
Increasing Role of Titin Mutations in Neuromuscular Disorders
Abstract
The TTN gene with 363 coding exons encodes titin, a giant muscle protein spanning from the Z-disk to the M-band within the sarcomere. Mutations in the TTN gene have been associated with different genetic disorders, including hypertrophic and dilated cardiomyopathy and several skeletal muscle diseases.
Before the introduction of next generation sequencing (NGS) methods, the molecular analysis of TTN has been laborious, expensive and not widely used, resulting in a limited number of mutations identified. Recent studies however, based on the use of NGS strategies, give evidence of an increasing number of rare and unique TTN variants. The interpretation of these rare variants of uncertain significance (VOUS) represents a challenge for clinicians and researchers.
The main aim of this review is to describe the wide spectrum of muscle diseases caused by TTN mutations so far determined, summarizing the molecular findings as well as the clinical data, and to highlight the importance of joint efforts to respond to the challenges arising from the use of NGS. An international collaboration through a clinical and research consortium and the development of a single accessible database listing variants in the TTN gene, identified by high throughput approaches, may be the key to a better assessment of titinopathies and to systematic genotype– phenotype correlation studies.
INTRODUCTION
With its 363 coding exons and a full-length transcript of more than 100 kb [1] TTN gene encodes titin, the by far longest known polypeptide in nature. The longest human theoretical isoform of TTN would produce a protein of 3,960 kDa containing 35,991 amino acids, although this isoform has not been observed [1].
Titin acts as a scaffold protein aiding in myofibrillar assembly during myogenesis [2], as a molecular spring determining the passive elasticity of the muscle [3, 4], and as a mechanosensor serving various signaling functions [5, 6].
TTN mutations have to date been reported to cause various cardiomyopathies [7, 8] and a range of skeletal muscle diseases and phenotypes listed below:
– Late-onset autosomal dominant tibial muscular dystrophy (TMD) (MIM #600334);
– Young or early adult onset recessive distal titinopathy;
– Limb-girdle muscular dystrophy type 2J (LGMD2J; MIM #608807);
– Congenital centronuclear myopathy (CNM; MIM #255200);
– Early-onset myopathy with fatal cardiomyopathy, EOMFC (MIM #611705);
– Multi-minicore disease with heart disease (MmDHD) including clinical variations;
– Childhood-juvenile onset Emery-Dreifuss-like phenotype without cardiomyopathy;
– Hereditary myopathy with early respiratory failure (HMERF; MIM #603689);
– Adult onset recessive proximal muscular dystrophy.
Mutations in titin will probably prove to be the cause of many additional phenotypes of muscular disorders in the coming years.
Due to its huge size, it has not been possible to sequence the entire TTN gene routinely in research and diagnostic laboratories until recently. Thus, before implementation of the next generation sequencing (NGS) methods, only a limited amount of TTN mutations were identified. NGS sequencing has enabled the rapid and thorough investigation of genetic material [9] and has resulted in an explosion in the identification of new TTN variants. However, their clinical interpretation is a challenge.
Here, we focus on the current understanding of the titin gene and protein from a human disease perspective. In particular, we provide an overview of the different neuromuscular disorders caused by mutations in the TTN gene, reviewing the molecular findings as well as the clinical data. Finally, we highlight the difficulties related to the interpretation of the clinical significance of TTN variations and the need for further functional studies and bioinformatics tools.
THE TITIN GENE, ISOFORMS AND PROTEIN
The titin gene (MIM #188840), is located on the short arm of chromosome 2 (chromosomal band q31.2). It contains 363 coding exons and an additional first non-coding exon [1]. The longest theoretical transcript (variant IC, NM_001267550.2), virtually obtained by the transcription of all the coding exons (excluding the alternative C-terminal Novex-3 exon) and called “meta isoform”, has been adopted as the gold standard for describing TTN variants, and will be used as reference for cDNA and protein numbering in this review. Exon numbering will be according to the HGVS recommendations [10] and to the current Leiden database (LOVD) numbering (modified on 11th October 2013, changing exon 47b to exon 48 and adding +1 to all subsequent exon numbers) [11].
The titin protein spans from the Z-disk to the M-band [12]. Its modular structure is composed of four main parts (Fig. 1): the amino-terminal Z-disc region, the I-band and A-band regions, and the carboxyl-terminal part spanning the M-band. Titin is composed of repeated immunoglobulin-like (Ig) and fibronectin type 3 -like (FN3) domains, interspersed by unique sequence regions [1]. It also contains the repetitive PEVK region, rich in proline (P), glutamate (E), valine (V), and lysine (K) residues, in the I band, and a serine/threonine kinase (TK) domain in the M-band.
More than 1 million splice variants could be generated theoretically by the TTN gene [13]. Indeed, extensive alternative splicing results in a remarkable diversity of titin isoforms that can be divided into three main classes based on the presence of the N2A and N2B elements in the I-band region [1, 14, 15] (Fig. 1). Skeletal muscles express so-called N2A isoforms, characterized by the inclusion of the N2A element and exclusion of the cardiac-specific N2B element. In the heart, N2BA isoforms include both the N2B and N2A elements, while N2B isoforms use the N2B element only. The aforementioned isoforms also differ in the lengths of the proximal tandem-Ig and PEVK regions, which are longest in the N2A isoforms and shortest in the N2B isoforms. Within each isoform class, the tandem-Ig and PEVK regions also show variable expression in different muscles, and across developmental and physiological states. Moreover, the second last TTN exon 363 (Mex5), coding for the is7 domain located in the M-band, is differentially spliced, producing is7–and is7+ isoforms [16]
The major isoform classes are represented in the NCBI RefSeq database by the entries NM_133378 (N2A; NP_596869:3,680 kDa and 33,423 aa), NM_001256850.1 (N2BA; NP_001243779:3,780 kDa and 34,350 aa), and NM_003319 (N2B; NP_003310:2,960 kDa and 26,926 aa) [1, 14, 15].
The Novex-1 (NM_133432; NP_597676) and Novex-2 (NM_133437; NP_597681) isoforms are similar to N2B, but they also include further 125 and 192 amino acids encoded by the Novex-1 and Novex-2 exons in the I-band. Finally, the much smaller Novex-3 isoform (NM_133379; NP_596870:616 kDa and 5604 aa) only contains the N-terminal part of the protein. This isoform, expressed on a low level in all striated muscles, results from inclusion of the Novex-3 exon encoding an alternative C-terminus [1].
The best characterized titin function is that of a scaffold protein aiding myofibrillar assembly during myogenesis [17]. However, it is also the backbone for the positioning of myosin filaments in the center of the sarcomere, and a molecular spring responsible for the passive elasticity of the muscle [3, 4]. The passive force of the muscle cells is, in fact, largely due to the elastic properties of I-band titin, allowing shortening of the sarcomere in contraction and extension when stretched. A crucial role in the myofibrillar signal transduction pathways has also been demonstrated [18]: titin seems to integrate or coordinate multiple signaling pathways related to gene activation and/or to protein folding, quality control and degradation [6, 19].
INTERACTIONS OF TTN WITH OTHER PROTEINS
The versatile roles played by titin in cardiac and skeletal muscles are enabled and facilitated by a high (or presumably very high) number of different protein ligands.
The search for TTN interactors in large public databases (PSICQUIC (20), IntAct [21], BioGRID [22]) results in a list of more than 170 putative ligands, as a product of large-scale studies of protein-protein interactions.
Even if a detailed discussion of titin interactions is not the main aim of this review, a summary of the best characterized ones is provided below and in Fig. 1.
Several reports have confirmed that telethonin (also named Titin-cap or T-cap) and α-actinin bind to the N-terminal portion of titin [23–27]. Moreover, Kontrogianni-Konstantopoulos et al. [28] demonstrated that the small ankyrin-1 (sAnk1) and the two most N-terminal Ig domains of titin form a three-way complex with telethonin.
Similarly, the actin binding proteins, filamin C and nebulin, have been shown to interact with titin in the Z-region [15, 29].
The central I-band region of titin has been widely studied, and several interactors identified, including tropomyosin [30], αB-crystallin [31], FHL1 [32] and FHL2 [33], two members of the four-and-a-half-LIM-only protein family, calpains 1 and 3 [34–36], and muscle ankyrin repeat proteins (MARPs) [37]. Protein kinases A and G (PKA and PKG) phosphorylate the N2B region, reducing the passive tension [38, 39]. Furthermore, PEVK region interacts with Ca2+, actin and S100A1, which is able to control the PEVK/actin interaction in a Ca2+-dependent manner [40].
The A-band region of titin, tightly associated with thick filaments, binds myosin heavy chain and MyBP-C [41].
The M-band region of titin has several interactors. The domains at the A-band/M-band boundary bind the ubiquitin ligases MURF1 and MURF2 (muscle RING finger 1 and 2) [42, 43]. The titin kinase (TK) domain, located at the M-band periphery, interacts with calmodulin [44], as well as with the signalosome composed of nbr1 p62, and MURF2 [33]. FHL2, expressed predominantly in the heart, binds to the is2 region [45]. The alternatively spliced is7 region binds calpain 3 (CAPN3) [35], the calcium-dependent protease involved in the pathogenesis of LGMD2A. The M10 domain interacts with the giant structural and signaling protein obscurin and its smaller homologue obscurin-like 1 (OBSL1) [46], and the A-kinase anchoring intermediate filament protein alpha-synemin [47]. Finally, several of the C-terminal titin domains can bind the multifunctional docking protein myospryn (CMYA5) [48].
