
Sarcomere Dysfunction in Nemaline Myopathy
Abstract
Nemaline myopathy (NM) is among the most common non-dystrophic congenital myopathies (incidence 1:50.000). Hallmark features of NM are skeletal muscle weakness and the presence of nemaline bodies in the muscle fiber. The clinical phenotype of NM patients is quite diverse, ranging from neonatal death to normal lifespan with almost normal motor function. As the respiratory muscles are involved as well, severely affected patients are ventilator-dependent. The mechanisms underlying muscle weakness in NM are currently poorly understood. Therefore, no therapeutic treatment is available yet.
Eleven implicated genes have been identified: ten genes encode proteins that are either components of thin filament, or are thought to contribute to stability or turnover of thin filament proteins. The thin filament is a major constituent of the sarcomere, the smallest contractile unit in muscle. It is at this level of contraction – thin-thick filament interaction – where muscle weakness originates in NM patients.
This review focusses on how sarcomeric gene mutations directly compromise sarcomere function in NM. Insight into the contribution of sarcomeric dysfunction to muscle weakness in NM, across the genes involved, will direct towards the development of targeted therapeutic strategies.
INTRODUCTION
Nemaline myopathy (NM) is among the most common non-dystrophic congenital myopathies [1] (eg. incidence 1:50.000 in Finland [2]). Hallmark features of NM are skeletal muscle weakness and the presence of nemaline bodies in the muscle fiber. The clinical phenotype of NM patients is quite diverse, ranging from neonatal death to normal lifespan with almost normal motor function [3]. As the respiratory muscles are involved as well, severely affected patients are ventilator-dependent [4]. NM is a progressive muscle disorder of which the mechanisms underlying weakness are currently poorly understood [5]. Knowledge of the underlying mechanisms may help determine treatment strategies.
The discovery of genes that are implicated in NM was an impetus to unravel the pathophysiology of muscle weakness in this debilitating disease. In 1995, it was discovered that a mutation in the gene encoding alpha-tropomyosin – a constituent of the sarcomeric thin filament- results in NM [6]. Currently, eleven implicated genes have been identified: ten genes encode proteins that are either components of the skeletal muscle thin filament (Fig. 1), including nebulin (NEB), skeletal muscle alpha-actin1 (ACTA1), beta-tropomyosin 2 (TPM2), alpha-tropomyosin 3 (TPM3), troponin T type 1 (TNNT1), cofilin-2 (CFL2), and leiomodin-3 (LMOD3), or are thought to contribute to stability or turnover of thin filament proteins, such as kelch repeat and BTB (POZ) Domain Containing 13 (KBTBD13), kelch-likefamily members 40 (KLHL40) and -41 (KLHL41) [3, 7–10]. The eleventh gene encodes myosin 18B, of which the function is unknown [11]. The thin filament is a major constituent of the sarcomere, the smallest contractile unit in muscle. It is at this level of contraction – thin-thick filament interaction – where muscle weakness originates in NM patients.
Fig.1
Schematic of skeletal muscle, from the level of the whole muscle to the sarcomeric thin filament. Note that the schematic of the thin filament illustrates the protein products of all thin filament genes implicated in nemaline myopathy.
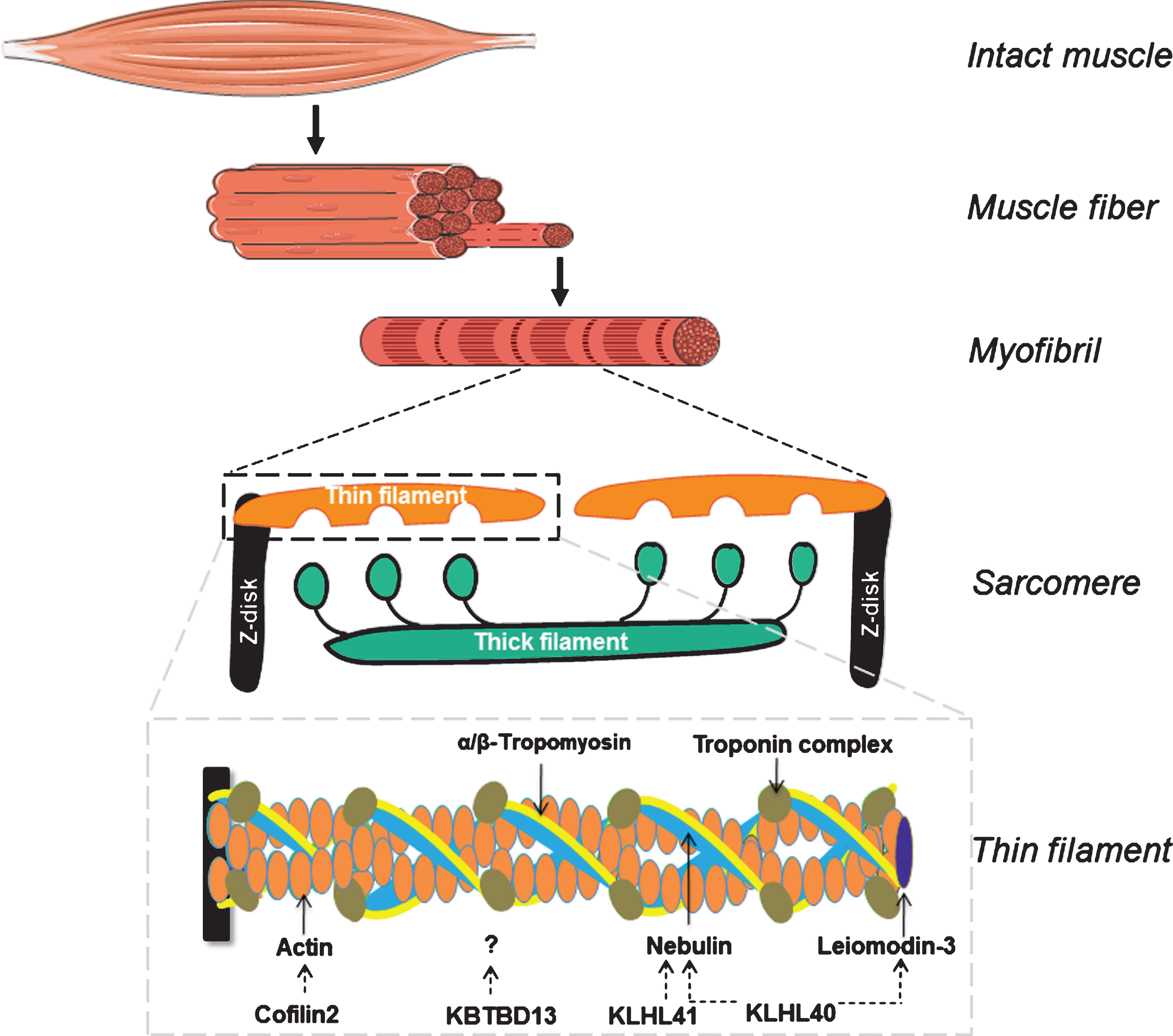
The sarcomere has four main structural components: the Z-disks, the thick filament, the thin filament and titin, a molecular spring that connects the three previous components. In between the Z-disks are the thick and the thin filament (Fig. 1). The thin filament is composed of an actin-based backbone that is decorated with proteins that are involved in (1) the regulation of thin filament length and in (2) the regulation of acto-myosin interaction. Actin monomers contain binding sites for the thick filament, which is mainly composed of myosin molecules. Myosin, a molecular motor, has a high affinity for actin and aims for interaction, i.e. cross-bridge formation. That way, myosin heads pull on the thin filaments, moving the Z-disks towards each other. Interaction between actin and myosin is strictly regulated by various proteins, of which the troponin complex, nebulin and tropomyosin are the key players [12, 13]. In addition to their role in the regulation of contraction, sarcomeric proteins are also involved in specifying the length of the thin filament and the thick filament. Note that thin-thick filament overlap is an important determinant of force generation: it determines the number of cross-bridges that can be formed. Thus, considering the important role of the proteins that they encode, mutations in sarcomere genes can have a major impact on muscle function.
A key question in the NM field is: what are the mechanisms underlying muscle weakness? Sarcomeric gene mutations result in altered gene expression and protein levels, which can directly compromise sarcomere function. Sarcomere dysfunction can lead to secondary changes in the muscle’s ultrastructure that further contribute to muscle weakness. Examples of changes in the ultrastructure of nemaline myopathic muscle are: the presence of nemaline bodies [14, 15], Z-disks that lose integrity (i.e. display streaming and widening) [16], abnormal myofibrillar and mitochondrial organization and muscle fiber atrophy [15]. This review focusses on how sarcomeric gene mutations directly compromise sarcomere function. To investigate to what extent sarcomere function is compromised and thus contributes to the observed muscle weakness in NM, it is crucial to investigate the contractile performance of nemaline myopathic muscle. In 2001 the first NM mouse model was generated by transgene expression of Tpm3M9R [17], which allowed the investigation of the effect of an NM mutation on intact muscle function. Next, techniques were developed to isolate individual muscle fibers from frozen patient biopsies and study their contractile function [18] (an overview of some of these experimental techniques is shown in Fig. 2). In addition, fish models have been engineered, that phenocopy observations from mouse and human tissue [19–21]. These studies revealed that sarcomere dysfunction contributes to muscle weakness in NM, and they provided insight into the mechanisms that are responsible for the loss of function.
