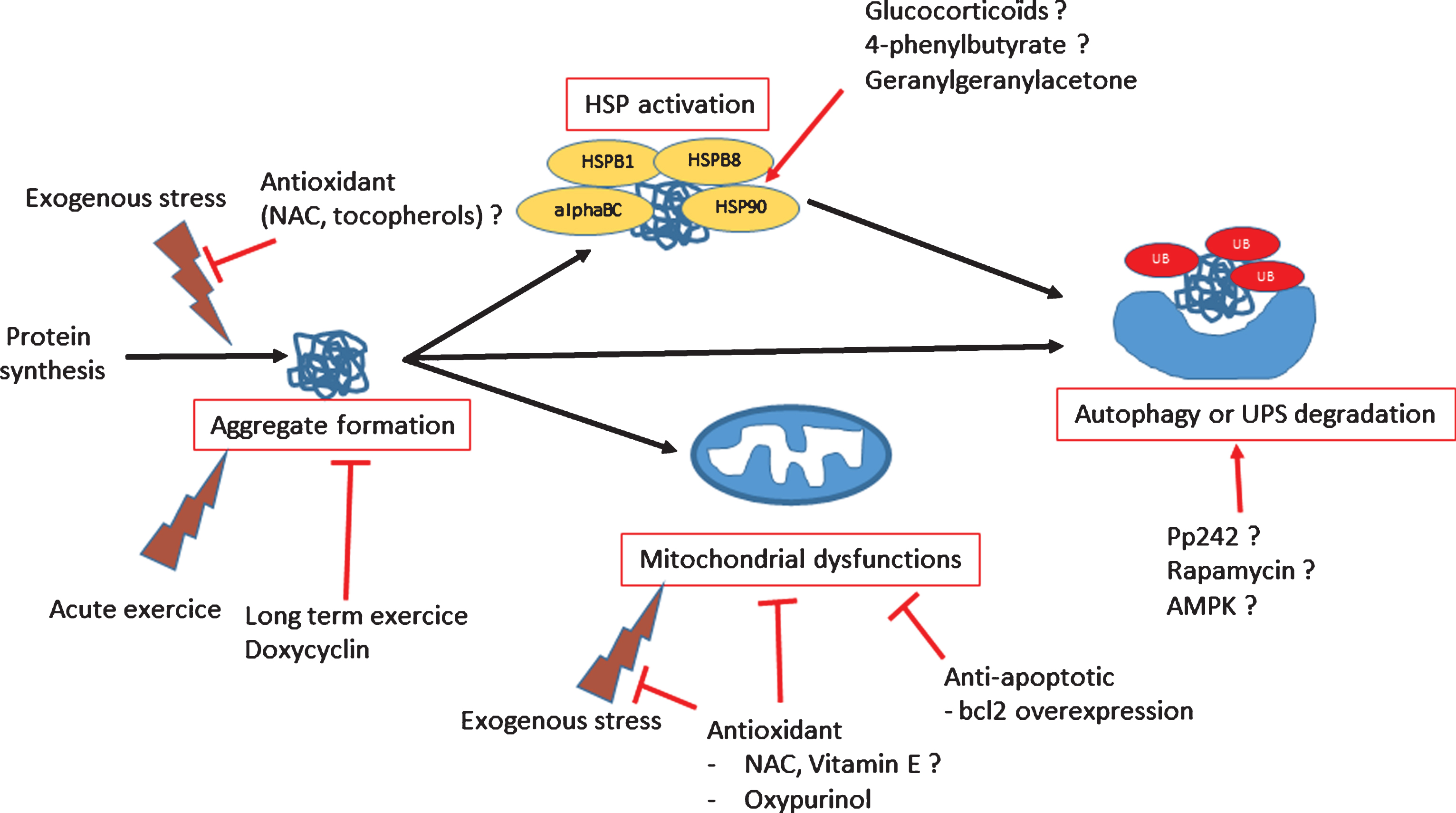
Table 1
Mammalian MFM models with skeletal, and cardiac phenotypes
| A/ SKELETAL MUSCLE | ||||||||||
| Models | Pathologies | Modification type | Expression | Aggregation | myofibrillar disorganization | contractile function | recovery from fatigue | Mitochondria | Nota bene | Publication |
| Mouse | DES | AAV-injection, p.R406W | skeletal muscle (high) | + (tibialis anterior) | + | decreased | N/D | N/D | 39 | |
| AAV-injection, p.E413K | skeletal muscle (high) | + (tibialis anterior) | + | decreased | N/D | N/D | 39 | |||
| AAV-injection, p.D399Y | skeletal muscle (high) | + (tibialis anterior) | + | N/D | N/D | N/D | unpublished | |||
| p.L345P-HA, promoter desmin | all muscle types (low) | – | decreased in soleus(not in EDL) | impaired | vacuolization, giant, decreased cristae density (Soleus) | 40 | ||||
| p.R405W-knock-In | all muscle types (low) | + (age dependent) | N/D | N/D | N/D | N/D | unpublished | |||
| p.R349P-knock-In | all muscle types (low) | + age dependent (soleus not in Quadriceps nor Gastrocnemius) | + (soleus not in Quadriceps nor Gastrocnemius) | Decreased in Soleus (not in EDL) | N/D | Accumulation or depletion, giant, Increased COX, SDH activities (soleus not in Quadriceps nor Gastrocnemius) | Homozygous only | 41 | ||
| FLNC | W2710X-knock-In | all muscle types (low) | lesions FLNC, different to classical MFM aggregates | + | decreased (grip strength..) | N/D | 42 | |||
| MYOT | T57I-transgenic | skeletal muscle (high) | + | + | Lost in EDL Soleus-Diaphragm not affected | N/D | 43 | |||
| CRYAB | p.R120G-knock-in | all muscle types (low) | + | + | Decreased | N/D | N/D | 60 | ||
| B/ CARDIAC FEATURES | ||||||||||||
| Models | Pathologies | Modification type | Expression | Aggregation | cardiomyopathy | conduction defects | fibrose | left ventricular wall hypertrophy | Mitochondria | Calcium levels | ER Stress | Publication |