TITIN VARIANTS AND DISEASES
Mutations in the TTN gene have been associated with several different muscle diseases, cardiomyopathies and combinations of these. The latter include dilated cardiomyopathy (DCM, MIM#604145), familial hypertrophic cardiomyopathy (HCM; MIM #613765), arrhythmogenic right ventricular cardiomyopathy (ARVC; MIM #602087) and monogenic restrictive cardiomyopathy (RCM). In this review, however, we focus on the large spectrum of skeletal muscle diseases caused by TTN mutations (Fig. 2), since other reviews provide a more detailed description of cardiac phenotypes linked to TTN variants [7, 8].
Late-onset autosomal dominant tibial muscular dystrophy (TMD)
Tibial muscular dystrophy (TMD; MIM #600334]) is a mild autosomal dominant distal myopathy involving the anterior compartment muscles of the lower legs but sparing of the short toe extensor digitorum brevis muscles [49].
It is characterized by a late onset (>35 years), a slow progression, normal or slightly increased values of serum creatine kinase (CK) and a myopathic EMG pattern [49, 50]. Biopsy findings in the target muscles include fiber size variability, central nuclei, necrosis, presence of fibroadipose tissue and rimmed vacuoles. Electron microscopy showed autophagic vacuoles without membrane and very rare inclusions of 15–18-nm filaments [51]. Muscle imaging (CT or MRI) is very informative with selective fatty replacement in the muscles of the anterior compartments of the lower legs starting in the anterior tibial muscle and representing a useful clinical tool to address the diagnosis.
In 2002, the first TTN mutation associated with human skeletal-muscle disease and causing TMD was reported [52] (Table 1). This dominant founder mutation, termed FINmaj, is responsible for the high prevalence (2/10.000) of TMD observed in Finland [49]. FINmaj is an 11-bp insertion-deletion in the last exon (exon 364 or Mex6) of the gene, changing four amino acids in the C-terminal Ig domain M10 of M-band titin. Three affected members of a French TMD family showed a different missense mutation in the same exon (c.107867T>C p.Leu35956Pro) [52].
One year later, a dominant missense variant in the exon 364 (c.107840T>A p.Ile35947Asn) was also identified in a Belgian family with a similar phenotype [51]. In 2008, three novel truncating variants (two deletions – c.107647delT p.Ser35883Glnfs*10 and c.107889delA p.Lys35963Asnfs*9 – and a nonsense mutation – c.107890C>T p.Gln35964*) were identified in two French families and a Spanish kindred [50] and a further missense mutation in exon 364 was then described in a large Italian family [53].
Young or early adult onset recessive distal titinopathy
More recently, Evilä et al. [54] described four patients with a more severe distal phenotype, resulting in a young or early adult-onset recessive distal titinopathy: all these patients were compound heterozygotes for described TMD mutations and novel frameshift variants (Table 1). Two French patients, previously reported with a more severe distal phenotype compared to TMD, had a second causative mutation that explains the peculiar phenotype. Similarly, a 36-year-old Spanish female with a similar distal phenotype and an early onset had the previously described Iberian TTN mutation (c.107889delA) combined with a second frameshift mutation. Homozygosity for the Iberian mutation was the cause of the early distal progressive disease observed in a female Portuguese patient.
In the reported patients, biopsy findings are highly variable, from mild myopathy to severe dystrophic changes usually with rimmed vacuoles, depending on the site of biopsy and the disease duration.
Muscle imaging shows an early (already at age 20) fatty degeneration of the anterior compartment, frequently combined with a similar degree of involvement of soleus, which is unusual in TMD.
Limb-girdle muscular dystrophy type 2J (LGMD2J)
Limb-girdle muscular dystrophies (LGMD) are Mendelian disorders affecting the voluntary muscles in proximal limbs of the hip and shoulder areas [55]. LGMDs includes more than 30 different diseases with different but often overlapping clinical pictures [56]. LGMD2J represents a recessive disease with an early age of onset (<12 y.o.) [50, 57]. The first Finnish patients described were homozygous for the FINmaj mutation, presenting with a very different and much more severe phenotype than TMD [50, 57]. In addition, in a French family with a dominant TMD phenotype due to a nonsense mutation in the last exon (c.107890C>T p.Gln35964*), one deceased patient with a more severe generalized muscle weakness proved in retrospect to be homozygous for the mutation [58].
More recently, Evilä et al. described three further LGMD2J patients [54]. Three Finnish patients, heterozygous for the FINmaj variant and presenting with an early onset LGMD or generalized muscle weakness phenotype, were clinically and molecularly re-evaluated. In two out of three patients, a second frameshift variant was detected in the other TTN allele (Table 1), and in the third patient the changes on the protein level were identical to FINmaj homozygous LGMD2L, suggesting an undetected truncating mutation on the other allele. Moreover, Zheng et al. identified, by exome sequencing, a homozygous missense mutation in the last exon (c.107788T>C p. Trp35930Arg) in a Chinese Han consanguineous family with a LGMD phenotype [59].
On the protein level LGMD2J shows a secondary CAPN3 defect [60] and loss of titin C-terminus on Western blots and immunofluorescence microscopy with antibodies against several C-terminal domains [50, 61]. Most biopsied muscles of patients homozygous for the FINmaj variant show dystrophic findings with end stage pathology without rimmed vacuoles [48]. However, rimmed vacuolar pathology was reported in a recently described case compound heterozygous for the FINmaj mutation and a truncating mutation [52].
Muscle imaging shows a progressive fatty degeneration of skeletal muscles. Muscles are relatively well preserved in young patients. A small degree of fatty degeneration can be observed 10 years after onset of muscle weakness, and fatty replacement is usually total after 40 years [48, 52].
Congenital centronuclear myopathy
Centronuclear myopathies (CNMs) are congenital myopathies characterized by the presence of centralized nuclei in the muscle fibers [62]. The term has also been used for myopathies with less specific increase of internalized nuclei. Mutations in four different genes have been reported to cause CNM: DNM2 causes an autosomal dominant form [63]; BIN1 and RYR1 mutations may cause autosomal dominant or recessive forms [64, 65]; and the X-linked myotubular myopathy (XLMTM) is due to mutations in the MTM1 gene [66]. Recently, Ceyhan-Birsoy et al. described five patients with generalized infantile muscle weakness and muscle biopsy findings compatible with CNM [67]. Using next generation sequencing of whole exomes and genomes, recessive truncating TTN mutations were identified in all the five patients (Table 2).
Fattori at al. described a further CNM patient with two TTN nonsense mutations (Table 2) in the TTN gene, resulting in a severe reduction of titin C-terminus at protein level [68]. The latter Italian case provided a further proof of the correlation between specific titin variants and the CNM.
Early-onset myopathy with fatal cardiomyopathy (EOMFC) and multi-minicore disease with heart disease (MmDHD)
In 2007, Carmignac et al. reported a novel recessive titinopathy involving both heart and skeletal muscle, in two consanguineous families of Moroccan and Sudanese origin [69]. The disease was characterized by early onset, slowly progressive, muscle weakness (1 y.o.); conversely, a severe dilated cardiomyopathy with rhythm disturbances developed later and resulted in a premature sudden death before adulthood. Skeletal muscle biopsies showed minicore-like lesions, centralized nuclei and type 1 fiber predominance.
In 2014, Chauveau and collagues described four other families with congenital core myopathy and primary heart disease associated with TTN mutations (Table 2) and suggested the inclusive name of multi-minicore disease with heart disease (MmDHD) for all these clinically heterogeneous congenital diseases [70].
Chauveau described a wide range of phenotypes, spanning from an Emery-Dreifuss-like form to an unusual, severe condition with distal arthrogryposis multiplex congenita (AMC), congenital muscle weakness, kyphosis, and neonatal cardiac failure. All of them are congenital or infantile muscle conditions, characterized by weakness with rigid spine, distal or elbow joint contractures, impaired respiratory function and mild hyperCKemia (<5x).
Childhood-juvenile onset Emery-Dreifuss-like phenotype without cardiomyopathy
Emery-Dreifuss muscular dystrophy has been associated with several genes: EMD and FHL1 for X-linked forms [71, 72], LMNA [73, 74] with both an autosomal dominant and recessive inheritance, and SYNE1 and SYNE2 both causing a dominant phenotype. Recently, De Cid et al. reported on three patients with a peculiar phenotype, including limb-girdle weakness, high CK levels, early-onset contractures and a progressive course with permanent loss of ambulation during adolescence or early adulthood [76]. The clinical phenotype of the patients resembled EDMD, albeit with no cardiac abnormality. Novel truncating mutations in the C-terminus of titin segregating with the disease in all the three unrelated families were identified (Table 2), and they all had a secondary CAPN3 defect indicating a novel recessive titinopathy phenotype.