Fig.2
Overview of experimental techniques employed on muscle fibers isolated from patients’ biopsies. A) Single muscle fibers are isolated from a muscle biopsy and treated with Triton-X, a detergent that permeabilizes the membraneous structures and leaves the sarcomeres within the muscle fibers intact. The permeabilized muscle fiber is mounted between a force transducer and a length motor, activated by exogenous calcium, and the generated force is measured. B) By activating a muscle fiber at incremental sarcomere lengths, the sarcomere length-dependence of force can be determined. The shape of the force-sarcomere length relation provides important information on the length of thin and thick filaments; SLopt: sarcomere length at which maximal force is generated. For instance, in patients with nebulin deficiency SLopt is shifted leftwards. C) By rapidly releasing and restretching the muscle fiber, while activated, the rate constant of force development is determined (left panel shows the imposed change in fiber length; right panel the force response). This rate constant, Ktr, reflects the kinetics of cross bridge attachment and detachment. D) By exposing the muscle fiber to incremental calcium solutions (pCa = the negative logarithm of [Ca2 +]), the calcium sensitivity of force is determined. A rightward shift of the force-pCa relation, i.e. a decreased pCa50, indicates a reduced calcium sensitivity of force.
![Overview of experimental techniques employed on muscle fibers isolated from patients’ biopsies. A) Single muscle fibers are isolated from a muscle biopsy and treated with Triton-X, a detergent that permeabilizes the membraneous structures and leaves the sarcomeres within the muscle fibers intact. The permeabilized muscle fiber is mounted between a force transducer and a length motor, activated by exogenous calcium, and the generated force is measured. B) By activating a muscle fiber at incremental sarcomere lengths, the sarcomere length-dependence of force can be determined. The shape of the force-sarcomere length relation provides important information on the length of thin and thick filaments; SLopt: sarcomere length at which maximal force is generated. For instance, in patients with nebulin deficiency SLopt is shifted leftwards. C) By rapidly releasing and restretching the muscle fiber, while activated, the rate constant of force development is determined (left panel shows the imposed change in fiber length; right panel the force response). This rate constant, Ktr, reflects the kinetics of cross bridge attachment and detachment. D) By exposing the muscle fiber to incremental calcium solutions (pCa = the negative logarithm of [Ca2 +]), the calcium sensitivity of force is determined. A rightward shift of the force-pCa relation, i.e. a decreased pCa50, indicates a reduced calcium sensitivity of force.](https://content.iospress.com:443/media/jnd/2017/4-2/jnd-4-2-jnd160200/jnd-4-jnd160200-g002.jpg)
Here, we provide an overview of the current findings pertaining to the key question postulated above. Insight into the contribution of sarcomeric dysfunction to muscle weakness in NM, across the genes involved, will direct towards the development of targeted therapeutic strategies.
NEBULIN
The most frequently affected gene in NM is NEB, encoding nebulin, a multifunctional giant (800 kDa) that was discovered in 1982 [22]. A single nebulin molecule runs along the thin filament, with its C-terminus anchoring in the Z-disk and the N-terminus close to the thin filament capping proteins tropomodulin and leiomodin [18, 23–25]. Nebulin has a highly modular structure that is organized in a super-repeat structure. The super-repeats, consisting of 7 repeats, each span 43 nanometers. Typically, with its 43 nanometers, one super-repeat spans one regulatory unit: seven actin monomers, one tropomyosin molecule and one troponin complex [26, 27]. Hence, nebulin’s structure suggests a close interaction between nebulin and these thin filament proteins. Therefore, a mutation in NEB can have severe impact on sarcomeric function. To investigate the role of nebulin in sarcomeric structure and function, nebulin-deficient mouse models have been generated [28–31]. These models reveal that muscle that lacks nebulin displays severe muscle weakness, both at the intact whole muscle level [28] – a level of contraction that involves both calcium-handling and sarcomeric performance - and in permeabilized muscle fibers – here force generation is mainly determined by the contractile performance of the sarcomeric proteins [29].
Note that permeabilized muscle fibers are a large collection of interconnected myofibrils. As the magnitude of muscle weakness in permeabilized fibers of nebulin-deficient mice is similar to both that of permeabilized myofibrils from these mice [30] and to that observed in intact whole mouse muscle, it is suggested that sarcomeric weakness is the main contributor to muscle weakness in NM patients with nebulin deficiency. In addition, it indicates that changes in calcium-handling in nebulin-deficient muscle do not have a major role in the pathogenesis of muscle weakness [32]. Over the past ten years, nebulin’s role in various aspects of sarcomeric structure and function have been studied [33, 34], roles which, if impaired, all can contribute to sarcomeric weakness in nebulin-based NM. Here, we review the pathomechanisms underlying nebulin-based sarcomeric dysfunction.
Thin filament length dysregulation
The amount of overlap between the thin and the thick filament determines the number of cross-bridges that can be formed. Accordingly, force depends on sarcomere length, with increasing force as the overlap between thick and thin filaments increases (up to a sarcomere length of ∼2,6 μm, i.e. the ascending limb of the force-sarcomere length relation), and with decreasing force at longer sarcomere lengths as the overlap between thick and thin filaments decreases (i.e., the descending limb). Hence, appropriate length of the thin filament is important for muscle strength. The length of the thick filament is conserved throughout species and muscle types at 1.6 μm. The length of the thin filament varies between 1.1 – 1.3 μm among muscles types and species, and is regulated by various proteins of which nebulin is one of the key players [35]. Nebulin-deficient mouse muscle has shorter thin filaments [28, 29], which – especially at a longer sarcomere length - results in less overlap between the thick and thin filament, i.e. a lower number of cross-bridges that can be formed. As a consequence, nebulin-deficient muscle has a narrower functional length range and operates at a shorter optimal length for force generation compared to wildtype muscle [36]. This observation was confirmed in various nebulin-deficient mouse models, including a model with the NebΔexon55 mutation [30], a frequently found mutation in NM [37], and in a conditional nebulin-deficient mouse model [31]. In line with this observation in nebulin-deficient mouse muscle, muscle fibers from NM patients with mutations in NEB, leading to nebulin deficiency, have shorter thin filament lengths and shorter optimal sarcomere length for force generation (Fig. 2B, leftward shift of SLopt) [18, 38]. It should be noted that some studies reported preserved force-sarcomere length relation in muscle fibers from NM patient with nebulin mutations [38, 39], and it was speculated that the extent to which thin filament length is affected in nebulin-NM is mutation-specific and/or depends on the level of healthy nebulin protein. As the muscle fibers from nebulin-based NM patients with unaffected thin filament lengths did display sarcomeric weakness, other mechanisms contribute to muscle weakness. An important aspect of sarcomere function that depends on nebulin is cross-bridge cycling kinetics.
Altered cross-bridge cycling kinetics
At a given overlap between thick and thin filaments, the amount of force that can be generated depends on the force generated per cross-bridge and the fraction of strongly attached cross-bridges. Cross-bridge cycling kinetics (i.e. the rate of acto-myosin attachment and detachment) determine both the fraction of strongly bound cross-bridges and the force per cross-bridge by modulating the time span of strongly bound cross-bridges. The fraction of strongly bound cross-bridges can be measured in permeabilized muscle fibers. The rate of acto-myosin detachment is proportional to the ATP consumption of permeabilized muscle fibers during force generation (i.e. tension cost), as for myosin to detach from actin one ATP molecule is required. Next, the rate of force redevelopment (Ktr) reflects both the acto-myosin attachment and detachment rate. Hence, changes in the attachment rate can be deduced when Ktr (attachment rate + detachment rate) and tension cost (detachment rate) are known. Studies in permeabilized nebulin-deficient mouse muscle revealed that Ktr is reduced and tension cost is increased, suggesting that the fraction of strongly bound cross-bridges is lower in nebulin-deficient mouse muscle, a reduction which contributes to the lower force generating capacity [31, 40–42]. The force generation per strongly bound cross-bridge is estimated by measuring the active stiffness of a permeabilized muscle fiber. The unaffected active tension/active stiffness ratio in nebulin-deficient mouse muscle suggests that the force per cross-bridge is not altered in the absence of nebulin [40, 41]. Studies in muscle fibers isolated from biopsies of NM patients with mutations in NEB provided similar results. Various studies revealed reductions in Ktr [15, 39, 43, 44] and an increase in tension cost [43, 44], indicating a lower fraction of strongly bound cross-bridges. Next a study that included one biopsy of an NM patient with nebulin mutations, reported no changes in the force generated per strongly bound cross-bridge. Hence, mutations in NEB result in alterations in the cross-bridge cycling kinetics, i.e. a lower number of strongly bound cross-bridges. Thus, alterations in cross-bridge cycling kinetics contribute to the observed lower maximal active tension in muscle fibers of NM patients with NEB mutations.
Lower calcium-sensitivity of force generation
Daily life activities such as walking and biking typically are performed at sub-maximal levels of activation. When permeabilized muscle fibers of nebulin-deficient mouse tissue are exposed to incremental calcium levels, lower forces (relative to maximum force) are generated compared to wildtype muscle fibers [30, 31, 40, 42]. Thus, more calcium is needed to generate a given level of force, meaning that nebulin-deficient mouse muscle is less sensitive to calcium (see also Fig. 2D). These observations are also reported in multiple studies on permeabilized muscle fibers that were isolated from biopsies of NM patients with mutations in NEB [43, 45, 46]. A study on one patient’s biopsy reported no change in the calcium-sensitivity of force [39]. Note that this patient had normal nebulin levels whereas the patients reported in the other studies had nebulin levels that were less than 25% of normal levels. The molecular mechanisms underlying nebulin’s effect on the calcium sensitivity of force likely include its interaction with tropomyosin and actin, which is important for proper muscle function [47]. Thus, nebulin plays an important role in the regulation of calcium-sensitivity of force. Both the location of the mutation in NEB and the nebulin protein level determine the magnitude and/or the direction of the effect on the calcium-sensitivity of force.
In conclusion, sarcomeric weakness contributes to muscle weakness in nebulin-based NM; the extent of the contribution depends on the specific mutation and nebulin protein level.
