
“Get the Balance Right”: Pathological Significance of Autophagy Perturbation in Neuromuscular Disorders
Abstract
Recent research has revealed that autophagy, a major catabolic process in cells, is dysregulated in several neuromuscular diseases and contributes to the muscle wasting caused by non-muscle disorders (e.g. cancer cachexia) or during aging (i.e. sarcopenia). From there, the idea arose to interfere with autophagy or manipulate its regulatory signalling to help restore muscle homeostasis and attenuate disease progression. The major difficulty for the development of therapeutic strategies is to restore a balanced autophagic flux, due to the dynamic nature of autophagy. Thus, it is essential to better understand the mechanisms and identify the signalling pathways at play in the control of autophagy in skeletal muscle. A comprehensive analysis of the autophagic flux and of the causes of its dysregulation is required to assess the pathogenic role of autophagy in diseased muscle. Furthermore, it is essential that experiments distinguish between primary dysregulation of autophagy (prior to disease onset) and impairments as a consequence of the pathology. Of note, in most muscle disorders, autophagy perturbation is not caused by genetic modification of an autophagy-related protein, but rather through indirect alteration of regulatory signalling or lysosomal function. In this review, we will present the mechanisms involved in autophagy, and those ensuring its tight regulation in skeletal muscle. We will then discuss as to how autophagy dysregulation contributes to the pathogenesis of neuromuscular disorders and possible ways to interfere with this process to limit disease progression.
Abbreviations
Atg | Autophagy-related gene |
AVM | Autophagic vacuolar myopathy |
AVSF | Autophagic vacuole with sarcolemmal features |
BM | Bethlem myopathy |
CASA | Chaperone-assisted selective autophagy |
CMA | Chaperone-mediated autophagy |
(AD-)CNM | (Autosomal dominant) Centronuclear myopathies |
COLVI-RM | Collagen VI-related myopathies |
DM | Distal myopathies |
DM1 | Myotonic Dystrophy type I |
DMD | Duchenne muscular dystrophy |
EM | Electronic microscopy |
ER | Endoplasmic reticulum |
ERAD | ER associated protein degradation |
FoxO3 | Forkhead box class O |
GAA | Acid α-glucosidase |
GSDII(b) | Glycogen storage disease type II(b) |
Hsp | Heat shock protein |
IBMPFD | Inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia |
LPD | Low protein diet |
3-MA | 3-methyladenine |
MAP1LC3 | Microtubule-associated protein light chain 3 |
MDC1A | Congenital muscular dystrophy Type 1 |
MiT/TFE | microphthalmia/transcription factor E |
MFM | Myofibrillar myopathies |
mTORC1 | mammalian Target of Rapamycin Complex1 |
OPMD | Oculopharyngeal Muscular Dystrophy |
PE | Phosphatidylethanolamine |
PI3P | Phosphatidylinositol-3-phosphate |
RV | Rimmed vacuole |
sIBM | sporadic Inclusion Body Myositis |
SQSTM1 | (p62) Sequestosome1 |
UCMD | Ullrich congenital muscular dystrophy |
UPR | Unfolding protein response |
UPS | Ubiquitin-proteasome system |
VCP | Valosin containing protein |
Vps34 | Vacuolar sorting protein 34 |
XMEA | X-linked myopathy with excessive autophagy |
XLMTM | X-linked myotubular myopathies |
Introduction
Muscle tissue accounts for approximately 40% of whole body mass. It displays an incredible plasticity, enabling it to adjust its size based on external stimuli. Numerous pathological conditions can threaten its maintenance and cause muscle dysfunction. Muscle mass relies heavily on the balance between catabolic and anabolic processes, and more specifically on the equilibrium between protein synthesis and degradation (i.e. proteostasis). While promoting protein synthesis increases muscle mass, blocking protein degradation is rather detrimental. Importantly, due to the central metabolic importance of muscle tissue, changes in muscle mass and defects in skeletal muscle proteostasis often affect global body metabolism [1, 2]. Although these integrated perturbations may significantly impact the general health of patients suffering from neuromuscular disorders, this phenomenon has been only poorly explored. By contrast, the literature reporting pathogenic defects in protein degradation in muscle diseases has grown exponentially over the last decade. There are two major contributors to protein degradation in cells: The ubiquitin-proteasome system (UPS) and the autophagy-lysosome process. While involving different machineries and potentially targeting distinct proteins, the two systems are tightly connected and dysfunction of one impinges on the other [3]. Altered activity of the UPS contributes to muscle atrophy in neuromuscular disorders, such as MDC1A (congenital muscular dystrophy type 1) [4] and DMD (Duchenne muscular dystrophy) [5, 6]. Signs of impaired UPS include accumulation of misfolded and/or ubiquitinated proteins [7]. In parallel, autophagy coupled with the endo-cytic/lysosomal compartments permits recycling of proteins and organelles under basal conditions (referred to as constitutive autophagy) and upon different cellular stresses (adaptive, induced autophagy) [8]. Autophagy is essential for cell homeostasis as highlighted by the tissue alterations arising from its dysregulation. The role of autophagy impairment in the pathogenesis of neurodegenerative diseases [9, 10], cancers [11] or infectious diseases [12], as well as in age-associated homeostasis decline [13, 14] is now clearly documented. Most neuromuscular disorders have been linked to insufficient autophagy-mediated degradation, although some, such as MDC1A, have been shown to have enhanced autophagic flux. Based on these observations, autophagy has often been considered as a promising target for therapeutic strategies. In this review, the mechanisms at play in autophagy and the signalling involved in its regulation will be briefly presented; for further details, the reader is referred to reviews regarding specific autophagy-associated molecular mechanisms [15–18]. In the following sections, the pathological significance of autophagy perturbation in neuromuscular disorders will be discussed.