Congenital centronuclear myopathy [67, 68], early-onset myopathy with fatal cardiomyopathy (EOMFC) [69], multi-minicore disease with heart disease (MmDHD) [70], and childhood-juvenile onset Emery-Dreifuss-like phenotype withoutcardiomyopathy [76] (Table 2) represent a group of TTN-related recessive disorders characterized by an early onset. Most of the patients described are homozygous or compound heterozygous for truncating variants. The causative mutations in CNM patients are mainly localized in I- or A-bands but frameshift variants in M-band titin have been found in 3/6 patients [67, 68]. On the contrary, almost all the patients with an EDMD-like phenotype or an EOMFC/MmDHD described so far have truncating mutations in the M-band, but these variants do not involve the last exon, previously associated to TMD/LGMD phenotypes [69, 70, 76]. Interestingly, despite the location of all these M-band truncating variants in proximity to each other, the clinical pictures and the histological findings are heterogeneous and, above all, a cardiac phenotype is only reported in a subset of patients.
Hereditary myopathy with early respiratory failure (HMERF)
Hereditary myopathy with early respiratory failure (HMERF) is an adult-onset autosomal dominant myopathy with respiratory muscle involvement that may lead to a fatal respiratory crisis if not treated [77].
In 2005, Lange et al. identified a TTN mutation (c.102271C>T p.Arg34091Trp, also known as R279W according to residue numbering of the isolated TK structure) in the TK domain of two Swedish families and a third unrelated Swedish patient sharing the same haplotype [33]. The variant affects the interaction between nrb1 and the TK domain, disrupting the signaling pathway that involves p62/SQSTM1, MURF2 and SRF.
With the exception of the TK-R279W mutation, all other mutations reported as being causative of HMERF are localized in the TTN exon 344 (Table 3) and most of them seem to hamper the correct folding of the A150 domain (119th Fn3 domain) [78–84]. The most common mutation (c.95134T>C p.Cys31712Arg, first reported as p.Cys30071Arg) has been found in more than 20 families [78, 79, 82], most of which share some markers in the haplotype, except one Indian [83] and one Chinese family [85]. The high number of mutations identified in exon 344 confirms the presence of a mutational hotspot region. Until recently, all the variants associated with HMERF seemed to be dominant and fully penetrant. In 2014, Palmio et al. reported a missense mutation (c.95195C>T p.Pro31732Leu, first published as p.Pro30091Leu) in three different families [82]. The variant has been defined “semidominant” or “semirecessive” since it is fully penetrant in homozygosity causing a more severe phenotype. The heterozygous carriers may have a subclinical disease or may manifest a less severe disease.
In the affected members of the original Swedish family described by Lange et al. [33], Hedberg et al. reported the presence of a second variant, the “recessive” p.Pro31732Leu change [86]. A suggestive hypothesis is that the co-inheritance of both p.Pro31732Leu and p.Arg34091Trp may cause the disease by a fully penetrant bi-mutational dominant allele [87].
Other titinopathies
In two families, one Finnish and one Italian, with well-known dominant TMD disease one individual in each family developed a different phenotype: adult onset proximal lower limb weakness without the normal ankle dorsiflexion weakness [54, 88]. Muscle MRI consistently showed significant dystrophic changes in the thigh muscles, and in the Finnish patient marked soleus muscle involvement. Both patients proved to have a second recessive mutation inherited from the healthy non-TMD parent. In particular, the Finnish patient showed a missense mutation (c.92167C>T p.Pro30723Ser) in the exon 340 [54] and in the Italian patient a missense mutation (c.60494A>G p.His20165Arg) was identified in the exon 305 [88].
Exome sequencing detected two variants in compound heterozygosity (c.45599C>G p.Ala15200Gly and c.106154 A > C p.Lys35385Thr) in a male with Romanian and Hungarian origin with adult onset proximal weakness and an initial clinical suspicion of inflammatory disease [89]. Western blots with C-terminal titin antibodies showed significant reduction of identified protein, suggesting truncating mutations rather than the identified missense variants.
ANIMAL MODELS OF TITINOPATHIES
Several spontaneous and induced animal models with titinopathy have been described so far.
“Runzel” (“ruz”) is a dystrophic zebrafish mutant with a reduced expression of certain TTN isoforms [90]. Poor swimming ability and decreased birefringence at 5 dpf are the first signs of a progressive myofibrillar disorganization, resulting in a premature death (10–12 dpf) for homozygous animals.
A spontaneous mouse model with a complex rearrangement causing the loss of 83 amino acids from the N2A region exhibits a recessive muscular dystrophy with myositis (mdm) [91]. The homozygous mice show a progressive muscle degeneration involving prominently distal skeletal muscles such as the tibialis and a reduced expression (50–60%) of CAPN3.
Mice carrying the FINmaj mutation [92] in homozygosity develop a progressive muscular dystrophy as well as a dilated cardiomyopathy, whereas heterozygotes only show a mild, later onset restricted phenotype. Interestingly, crossing the FINmaj model with CAPN3-deficient mice attenuates the muscular disorder in double heterozygotes, although not in the FINmaj homozygotes, suggesting a role for CAPN3 in the pathogenesis.
Similarly, other models were characterized and stu-died to focus on the TTN-related cardiomyopathies.
“Pickwick” (“pik”) is a zebrafish mutant, carrying a TTN variant causing an alternative N2B exon splicing [93]. Pik heart is thin-walled, dilated and poorly contractile, resembling the human DCM phenotype.
A mouse lacking the cardiac N2B element was generated to study the role of this element in systole and diastole [94]. The shorter protein is correctly integrated into the sarcomere, but causes a restrictive diastolic dysfunction.
A murine conditional knock-out for the first two M-line exons (exons 359-360 or Mex1 and Mex2) has been produced to study the role of these domains during heart development [95, 96].
Finally, Drosophila melanogaster mutants have been characterized to study the role of D-TTN gene that shows a homology to vertebrate TTN. In particular, D-Titin plays a crucial role into the formation of multi-nucleate syncytia and the organization of actin-myosin filaments in the skeletal muscle.
GENOTYPE-PHENOTYPE CORRELATION
As described above, variants in the TTN gene may cause different diseases, including several muscular disorders. Deducing a genotype– phenotype correlation has, however, so far been possible only to a limited extent, although the concentration of the currently confirmed mutations in the C-terminal (M-band) part of the gene is apparent. For instance, dominant late onset tibial muscular dystrophy is caused by mutations in the last exon 364 (Mex6) [51–53, 98]. Young or early adult onset recessive distal titinopathy is based either on one mutation in exon 363 or 364 (Mex5 or Mex6) combined with a truncating mutation on the other allele [54], or both mutations in exons 363 or 364 (Mex5–6). LGMD2J presentation may occur from similar mutational sites but is more typical with FINmaj on one or both alleles (Table 1) [50, 57].
Homozygous or compound recessive truncating mutations in the first four M-line exons (exons 359–362 or Mex1–4) cause a range of severe congenital or very early onset muscle diseases with or without cardiomyopathy [67–70, 76]. The reason why some truncations in the same exon cause cardiomyopathy and others not is unexplained. All truncated transcripts do not undergo nonsense-mediated decay and some read-through occurs, which may lead to very variable amounts of titin protein available. Cardiomyopathy could be associated with lower amounts of protein, but this has not been conclusively confirmed. Compound heterozygosity including a missense mutation with a truncating change leads to functional homozygosity of the missense transcript due to nonsense-mediated decay of the truncated transcript. Moreover, a compound heterozygous patient (patient IV in ref [54]), carrying the FINmaj variant and a missense change in the A-band (p.Pro30723Ser), shows an atypical, adult onset, proximal lower limb titinopathy that spares the anterior tibial muscle [54]. An identical phenotype occurs with compound heterozygosity of the Italian TMD mutation (p.His35946Pro) and another missense A-band mutation (p.His20165Arg) [88].
The different molecular mechanisms underlying LGMD2J, the recessive distal titinopathy, or specific atypical phenotypes have not been clarified so far.
Finally, HMERF represents a unique, well recognized, phenotype (Table 3). Even if mutations in the exon 344 are the only changes confirmed to cause fully dominant HMERF, the recent finding of the more recessive change (p.Pro31732Leu) [82] highlights the possibility that a a second variant in cis may play an important role in the pathogenesis of a clinical phenotype [82, 87].
In the pre-NGS era, the low number of described patients, as well as the positional bias caused by the extensive scanning of M-band exons as compared to other TTN portions have prevented dissection of the genotype– phenotype correlations. Novel data arising from NGS projects, together with further functional studies, will be useful to get a more clear picture of TTN mutations and their associated phenotypes.
CHALLENGES ARISING FROM NGS PROJECTS
In the last few years, the next generation sequencing approaches have demonstrated to be extremely useful in research and diagnostic testing for various hereditary conditions, including neuromuscular disorders [99]. Whole exome (WES) [100], whole genome (WGS) [9], and targeted sequencing approaches [101] have been utilized to identify causative mutations in already known or novel disease genes. All these strategies are revealing a high number of novel and rare variants in the TTN gene. More than three rare non-synonymous titin variants are identified in any individual and this is of course partly due to the mere size of the gene.
In 2012, Herman et al. developed an affinity capture for the sequencing of the titin exons [102]. In this way, they identified 72 loss-of-function variants, indicating the important role of titin in the development of dominant dilated cardiomyopathy. Later studies have shown many truncating variants to be too common for fully penetrant dominant effects [103].
Several custom enrichment assays, including MyoCap [88], MotorPlex [104, 105], and others [106–108], have been developed to sequence specific genes of interest related to neuromuscular disorders.
The use of comprehensive NGS tools allows the analysis of almost all the coding regions of TTN gene, overcoming challenges related to its size.