SKELETAL MUSCLE ALPHA-ACTIN1
Skeletal muscle alpha actin-1 (actin) is a molecule of 42 kD. Actin monomers polymerize to filamentous actin, the backbone of the thin filament – note that actin is Greek for ‘ray’. Actin possesses binding spots for myosin heads to enable cross-bridge formation. The generation of two mouse models improved the understanding of the sarcomeric basis of weakness in actin-based NM [48, 49]. Both mouse models harbor a mutation that resembles a mutation found in severely affected NM patients: (1) Acta1H40Y and (2) Acta1D286G. Treadmill studies reveal that t Acta1H40Y mice exhibit significant deficits in the average distance daily travelled, the average speed and the maximal speed compared to wildtype mice [49]. Fore-arm grip strength was reduced in Acta1H40Y mice [48, 50]. In vivo force measurements confirmed a decrease in the absolute and the specific force production – i.e. the absolute force production normalized to the cross-sectional area of the muscle - in the gastrocnemius muscle of Acta1H40Y mice. In addition, t Acta1H40Y mouse muscle had an increased energy cost, which was assumed to be either related to disturbed mitochondrial function or sarcomeric dysfunction [51].
To further investigate the origin of muscle weakness, intact muscles were isolated and activated in vitro by field stimulation. The oxidative soleus muscle and the glycolytic extensor digitorum longus (EDL) muscle of Acta1H40Y mice show large force deficits both in the maximal absolute and the maximal specific force production [48]. No changes in excitation-contraction coupling were observed in Acta1H40Y mice, thus the force deficit is sarcomere-based [52].
Similar studies were performed in Acta1D286G mice. Reduced maximal specific force in permeabilized muscle fibers of Acta1D286G mice indicate that sarcomere dysfunction contributes to muscle weakness in these animals [49]. But the magnitude of sarcomere dysfunction was less than in Acta1H40Y mice and its effect on in vivo muscle strength and exercise capacity was less pronounced [49, 51].
Altered cross-bridge cycling kinetics
The observed sarcomeric weakness at saturating calcium levels can either arise from a lower number of strongly bound cross-bridges and/or from a depressed force generation per individual cross-bridge. As studies on permeabilized muscle fibers of both Acta1H40Y and Acta1D286G mice revealed that the strain of per myosin head was not affected, it was hypothesized that a lower number of strongly bound cross-bridges is responsible for sarcomere dysfunction in both mouse models [53, 54]. In line with findings of permeabilized nebulin-deficient muscle fibers of NM patients and mice, it is speculated that the acto-myosin attachment rate is reduced in permeabilized muscle fibers of both Acta1H40Y and Acta1D286G mice, and that the detachment rate is increased (note that an increased detachment rate is supported by the increased tension cost that was found in vivo). A decreased acto-myosin attachment rate combined with an increased detachment rate result in a shorter time span for the myosin head to generate a forceful power stroke, thereby depressing maximal contractile performance.
Surprisingly, similar studies in permeabilized muscle fibers from patients with a different mutation in actin, i.e. ACTA1P352S revealed that sarcomere function is increased compared to muscle fibers of healthy subjects. The origin of this force increase is that this specific mutation on actin enhances the strain of individual cross-bridges [52]. Note that this patient displays muscle weakness, which was speculated to be caused by atrophy and/or changes in excitation-contraction coupling. Similarly, a case report describes an NM patient harboring an ACTAL328A mutation but with a hypertonic phenotype. Although speculative, this specific mutation induces a conformational charge change in actin, thereby favoring the acto-myosin equilibrium towards the ‘on’ state [55]. Hence, changes in cross-bridge cycling kinetics caused by ACTA1 mutations can cause either a loss or a gain of function.
Thin filament length dysregulation
To date, one study investigated the force-sarcomere length relationship in muscle fibers of NM patients with various mutations in the ACTA1 gene [38]. This study reported sarcomeric weakness in muscle fibers of all 14 actin-based NM patients that were studied, a weakness which in 10 patients was partly explained by shorter thin filament lengths. Thus, thin filament length dysregulation is a cause of muscle weakness in actin-based NM, but the extent of the contribution is mutation-specific. Note that the lack of a distinct genotype-phenotype correlation in actin-based myopathies has been reported previously [56].
ALPHA- AND BETA-TROPOMYOSIN
Tropomyosin rotates along the axis of the actin backbone, thereby controlling the availability of myosin binding sites on actin – note that ‘tropomyosin’ in Greek means: that which is turned in muscle. Mutations in the TPM2 and -3 genes, both encoding skeletal muscle tropomyosins, can result in congenital myopathies, such as nemaline myopathy, core-rod myopathies, cap disease, congenital fiber-type disproportion and distal arthrogryposes. Three states of tropomyosin are described, corresponding to the position of tropomyosin along the thin filament. These states depend on the presence of cytosolic calcium. In the blocked state no calcium is present and tropomyosin sterically blocks myosin heads binding to actin. In the closed state, calcium has entered the cytosol and has bound to the troponin complex. As a consequence, tropomyosin unlocks and partially unblocks the binding of myosin to actin. In the open state, tropomyosin shifts further away through initial binding of myosin thereby exposing adjacent binding sites on actin to enable cross-bridge formation. This way, the thin filament is cooperatively activated by cross-bridge-induced cross-bridge formation [12, 57]. To induce muscle relaxation, calcium is pumped back into the sarcoplasmic reticulum, which requires ATP. As tropomyosin is an important player in directing the acto-myosin concert, mutations in tropomyosin can significantly impact muscle contractility.
Altered cross-bridge cycling kinetics
Studies on permeabilized muscle fibers from patients harboring mutations in TPM3 reported lower maximal active tension and slower cross-bridge cycling kinetics [58–62]. In addition, tension cost was increased in these muscle fibers [60]. Slower cross-bridge cycling kinetics (lower Ktr) combined with an increase in tension cost, indicate a decreased acto-myosin attachment rate and an increased acto-myosin detachment rate. In line with this, X-ray diffraction studies on a muscle biopsy of a patient harboring the beta-tropomyosinR133W showed that during activation, beta-tropomyosin movement is limited, reducing acto-myosin interaction [63]. Thus, a lower number of strongly bound cross-bridges contributes to sarcomeric muscle weakness in patients with mutations in alpha- and beta-tropomyosin.
Thin filament length regulation
No changes in thin filament length have been reported thus far: an in vivo study on the first NM mouse model [17] – a mouse carrying the Tpm3M9R mutation -, found no changes in the force-muscle length relation [64], findings which were confirmed in permeabilized muscle fibers from this mouse model [65]. Two studies on biopsies of patients with mutations in TPM3 and TPM2, revealed no changes in thin filament length and in the force-sarcomere length relation [38, 60]. Thus, changes in thin filament length are unlikely to contribute to muscle weakness in tropomyosin-based myopathies.
Changes in the calcium-sensitivity of force generation
Whereas in muscle fibers from patients with mutations in NEB only reductions in the calcium-sensitivity of force generation have been observed, in muscle fibers from patients with mutations in TPM2 and TPM3 both increased and decreased calcium-sensitivity of force generation have been reported [59–62, 66, 67]. The increased calcium-sensitivity of force generation provides a mechanical basis for the muscle stiffness that some patients experience [62, 67, 68]. In vitro motility assays and biochemical assays confirm the existence of mutation-specific effects on the direction of the calcium-sensitivity of force generation. In brief, mutation-specific effects that favor the open state of tropomyosin by destabilizing the blocked state result in an increase in the calcium-sensitivity of force generation, and those effects that impede tropomyosin movement decrease the calcium-sensitivity of force [69–72].
Thus, sarcomeric dysfunction contributes to the muscles weakness in NM patients that harbor mutations in TPM2 and TPM3. The diversity in clinical phenotypes - hypotonic versus hypertonic - is at least partly explained by mutation-specific effects on tropomyosin movement on the thin filament.
TROPONIN
The troponin complex is composed of three troponin subunits: troponin-C (TnC), troponin-I (TnI) and troponin-T (TnT). These subunits interact with each other and have specific tasks: TnC binds calcium, TnI is primarily responsible for maintaining tropomyosin in the blocked state and TnT modulates the closed and open state of tropomyosin [12]. To date, mutations in TNNT1 (encoding slow skeletal TnT) have been implicated in NM [73–77] and TNNT3 mutations (fast skeletal TnT) and TNNI2 (fast skeletal TnI) in distal arthrogryposis [78–82]. NM patients with TNNT1 mutations have severe muscle weakness. Intact whole muscle of slow-skeletal TnT-deficient mice are less tolerant to fatigue [83]. This is mainly due to a fiber-type shift towards fast glycolytic fibers. Intrinsic changes in contractile protein function in slow-skeletal muscle fibers of these mice have not been reported. One study has investigated the contractile function of permeabilized muscle fibers of a NM patient with a TNNT1 mutation [38]. This study revealed sarcomeric weakness in TNNT1-NM muscle fibers, weakness which was not caused by changes in thin filament length. Thus, other mechanisms are at play, such as a reduced ability to bind totropomyosin [84].
KELCH PROTEINS: KBTBD13, KLHL40 AND KLHL41
Recently, a new class of genes was discovered to be implicated in NM: genes from the kelch family encoding kelch-like family members 40 (KLHL40) and -41 and kelch repeat and BTB (POZ) Domain Containing 13 (KBTBD13) [7–9]. Their protein products interact with the Cullin E3 ubiquitin-ligase, a core component of the ubiquitin-proteasome pathway [85]. This pathway is involved in the regulation of protein turnover. Kelch proteins might function as a substrate adaptor for specific (sarcomeric) proteins to form a functional complex with the Cullin E3 ubiquitin-ligase. KLHL40 binds to the thin filament proteins nebulin and leiomodin-3 and to Cullin E3, thereby stabilizing them [86, 87]. KLHL41 also interacts with nebulin and Cullin E3 [9]. Although conclusive evidence is lacking, it is presumed that also KLHL41 plays a role in stabilizing nebulin by forming a functional ubiquitin-complex through interaction with Cullin E3 [9]. For KBTBD13, the specific target proteins for Cullin E3 interaction are not known yet. Insight into the contribution of sarcomeric weakness to muscle weakness in patients harboring kelch mutations has just started to evolve. Here, we summarize the current knowledge on sarcomeric weakness caused by mutations in these genes.