Autophagy at a glance
Autophagy is an evolutionary conserved homeostatic process, associated with unfavourable and conditions of stress (Fig. 1) [19]. Constitutive autophagy ensures basal turnover of proteins and organelles and is a key element of the quality control program in cells [20]. Induced autophagy is triggered by various stimuli, such as nutrient deprivation, energy depletion or growth factor withdrawal, as well as oxidative or endoplasmic reticulum (ER) stress. Autophagy is coupled to lysosomes, constituting the end point of the degradative process. There are three different types of autophagy: Macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), which each differ by their mechanism of cargo delivery to the lysosomes [21, 22] (Fig. 1). Microautophagy involves direct invagination of a small cytosolic portion by lysosomes and may contribute to glycogen loading in these organelles. CMA relies on the selective translocation of soluble, unfolded proteins into lysosomes, after multimerization of the lysosomal membrane receptor LAMP2A [23, 24]. The KFERQ motif of the target proteins is recognized by chaperones (e.g. Hsc70) and co-chaperones [25]. Macroautophagy, also simply referred to as autophagy, proceeds through the formation of double membrane vesicles, called autophagosomes. They engulf large cytoplasmic portions and organelles, which are degraded after fusion with lysosomes [20, 21]. Autophagosome formation involves more than 30 Atg (autophagy-related gene) proteins (Fig. 1).
Autophagy induction
Nutrient sensing in cells is controlled by the large multiprotein complex mTORC1 (mammalian Target of Rapamycin Complex 1), which includes, as major constituents, mTOR and raptor. Amino acid-dependent activation of mTORC1 by the GTPase Rheb occurs at the lysosomal surface and is tightly controlled by Rag GTPases and the Ragulator complex [26–28]. Rheb is inhibited by the TSC1/TSC2 complex, which integrates energetic and growth factor signals through its phosphorylation by AMPK and Akt/PKB, respectively [29] (Fig. 2). In most cell types, autophagy induction is determined by mTORC1 inhibition, as seen upon treatment with rapamycin, a pharmacologic mTORC1 inhibitor [30]. mTORC1 inhibits autophagy by phosphorylating Ulk1 (Ser 757) and Atg13, in the Ulk1-Atg13-Fip200-Atg101 complex [31, 32]. mTORC1 activity disrupts the interaction of Ulk1 and AMPK [33, 34]. Upon energy deprivation, this interaction is restored, leading to Ulk1 phosphorylation at Ser317 and Ser777 and thereby to autophagy induction [34–38] (Fig. 1). Ulk1 stabilization further depends on its ubiquitination by the E3 ubiquitin ligase Traf6. Inhibitory phosphorylation of Ambra1, a positive kinase for Traf6, by mTORC1 limits Ulk1 activation [39] (Fig. 1). ER stress, reactive oxygen species (ROS) and hypoxia induce autophagy by modulating mTORC1 and AMPK signalling, although their effect also depends on PERK/eIF2α, IRE1/JNK, and HIF1/Bnip3 pathways [40].
Autophagy initiation
Activation of the Ulk1-Atg13-Fip200-Atg101 complex triggers assembly of the nucleation machinery at multiple sites, initiating de novo vesicle formation. Membranes to build the initial autophagy compartment (also called phagophore) are provided by ER, Golgi, mitochondria, and plasma membrane [41–46]. Nucleation requires production of phosphatidylinositol-3-phosphate (PI3P), which is reliant on the class III PI3 kinase, Vps34 (Vacuolar Sorting Protein), complexed with Vps15-Beclin1-Atg14L proteins [47, 48]. Activation of Vps34 complex is mediated by AMPK and Ulk1, through phosphorylation of Beclin1 and/or Ambra1, which binds to Bcl2 and prevents it from sequestering Beclin1 [49–53] (Fig. 1). PI3P formation permits the recruitment of Atg complexes at the phagophore [54].
Autophagosome maturation
Expansion and closure of autophagic vesicles involve transient attachment of the ubiquitin-like conjugation complex Atg12-Atg5 at the phagophore, following Atg12 activation by Atg7 (E1 activating enzyme) and its transfer to Atg10 (E2 conjugatingenzyme) (Fig. 1). Interaction of Atg12-Atg5 complex with Atg16L facilitates the conjugation of LC3 (MAP1LC3 for microtubule-associated protein 1 light chain 3) to the lipid phosphatidylethanolamine (PE), after its activation by Atg7 and its transfer to Atg3 (E2 like enzyme) [54, 55]. LC3-PE (or LC3II) is inserted into autophagosome membranes and controls vesicle expansion and size [56]. Fusion between the lysosome and autophagosome, resulting in formation of the autolysosome, requires LAMP2 and Rab7 proteins and marks the final step of vesicle maturation. It correlates with autophagic vesicle acidification conferred by the lysosomal environment [57] (Fig. 1). Notably, both autophagosome maturation and lysosome biogenesis involve a tight interaction with the endocytic system, highly dynamic membrane remodelling and microtubule-dependent vesicle and cargo movements [58–60]. In particular, endosomes can fuse with autophagosomes to form amphisomes before fusing with lysosomes, which would facilitate aggregate removal [61]. Lastly, lysosome recycling from the autolysosome is essential to maintain autophagy and may depend on mTOR activity [62].
Cargo recognition and delivery
It was recently recognized that autophagy canproceed via selective degradation of cargoes. Clearance of specific substrates, including damaged organelles or ubiquitinated proteins, is mediated by receptors, such as p62/SQSTM1 or Nbr1, which recognize and bind to unfolded protein regions or poly/mono-ubiquitin chains of the cargoes and allow their translocation to the autophagy machinery by interacting with LC3/Gabarap proteins [63–65] (Fig. 1). Notably, chaperones of the heat shock protein family (Hsp70 and HspB8) and the BAG3 co-chaperone coordinate selective degradation of ubiquitinated substrates upon autophagosome formation through a process called chaperone-assisted selective autophagy (CASA) [66, 67]. Moreover, p62, BAG3 and also HDAC6 participate in aggregate clearance by targeting misfolded proteins, which are clustered in inclusion bodies and large aggresomes, to autophagosomes (so called aggrephagy) [68–72] (Fig. 1).