On the other hand, novel challenges arise from the NGS data. TTN variants identified in NGS studies are rarely already known and characterized. Moreover, recent papers underline that a single heterozygous truncating mutation in the titin gene usually does not cause any relevant muscular phenotype, and a second mutation is necessary to generate a recessive condition [54, 59, 70, 76, 89].
Most of the patients analyzed by NGS strategies show previously undescribed rare missense variants. The clinical interpretation of missense variants in the TTN gene represents one of the most significant challenges related to NGS investigation in the field of neuromuscular disorders. In silico predictions are questionable and a careful approach in the interpretation of missense variants should include a comprehensive segregation analysis and mandatory functional assays. Unfortunately, functional validations of missense changes in the TTN gene are notoriously difficult, as the huge size of titin prevents the cloning and expression of the full-length protein in in vitro systems. So far, for in vitro experimentation, titin has been dissected into more manageable protein constructs, which have been used for testing the effects of mutations on protein– protein interactions in various assays, or on structural stability of the protein. For example, the missense variant p.Trp34072Arg has been proven to abolish the interactions of titin’s kinase domain (TK) with its known ligands and to reduce the TK stability, providing a robust proof of its pathogenicity [70]. Similarly, some of the TMD-causing missense mutations have been shown to destabilize the titin M10 domain and to affect its binding to obscurin and/or obscurin-like 1 [109–111].
According to a recent large-scale study, most disease-associated variants perturb protein– protein interactions without causing misfolding [112]. Moreover, each variant may affect only specific interactions while leaving most other interactions unperturbed [112], explaining how different variants in the same gene can cause different phenotypes. These notions likely hold true also for TTN variants, and for the related phenotypes. For most of titin’s domains, interaction partners and biological functions remain unknown. Their elucidation will be a prerequisite for the correct interpretation of rare missense changes identified as well as for the identification of therapeutic targets for innovative drug therapies for titinopathies.
Meanwhile, the research community has responded to the challenges arising from the use of NGS through the formation of clinical and research consortia [113, 114]. These collaborations take advantage of the power of shared resources and expertise, and particularly the benefit of combining cohorts of patients into larger groups. This greatly increases the likelihood of success of NGS projects and enhances the impact of these projects in terms of the clinically relevant data that is associated with them. To reach this goal, there is an urgent need to collect all reported, novel detected and rare TTN variants from patients all over the world and combine them into a single accessible database, in order to better be able to compare the results and draw conclusions on genotype– phenotype correlations. As discussed in the recent 219th ENMC workshop on titinopathies, an international database of TTN mutations, variation and their clinical phenotypes, could help all the researchers and clinicians to assess the pathogenicity of found variants in NGS analyses and would promote significant advances in the understanding of titinopathies (Ms in preparation).
ACKNOWLEDGMENTS
This study was supported by the Folkhälsan Research Foundation, the Jane and Aatos Erkko Foundation, Association Française contre les Myopathies (SB/CP2013-0106, B.U. and PF2016-19522, M.S.), the Academy of Finland (no. 138491, B.U.), the Sigrid Jusélius Foundation, the Liv och Hälsa Foundation, The Finnish Cultural Foundation, the Austrian Science Fund (FWF, P27634FW), and Tampere University Hospital Research Funds (B.U.).
REFERENCES
[1] | Bang M-L , Centner T , Fornoff F , Geach AJ , Gotthardt M , McNabb M , et al. The Complete Gene Sequence of Titin, Expression of an Unusual â‰^700-kDa Titin Isoform, and Its Interaction With Obscurin Identify a Novel Z-Line to I-Band Linking System. Circulation Research. (2001) ;89: (11):1065–72. |
[2] | Kontrogianni-Konstantopoulos A , Ackermann MA , Bowman AL , Yap SV , Bloch RJ . Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiological Reviews. (2009) ;89: (4):1217–67. |
[3] | Linke WA , Stockmeier MR , Ivemeyer M , Hosser H , Mundel P . Characterizing titin’s I-band Ig domain region as an entropic spring. Journal of Cell Science. (1998) ;111: (Pt 11):1567–74. |
[4] | Linke WA , Kulke M , Li H , Fujita-Becker S , Neagoe C , Manstein DJ , et al. PEVK domain of titin An entropic spring with actin-binding properties. Journal of Structural Biology. (2002) ;137: (1-2):194–205. |
[5] | Puchner EM , Alexandrovich A , Kho AL , Hensen U , Schafer LV , Brandmeier B , et al. Mechanoenzymatics of titin kinase. Proc Natl Acad Sci U S A. (2008) ;105: (36):13385–90. |
[6] | Kruger M , Linke WA . The giant protein titin: A regulatory node that integrates myocyte signaling pathways. The Journal of Biological Chemistry. (2011) ;286: (12):9905–12. |
[7] | LeWinter MM , Granzier HL . Titin is a major human disease gene. Circulation. (2013) ;127: (8):938–44. |
[8] | Gerull B . The Rapidly Evolving Role of Titin in Cardiac Physiology and Cardiomyopathy. The Canadian Journal of Cardiology. (2015) ;31: (11):1351–9. |
[9] | Biesecker LG , Green RC . Diagnostic clinical genome and exome sequencing. N Engl J Med. (2014) ;371: (12):1170. |
[10] | den Dunnen JT , Dalgleish R , Maglott DR , Hart RK , Greenblatt MS , McGowan-Jordan J , et al. HGVS Recommendations for the Description of Sequence Variants: Update. Hum Mutat. (2016) . |
[11] | Fokkema IF , Taschner PE , Schaafsma GC , Celli J , Laros JF , den Dunnen JT . LOVD v.2.0: The next generation in gene variant databases. Hum Mutat. (2011) ;32: (5):557–63. |
[12] | Linke WA , Hamdani N . Gigantic business: Titin properties and function through thick and thin. Circulation Research. (2014) ;114: (6):1052–68. |
[13] | Guo W , Bharmal SJ , Esbona K , Greaser ML . Titin diversity–alternative splicing gone wild. Journal of Biomedicine & Biotechnology. (2010) ;2010: :753675. |
[14] | Freiburg A , Trombitas K , Hell W , Cazorla O , Fougerousse F , Centner T , et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circulation Research. (2000) ;86: (11):1114–21. |
[15] | Labeit S , Lahmers S , Burkart C , Fong C , McNabb M , Witt S , et al. Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. Journal of Molecular Biology. (2006) ;362: (4):664–81. |
[16] | Kolmerer B , Olivieri N , Witt CC , Herrmann BG , Labeit S . Genomic organization of M line titin and its tissue-specific expression in two distinct isoforms. Journal of Molecular Biology. (1996) ;256: (3):556–63. |
[17] | Labeit S , Gautel M , Lakey A , Trinick J . Towards a molecular understanding of titin. The EMBO Journal. (1992) ;11: (5):1711–6. |
[18] | Kruger M , Linke WA . Titin-based mechanical signalling in normal and failing myocardium. Journal of Molecular and Cellular Cardiology. (2009) ;46: (4):490–8. |
[19] | Kravchenko IV , Furalyov VA , Chatziefthimiou S , Wilmanns M , Popov VO . Induction of insulin-like growth factor 1 splice forms by subfragments of myofibrillar proteins. Molecular and Cellular Endocrinology. (2015) ;399: :69–77. |
[20] | del-Toro N , Dumousseau M , Orchard S , Jimenez RC , Galeota E , Launay G , et al. A new reference implementation of the PSICQUIC web service. Nucleic Acids Res. (2013) ;41: (Web Server issue):W601–6. |
[21] | Orchard S , Ammari M , Aranda B , Breuza L , Briganti L , Broackes-Carter F , et al. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. (2014) ;42: (Database issue):D358–63. |
[22] | Stark C , Breitkreutz BJ , Reguly T , Boucher L , Breitkreutz A , Tyers M . BioGRID: A general repository for interaction datasets. Nucleic Acids Res. (2006) ;34: (Database issue):D535–9. |
[23] | Gregorio CC , Trombitas K , Centner T , Kolmerer B , Stier G , Kunke K , et al. The NH2 terminus of titin spans the Z-disc: Its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. The Journal of Cell Biology. (1998) ;143: (4):1013–27. |
[24] | Mues A , van der Ven PF , Young P , Furst DO , Gautel M . Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Letters. (1998) ;428: (1-2):111–4. |
[25] | Pinotsis N , Petoukhov M , Lange S , Svergun D , Zou P , Gautel M , et al. Evidence for a dimeric assembly of two titin/telethonin complexes induced by the telethonin C-terminus. Journal of Structural Biology. (2006) ;155: (2):239–50. |
[26] | Sorimachi H , Freiburg A , Kolmerer B , Ishiura S , Stier G , Gregorio CC , et al. Tissue-specific expression and alpha-actinin binding properties of the Z-disc titin: Implications for the nature of vertebrate Z-discsJournal of Molecular Biology. (1997) ;270: (5):688–95 . |
[27] | Young P , Ferguson C , Banuelos S , Gautel M . Molecular structure of the sarcomeric Z-disk: Two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. The EMBO Journal. (1998) ;17: (6):1614–24. |
[28] | Kontrogianni-Konstantopoulos A , Bloch RJ . The hydrophilic domain of small ankyrin-1 interacts with the two N-terminal immunoglobulin domains of titin. The Journal of Biological Chemistry. (2003) ;278: (6):3985–91. |