Two Klhl40-deficient mouse models have been generated [86, 88]. Maximal active tension was severely reduced at the whole intact muscle level. The ultrastructure of Khlh40-deficient muscle is compromised, without changes in thin filament length. Similar findings were observed in permeabilized muscle fibers from an NM patient with a mutation in KLHL40 [38]. Thus, other sarcomeric changes account for the weakness. A Klhl41-deficient zebrafish revealed impaired motor function accompanied by sarcomeric misalignment [9]. In line with these findings, contractile studies on human muscle fibers from a patient harboring a mutation in KLHL40 revealed contractile weakness and sarcomeric disorganization. However, thin filament length regulation was not affected in this patient [38]. Note that caution should be warranted interpreting the results on thin filament length assessment, as only one biopsy for both gene cohorts was studied. For KBTBD13 no animal models have been published yet. Muscle weakness in NM patients with mutations in KBTBD13 [89] is at least partly caused by decreased contractility of sarcomeres without changes in thin filament length [38].
COFILIN-2
In 2007, CFL2, the gene encoding cofilin-2 was first implicated in NM [90]. A homozygous missense mutation was identified in two affected siblings in a large family of Middle Eastern origin. In 2012 [91], a second family of Middle Eastern origin with NM and a CFL2 mutation was identified. CFL2 encodes a skeletal muscle–specific isoform of cofilin, which localizes to the thin filaments. Cofilin-2 has been postulated to be essential for the regulation of the dynamics of actin polymerization. The mechanisms underlying muscle weakness in patients with CFL2 mutations are unknown. Studies on Cfl2-deficient mice show that sarcomeric ultrastructure and thin filament length is preserved at birth. However, postnatally, sarcomeric structure was severely disrupted, a process that originated from the Z-line and was accompanied by actin aggregation and lack of actin depolymerization activity [92]. In addition, sarcomeric integrity worsened by reduced interlinking of adjacent myofibrils [93]. Thus, the identified CFL2-mutations in NM patients might reduce the protein levels of cofilin-2, resulting in reduced depolymerization of actin filaments. This will have significant impact on muscle fiber contractility and induce thin filament breakdown. Indeed, muscle fibers of patients with CFL2 mutations display aggregates of thin filaments. Unfortunately, so far no studies have reported the effects of mutations in CFL2 on muscle fiber contractility.
LEIOMODIN-3
Recently, mutations in L MOD3 were identified as a new cause of NM: affected children have severe weakness at birth, requiring mechanical ventilation and tube feeding to survive and usually die soon after birth. LMOD3 encodes leiomodin-3, a protein expressed in cardiac and skeletal muscle, and whose function is unknown. Mutations in LMOD3 most often result in absence of leiomodin-3 from patient muscle, which is associated with myofiber atrophy and replacement of muscle tissue with connective tissue. Loss of leiomodin-3 in patient muscle resulted in shortening and disorganization of thin filaments, and in weakness that is more pronounced at longer muscle fiber lengths [10]. Functional effects of loss of leiomodin-3 on myosin-actin interactions are unknown yet. Two leiomodin-3-defiecient mouse models are available to further investigate [87, 94] sarcomeric function in the absence of leiomodin-3.
DISCUSSION
NM is a debilitating muscle disease, for which currently no treatment exists. This paper’s aim was to review the current knowledge on sarcomeric weakness in NM. Below, we summarize our findings and propose therapeutic strategies that target thesarcomere.
Genotype-phenotype correlations in NM?
NM is a disease that affects the skeletal muscle thin filament, either by changes in its length and/or its interaction with the thick filament. The nature of the sarcomeric dysfunction depends on the implicated gene. For instance, contribution of thin filament length dysregulation to muscle weakness occurs in muscle harboring mutations in NEB, ACTA1 and LMOD3, but not in the other implicated genes [10, 18, 38]. Muscle of patients with NEB mutations have slower cross-bridge cycling kinetics, whereas muscle of patients with TPM2 mutations can display faster cross-bridge cycling kinetics. Furthermore, muscle of NM patients with mutations in ACTA1 and TPM3 have faster or slower cross-bridge cycling kinetics depending on the location of the mutation. Similar unequivocal observations were reported regarding the calcium-sensitivity of force generation: mutations in TPM3 can both increase and decrease the calcium-sensitivity of force generation have been reported [60–62, 67]. Thus, the concept arises that sarcomeric weakness contributes to muscle weakness in NM and that the nature of the contribution depends on the specific gene as well as the specific mutation, and the resulting effect at the protein level.
The sarcomere as a therapeutic target
The establishment of sarcomeric weakness as an important contributor to muscle weakness in NM provided an impetus to test the ability of drugs that target the sarcomere to augment muscle force in NM. Troponin activators slow the dissociation of calcium from the troponin complex, thereby increasing the calcium-sensitivity of force generation. Studies on nebulin-deficient muscle fibers of NM patients and mice show that a fast skeletal troponin activator increases muscle force at submaximal calcium levels [30, 42, 45]. This finding is of clinical relevance, as these submaximal calcium levels reflect physiological activity levels: i.e. levels at which daily life activities such as breathing and walking occur. It is hypothesized that administration of a fast skeletal troponin activator will lower fatigue during daily life activities and thus contribute to the quality of life in NM patients with NEB mutations. Recent studies in healthy controls reveal that a fast skeletal troponin activator amplifies the force response of the sarcomere in vivo [95]. Furthermore, positive effects on respiratory function are reported in patients with amyotrophic lateral sclerosis and myasthenia gravis during administration of a fast skeletal troponin activator [96, 97]. As skeletal muscle is composed of both slow and fast fibers, targeting both fast and slow skeletal muscle fibers should benefit NM patients. Unfortunately, no gain of function was observed with Levosimendan – a clinically-approved slow troponin activator- in muscle fibers of NM patients with mutations in NEB [46]. Besides Levosimendan, other slow troponin activators are available for pre-clinical and clinical use [98]. Therefore, future studies should investigate whether these slow troponin activators have the potential to augment muscleforce in NM.
Next to troponin, also myosin is a candidate to augment muscle force in NM. A recent study revealed that increasing the expression of the MYL4 gene in a NM mouse model harboring the ActaH40Ymutation induced the expression of MyLC1a/emb, an embryonic myosin light chain isoform [99]. This isoform increases the force-generating capacity of myosin. Induction of MYL4 was achieved by transgene delivery. Increased expression of MyLC1a/emb increased maximal force generation in permeabilized muscle fibers.
A drug called omecamtiv mecarbil directly targets myosin function and increases force production [100]. This drug augments the activity of the myosin motor by inducing a faster rate of weakly to strongly bound cross-bridges. As slow skeletal myosin isoforms are analogous to cardiac muscle isoforms, modulating myosin motor activity by omecamtiv mecarbil has potential to increase sarcomeric performance in skeletal muscle as well.
A novel therapeutic target is modulating the structural organization of sarcomeres. Conditional nebulin knock-out mice compensate for shorter thin filament lengths by increasing the number of sarcomeres in series [38]. As a result, the optimal muscle length for force generation in intact muscle was comparable to wild-type mice. Thus, the addition of sarcomeres in series allowed the muscles to operate at a shorter sarcomere length, a length closer to their optimal sarcomere length. Adapting the number of sarcomeres in series to compensate for changes in thin filament length constitutes a novel control mechanism in muscle. Whether this control mechanism is also at play in human diseased muscle is unknown, but the elucidation of this control mechanism might have implications for treatment strategies. In rodents, active stretching of muscle stimulates the addition of sarcomeres in series [101]. Thus, active stretching of muscles of patients with shorter thin filament lengths might be an interesting direction to explore to stimulate the addition of sarcomeres in series and alleviate muscle weakness.
Finally, an interesting approach to restore sarcomeric performance caused by reduced sarcomeric protein levels is to express the cardiac isoform of the implicated protein in skeletal muscle. Nowak and colleagues succeeded to express cardiac actin in postnatal skeletal muscle of Acta1-deficient mice, which resulted in restoration of grip strength and locomotor activity [102]. Note that Acta1-deficient mice normally die by day 9 after birth. Expressing cardiac actin also improved life span in the Acta1D286G mice [103]. However, in the Acta1H40Y mouse model, expression of cardiac actin in postnatal skeletal muscle did not improve survival. Thus, this therapeutic approach is promising, at least for specific mutations in the ACTA1 gene.
In addition to strategies that target the sarcomeres, strategies aimed at targeting other muscle structures might restore muscle strength in NM. Positive effects on muscle strength and endurance have been reported upon dietary L-tyrosine supplementation in an NM mouse model and in patients [104, 105]. Although promising, the mechanism responsible for the improvement in muscle performance upon L-tyrosine supplementation is not known yet. Second, a recent study in NM mice revealed that administration of an inhibitor of myostatin signaling – note that myostatin is a suppressor of muscle hypertrophy – resulted in improved muscle size and a prolonged lifespan [50]. Third, as NM is a genetic disease, strategies aimed at delivery of the cDNA of NM genes that might be amenable to viral delivery, or those that aim at correcting the mutations themselves are of high interest for the NM field [106, 107]. However, these techniques require further optimization before safety and efficacy is guaranteed forclinical use.