| Mouse | DES | p.L345P-HA, promoter desmin | all muscle types (low) | – | + | N/D | + | + | vacuolization, giant, decreased cristae density | Increased | 40 | |
| p.R405W-knock-In | all muscle types (low) | N/D | N/D | N/D | N/D | N/D | N/D | N/D | N/D | unpublished | ||
| p.R349P-knock-In | all muscle types (low) | + age dependent | + | + | + | N/D | N/D | N/D | N/D | 41 | ||
| p.I451M-transgenic | Heart (low) | – but desmin mutant mislocalization | + | N/D | few | N/D | N/D | N/D | N/D | 45 | ||
| D7-Des, transgenic (alphaMHC) | Heart (high) | + | + | + | none | + | N/D | N/D | N/D | 44;67 | ||
| FLNC | W2710X-knock-In | all muscle types (low) | N/D | N/D | N/D | N/D | N/D | N/D | N/D | N/D | 42 | |
| CRYAB | p.R120G-transgenic | Heart (mild) | + | + | N/D | N/D | N/D | impaired | calcium regulation impaired | + | 46; 48; 51; 52 | |
| CRYAB | p.R120G-knock-in | all muscle types (low) | N/D | N/D | N/D | N/D | N/D | N/D | N/D | N/D | 60 | |
| ZASP | p.S196L-transgenic | Heart (high) | N/D | + | + | mild | N/D | N/D | impaired | N/D | 47 | |
| C/ OTHER FUNCTIONS | ||||||||
| Models | Pathologies | Modification type | Expression | PQC | other dysfunctions | Publication | ||
| UPS | Autophagy | Apoptosis | Cataract | |||||
| Mouse | DES | D7-Des, transgenic (alphaMHC) | Heart (high) | impaired | none | 49 | ||
| CRYAB | p.R120G-transgenic | Heart (mild) | impaired | flux increased | up-regulation cyt-C/BAX3 activated caspase3, down-regulation BCL-2 | none | 52 | |
| CRYAB | p.R120G-knock-in | all muscle types (low) | N/D | N/D | N/D | + | 59 | |
| Rat | BAG3 | R128W et L462-overexpression | Neonatal cardiomyocytes | N/D | N/D | increased sensitivity to serum depletion | none | 54 |