[29] | Witt CC , Burkart C , Labeit D , McNabb M , Wu Y , Granzier H , et al. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo . EMBO J. (2006) ;25: (16):3843–55. |
[30] | Raynaud F , Astier C , Benyamin Y . Evidence for a direct but sequential binding of titin to tropomyosin and actin filaments. Biochimica et Biophysica Acta. (1700) 2 171–8. |
[31] | Golenhofen N , Arbeiter A , Koob R , Drenckhahn D . Ischemia-induced association of the stress protein alpha B-crystallin with I-band portion of cardiac titin. J Mol Cell Cardiol. (2002) ;34: (3):309–19. |
[32] | Sheikh F , Raskin A , Chu PH , Lange S , Domenighetti AA , Zheng M , et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. The Journal of Clinical Investigation. (2008) ;118: (12):3870–80. |
[33] | Lange S , Xiang F , Yakovenko A , Vihola A , Hackman P , Rostkova E , et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science (New York, NY). 308. (5728) 1599–603. |
[34] | Raynaud F , Fernandez E , Coulis G , Aubry L , Vignon X , Bleimling N , et al. Calpain 1-titin interactions concentrate calpain 1 in the Z-band edges and in the N2-line region within the skeletal myofibril. The FEBS Journal. (2005) ;272: (10):2578–90. |
[35] | Sorimachi H , Kinbara K , Kimura S , Takahashi M , Ishiura S , Sasagawa N , et al. Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. The Journal of Biological Chemistry. (1995) ;270: (52):31158–62. |
[36] | Sorimachi H , Kimura S , Kinbara K , Kazama J , Takahashi M , Yajima H , et al. Structure and physiological functions of ubiquitous and tissue-specific calpain species. Muscle-specific calpain, p94, interacts with connectin/titin. Advances in Biophysics. (1996) ;33: :101–22. |
[37] | Miller MK , Bang ML , Witt CC , Labeit D , Trombitas C , Watanabe K , et al. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. Journal of Molecular Biology. (2003) ;333: (5):951–64. |
[38] | Kruger M , Kotter S , Grutzner A , Lang P , Andresen C , Redfield MM , et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circulation Research. (2009) ;104: (1):87–94. |
[39] | Yamasaki R , Wu Y , McNabb M , Greaser M , Labeit S , Granzier H . Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circulation Research. (2002) ;90: (11):1181–8. |
[40] | Yamasaki R , Berri M , Wu Y , Trombitas K , McNabb M , Kellermayer MS , et al. Titin-actin interaction in mouse myocardium: Passive tension modulation and its regulation by calcium/S100A1. Biophysical Journal. (2001) ;81: (4):2297–313. |
[41] | Freiburg A , Gautel M . A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem. (1996) ;235: (1-2):317–23. |
[42] | Centner T , Yano J , Kimura E , McElhinny AS , Pelin K , Witt CC , et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. (2001) ;306: (4):717–26. |
[43] | Pizon V , Iakovenko A , Van Der Ven PF , Kelly R , Fatu C , Furst DO , et al. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci. (2002) ;115: (Pt 23):4469–82. |
[44] | Gautel M , Castiglione Morelli MA , Pfuhl M , Motta A , Pastore A . A calmodulin-binding sequence in the C-terminus of human cardiac titin kinase. European Journal of Biochemistry/FEBS. (1995) ;230: (2):752–9. |
[45] | Lange S , Auerbach D , McLoughlin P , Perriard E , Schafer BW , Perriard JC , et al. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. Journal of Cell Science. (2002) ;115: (Pt 24):4925–36. |
[46] | Fukuzawa A , Lange S , Holt M , Vihola A , Carmignac V , Ferreiro A , et al. Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band: Implications for hereditary myopathies. Journal of Cell Science. (2008) ;121: (11):1841–51. |
[47] | Prudner BC , Roy PS , Damron DS , Russell MA . alpha-Synemin localizes to the M-band of the sarcomere through interaction with the M10 region of titin. FEBS Lett. (2014) ;588: (24):4625–30. |
[48] | Sarparanta J , Blandin G , Charton K , Vihola A , Marchand S , Milic A , et al. Interactions with M-band titin and calpain 3 link myospryn (CMYA5) to tibial and limb-girdle muscular dystrophies. The Journal of Biological Chemistry. (2010) ;285: (39):30304–15. |
[49] | Udd B , Partanen J , Halonen P , Falck B , Hakamies L , Heikkila H , et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Archives of Neurology. (1993) ;50: (6):604–8. |
[50] | Hackman P , Marchand S , Sarparanta J , Vihola A , Penisson-Besnier I , Eymard B , et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscular disorders: NMD. (2008) ;18: (12):922–8. |
[51] | Van den Bergh PYK , Bouquiaux O , Verellen C , Marchand S , Richard I , Hackman P , et al. Tibial muscular dystrophy in a Belgian family. Annals of Neurology. (2003) ;54: (2):248–51. |
[52] | Hackman P , Vihola A , Haravuori H , Marchand S , Sarparanta J , De Seze J , et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. American Journal of Human Genetics. (2002) ;71: (3):492–500. |
[53] | Pollazzon M , Suominen T , Penttila S , Malandrini A , Carluccio MA , Mondelli M , et al. The first Italian family with tibial muscular dystrophy caused by a novel titin mutation. Journal of Neurology. (2010) ;257: (4):575–9. |
[54] | Evila A , Vihola A , Sarparanta J , Raheem O , Palmio J , Sandell S , et al. Atypical phenotypes in titinopathies explained by second titin mutations. Annals of Neurology. (2014) ;75: (2):230–40. |
[55] | Guglieri M , Straub V , Bushby K , Lochmuller H . Limb-girdle muscular dystrophies. Current Opinion in Neurology. (2008) ;21: (5):576–84. |
[56] | Nigro V , Savarese M . Genetic basis of limb-girdle muscular dystrophies: The update. Acta myologica: Myopathies and cardiomyopathies: Official journal of the Mediterranean Society of Myology/edited by the Gaetano Conte Academy for the Study of Striated Muscle Diseases. (2014) ;33: (1):1–12. |
[57] | Udd B , Kaarianen H , Somer H . Muscular dystrophy with separate clinical phenotypes in a large family. Muscle & Nerve. (1991) ;14: (11):1050–8. |
[58] | Penisson-Besnier I , Hackman P , Suominen T , Sarparanta J , Huovinen S , Richard-Cremieux I , et al. Myopathies caused by homozygous titin mutations: Limb-girdle muscular dystrophy 2J and variations of phenotype. Journal of Neurology, Neurosurgery, and Psychiatry. (2010) ;81: (11):1200–2. |
[59] | Zheng W , Chen H , Deng X , Yuan L , Yang Y , Song Z , et al. Identification of a Novel Mutation in the Titin Gene in a Chinese Family with Limb-Girdle Muscular Dystrophy 2J. Molecular Neurobiology. (2015) . |
[60] | Haravuori H , Vihola A , Straub V , Auranen M , Richard I , Marchand S , et al. Secondary calpain3 deficiency in 2q-linked muscular dystrophy: Titin is the candidate gene. Neurology. (2001) ;56: (7):869–77. |
[61] | Charton K , Sarparanta J , Vihola A , Milic A , Jonson PH , Suel L , et al. CAPN3-mediated processing of C-terminal titin replaced by pathological cleavage in titinopathy. Hum Mol Genet. (2015) ;24: (13):3718–31. |
[62] | North KN , Wang CH , Clarke N , Jungbluth H , Vainzof M , Dowling JJ , et al. Approach to the diagnosis of congenital myopathies. Neuromuscular disorders: NMD. (2014) ;24: (2):97–116. |
[63] | Bitoun M , Romero NB , Guicheney P . Mutations in dynamin 2 cause dominant centronuclear myopathy. Medecine Sciences: M/S. (2006) ;22: (2):101–2. |
[64] | Bohm J , Biancalana V , Malfatti E , Dondaine N , Koch C , Vasli N , et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain: A Journal of Neurology. (2014) ;137: (Pt 12):3160–70. |
[65] | Wilmshurst JM , Lillis S , Zhou H , Pillay K , Henderson H , Kress W , et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Annals of Neurology. (2010) ;68: (5):717–26. |
[66] | Laporte J , Hu LJ , Kretz C , Mandel JL , Kioschis P , Coy JF , et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nature Genetics. (1996) ;13: (2):175–82. |
[67] | Ceyhan-Birsoy O , Agrawal PB , Hidalgo C , Schmitz-Abe K , DeChene ET , Swanson LC , et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. (2013) ;81: (14):1205–14. |
[68] | Fattori F , Maggi L , Bruno C , Cassandrini D , Codemo V , Catteruccia M , et al. Centronuclear myopathies: Genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. Journal of Neurology. (2015) ;262: (7):1728–40. |
[69] | Carmignac V , Salih MA , Quijano-Roy S , Marchand S , Al Rayess MM , Mukhtar MM , et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Annals of Neurology. (2007) ;61: (4):340–51. |
[70] | Chauveau C , Bonnemann CG , Julien C , Kho AL , Marks H , Talim B , et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Human Molecular Genetics. (2014) ;23: (4):980–91. |
[71] | Bione S , Maestrini E , Rivella S , Mancini M , Regis S , Romeo G , et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nature Genetics. (1994) ;8: (4):323–7. |
[72] | Gueneau L , Bertrand AT , Jais JP , Salih MA , Stojkovic T , Wehnert M , et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. American Journal of Human Genetics. (2009) ;85: (3):338–53. |
[73] | Bonne G , Di Barletta MR , Varnous S , Becane HM , Hammouda EH , Merlini L , et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nature Genetics. (1999) ;21: (3):285–8. |
[74] | Raffaele Di Barletta M , Ricci E , Galluzzi G , Tonali P , Mora M , Morandi L , et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. American Journal of Human Genetics. (2000) ;66: (4):1407–12. |
[75] | Zhang Q , Bethmann C , Worth NF , Davies JD , Wasner C , Feuer A , et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Human Molecular Genetics. (2007) ;16: (23):2816–33. |