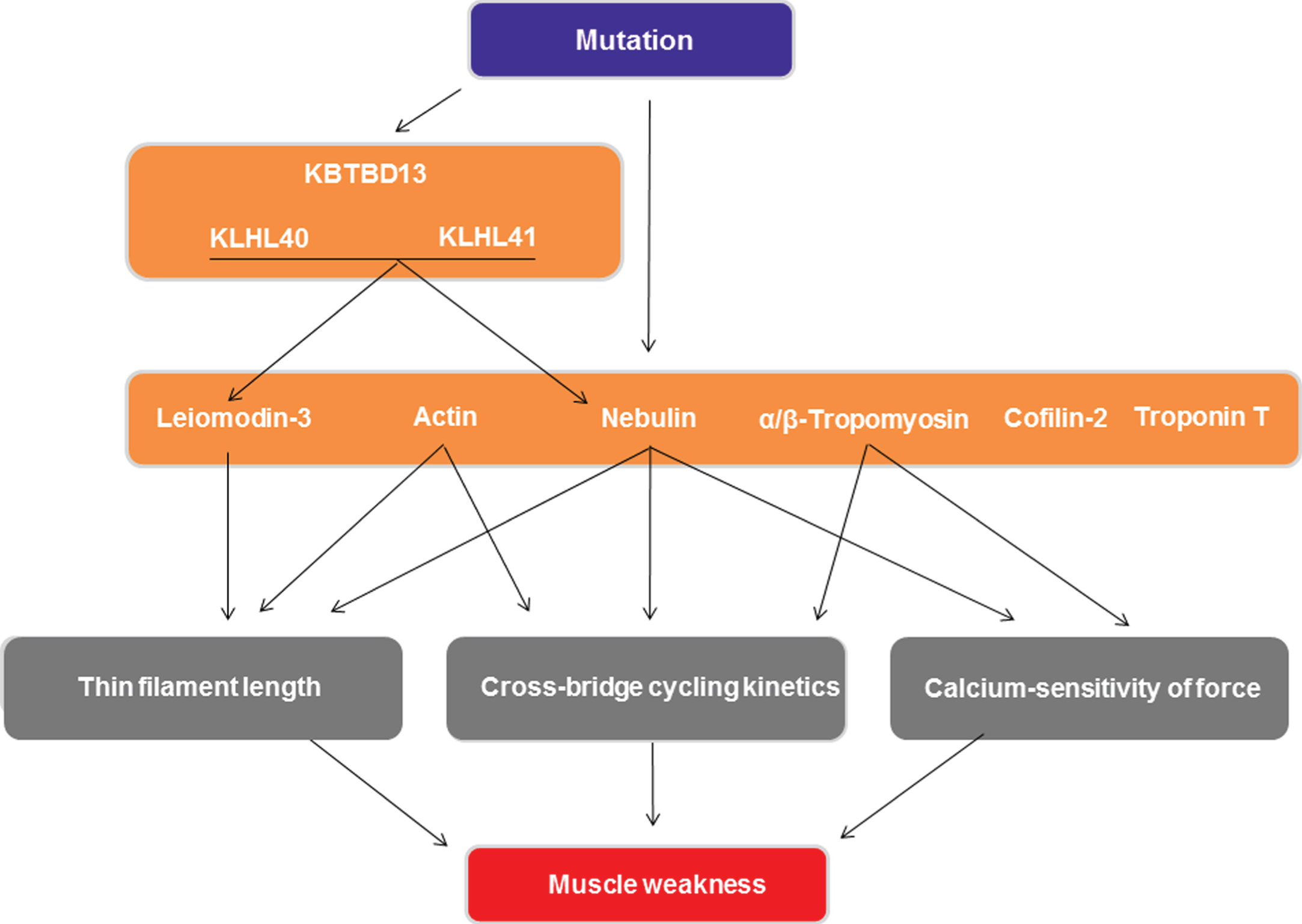
In conclusion, this review focused on the various aspects of sarcomeric weakness that contribute to muscle weakness in NM. Mechanisms that can contribute to muscle weakness are changes in thin filament length, in cross-bridge cycling kinetics and in the calcium-sensitivity of force generation (see Fig. 3). The magnitude and direction of these changes depend on the specific mutation and on the healthy protein level. As the sarcomere is primarily involved in the pathomechanisms underlying NM muscle weakness, it is an interesting therapeutic target to combat this weakness. Promising therapeutic targets to augment muscle strength in NM are troponin activation, myosin modulation, the addition of sarcomeres in series and expressing cardiac proteins in postnatal skeletal muscle.
Fig.3
Schematic overview of the mechanisms by which mutations in the genes implicated in nemaline myopathy cause muscle weakness. Only mechanisms involving sarcomere contractility are shown.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
Grant support: CACO: NWO-VIDI grant (016.126.319).
REFERENCES
[1] | Colombo I , Scoto M , Manzur AY , Robb SA , Maggi L , Gowda V , Cullup T , Yau M , Phadke R , Sewry C , Jungbluth H , Muntoni F . Congenital myopathies: Natural history of a large pediatric cohort. Neurology. (2015) ;84: (1):28–35. |
[2] | Wallgren-Pettersson C . Congenital nemaline myopathy: A longitudinal study, vol. 30. Academic dissertation. Helsinki: The Finnish Society of Sciences and Letters, Commentationes Physico-Mathematicae 111/Dissertationes; (1990) . |
[3] | Sanoudou D , Beggs AH . Clinical and genetic heterogeneity in nemaline myopathy-a disease of skeletal muscle thin filaments. Trends Mol Med. (2001) ;7: (8):362–8. |
[4] | North KN , Laing NG , Consortium I . Nemaline myopathy: Currentconcepts. The ENMC International Consortium and Nemaline Myopathy. J Med Genet. (1997) ;34: (9):705–13. |
[5] | North KN , Laing NG , Wallgren-Pettersson C . Nemaline myopathy: Current concepts. The ENMC International Consortium and Nemaline Myopathy. J Med Genet. (1997) ;34: (9):705–13. |
[6] | Laing NG , Wilton SD , Akkari PA , Dorosz S , Boundy K , Kneebone C , Blumbergs P , White S , Watkins H , Love DR . A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy. Nat Genet. (1995) ;9: (1):75–9. |
[7] | Sambuughin N , Yau KS , Olivé M , Duff RM , Bayarsaikhan M , Lu S , Gonzalez-Mera L , Sivadorai P , Nowak KJ , Ravenscroft G , Mastaglia FL , North KN , Ilkovski B , Kremer H , Lammens M , van Engelen BGM , Fabian V , Lamont P , Davis MR , Laing NG , Goldfarb LG . Dominantmutations in KBTBD13, a member of the BTB/Kelch family, causenemaline myopathy with cores. Am J Hum Genet. (2010) ;87: (6):842–7. |
[8] | Ravenscroft G , Miyatake S , Lehtokari V-L , Todd EJ , Vornanen P , Yau KS , Hayashi YK , Miyake N , Tsurusaki Y , Doi H , Saitsu H , Osaka H , Yamashita S , Ohya T , Sakamoto Y , Koshimizu E , Imamura S , Yamashita M , Ogata K , Shiina M , Bryson-Richardson RJ , Vaz R , Ceyhan O , Brownstein CA , Swanson LC , Monnot S , Romero NB , Amthor H , Kresoje N , Sivadorai P , Kiraly-Borri C , Haliloglu G , Talim B , Orhan D , Kale G , Charles AK , Fabian VA , Davis MR , Lammens M , Sewry CA , Manzur A , Muntoni F , Clarke NF , North KN , Bertini E , Nevo Y , Willichowski E , Silberg IE , Topaloglu H , Beggs AH , Allcock RJN , Nishino I , Wallgren-Pettersson C , Matsumoto N , Laing NG . Mutations in KLHL40 Are a Frequent Cause of Severe Autosomal-Recessive Nemaline Myopathy. Am J Hum Genet. (2013) ;93: (1):6–18. |
[9] | Gupta VA , Ravenscroft G , Shaheen R , Todd EJ , Swanson LC , Shiina M , Ogata K , Hsu C , Clarke NF , Darras BT , Farrar MA , Hashem A , Manton ND , Muntoni F , North KN , Sandaradura SA , Nishino I , Hayashi YK , Sewry CA , Thompson EM , Yau KS , Brownstein CA , Yu TW , Allcock RJN , Davis MR , Wallgren-Pettersson C , Matsumoto N , Alkuraya FS , Laing NG , Beggs AH . Identification of KLHL41 Mutations Implicates BTB-Kelch-Mediated Ubiquitination as an Alternate Pathway to Myofibrillar Disruption in Nemaline Myopathy. Am J Hum Genet. (2013) ;93: (6):1108–17. |
[10] | Yuen M , Sandaradura SA , Dowling JJ , Kostyukova AS , Moroz N , Quinlan KG , Lehtokari V-L , Ravenscroft G , Todd EJ , Ceyhan-Birsoy O , Gokhin DS , Maluenda J , Lek M , Nolent F , Pappas CT , Novak SM , D’Amico A , Malfatti E , Thomas BP , Gabriel SB , Gupta N , Daly MJ , Ilkovski B , Houweling PJ , Davidson AE , Swanson LC , Brownstein CA , Gupta VA , Medne L , Shannon P , Martin N , Bick DP , Flisberg A , Holmberg E , Van den Bergh P , Lapunzina P , Waddell LB , Sloboda DD , Bertini E , Chitayat D , Telfer WR , Laquerrière A , Gregorio CC , Ottenheijm CAC , Bönnemann CG , Pelin K , Beggs AH , Hayashi YK , Romero NB , Laing NG , Nishino I , Wallgren-Pettersson C , Melki J , Fowler VM , MacArthur DG , North KN , Clarke NF . Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. (2014) ;124: (11). |
[11] | Malfatti E , Böhm J , Lacène E , Beuvin M , Guy Brochier NB , Romero NB , Laporte J . A Premature Stop Codon in MYO18B isAssociated with Severe Nemaline Myopathy with Cardiomyopathy. J Neuromuscul Dis. (2015) ;2: (3):219–27. |
[12] | Gordon AM , Homsher E , Regnier M . Regulation of Contraction in Striated Muscle. Physiol Rev. (2000) ;80: (2):853–924. |
[13] | Lukoyanova N , VanLoock MS , Orlova A , Galkin VE , Wang K , Egelman EH . Each actin subunit has three nebulin binding sites: Implications for steric blocking. Curr Biol. (2002) ;12: (5):383–8. |
[14] | Ryan MM , Ilkovski B , Strickland CD , Schnell C , Sanoudou D , Midgett C , Houston R , Muirhead D , Dennett X , Shield LK , De Girolami U , Iannaccone ST , Laing NG , North KN , Beggs a H . Clinical course correlates poorly with muscle pathology in nemaline myopathy. Neurology. (2003) ;60: (4):665–73. |
[15] | Malfatti E , Lehtokari V-L , Böhm J , De Winter JM , Schäffer U , Estournet B , Quijano-Roy S , Monges S , Lubieniecki F , Bellance R , Viou MT , Madelaine A , Wu B , Taratuto AL , Eymard B , Pelin K , Fardeau M , Ottenheijm CA , Wallgren-Pettersson C , Laporte J , Romero NB . Muscle histopathology in nebulin-related nemaline myopathy:Ultrastructural findings correlated to disease severity andgenotype. Acta Neuropathol Commun. (2014) ;2: (1):44. |
[16] | Tonino P , Pappas CT , Hudson BD , Labeit S , Gregorio CC , Granzier H . Reduced myofibrillar connectivity and increased Z-disk width in nebulin-deficient skeletal muscle. J Cell Sci. (2010) ;123: (Pt 3):384–91. |
[17] | Corbett MA . A mutation in alpha-tropomyosinslow affects muscle strength, maturation and hypertrophy in a mouse model for nemaline myopathy. Hum Mol Genet. (2001) ;10: (4):317–28. |
[18] | Ottenheijm CAC , Witt CC , Stienen GJ , Labeit S , Beggs AH , Granzier H . Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet. (2009) ;18: (13):2359–69. |
[19] | Sztal TE , Zhao M , Williams C , Oorschot V , Parslow AC , Giousoh A , Yuen M , Hall TE , Costin A , Ramm G , Bird PI , Busch-Nentwich EM , Stemple DL , Currie PD , Cooper ST , Laing NG , Nowak KJ , Bryson-Richardson RJ . Zebrafish models for nemaline myopathy reveal a spectrum of nemaline bodies contributing to reduced muscle function. Acta Neuropathol. (2015) ;130: (3):389–406. |