Degradation steps
Lysosomal hydrolases, including cathepsin D and L, ensure degradation of the autolysosomal content and of the substrates of microautophagy and CMA. The resulting products (i.e. amino acids, free fatty acids and glucose) are released into the cytosol to be reused for energy production or in biosynthesis pathways (e.g. protein synthesis).
Balanced autophagy is required for muscle homeostasis
How to monitor autophagy in skeletal muscle
To best identify autophagy perturbation and determine its role in muscle disorders, one would need to combine in vitro analyses in human muscle cells, in vivo procedures in animal models of the pathology, as well as evaluation of autophagy markers in muscle biopsies from patients. However, the latter is often difficult as sampling site and nutritive status of the patients at the time of biopsy are usually not defined. Further, heterogeneity in the results may arise from variability in the disease stages and quality of the muscle biopsy.
Quantification of LC3 and p62 protein levels constitutes the initial experiment when investigating autophagy. The amount of LC3II mirrors autophagosome accumulation in cells. Normalization of LC3II levels to the expression of housekeeping proteins is preferred over determination of LC3II/LC3I ratio, as the latter may be biased by LC3 transcriptional changes [16, 73]. Similarly, p62 accumulation is often considered as a relevant sign for autophagy impairment, but it may also be caused by transcriptional changes in the absence of autophagy blockade [74, 75]. Hence, transcript quantification of autophagy genes, including Sqstm1/p62 and Map1Lc3 (Lc3) needs to complement protein evaluation, but is not sufficient per se to assess the autophagic flux. Indeed, autophagy can be blocked at the induction or degradation steps, irrespective of any changes in gene expression [76]. Autophagosome accumulation can also be evaluated by quantifying the number of LC3-positive vesicles, an experiment that may require the use of GFP-LC3 transgenics or transfection [77].
A major drawback, often not considered in autophagy studies, is that LC3II levels or the autophagosome number are not sufficient to judge whether the autophagic flux is increased or blocked. Actually, accumulation of autophagic vesicles in muscle cells is more frequently associated with impaired autophagic flux than with increased autophagy induction. To characterize the flux, one needs to monitor the changes in autophagosome number or LC3II levels when lysosomal degradation is blocked. Such procedures should also allow discrimination between alterations in induction vs. degradation steps. While chloroquine and bafilomycin (both inhibitors of lysosomal acidification) have shown efficacy in vitro, colchicine, a microtubule depolarizing drug, is preferred for the targeting of muscle in mouse models [78, 79]. Complementary studies include electron microscopy (EM), which may clarify the nature of autophagic vacuoles. When possible, confocal microscopy monitoring - in cells or muscle - using the RFP/mCherry-GFP-LC3 reporter allows distinction between neutral, non-degradative vesicles (RFP+, GFP+) from acidic, functional autolysosomes (RFP+ only) [73]. Lastly, autophagy-mediated proteolysis can be evaluated in cells and mice by measuring the degradation rate of labelled proteins in the presence of an autophagy and/or an UPS inhibitor [77, 80].
Of note, upon treatment of cell lines, mouse models or patients with candidate drugs that affect autophagy, evaluation of the effect of the treatment on autophagic flux is required in addition to the monitoring of phenotypic changes. In clinical trials, the nutritive status of patients should be clearly defined and ideally standardised at the time points of investigation. Furthermore, immunostaining and biochemical analyses of autophagy and lysosome markers should be combined to reduce the inherent limitations of sampling. Autophagy may be analysed in other cell types, such as melanocytes from patients, if those cells reproduce the autophagic perturbations observed in muscle [81, 82].
Conditions increasing autophagic flux in muscle
Exercise has been shown to induce autophagy in muscle and is thought to enable the tissue to dispose of altered contractile proteins and/or to adapt to increased energetic demand [83–87]. Nevertheless, the beneficial effect of autophagy induction in muscle on global glucose and lipid metabolism remains controversial [2, 83]. Similarly, fasting triggers autophagy in muscle (particularly in fast-twitch muscle) in response to nutrient or energy depletion [88, 89]. In these conditions, increased muscle breakdown not only ensures energetic maintenance in the tissue but also preserves global body metabolism by supplying amino acids to non-muscle tissues. Autophagy has also been implicated in denervation-induced muscle atrophy [78, 90–93] although mTORC1-dependent autophagy inhibition was suggested to limit muscle wasting upon nerve injury [94]. Additionally, in humans, the muscle wasting associated with sepsis [95, 96], cancer [97, 98], disuse [99] or cirrhosis [100] has been related to increased autophagic flux.
Impaired autophagic flux and atrophy
The development of mouse models deficient for autophagy-related proteins in muscle has revealed the deleterious effect of autophagy blockade on this tissue. In muscle depleted of Atg7 or Atg5, autophagy is blunted and mutant mice develop a progressive myopathy, characterised by muscle atrophy and weakness. Further, the loss of mass upon fasting and denervation is exacerbated in Atg7-deficient mice compared to controls [101]. Autophagy impairment causes fibre vacuolization and intracellular accumulation of abnormal organelles, ubiquitinated proteins and p62-positive aggregates. In addition, increased oxidative and ER stresses were observed, which were likely responsible for muscle degeneration and dysfunction. Of note, Vps15 deficiency in mouse muscle does not alter autophagy induction but impinges on maturation steps and causes a severe myopathy. Pathological features in these mice include p62-positive aggregates and abnormal organelles, as seen in Atg7-deficient muscle, but also necrosis, inflammation, glycogen accumulation, autophagosome build-up and vacuoles lined with sarcolemmal proteins [102]. These alterations are reminiscent of those described in toxic vacuolar myopathies in humans, which can develop after long-term treatment with drugs interfering with autophagy, such as chloroquine (anti-malarial agent) or colchicine (used to treat gout) [103, 104]. Although recovery is obtained by ceasing treatment, adverse effects may come from the simultaneous administration of drugs promoting autophagy induction, such as rapamycin, which could dramatically aggravate muscle damage [105]. These pathological conditions are further evidence that autophagy imbalance is detrimental for skeletal muscle. Accordingly, a decline in autophagy capacity may contribute to aging-related muscle alterations, by reducing clearance of toxic organelles and products [106, 107]. Importantly, autophagy restriction in satellite cells also causes quiescence loss with age and thereby a decline in muscle regenerative capacity [108]. Together with the aforementioned data, these observations indicate that a fine-tuned balance of autophagy is required for muscle homeostasis.