[76] | De Cid R , Ben Yaou R , Roudaut C , Charton K , Baulande S , Leturcq F , et al. A new titinopathy: Childhood-juvenile onset Emery-Dreifuss-like phenotype without cardiomyopathy. Neurology. (2015) . |
[77] | Edstrom L , Thornell LE , Albo J , Landin S , Samuelsson M . Myopathy with respiratory failure and typical myofibrillar lesions. Journal of the Neurological Sciences. (1990) ;96: (2-3):211–28. |
[78] | Pfeffer G , Elliott HR , Griffin H , Barresi R , Miller J , Marsh J , et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain: A Journal of Neurology. (2012) ;135: (Pt 6):1695–713. |
[79] | Ohlsson M , Hedberg C , Bradvik B , Lindberg C , Tajsharghi H , Danielsson O , et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain: A Journal of Neurology. (2012) ;135: (Pt 6):1682–94. |
[80] | Toro C , Olive M , Dalakas MC , Sivakumar K , Bilbao JM , Tyndel F , et al. Exome sequencing identifies titin mutations causing hereditary myopathy with early respiratory failure (HMERF) in families of diverse ethnic origins. BMC Neurology.29–. (2377) ;13: :13–29. |
[81] | Izumi R , Niihori T , Aoki Y , Suzuki N , Kato M , Warita H , et al. Exome sequencing identifies a novel TTN mutation in a family with hereditary myopathy with early respiratory failure. Journal of Human Genetics. (2013) ;58: (5):259–66. |
[82] | Palmio J , Evila A , Chapon F , Tasca G , Xiang F , Bradvik B , et al. Hereditary myopathy with early respiratory failure: Occurrence in various populations. Journal of Neurology, Neurosurgery, and Psychiatry. (2014) ;85: (3):345–53. |
[83] | Pfeffer G , Barresi R , Wilson IJ , Hardy SA , Griffin H , Hudson J , et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. Journal of Neurology, Neurosurgery, and Psychiatry. (2014) ;85: (3):331–8. |
[84] | Hedberg C , Toledo AG , Gustafsson CM , Larson G , Oldfors A , Macao B . Hereditary myopathy with early respiratory failure is associated with misfolding of the titin fibronectin III 119 subdomain. Neuromuscular Disorders: NMD. (2014) ;24: (5):373–9. |
[85] | Yue D , Gao M , Zhu W , Luo S , Xi J , Wang B , et al. New disease allele and de novo mutation indicate mutational vulnerability of titin exon 343 in hereditary myopathy with early respiratory failure. Neuromuscular Disorders: NMD. (2015) ;25: (2):172–6. |
[86] | Hedberg C , Melberg A , Dahlbom K , Oldfors A . Hereditary myopathy with early respiratory failure is caused by mutations in the titin FN3 119 domain. Brain: A Journal of Neurology. (2014) ;137: (Pt 4):e270. |
[87] | Lange S , Edstrom L , Udd B , Gautel M . Reply: Hereditary myopathy with early respiratory failure is caused by mutations in the titin FN3 119 domain. Brain: A Journal of Neurology. (2014) ;137: (Pt 6):e279. |
[88] | Evila A , Arumilli M , Udd B , Hackman P . Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscular Disorders: NMD. (2015) . |
[89] | Dabby R , Sadeh M , Hilton-Jones D , Plotz P , Hackman P , Vihola A , et al. Adult onset limb-girdle muscular dystrophy - a recessive titinopathy masquerading as myositis. Journal of the Neurological Sciences. (2015) ;351: (1-2):120–3. |
[90] | Steffen LS , Guyon JR , Vogel ED , Howell MH , Zhou Y , Weber GJ , et al. The zebrafish runzel muscular dystrophy is linked to the titin gene. Developmental Biology. (2007) ;309: (2):180–92. |
[91] | Garvey SM , Rajan C , Lerner AP , Frankel WN , Cox GA . The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics. (2002) ;79: (2):146–9. |
[92] | Charton K , Daniele N , Vihola A , Roudaut C , Gicquel E , Monjaret F , et al. Removal of the calpain 3 protease reverses the myopathology in a mouse model for titinopathies. Human Molecular Genetics. (2010) ;19: (23):4608–24. |
[93] | Xu X , Meiler SE , Zhong TP , Mohideen M , Crossley DA , Burggren WW , et al. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nature Genetics. (2002) ;30: (2):205–9. |
[94] | Radke MH , Peng J , Wu Y , McNabb M , Nelson OL , Granzier H , et al. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proceedings of the National Academy of Sciences of the United States of America. (2007) ;104: (9):3444–9. |
[95] | Gotthardt M , Hammer RE , Hubner N , Monti J , Witt CC , McNabb M , et al. Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. The Journal of Biological Chemistry. (2003) ;278: (8):6059–65. |
[96] | Peng J , Raddatz K , Labeit S , Granzier H , Gotthardt M . Muscle atrophy in titin M-line deficient mice. Journal of Muscle Research and Cell Motility. (2005) ;26: (6-8):381–8. |
[97] | Zhang Y , Featherstone D , Davis W , Rushton E , Broadie K . Drosophila D-titin is required for myoblast fusion and skeletal muscle striation. Journal of Cell Science. (2000) 113: (Pt 71)(Pt 17):3103–15. |
[98] | de Seze J , Udd B , Haravuori H , Sablonniere B , Maurage CA , Hurtevent JF , et al. The first European family with tibial muscular dystrophy outside the Finnish population. Neurology. (1998) ;51: (6):1746–8. |
[99] | Vasli N , Laporte J . Impacts of massively parallel sequencing for genetic diagnosis of neuromuscular disorders. Acta Neuropathologica. (2013) ;125: (2):173–85. |
[100] | Gilissen C , Hoischen A , Brunner HG , Veltman JA . Unlocking Mendelian disease using exome sequencing. Genome Biol. (2011) ;12: (9):228. |
[101] | Mamanova L , Coffey AJ , Scott CE , Kozarewa I , Turner EH , Kumar A , et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. (2010) ;7: (2):111–8. |
[102] | Herman DS , Lam L , Taylor MR , Wang L , Teekakirikul P , Christodoulou D , et al. Truncations of titin causing dilated cardiomyopathy. The New England Journal of Medicine. (2012) ;366: (7):619–28. |
[103] | Akinrinade O , Koskenvuo JW , Alastalo TP . Prevalence of Titin Truncating Variants in General Population. PLoS One. (2015) ;10: (12):e0145284. |
[104] | Savarese M , Di Fruscio G , Mutarelli M , Torella A , Magri F , Santorelli FM , et al. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathologica Communications. (2014) ;2: :100-014-0100-3. |
[105] | Savarese M , Di Fruscio G , Torella A , Fiorillo C , Magri F , Fanin M , et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology. (2016) ;87: (1):71–6. |
[106] | Vasli N , Bohm J , Le Gras S , Muller J , Pizot C , Jost B , et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathologica. (2012) ;124: (2):273–83. |
[107] | Sevy A , Cerino M , Gorokhova S , Dionnet E , Mathieu Y , Verschueren A , et al. Improving molecular diagnosis of distal myopathies by targeted next-generation sequencing. Journal of Neurology, Neurosurgery, and Psychiatry. (2015) |
[108] | Dai Y , Wei X , Zhao Y , Ren H , Lan Z , Yang Y , et al. A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next-generation sequencing. Neuromuscular Disorders: NMD. (2015) ;25: (8):617–24. |
[109] | Pernigo S , Fukuzawa A , Bertz M , Holt M , Rief M , Steiner RA , et al. Structural insight into M-band assembly and mechanics from the titin-obscurin-like-1 complex. Proceedings of the National Academy of Sciences of the United States of America. (2010) ;107: (7):2908–13. |
[110] | Sauer F , Vahokoski J , Song YH , Wilmanns M . Molecular basis of the head-to-tail assembly of giant muscle proteins obscurin-like 1 and titin. EMBO Reports. (2010) ;11: (7):534–40. |
[111] | Rudloff MW , Woosley AN , Wright NT . Biophysical characterization of naturally occurring titin M10 mutations. Protein science: A publication of the Protein Society. 2015. |
[112] | Sahni N , Yi S , Taipale M , Fuxman Bass JI , Coulombe-Huntington J , Yang F , et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. (2015) ;161: (3):647–60. |
[113] | Beaulieu CL , Majewski J , Schwartzentruber J , Samuels ME , Fernandez BA , Bernier FP , et al. FORGE Canada Consortium: Outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet. (2014) ;94: (6):809–17. |
[114] | Thompson R , Johnston L , Taruscio D , Monaco L , Beroud C , Gut IG , et al. RD-Connect: An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. J Gen Intern Med.S. (2014) ;29: (Suppl 3):780–7. |
Figures and Tables
Fig.1
Top: A schematic view of the sarcomere, with titin filaments shown in red. One titin molecule, extending from the Z-disc to M-band, is highlighted. Middle: The modular structure of the titin protein (theoretical meta isoform). Titin is mostly comprised of repeated immunoglobulin-like (Ig; red) and fibronectin type 3-like (FN3; white) domains. Selected domains are labeled above the diagram with the classical titin nomenclature (sarcomere region Z/I/A/M+domain number; Bang et al. 2001), followed in parentheses by the alternative numbering scheme (domain type Ig/FN3 + domain number), and with the corresponding exon number. Also indicated are other structural features: the Z-repeats, the Novex-1 and Novex-2 exons, the N2B and N2A elements, the PEVK (proline/glutamate/valine/lysine-rich) region, and the alternatively spliced M-band is7 (M-is7) region. Protein interactions of the different parts of titin are summarized below the diagram. Bottom: Exon inclusion in NCBI RefSeq database sequences representing the main titin isoform classes. The bars align to the protein diagram of the meta isoform above, except for the alternative C-terminal exon of the Novex-3 isoform (grey). Note that within each class there is further developmental, anatomical, and physiological variation in exon inclusion, mostly in the proximal tandem-Ig and PEVK regions.