[20] | Berger J , Tarakci H , Berger S , Li M , Hall TE , Arner A , Currie PD . Loss of Tropomodulin4 in the zebrafish mutant träge causes cytoplasmic rod formation and muscle weakness reminiscent of nemaline myopathy. Dis Model Mech. (2014) ;7: (12):1407–15. |
[21] | Telfer WR , Nelson DD , Waugh T , Brooks SV , Dowling JJ . Neb: A zebrafish model of nemaline myopathy due to nebulin mutation. Dis Model Mech. (2012) ;5: (3):389–96. |
[22] | Wang K . Structural and Contractile Proteins Part B: The Contractile Apparatus and the Cytoskeleton. Methods in enzymology. Elsevier; (1982) . 264–74. (Methods in Enzymology; vol. 85). |
[23] | Labeit S , Kolmerer B . The complete primary structure of humannebulinand its correlation to muscle structure. J Mol Biol. (1995) ;248: (2):308–15. |
[24] | Wang K , Knipfer M , Huang Q-Q , van Heerden A , Hsu LC-L , Gutierrez G , Quian X-L , Stedman H . Human Skeletal Muscle Nebulin Sequence Encodes a Blueprint for Thin Filament Architecture: Sequence Motifs and Affinity Profiles of Tandem Repeats and Terminal SH3. J Biol Chem. (1996) ;271: (8):4304–14. |
[25] | Castillo A , Nowak R , Littlefield KP , Fowler VM , Littlefield RS . A nebulin ruler does not dictate thin filament lengths. Biophys J. (2009) ;96: (5):1856–65. |
[26] | Ogut O , Hossain MM , Jin J-P . Interactions between nebulin-like motifs and thin filament regulatory proteins. J Biol Chem. (2003) ;278: (5):3089–97. |
[27] | Jin JP , Wang K . Nebulin as a giant actin-binding template protein in skeletal muscle sarcomere Interaction of actin and cloned human nebulin fragments. FEBS Lett. (1991) ;281: (1-2):93–6. |
[28] | Bang M-L , Li X , Littlefield R , Bremner S , Thor A , Knowlton KU , Lieber RL , Chen J . Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J Cell Biol. (2006) ;173: (6):905–16. |
[29] | Witt CC , Burkart C , Labeit D , McNabb M , Wu Y , Granzier H , Labeit S . Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J. (2006) ;25: (16):3843–55. |
[30] | Ottenheijm CAC , Buck D , de Winter JM , Ferrara C , Piroddi N , Tesi C , Jasper JR , Malik FI , Meng H , Stienen GJM , Beggs AH , Labeit S , Poggesi C , Lawlor MW , Granzier H . Deleting exon 55 from the nebulin gene induces severe muscle weakness in a mouse model for nemaline myopathy. Brain. (2013) ;136: (Pt 6):1718–31. |
[31] | Li F , Buck D , De Winter J , Kolb J , Meng H , Birch C , Slater R , Escobar YN , Smith JE , Yang L , Konhilas J , Lawlor MW , Ottenheijm C , Granzier HL . Nebulin deficiency in adult muscle causes sarcomere defects and muscle-type dependent changes in trophicity–novel insights in nemaline myopathy. Hum Mol Genet. (2015) ;24: (18):5219–33. |
[32] | Ottenheijm CAC , Fong C , Vangheluwe P , Wuytack F , Babu GJ , Periasamy M , Witt CC , Labeit S , Granzier H . Sarcoplasmic reticulum calcium uptake and speed of relaxation are depressed in nebulin-free skeletal muscle. FASEB J. (2008) ;22: (8):2912–9. |
[33] | Labeit S , Ottenheijm CAC , Granzier H . Nebulin, a major player in muscle health and disease. FASEB J. (2011) ;25: (3):822–9. |
[34] | Ottenheijm CAC , Granzier H , Labeit S . The sarcomeric protein nebulin: Another multifunctional giant in charge of muscle strength optimization. Front Physiol. (2012) ;3: :37. |
[35] | Littlefield R , Fowler VM . Measurement of thin filament lengths by distributed deconvolution analysis of fluorescence images. Biophys J. (2002) ;82: (5):2548–64. |
[36] | Gokhin D , Bang M . Reduced thin filament length in nebulin-knockout skeletal muscle alters isometric contractile properties. Am J Resp Crit Care. (2009) ;296: :1123–32. |
[37] | Anderson S , Ekstein J , Donnelly M , Keefe E , Toto N , LeVoci L , Rubin B . Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet. (2004) ;115: (3):185–90. |
[38] | Winter JM de , Joureau B , Lee E-J , Kiss B , Yuen M , Gupta VA , Pappas CT , Gregorio CC , Stienen GJM , Edvardson S , Wallgren-Pettersson C , Lehtokari V-L , Pelin K , Malfatti E , Romero NB , Engelen van BG , Voermans NC , Donkervoort S , Bönnemann CG , Clarke NF , Beggs AH , Granzier H , Ottenheijm CAC . Mutation-specific effects on thin filament length in thin filament myopathy. Ann Neurol. (2016) ;79: (6):959–69. |
[39] | Ochala J , Lehtokari V-L , Iwamoto H , Li M , Feng H-Z , Jin J-P , Yagi N , Wallgren-Pettersson C , Pénisson-Besnier I , Larsson L . Disrupted myosin cross-bridge cycling kinetics triggers muscleweakness in nebulin-related myopathy. FASEB J. (2011) ;25: (6):1903–13. |
[40] | Chandra M , Mamidi R , Ford S , Hidalgo C , Witt CC , Ottenheijm CA , Labeit S , Granzier H . Nebulin alters cross-bridge cycling kineticsand increases thin filament activation: A novel mechanism forincreasing tension and reducing tension cost. J Biol Chem. (2009) ;284: (45):30889–96. |
[41] | Bang M-L , Caremani M , Brunello E , Littlefield R , Lieber RL , Chen J , Lombardi V , Linari M . Nebulin plays a direct role in promoting strong actin-myosin interactions. FASEB J. (2009) ;23: (12):4117–25. |
[42] | Lee E-J , De Winter JM , Buck D , Jasper JR , Malik FI , Labeit S , Ottenheijm CA , Granzier H . Fast skeletal muscle troponin activation increases force of mouse fast skeletal muscle and ameliorates weakness due to nebulin-deficiency. Ervasti JM , editor. PLoS One. (2013) ;8: (2):e55861. |
[43] | Ottenheijm CAC , Hooijman P , DeChene ET , Stienen GJ , Beggs AH , Granzier H . Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol. (2010) ;170: (2):334–43. |
[44] | Lawlor MW , Ottenheijm CA , Lehtokari V-L , Cho K , Pelin K , Wallgren-Pettersson C , Granzier H , Beggs AH . Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet Muscle. (2011) ;1: (1):23. |
[45] | de Winter JM , Buck D , Hidalgo C , Jasper JR , Malik FI , Clarke NF , Stienen GJM , Lawlor MW , Beggs AH , Ottenheijm CAC , Granzier H . Troponin activator augments muscle force in nemaline myopathy patients with nebulin mutations. J Med Genet. (2013) ;50: (6):383–92. |
[46] | de Winter JM , Joureau B , Sequeira V , Clarke NF , van der Velden J , Stienen GJ , Granzier H , Beggs AH , Ottenheijm CA . Effect of levosimendan on the contractility of muscle fibers from nemaline myopathy patients with mutations in the nebulin gene. Skelet Muscle. (2015) ;5: (1):12. |
[47] | Marttila M , Hanif M , Lemola E , Nowak KJ , Laitila J , Grönholm M , Wallgren-Pettersson C , Pelin K . Nebulin interactions with actin and tropomyosin are altered by disease-causing mutations. Skelet Muscle. (2014) ;4: :15. |
[48] | Nguyen M-AT , Joya JE , Kee AJ , Domazetovska A , Yang N , Hook JW , Lemckert FA , Kettle E , Valova VA , Robinson PJ , North KN , Gunning PW , Mitchell CA , Hardeman EC . Hypertrophy and dietary tyrosine ameliorate the phenotypes of a mouse model of severe nemaline myopathy. Brain. (2011) ;134: (Pt 12):3516–29. |
[49] | Ravenscroft G , Jackaman C , Sewry CA , McNamara E , Squire SE , Potter AC , Papadimitriou J , Griffiths LM , Bakker AJ , Davies KE , Laing NG , Nowak KJ . Actin nemaline myopathy mouse reproduces disease, suggests other actin disease phenotypes and provides cautionary note on muscle transgene expression. PLoS One. (2011) ;6: (12):e28699. |
[50] | Tinklenberg J , Meng H , Yang L , Liu F , Hoffmann RG , Dasgupta M , Allen KP , Beggs AH , Hardeman EC , Pearsall RS , Fitts RH , Lawlor MW . Treatment with ActRIIB-mFc Produces Myofiber Growth and Improves Lifespan in the Acta1 H40Y Murine Model of Nemaline Myopathy. Am J Pathol. (2016) ;186: (6):1568–81. |