Importance of mitophagy in muscle
Mitophagy refers to the selective degradation of mitochondria by autophagy and is essential to avoid accumulation of damaged mitochondria and excessive ROS generation during oxidative phosphorylation [109] (Fig. 1). It involves specific receptors, such as Bnip3, that are able to interact with depolarized mitochondria and the autophagic machinery [110, 111]. The E3 ubiquitin ligase Parkin and the kinase Pink1 also contribute to ubiquitin-dependent mitochondrial breakdown [112–115]. Mitophagy is induced in catabolic conditions and leads to remodelling of the mitochondria network, together with the fusion and fission machineries [116]. Importantly, mitochondria fission is necessary and sufficient for muscle atrophy induction [117]. Conversely, accumulation of damaged mitochondria related to impaired mitophagy is detrimental for muscle tissue [118]. Hence, a tight regulation of mitophagy is also essential for muscle homeostasis, and to date, its pathogenic involvement in neuromuscular disorders probably remains underestimated [119].
AUTOPHAGY REGULATION in skeletal muscle
Transcriptional regulation of autophagy
In 2009, Mammucari et al. hypothesized that muscle ability to maintain stress-induced autophagy for a prolonged period requires autophagy gene up-regulation. By using transcriptomic analyses, they established that autophagy genes, such as Map1lc3, Gabarapl1, Bnip3 and Sqstm1, are induced upon fasting and denervation, and that their regulation is driven by the FoxO3 (Forkhead box class O) transcription factor [120]. FoxO1 up-regulates Cathepsin L expression [121]. In mouse muscle, gene expression was increased after as little as 12 hrs of starvation, but remained unchanged from fed to basal conditions when constitutive autophagy was induced [76]. Interestingly, overexpression of Bnip3 or of a constitutively active form of FoxO3 enhanced autophagosome formation, whereas expression of a dominant-negative form of FoxO3 or downregulation of either FoxO3 or Bnip3 by shRNA reduced autophagy induction upon fasting [120, 122]. FoxO3 phosphorylation by Akt/PKB, in response to growth factors, inhibits its nuclear import, while, upon energy deprivation, AMPK-mediated phosphorylation of FoxO3 activates its translocation [123–125]. In parallel, transcription factors of the MiT/TFE family, including TFEB, promote expression of both lysosomal (e.g. encoding V-ATPases, Cathepsin) and autophagy (e.g. Map1lc3, Sqstm1, Atg9) genes [126, 127]. mTORC1 regulates TFEB activity, and thus autophagy gene transcription, by phosphorylating TFEB and thereby modifying its nuclear translocation [128–130]. Lastly, epigenetic modulations likely contribute to autophagy gene regulation. For example, HDAC1/2 depletion in mouse muscle blunts autophagy gene expression and causes a myopathy in the surviving mutant mice. Whether this effect is independent of FoxO remains to be determined [131].
Role of mTORC1 in autophagy induction
In contrast to the well-established role of mTORC1 in autophagy regulation in most cell types, autophagy was suggested to be independent of mTORC1 inskeletal muscle [88, 132, 133]. In particular, rapamycin treatment and shRNA directed against Mtor failed to promote autophagy induction in muscle cells [120, 122, 133]. Further, acute Akt/PKB activation blocked LC3 lipidation and autophagy gene expression and caused severe muscle degeneration characterized by p62 aggregates and myofibre vacuolization [118, 120, 122]. Rapamycin did not restore autophagy induction in those conditions, suggesting that FoxOs were the determinant factors controlling autophagy. Nonetheless, TSCmKO mice, specifically depleted for TSC1 in muscle and characterized by constant activation of mTORC1 in the tissue, revealed new insights into the role of mTORC1 in autophagy control. Indeed, constitutive and fasting-induced autophagy is blocked in TSCmKO muscle and the mice show an age-dependent accumulation of autophagic vacuoles and debris [76]. Importantly, autophagy impairment occurs despite simultaneous FoxO-dependent up-regulation of autophagy genes and is mediated by mTORC1-dependent inhibition of Ulk1. In control muscle, mTORC1 inhibition correlates with induction of constitutive autophagy (from feeding to non-feeding time), while FoxO activation matches with starvation-induced autophagy. Such distinction between constitutive and stress-induced autophagy likely contributed to the initial oversight of mTORC1 involvement [134]. It is now clear that autophagy involves a dual regulation in muscle: While mTORC1 activation locks autophagy at the induction step, FoxOs may ensure prolonged, enhanced flux via transcriptional regulation of autophagy genes. Consistently, mTORC1 inactivation in raptor-depleted muscle increases autophagy induction, but concomitant FoxO inactivation seems to impinge on autophagosome processing [76]. It is worth noting that Fyn/STAT signalling was suggested to block the Vps34/Beclin1 complex and thereby autophagy, independently of mTORC1 [135]. However, mTORC1 is activated in Fyn-overexpressing transgenic muscle and likely contributes to autophagy-related atrophy of glycolytic muscle, as observed in TSCmKO muscle [135]. Autophagy regulation was also attributed to mTORC2 (a second mTOR complex characterized by its associated protein rictor) as rictor knock-down induced autophagy in an Akt/PKB- and FoxO-dependent, but mTORC1-independent manner in muscle fibres [120]. However, perturbation in autophagic flux was detected in mice deficient for rictor in skeletal muscle, suggesting that mTORC2 does not contribute to autophagy control in muscle [76]. Lastly, it should be noted that differences exist between muscles regarding theirsensitivity to autophagy and the corresponding regulatory signalling [76, 118, 136].