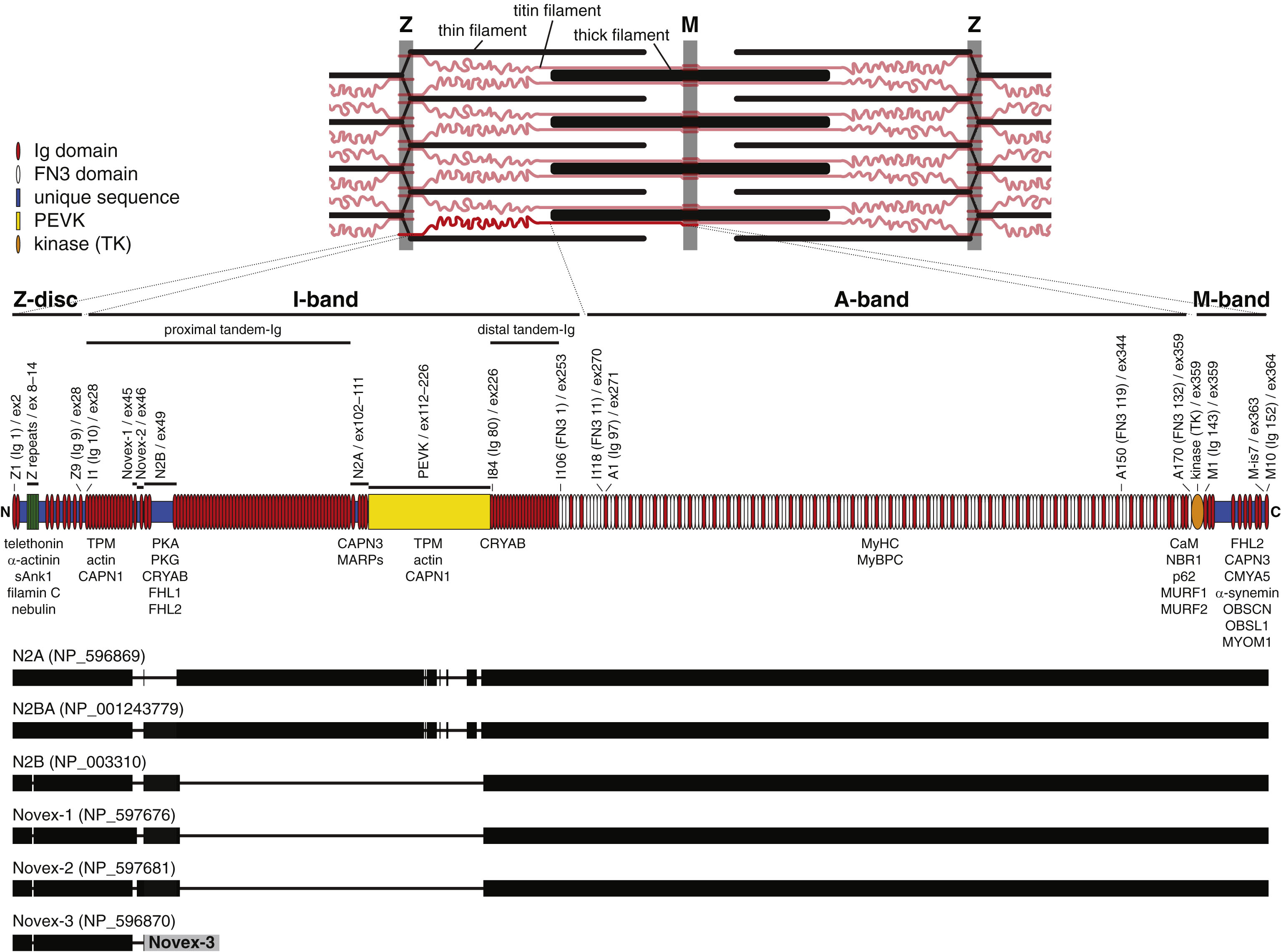
Fig.2
Skeletal muscle disease mutations in titin. Symbols below the diagram depict mutations associated with neuromuscular diseases, with the symbol shape indicating mutation type and symbol color indicating the predominant clinical phenotype.
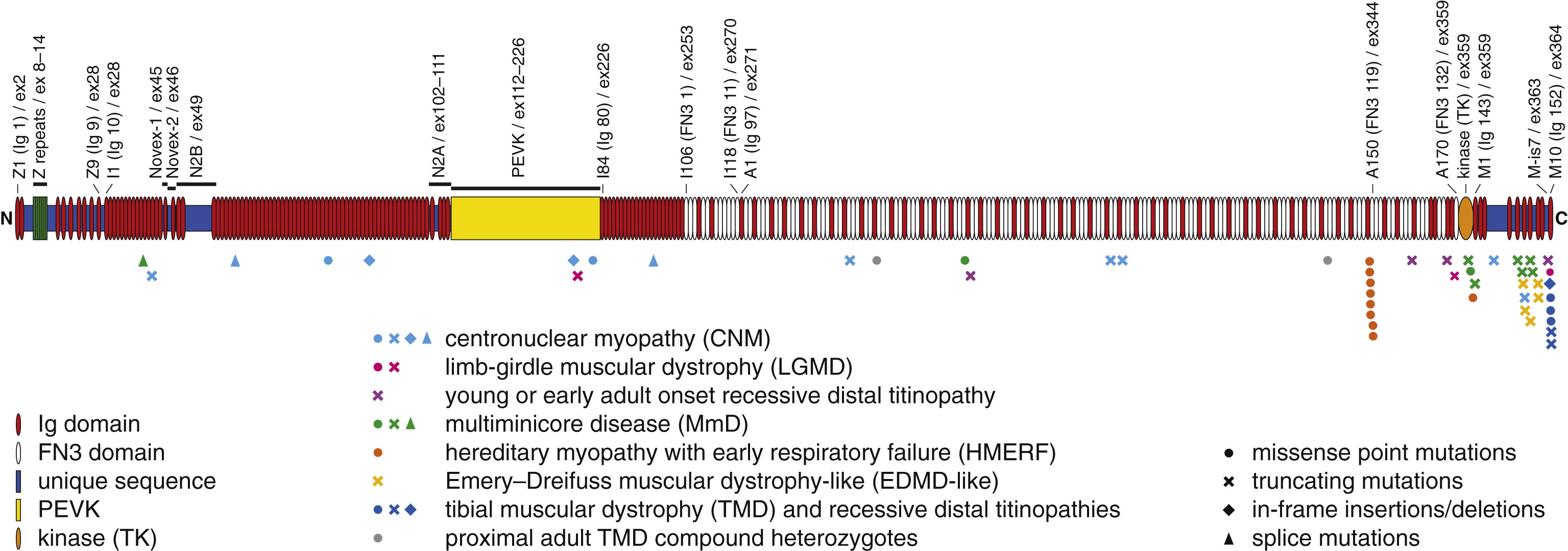
Table 1
Mutations causing TMD, young or early adult onset recessive distal titinopathy, LGMD or adult proximal phenotype
| Allele 1 | |||||||
| Mutation§ | Exon† | Domain | |||||
| TMD | Finnish TMD | FINmaj mutation | 364 (363#) | M10 (Ig 152) | |||
| de Seze 1998/Hackman 2002 (French family A) | c.107867T>C (p.Leu35956Pro) | 364 (363#) | M10 (Ig 152) | ||||
| Hackman 2008 (French family B) | c.107890C>T (p.Gln35964*) | 364 (363#) | M10 (Ig 152) | ||||
| Hackman 2008 (Albacete family) | c.107889delA (p.Lys35963Asnfs*9) | 364 (363#) | M10 (Ig 152) | ||||
| Hackman 2008 (Barcelona family) | c.107889delA (p.Lys35963Asnfs*9) | 364 (363#) | M10 (Ig 152) | ||||
| Van den Bergh 2003 (Belgian family) | c.107840T>A (p.Ile35947Asn) | 364 (363#) | M10 (Ig 152) | ||||
| Pollazzon 2010 (Italian family) | c.107837A>C (p.His35946Pro) | 364 (363#) | M10 (Ig 152) | ||||
| Allele 1 | Allele 2 | ||||||
| Mutation§ | Exon† | Domain | Mutation§ | Exon† | Domain | ||
| Young or early adult onset recessive distal titinopathy | Hackman 2008 (French family C - Proband) and Evila2015 (pt.5b) | c.100558-100561dup (p.Gly33521Aspfs*25) | 358 (357#) | A169 (Ig 141) | c.107647delT (p.Ser35883Glnfs*10) | 363 (362#) | M-is7 |
| Hackman 2008 (French family C - Mother) and Evila2014 (pt.5a) | c.98105delC (p.Pro32702Leufs*15) | 353 (352#) | A160 (Ig 139) | c.107647delT (p.Ser35883Glnfs*10) | 363 (362#) | M-is7 | |
| Evila2014 (pt.6) | c.67089delT (p.Lys22364Argfs*24) | 319 (318#) | A55 (FN3 50) | c.107889delA (p.Lys35963Asnfs*9) | 364 (363#) | M10 (Ig 152) | |
| Evila2014 (pt.7) | c.107889delA (p.Lys35963Asnfs*9) | 364 (363#) | M10 (Ig 152) | c.107889delA (p.Lys35963Asnfs*9) | 364 (363#) | M10 (Ig 152) | |
| LGMD | Finnish original cases | FINmaj | 364 (363#) | M10 (Ig 152) | FINmaj | 364 (363#) | M10 (Ig 152) |
| Penisson-Besnier 2010 (French family B - pt.IV-5) | c.107890C>T (p.Gln35964*) | 364 (363#) | M10 (Ig 152) | c.107890C>T (p.Gln35964*) | 364 (363#) | M10 (Ig 152) | |
| Evila 2014 (pt.1) | c.101113delT (p.Ser33705Leufs*4) | 359 (358#) | A170 (FN3 132) | FINmaj | 364 (363#) | M10 (Ig 152) | |
| Evila 2014 (pt.2) | c.39492dupT (p.Glu13165*) | 208 (207#) | PEVK | FINmaj | 364 (363#) | M10 (Ig 152) | |
| Evila 2014 (pt.3) | ? | FINmaj | 364 (363#) | M10 (Ig 152) | |||
| Zheng 2015 | c.107788T>C (p.Trp35930Arg) | 364 (363#) | M10 (Ig 152) | c.107788T>C (p.Trp35930Arg) | 364 (363#) | M10 (Ig 152) | |
| Proximal adult TMD compound heterozygotes | Evila 2014 (pt4) | c.92167C>T (p.Pro30723Ser) | 340 (339#) | A140 (FN3 112) | FINmaj | 364 (363#) | M10 (Ig 152) |
| Evila 2015 (pt t13) | c.60494A>G (p.His20165Arg) | 305 (304#) | A32 (Ig 106) | c.107837A>C (p.His35946Pro) | 364 (363#) | M10 (Ig 152) | |
§Reported according to the longest theoretical transcript (NM_001267550). †Numbered according to the HGVS recommendations (ref.10) and to the current Leiden database (LOVD) numbering (modified on 11th October 2013) (ref.11). #Numbered according to the old numbering (before 11th October 2013).
Table 2
Mutations causing congenital centronuclear myopathy, multiminicore disease and an Emery-Dreifuss-like phenotype
| Allele 1 | Allele 2 | ||||||
| Mutation§ | Exon† | Domain | Mutation§ | Exon† | Domain | ||
| Congenital centronuclear myopathy | Ceyhan-Birsoy 2013 (pt.314-1) | c.40558G>C (p.Val13520Leu?) | 220 (219#) | PEVK | c.44816-1G>A (p.[?]) | 243i (242i#) | – |