[51] | Gineste C , Duhamel G , Le Fur Y , Vilmen C , Cozzone PJ , Nowak KJ , Bendahan D , Gondin J . Multimodal MRI and (31)P-MRS investigations of the ACTA1 (Asp286Gly) mouse model of nemaline myopathy provide evidence of impaired in vivo muscle function, altered muscle structure and disturbed energy metabolism. PLoS One. (2013) ;8: (8):e72294. |
[52] | Lindqvist J , Pénisson-Besnier I , Iwamoto H , Li M , Yagi N , Ochala J . A myopathy-related actin mutation increases contractilefunction. Acta Neuropathol. (2012) ;123: (5):739–46. |
[53] | Ochala J , Ravenscroft G , Laing NG , Nowak KJ . Nemaline Myopathy-Related Skeletal Muscle α-Actin (ACTA1) Mutation, Asp286Gly, Prevents Proper Strong Myosin Binding and Triggers Muscle Weakness. Kellermayer MS , editor. PLoS One. (2012) ;7: (9):e45923. |
[54] | Lindqvist J , Cheng AJ , Renaud G , Hardeman EC , Ochala J . DistinctUnderlying Mechanisms of Limb and Respiratory Muscle FiberWeaknesses in Nemaline Myopathy. J Neuropathol Exp Neurol. (2013) ;72: (6):472–81. |
[55] | Orzechowski M , Fischer S , Moore JR , Lehman W , Farman GP . Energy landscapes reveal the myopathic effects of tropomyosin mutations. Arch Biochem Biophys. (2014) ;564: :89–99. |
[56] | Feng J-J , Marston S . Genotype-phenotype correlations in ACTA1 mutations that cause congenital myopathies. Neuromuscul Disord. (2009) ;19: (1):6–16. |
[57] | McKillop DF , Geeves MA . Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys J. (1993) ;65: (2):693–701. |
[58] | Ochala J , Li M , Tajsharghi H , Kimber E , Tulinius M , Oldfors A , Larsson L . Effects of a R133W β-tropomyosin mutation on regulation of muscle contraction in single human muscle fibres. J Physiol. (2007) ;581: (3):1283–92. |
[59] | Ochala J , Li M , Ohlsson M , Oldfors A , Larsson L . Defective regulation of contractile function in muscle fibres carrying an E41K beta-tropomyosin mutation. J Phys. (2008) ;586: (Pt 12):2993–3004. |
[60] | Ottenheijm CAC , Lawlor MW , Stienen GJM , Granzier H , Beggs AH . Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy. Hum Mol Genet. (2011) ;20: (10):2015–25. |
[61] | Yuen M , Cooper ST , Marston SB , Nowak KJ , McNamara E , Mokbel N , Ilkovski B , Ravenscroft G , Rendu J , de Winter JM , Klinge L , Beggs AH , North KN , Ottenheijm CAC , Clarke NF . Muscle weakness in TPM3-myopathy is due to reduced Ca2+-sensitivity and impaired acto-myosin cross-bridge cycling in slow fibres. Hum Mol Genet. (2015) ;24: (22):6278–92. |
[62] | Donkervoort S , Papadaki M , de Winter JM , Neu MB , Kirschner J , Bolduc V , Yang ML , Gibbons MA , Hu Y , Dastgir J , Leach ME , Rutkowski A , Foley AR , Krüger M , Wartchow EP , McNamara E , Ong R , Nowak KJ , Laing NG , Clarke NF , Ottenheijm CAC , Marston SB , Bönnemann CG . TPM3 deletions cause a hypercontractilecongenital muscle stiffness phenotype. Ann Neurol. (2015) ;78: (6):982–94. |
[63] | Ochala J , Iwamoto H , Larsson L , Yagi N . A myopathy-linked tropomyosin mutation severely alters thin filament conformational changes during activation. Proc Natl Acad Sci. (2010) ;107: (21):9807–12. |
[64] | de Haan A , van der Vliet M , Gommans IM , Hardeman E , van Engelen BG . Skeletal muscle of mice with a mutation in slow α-tropomyosin is weaker at lower lengths. Neuromuscul Disord. (2002) ;12: (10):952–7. |
[65] | Gineste C , Ottenheijm C , Le Fur Y , Banzet S , Pecchi E , Vilmen C , Cozzone PJ , Koulmann N , Hardeman EC , Bendahan D , Gondin J . Alterations at the cross-bridge level are associated with a paradoxical gain of muscle function in vivo in a mouse model of nemaline myopathy. Ervasti JM , editor. PLoS One. (2014) ;9: (9):e109066. |
[66] | Ochala J , Gokhin DS , Pénisson-Besnier I , Quijano-Roy S , Monnier N , Lunardi J , Romero NB , Fowler VM . Congenitalmyopathy-causing tropomyosin mutations induce thin filamentdysfunction via distinct physiological mechanisms. Hum Mol Genet. (2012) ;21: (20):4473–85. |
[67] | Mokbel N , Ilkovski B , Kreissl M , Memo M , Jeffries CM , Marttila M , Lehtokari V-L , Lemola E , Gronholm M , Yang N , Menard D , Marcorelles P , Echaniz-Laguna A , Reimann J , Vainzof M , Monnier N , Ravenscroft G , McNamara E , Nowak KJ , Laing NG , Wallgren-Pettersson C , Trewhella J , Marston S , Ottenheijm C , North KN , Clarke NF . K7delis a common TPM2 gene mutation associated with nemaline myopathyand raised myofibre calcium sensitivity. Brain. (2013) ;136: (2):494–507. |
[68] | Marttila M , Lehtokari V-L , Marston S , Nyman TA , Barnerias C , Beggs AH , Bertini E , Ceyhan-Birsoy O , Cintas P , Gerard M , Gilbert-Dussardier B , Hogue JS , Longman C , Eymard B , Frydman M , Kang PB , Klinge L , Kolski H , Lochmüller H , Magy L , Manel V , Mayer M , Mercuri E , North KN , Peudenier-Robert S , Pihko H , Probst FJ , Reisin R , Stewart W , Taratuto AL , de Visser M , Wilichowski E , Winer J , Nowak K , Laing NG , Winder TL , Monnier N , Clarke NF , Pelin K , Grönholm M , Wallgren-Pettersson C . Mutation update andgenotype-phenotype correlations of novel and previously describedmutations in TPM2 and TPM3 causing congenital myopathies. HumMutat. (2014) ;35: (7):779–90. |
[69] | Robaszkiewicz K , Dudek E , Kasprzak AA , Moraczewska J . Functional effects of congenital myopathy-related mutations in gamma-tropomyosin gene. Biochim Biophys Acta. (1822) 10 1562–9. |
[70] | Marttila M , Lemola E , Wallefeld W , Memo M , Donner K , Laing NG , Marston S , Grönholm M , Wallgren-Pettersson C . Abnormal actinbinding of aberrant β-tropomyosins is a molecular cause ofmuscle weakness in TPM2-related nemaline and cap myopathy. Biochem J. (2012) ;442: (1):231–9. |
[71] | Memo M , Marston S . Skeletal muscle myopathy mutations at the actin tropomyosin interface that cause gain- or loss-of-function. J Muscle Res Cell Motil. (2013) ;34: (3-4):165–9. |
[72] | Marston S , Memo M , Messer A , Papadaki M , Nowak K , McNamara E , Ong R , El-Mezgueldi M , Li X , Lehman W . Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Hum Mol Genet. (2013) ;22: (24):4978–87. |
[73] | Johnston JJ , Kelley RI , Crawford TO , Morton DH , Agarwala R , Koch T , Schäffer AA , Francomano CA , Biesecker LG . A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet. (2000) ;67: (4):814–21. |
[74] | Marra JD , Engelstad KE , Ankala A , Tanji K , Dastgir J , De Vivo DC , Coffee B , Chiriboga CA . Identification of a Novel Nemaline Myopathy-causing Mutation in the Troponin T1 (TNNT1) Gene: A case outside of the Old Order Amish. Muscle Nerve. (2015) ;51: (5):767–72. |
[75] | Abdulhaq UN , Daana M , Dor T , Fellig Y , Eylon S , Schuelke M , Shaag A , Elpeleg O , Edvardson S . Nemaline body myopathy caused by a novel mutation in Troponin T1 (TNNT1). Muscle Nerve. (2016) ;53: (4);564–9. |
[76] | Jin J-P , Brotto MA , Hossain MM , Huang Q-Q , Brotto LS , Nosek TM , Morton DH , Crawford TO . Truncation by Glu180 Nonsense Mutation Results in Complete Loss of Slow Skeletal Muscle Troponin T in a Lethal Nemaline Myopathy. J Biol Chem. (2003) ;278: (28):26159–65. |
[77] | van der Pol WL , Leijenaar JF , Spliet WGM , Lavrijsen SW , Jansen NJG , Braun KPJ , Mulder M , Timmers-Raaijmakers B , Ratsma K , Dooijes D , van Haelst MM . Nemaline myopathy caused by TNNT1 mutations in aDutch pedigree. Mol Genet genomic Med. (2014) ;2: (2):134–7. |