NEUROMUSCULAR disorders RELATED TO ABNORMAL AUTOPHAGY INDUCTION
In the last decade, growing evidence has pointed to a converging role of autophagy perturbation in muscle diseases. Suspicion of autophagic defects was guided by characteristic histological features, including vacuoles, protein inclusions and damaged organelles in muscle fibres, although less specific alterations, such as fibre degeneration, can also arise from abnormal flux. In the following section we will discuss the pathologies related to Akt/PKB, mTORC1 and/or FoxO dysregulation with potential increased autophagic flux or impaired autophagy induction (Table 1).
Muscle pathologies with increased autophagic flux
Congenital Muscular Dystrophy type I –MDC1A is caused by mutations in the LAMA2 gene, leading to deficiency in the laminin-α2 chain, which assembles with the β1 and γ1 chains to form laminin-211 [137] (Fig. 2). Utilising the severely affected, laminin-α2-deficient, dy3K/dy3K mouse model, Carmignac et al. (2011) suggested that muscle atrophy and degeneration are related to increased autophagic flux, caused by Akt/PKB inhibition in diseased muscle (Fig. 2). Consistently, expression of the autophagy genes was increased in dy3K/dy3K muscle. The authors further showed that two successive injections of3-methyladenine (3-MA), an inhibitor of Vps34, were sufficient to ameliorate muscle morphology, locomotion and lifespan of the mice [138]. However, the efficacy of 3-MA to inhibit autophagy induction was not assessed in the study. In fact, others reported that 3-MA may induce autophagy and inhibit Akt/PKB by interfering with class I PI3 kinase activity [139, 140]. Moreover, questions remain as to how laminin-211 deficiency leads to Akt/PKB dysregulation, and as to how 3-MA administration normalizes Akt/PKB activation and autophagy gene expression.
Myotonic Dystrophy type I – DM1 is a multisystemic neuromuscular disorder, characterized - amongst other symptoms - by pronounced muscle atrophy. It is caused by a CTG repeat expansion in the DMPK gene; the expanded RNA leads to mis-splicing of numerous genes due to the nuclear sequestration of splicing factors [141, 142] (Fig. 2). Muscle wasting in DM1 was recently related to enhanced autophagy, based on the accumulation of LC3II and/or of autophagic vesicles in primary DM1 myoblasts [143–145], in muscle biopsies from patients [146–149] and in flight muscles of a DM1 Drosophila model [145]. However, it is still unclear whether these features arose from increased autophagy induction or from a defect in the degradation steps. Interestingly, altered expression of the insulin receptor, which correlates with glucose intolerance in DM1 patients [150–152], may cause Akt/PKB-mTORC1 dysregulation in diseased muscle. Akt/PKB down-regulation and/or mTORC1 inhibition were reported in muscle from Dmpk–/– mice [152] and DM1 flies [145], as well as in DM1 human ES-derived neural stem cells [153]. In muscle biopsies and myoblasts from DM1 patients, results have not been consistent [144, 145, 154, 155]. Of note, approaches aiming at inhibiting autophagic flux tended to limit muscle atrophy in DM1 flies [145]. However, these results need to be reproduced in DM1 mouse models and in human muscle cells. Whether autophagy perturbation in DM1 only depends on Akt/PKB changes or involves other splicing-dependent and -independent events (such as p53 [143] or Tweak/Fn14 [156]) still needs to be addressed.
Muscle pathologies with impaired autophagy induction
Collagen VI-related myopathies – COLVI-RM are caused by mutations in genes encoding one of the three collagen VI α chains, and range from a severe, congenital form (Ullrich congenital muscular dystrophy - UCMD) to milder forms (Bethlem myopathy - BM) [157]. In 2010, Grumati et al. established that the autophagic flux is blocked at the induction step in muscle of Col6a1–/– mice, which would contribute to pathological accumulation of abnormal organelles (e.g. dysfunctional mitochondria), increased fibre apoptosis and eventually muscle degeneration [118]. The autophagy defect relied, at least in part, on the restricted, FoxO3-dependent expression of Beclin1 and correlated with an abnormal Akt/PKB activation (Fig. 2). Although Beclin1 expression was shown to be necessary and sufficient to restore autophagy in mutant muscle, the role of Akt/PKB dysregulation remains to be clarified. Indeed, while prolonged starvation normalized Beclin1 expression and the autophagic flux, Akt/PKB activation was only slightly changed in mutant muscle. As AMPK signalling was also altered in Col6a1–/– muscle, further investigations on the activation state and pathogenic involvement of FoxO, mTORC1 and Ulk1 would be of interest. Although similar reduction in Beclin1 protein levels has been reported in muscle biopsies from patients, analysis of the autophagic flux in human muscle cells would provide a better insight into the pathogenic role of autophagy. Interestingly, autophagy induction also seems to be impaired in COLVI-deficient zebrafish, but was not related to abnormal Akt activation [158].