| Ceyhan-Birsoy 2013 (pt.966-1) | c.24863_24877del | 87 (86#) | I65 (Ig 62) | c.103846_103849dup | 359 (358#) | M-is2 | |
| (p.Asp8288_Ile8293delinsVal) | (p.Pro34617Glnfs*3) | ||||||
| c.39201_39203dup (p.Pro13068dup) | 204 (203#) | PEVK | |||||
| Ceyhan-Birsoy 2013 (pt.979-1) | c.15496 + 1 G>A (p.[?]) | 53i (52i#) | – | c.76393_76396del (p.Asn25465*) | 327 (326#) | A86 (FN3 73) | |
| c.106019delG (p.Gly35340Valfs*65) | 359 (358#) | M-is4 | |||||
| Ceyhan-Birsoy 2013 (pt.1044-1) | c.77764C>T (p.Gln25922*) | 327 (326#) | A91 (FN3 76) | c.107889delA (p.Lys35963Asnfs*9) | 364 (363#) | M10 (Ig 152) | |
| Ceyhan-Birsoy 2013 (pt.1093-1) | c.21961G>A (p.Glu7321Lys) | 76 (75#) | I54– I55 (Ig 51– 52) | c.58620delA (p.Val19541Phefs*22) | 299 (298#) | A26 (FN3 29) | |
| Fattori 2015 (pt.38) | c.9577C>T (p. Arg3193*) | 41 | I13 | c.105832C>T (p.Gln35278*) | 359 (358#) | M-is4 | |
| Multiminicore disease | Carmignac 2007 (family 1) | c.106571delA (p.Lys35524Argfs*22) | 361 (360#) | M8 (Ig 150) | c.106571delA (p.Lys35524Argfs*22) | 361 (360#) | M8 (Ig 150) |
| Carmignac 2007 (family 2) | c.105528_105535del | 359 (358#) | M5 (Ig 147) | c.105528_105535del | 359 (358#) | M5 (Ig 147) | |
| (p.Gln35175Hisfs*9) | (p.Gln35175Hisfs*9) | ||||||
| Chauveau 2014 (family 1) | c.106407_106408del | 360 (359#) | M7 (Ig 149) | c.106407_106408del | 360 (359#) | M7 (Ig 149) | |
| (p.Glu35470Argfs*11) | (p.Glu35470Argfs*11) | ||||||
| Chauveau 2014 (family 2) | c.102523C>T (p.Arg34175*) | 359 (358#) | M1 (Ig 143) | c.105832C>T (p.Gln35278*) | 359 (358#) | M-is4 | |
| Chauveau 2014 (family 3) | c.66695T>A (p.Val22232Glu) | 317 (316#) | A53 (FN3 49) | c.102057delT (p.Asn34020Thrfs*9) | 359 (358#) | TK | |
| Chauveau 2014 (family 4) | c.9163 + 1 G>C (p.?) | 38i | – | c.102214T>C (p.Trp34072Arg) | 359 (358#) | TK | |
| Emery-Dreifuss-like phenotype | De Cid 2015 (pt.1) | c.106959T>A (p.Tyr35653*) | 361 (360#) | M8 (Ig 150) | c.106959T>A (p.Tyr35653*) | 361 (360#) | M8 (Ig 150) |
| De Cid 2015 (pt.2) | c.106051delT (p.Glu35351Asnfs*54) | 359 (358#) | M6 (Ig 148) | c.106978C>T (p.Gln35660*) | 361 (360#) | M8 (Ig 150) | |
| De Cid 2015 (pt.3) | c.105910_105914del | 359 (358#) | M-is4 | c.106422delG (p.Phe35475Serfs*4) | 360 (359#) | M7 (Ig 149) | |
| (p.Thr35304Cysfs*3) | |||||||
§Reported according to the longest theoretical transcript (NM_001267550). †Numbered according to the HGVS recommendations (ref.10) and to the current Leiden database (LOVD) numbering (modified on 11th October 2013) (ref.11). #Numbered according to the old numbering (before 11th October 2013).
Table 3
Mutations causing hereditary myopathy with early respiratory failure (HMERF)
| Mutations§ | ||
| Palmio 2014 (fam A) | c.95126C>G (p.Pro31709Arg) | |
| Pfeffer 2012 (fam A/B/C) | c.95134T>C (p.Cys31712Arg) | |
| Ohlsson 2012 (fam A/B/C) | c.95134T>C (p.Cys31712Arg) | |
| Pfeffer 2013 (fam 1/2/3/4/5) | c.95134T>C (p.Cys31712Arg) | |
| Toro 2013 (fam B/C) | c.95134T>C (p.Cys31712Arg) | |
| Palmio 2014 (fam B/C/D/E/F/G) | c.95134T>C (p.Cys31712Arg) | |
| Yue 2015 (pt 2) | c.95134T>C (p.Cys31712Arg) | |
| Izumi 2013 | c.95186G>T (p.Trp31729Leu) | |
| Palmio 2014 (fam H) | c.95185T>C (p.Trp31729Arg) | |
| Palmio 2014 (fam I) | c.95187G>C (p.Trp31729Cys) | |
| Pfeffer 2013 (fam 6) | c.95195C>T (p.Pro31732Leu) | |
| Palmio 2014 (fam L) | c.95195C>T (p.Pro31732Leu) | |
| Yue 2015 (pt 1) | c.95195C>T (p.Pro31732Leu) | |
| Palmio 2014 (fam J/K) | c.95195C>T (p.Pro31732Leu) | c.95195C>T (p.Pro31732Leu) |
| Pfeffer 2013 (fam 7) | c.95358C>G (p.Asn31786Lys) | |
| Toro 2013 (fam A) | c.95372G>A (p.Gly31791Asp) | |
§Reported according to the longest theoretical transcript (NM_001267550).