[78] | Kimber E , Tajsharghi H , Kroksmark A-K , Oldfors A , Tulinius M . A mutation in the fast skeletal muscle troponin I gene causes myopathy and distal arthrogryposis. Neurology. (2006) ;67: (4):597–601. |
[79] | Robinson P , Lipscomb S , Preston LC , Altin E , Watkins H , Ashley CC , Redwood CS . Mutations in fast skeletal troponin I, troponin T, and -tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. (2007) ;21: (3):896–905. |
[80] | Zhao N , Jiang M , Han W , Bian C , Li X , Huang F , Kong Q , Li J . A novel mutation in TNNT3 associated with Sheldon-Hall syndrome in a Chinese family with vertical talus. Eur J Med Genet. (2011) ;54: (3):351–3. |
[81] | Beck AE , McMillin MJ , Gildersleeve HIS , Kezele PR , Shively KM , Carey JC , Regnier M , Bamshad MJ . Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am J Med Genet Part A. (2013) Mar;161: (3):550–5. |
[82] | Zhu X , Wang F , Zhao Y , Yang P , Chen J , Sun H , Liu L , Li W , Pan L , Guo Y , Kou Z , Zhang Y , Zhou C , He J , Zhang X , Li J , Han W , Li J , Liu G , Gao S , Yang Z . A Gain-of-Function Mutation in Tnni2 Impeded Bone Development through Increasing Hif3a Expression in DA2B Mice. Hunter KW, editor. PLoS Genet. (2014) ;10: (10):e1004589. |
[83] | Wei B , Lu Y , Jin J-P . Deficiency of slow skeletal muscle troponin T causes atrophy of type I slow fibres and decreases tolerance to fatigue. J Physiol. (2014) ;592: (6):1367–80. |
[84] | Amarasinghe C , Hossain MM , Jin J-P . Functional Basis of Three New Recessive Mutations of Slow Skeletal Muscle Troponin T Found in Non-Amish TNNT1 Nemaline Myopathies. Biochemistry. (2016) ;55: (32):4560–7. |
[85] | Gupta VA , Beggs AH . Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet Muscle. (2014) ;4: :11. |
[86] | Garg A , O’Rourke J , Long C , Doering J , Ravenscroft G , Bezprozvannaya S , Nelson BR , Beetz N , Li L , Chen S , Laing NG , Grange RW , Bassel-Duby R , Olson EN . KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J Clin Invest. (2014) ;124: (8):3529–39. |
[87] | Cenik BK , Garg A , McAnally JR , Shelton JM , Richardson JA , Bassel-Duby R , Olson EN , Liu N . Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J Clin Invest. (2015) ;125: (4):1569–78. |
[88] | Gong W , Gohla RM , Bowlin KM , Koyano-Nakagawa N , Garry DJ , Shi X . Kelch Repeat and BTB Domain Containing Protein 5 (Kbtbd5) Regulates Skeletal Muscle Myogenesis through the E2F1-DP1 Complex. J Biol Chem. (2015) ;290: (24):15350–61. |
[89] | Pauw-Gommans , Gerrits , de Haan A , van Engelen BGM . Muscle slowness in a family with nemaline myopathy. Neuromuscul Disord. (2006) ;16: (8):477–80. |
[90] | Agrawal PB , Greenleaf RS , Tomczak KK , Lehtokari V-L , Wallgren-Pettersson C , Wallefeld W , Laing NG , Darras BT , Maciver SK , Dormitzer PR , Beggs AH . Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet. (2007) ;80: (1):162–7. |
[91] | Ockeloen CW , Gilhuis HJ , Pfundt R , Kamsteeg EJ , Agrawal PB , Beggs AH , Dara Hama-Amin A , Diekstra A , Knoers NVAM , Lammens M , van Alfen N . Congenital myopathy caused by a novel missense mutation in the CFL2 gene. Neuromuscul Disord. (2012) ;22: (7):632–9. |
[92] | Agrawal PB , Joshi M , Savic T , Chen Z , Beggs AH . Normalmyofibrillar development followed by progressive sarcomericdisruption with actin accumulations in a mouse Cfl2 knockoutdemonstrates requirement of cofilin-2 for muscle maintenance. HumMol Genet. (2012) ;21: (10):2341–56. |
[93] | Gurniak CB , Chevessier F , Jokwitz M , Jönsson F , Perlas E , Richter H , Matern G , Boyl PP , Chaponnier C , Fürst D , Schröder R , Witke W . Severe protein aggregate myopathy in a knockout mouse model points to an essential role of cofilin2 in sarcomeric actin exchange and muscle maintenance. Eur J Cell Biol. (2014) ;93: (5-6):252–66. |
[94] | Tian L , Ding S , You Y , Li T , Liu Y , Wu X , Sun L , Xu T . Leiomodin-3-deficient mice display nemaline myopathy with fast-myofiber atrophy. Dis Model Mech. (2015) ;8: (6):635–41. |
[95] | Hansen R , Saikali KG , Chou W , Russell AJ , Chen MM , Vijayakumar V , Stoltz RR , Baudry S , Enoka RM , Morgans DJ , Wolff AA , Malik FI . Tirasemtiv amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. (2014) ;50: (6):925–31. |
[96] | Shefner J , Cedarbaum JM , Cudkowicz ME , Maragakis N , Lee J , Jones D , Lou Watson M , Mahoney K , Chen M , Saikali K , Mao J , Russell AJ , Hansen RL , Malik F , Wolff A a . Safety, tolerability and pharmacodynamics of a skeletal muscle activator in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2012) ;13: (5):430–8. |
[97] | Sanders DB , Rosenfeld J , Dimachkie MM , Meng L , Malik FI . A Double-Blinded, Randomized, Placebo-Controlled Trial to Evaluate Efficacy, Safety, and Tolerability of Single Doses of Tirasemtiv in Patients with Acetylcholine Receptor-Binding Antibody-Positive Myasthenia Gravis. Neurotherapeutics. (2015) ;12: (2):455–60. |
[98] | Schlecht W , Li K-L , Hu D , Dong W . Fluorescence Based Characterization of Calcium Sensitizer Action on the Troponin Complex. Chem Biol Drug Des. (2016) ;87: (2):171–81. |
[99] | Lindqvist J , Levy Y , Pati-Alam A , Hardeman EC , Gregorevic P , Ochala J . Modulating myosin restores muscle function in a mouse model of nemaline myopathy. Ann Neurol. (2016) ;79: (5):717. |
[100] | Malik FI , Hartman JJ , Elias KA , Morgan BP , Rodriguez H , Brejc K , Anderson RL , Sueoka SH , Lee KH , Finer JT , Sakowicz R , Baliga R , Cox DR , Garard M , Godinez G , Kawas R , Kraynack E , Lenzi D , Lu PP , Muci A , Niu C , Qian X , Pierce DW , Pokrovskii M , Suehiro I , Sylvester S , Tochimoto T , Valdez C , Wang W , Katori T , Kass DA , Shen Y-T , Vatner SF , Morgans DJ . Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science. (2011) ;331: (6023):1439–43. |
[101] | Riley DA , Van Dyke JM . The effects of active and passive stretching on muscle length. Phys Med Rehabil Clin N Am. (2012) ;23: (1):51–7. |
[102] | Nowak KJ , Ravenscroft G , Jackaman C , Filipovska A , Davies SM , Lim EM , Squire SE , Potter AC , Baker E , Clément S , Sewry CA , Fabian V , Crawford K , Lessard JL , Griffiths LM , Papadimitriou JM , Shen Y , Morahan G , Bakker AJ , Davies KE , Laing NG . Rescue of skeletal muscle α-actin-null mice bycardiac (fetal) α-actin. J Cell Biol. (2009) ;185: (5):903–15. |
[103] | Ravenscroft G , McNamara E , Griffiths LM , Papadimitriou JM , Hardeman EC , Bakker AJ , Davies KE , Laing NG , Nowak KJ . Cardiac -actin over-expression therapy in dominant ACTA1 disease. Hum Mol Genet. (2013) ;22: (19):3987–97. |
[104] | Ryan MM , Sy C , Rudge S , Ellaway C , Ketteridge D , Roddick LG , Iannaccone ST , Kornberg AJ , North KN . Dietary L-tyrosine supplementation in nemaline myopathy. J Child Neurol [Internet]. (2008) Jun [cited 2012 Jul 20];23: (6):609–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18079309 |
[105] | Nguyen M-AT , Joya JE , Kee AJ , Domazetovska A , Yang N , Hook JW , Lemckert FA , Kettle E , Valova VA , Robinson PJ , North KN , Gunning PW , Mitchell CA , Hardeman EC . Hypertrophy and dietary tyrosine ameliorate the phenotypes of a mouse model of severe nemaline myopathy. Brain. (2011) ;134: (12):3516–29. |
[106] | Long C , Amoasii L , Bassel-Duby R , Olson EN . Genome Editing of Monogenic Neuromuscular Diseases. JAMA Neurol. (2016) ;73: (11):1349. |
[107] | Long C , McAnally JR , Shelton JM , Mireault AA , Bassel-Duby R , Olson EN . Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. (2014) ;345: (6201):1184–8. |