Importantly, forcing autophagy induction with prolonged starvation, long-term low protein diet (LPD), or treatments with rapamycin, cyclosporin or spermidine ameliorated the phenotype of mutant muscle. Improvements included normalization of muscle ultrastructure, decreased apoptosis, increased muscle strength and/or reduced muscle degeneration [118, 159]. By contrast, physical exercise failed to promote autophagic flux in mutant mice and led to major aggravation of muscle pathology [87]. Notwithstanding, a clinical trial in which 8 BM and UCMD patients were treated with LPD over 12 months has been initiated in 2011 (Merlini et al., NCT01438788). A preliminary report indicates that autophagy seems to be re-induced based on the comparison of autophagy markers in two patients before and after treatment [160].
Duchenne muscular dystrophy – Mutations in the gene encoding dystrophin, a cytoskeletal scaffolding protein, cause Duchenne or Becker muscular dystrophies (DMD and BMD, for the severe and milder forms, respectively) (Fig. 2). DMD is characterized by severe muscle atrophy and weakness, caused by progressive muscle degeneration, which is insufficiently compensated for by regeneration. Hence, with time, muscle is progressively replaced by connective tissue and fat [161]. Similar features are observed in mdx and mdx/utrophin–/– mouse models [162–164]. The presence of damaged organelles and protein aggregates in DMD and mdx muscle suggest impaired autophagic flux; vacuoles could also be detected in muscles from BMD patients [165]. A detailed analysis of the autophagic flux was conducted by De Palma et al. [166] in mdx mice and later confirmed by others [167–170]. In these studies, autophagy induction was altered in adult mdx mice as indicated by increased p62 levels and impaired LC3II formation in basal and fasted conditions. These autophagy markers were similarly affected in DMD biopsies [166]. One report also suggested a potential defect in lysosomal biogenesis in mdx muscle, based on LAMP1 downregulation [169].
Interestingly, as for COLVI-RM, autophagy blockade was related to Akt/PKB activation in mdx muscle, in mouse primary muscle cells and in DMD muscle biopsies [166, 167, 169, 171, 172]. Akt/PKB dysregulation was detected prior to necrosis and peaked during the necrotic and hypertrophic phases [166]. Consecutively, mTORC1 hyperactivation and restricted FoxO3-dependent autophagy gene expression could impair autophagy induction [166–168] (Fig. 2). Whether phosphorylation of FoxO3 and Ulk1 was altered in mdx mice was not described. There is evidence that imbalance in Akt/PKB-mTORC1 signalling in dystrophin-deficient muscle relies on Nox2 (NADPH oxidase)-dependent oxidative stress and activation of the associated Src kinase. Consistently, Src or Nox2 inhibition normalized Akt/PKB-mTORC1 activation and restored autophagosome formation and lysosome biogenesis [169]. It is important to note that other authors reported that Akt/PKB-mTORC1 activation is unchanged in mdx mice [173, 174].
Increased Akt/PKB activation may constitute an adaptive compensation to muscle damage in young mice [175]; this would promote regeneration, cause muscle hypertrophy and reduce myofibre death. Accordingly, Akt/PKB-mTORC1 stimulation improved muscle histology and/or function in mdx mice [173, 176–181], which may, at least partly, rely on the compensatory expression of cytoskeleton molecules, such as utrophin [182, 183]. Conversely, Akt/PKB-mTORC1 hyperactivation in mdx muscle may in fact be detrimental, and strategies that aim to normalize the pathways, such as rapamycin treatment, long-term LPD or Nox2 genetic inhibition, are sufficient to restore autophagic flux and ameliorate the dystrophic phenotype in mdx muscle [166, 169, 174, 184]. Similarly, AMPK activation in mdx muscle may constitute an adaptive response to the energetic stress associated with the opening of the mitochondrial permeability transition pore; reinforcing its activation with the AMP analog, AICAR, improved diaphragm muscle structure and force, likely by increasing mitochondria integrity and autophagic flux [168]. Recently, treatment with the lipophilic statin, simvastatin, also improved muscle pathology in adult and old mdx mice, at least partly by re-inducing autophagic flux, although its effect on Akt/PKB-mTORC1 and AMPK signalling was not tested [185]. In 2013, Hindi et al. reported a dual effect of Traf6, depending on disease stage, in dystrophin-deficient muscle. Increased Traf6 levels, detected early in mdx muscle, may contribute to the massive inflammatory response in the diseased muscle. Consistently, Traf6 depletion improved muscle function in young mdx mice, likely by limiting tissue inflammation and promoting its regeneration [170]. Conversely, with age, Traf6 inhibition worsened muscle degeneration: This deleterious effect was associated with autophagic flux reduction, despite Akt/PKB-mTORC1 inhibition in mdx:Traf6–/– mice [170]. Traf6-dependent autophagy induction would thus have a predominant, protective effect during late stages of the disease by promoting the clearance of by-products and damaged organelles. Hence, therapeutic strategies targeting autophagy in DMD muscle may be promising, especially during advanced stages of the disease. In contrast, a proper balance would be required in earlier phases to equilibrate muscle homeostasis, regeneration, growth, and to limit tissue inflammation [186].
X-linked myotubular myopathies – Congenital XLMTM are caused by mutations in the MTM1 gene, encoding myotubularin 1. They are characterized by the predominance of centronucleated myofibres and thus part of the centronuclear myopathies (CNM). Pathological features observed in muscle biopsies from patients and/or muscle from Mtm1-deficient mice and dogs include abnormal mitochondria, ubiquitinated proteins, p62-positive aggregates, glycogen accumulation and/or autophagic vesicles [187–190]. While LC3 levels were increased in Mtm1-null mouse muscle, LC3II lipidation was impaired upon starvation of the mutant mice [187], suggesting that autophagy induction is blocked. Akt/PKB-mTORC1 activation, which was detected in Mtm1-deficient muscle as well as upon acute AAV-induced Mtm1 depletion, may contribute to autophagy blockade in the diseased muscle, as indicated by increased phosphorylation of Ulk1 at Ser757 [187, 189, 191]. In contrast to COLVI-RM, simultaneous FoxO3 activation was associated with increased expression of autophagy genes (e.g. Map1lc3 or Sqstm1), which may participate in their accumulation in mutant muscle [189]. Intriguingly, while treatment with an mTORC1 inhibitor restored autophagy and improved the muscle phenotype in Mtm1-deficient mice, AAV-mediated delivery of Mtm1 normalized autophagy markers independently of the FoxO and mTORC1 pathways [187, 191]. Moreover, the pre-symptomatic accumulation of autophagosomes in Mtm1-deficient mouse muscle, in the absence of FoxO/mTORC1 alteration, suggests that Mtm1 depletion may primarily cause an abnormal induction of the autophagic flux [189]. This hypothesis is consistent with the role of Mtmr14 (Myotubularin-related protein 14 - Jumpy) to negatively regulate autophagy. The PI3P phosphatase activity of Mtm1 and Mtmr14 is supposed to counteract the function of Vps34 at the phagophore stage. Mtmr14 knock-down in C2C12 cells resulted in a loss of autophagy control and increased proteolysis [192]. Since mutations in the MTMR14 gene were associated with CNM, one can hypothesize that uncontrolled, excessive autophagic flux may be part of the pathomechanisms leading to myotubularin-related disease [193]. Notwithstanding, Mtmr14 deficiency in mice or zebrafish only led to a slight perturbation of autophagy although combined knock-down of mtm1 and mtmr14 in zebrafish caused a severe phenotype marked by increased autophagic flux [194, 195]. Hence, the role of myotubularins and their potential redundancy need to be further analysed to better understand the consequences of their deficiency in patients.
Laminopathies – Mutations in the LMNA gene, encoding A-type lamins, are responsible for several disorders (e.g. Emery-Dreifuss Muscular Dystrophy or Limb-Girdle Muscular Dystrophy type 1B) affecting, among other tissues, skeletal muscle and heart (Fig. 2). Growing evidence suggests that A-type lamins have distinct functions besides their structural role in the nuclear lamina [196]. Autophagic vacuoles described in muscle biopsies of patients and in skeletal muscle of lamin A/C-deficient mice first suggested that increased autophagy (i.e. nucleophagy) may constitute an adaptive mechanism to remove abnormal nuclei in diseased myofibres [197]. Alternatively, such vacuolar structures may arise from impaired autophagy. As for COLVI-RM, DMD and XLMTM, mTORC1 activation was reported in skeletal muscle and heart from lamin A/C-deficient mice [198, 199]. While this correlates with Akt/PKB hyperactivation in cardiac muscle of patients and mutant mice, the status of Akt/PKB and FoxO signalling has not yet been analysed in the skeletal muscle of Lmna-knock-in (KI) or knockout (KO) mice. Similarly, although results obtained in diseased heart indicate that autophagy induction is impaired [199], few data have been collected in skeletal muscle. Levels of some autophagy markers (e.g. LC3 or Atg7) are increased, but only Ulk1 inhibition, as shown by its elevated mTORC1-dependent phosphorylation in muscle from mutant mice, suggests that autophagy induction may be hampered in skeletal muscle [198] (Fig. 2). Treatment with rapamycin or temsirolimus, which both inhibit mTORC1, increased lifespan of lamin A/C-deficient mice and improved heart function, likely by restoring autophagic flux [198, 199]. Despite the failure of dietary administration of rapamycin to reduce mTORC1 in mutant skeletal muscle and consequently to normalize autophagy, intriguingly, the treatment reduced desmin accumulation in the tissue and increased motor coordination (assessed by a rotarod test) of the mice. Hence, whether autophagy dysregulation contributes to pathogenic alterations of the tissue remains to be investigated in more detail.
pathogenic ROLE OF AUTOPHAGY IN autophagic vacuolar myopathies
Muscle disorders discussed in the following sections belong to the group of AVM, characterized by the accumulation of autophagic vacuoles in muscle biopsies from patients. AVM include i) lysosomal storage diseases (LSD; e.g. Danon disease), in which vacuoles result from primary lysosomal dysfunction, and ii) myopathies with rimmed vacuoles (RV; e.g. IBMPFD), interpreted as autophagic vacuoles due to their reactivity with lysosomal markers. Although the mechanisms underlying their formation are not known, RV may only secondarily arise from lysosomal defects, which may lead to autophagosome proliferation and enlargement. An alternative, non-exclusive hypothesis is that RV derives from defective nuclear breakdown [200].
Muscle pathologies related to impaired maturation of autophagic vesicles
Glycogen storage disease type IIb – GSDIIb (or Danon disease) is an X-linked multisystemic disorder characterized by severe hypertrophic cardiomyopathy, normally alongside a mild myopathy with predominant involvement of proximal muscle and potential mental retardation [201]. Although dominant mutations in LAMP2 were first shown to cause the disease in 2000, there is still little data on the pathomechanism underlying skeletal muscle alterations [202]. LAMP2 is a transmembrane protein mainly found in lysosomes and late endosomes, suggesting that its deficiency may affect lysosome-related processes, such as autophagy. Indeed, vacuoles containing glycogen and autophagic debris, surrounding acid phosphatase-positive basophilic granules recognized as lysosomes, accumulate in muscle biopsies from Danon patients [202]. Vacuoles display a single membrane with sarcolemmal and extracellular matrix proteins, forming at their inner surface a layer of basal lamina (so called AVSF for autophagic vacuole with sarcolemmal features); their occurrence increases with age but their formation remains poorly understood (Fig. 3). It is noteworthy that such vacuoles are also found in cardiac muscle from Danon patients, in muscle and non-muscle tissues from LAMP2-deficient mice, and in Lamp1/2-double KO embryonic cells [203–205]. LAMP2 depletion in hepatocytes and cardiomyocytes delays autophagosome maturation, restricts long-lived protein degradation and increases the half-life of autophagic vesicles [203]. Accordingly, LAMP2 and/or LAMP1 seem to be required for the efficient recruitment of Rab7 and the fusion of (auto)phagosomes with lysosomes in macrophages, hepatocytes, and mouse embryonic fibroblasts (MEFs) [206, 207]. Nonetheless, the proteolysis rate in Lamp1/2-KO MEFs was unaffected, suggesting that the role and importance of LAMP2 differ between cell types [204]. Further, LAMP2 deficiency in hepatocytes altered the recycling of the mannose-6-phosphate receptor from endosomes to the trans-Golgi network, which would cause mis-targeting and secretion of lysosomal enzymes (e.g. Cathepsin D) [208]. It remains unsolved whether autophagic vacuoles accumulate in muscle fibres because of defective fusion with lysosome or restricted lysosome degradative capacity. Interestingly, overexpression of Vps15/Vps34 in Danon human myoblasts reduced glycogen and LC3 accumulation, suggesting that compensation by other functional proteins may constitute a therapeutic option [102]. Lastly, although the LAMP2B splice isoform is predominantly expressed in skeletal muscle, LAMP2A deficiency may impair the degradation of specific cytosolic substrates targeted for chaperone-mediated autophagy (CMA). No major alteration of CMA was detected in LAMP1/2-deficient MEFs [205] or neuronal cells [209].
Glycogen storage disease type II – As for Danon disease, GSDII (or Pompe disease) is characterized by glycogen accumulation in lysosomes of muscle fibres. The defect arises from genetic deficiency in acid α-glucosidase (GAA), the enzyme responsible for glycogen hydrolysis. While earlier hypotheses proposed that glycogen-filled lysosome enlargement and rupture led to muscle alterations, recent observations suggest that autophagy impairment may contribute to the loss of muscle homeostasis in GSDII [210, 211]. Autophagic vacuoles and debris, protein aggregates, as well as increased levels of LC3II, p62 and ubiquitinated proteins have been described in muscle biopsies from GAA-deficient patients [74] and GAA-KO mice [212–214] (Fig. 3). Confocal microscopy further confirmed the presence of large areas of autophagic build-up in the central region of muscle fibres of mutant mice, suggesting inefficient autophagosome disposal [215]. Elegant time-lapse analysis revealed defective autophagosome-lysosome fusion in GAA-KO fibres, which could be responsible for the observed defects [216] (Fig. 3). It remains to be answered whether altered fusion and concomitant restriction in lysosome biogenesis are caused by glycogen accumulation, lysosome rupture or insufficient acidification of endocytic/lysosomal vesicles [213, 217]. When glycogen accumulation was restricted by knocking-down or depleting glycogen synthase in GAA-deficient muscle, autophagic build-up was reduced and muscle function improved [218, 219]. In light of the increased levels of autophagy-related proteins, including LC3, Atg7 or Beclin1, in GAA-deficient muscle, some authors suggested that simultaneous enhanced autophagy induction, related to GSK3β activation, may aggravate autophagic build-up [215, 220]. However, in the few experiments that tested the autophagic flux using lysosomal inhibitors, no major difference was detected between control and mutant cells [74, 221]. Importantly, numerous reports have established that resistance of skeletal muscle to enzyme replacement therapy (ERT) with recombinant human (rh) GAA is caused by autophagic build-up, which restricts trafficking and processing of rhGAA [222]. ERT was shown to be efficacious in heart tissue of severely affected patients but did not reduce skeletal muscle alterations, specifically because of its restricted effect on type II fibres [211]. Confocal microscopy revealed that rhGAA enzyme was trapped in autophagic vesicles in GAA-KO single fibres, rather than efficiently translocated into lysosomes as seen in control cells [223].
Attempts to modulate autophagy in order to limit muscle alterations have led to divergent results. TFEB or TFE3 overexpression in GSDII myotubes and GAA-KO muscle was sufficient to restore autophagosome-lysosome fusion, promote exocytosis and clear mutant fibres from enlarged lysosomes, glycogen and vacuoles [216, 224, 225]. Functional autophagy constituted a prerequisite for TFEB-mediated lysosome exocytosis therapy [216]. Similarly, PGC1α overexpression cleared autophagic build-up, likely by improving lysosome biogenesis, but did not impact on glycogen accumulation [226]. Moreover, rapamycin-treated primary myotubes from patients displayed reduced vacuolization [74], whereas addition of torin1/2, another inhibitor of mTOR, failed to reduce lysosome enlargement despite autophagy induction [216]. Conversely, genetic suppression of autophagy by depleting Atg7 prevented glycogen accumulation and autophagic build-up in GAA-KO mice, while Atg5 depletion in mutant mice [214] and Atg7 knock-down in primary mouse myotubes [213] had no effect on glycogen storage. Moreover, Atg7-GAA double KO mice displayed no major change in their overall phenotype [220] and Atg5 loss even worsened the muscle phenotype of GAA-KO mice. Combining ERT with autophagy inhibition led to significant improvements in glycogen removal and lysosomal function, which were related to decreased autophagic build-up with Atg7 depletion, but not Atg5 suppression [220]. However, there was no evidence for improvement of muscle function. Hence, although the pathogenic role of autophagy blockade is well established in GSDII, whether strategies should target or limit its induction in combination with other therapeutic options remains to be discussed and functionally analysed.
Vici syndrome – Vici syndrome is a rare disorder including neurologic, ocular, cardiac and muscular symptoms; mutations in the EPG5 gene (ectopic P-granules autophagy protein 5) were reported in 2013 [227]. Epg5-deficient mice reproduced, among other alterations, the myopathy described in Vici patients, with fibre atrophy and degeneration [228]. Interestingly, accumulation of vacuoles, enlarged mitochondria and glycogen, as well as increased levels of LC3II, p62 and Nbr1 were detected in muscle from patients and/or Epg5-KO mice [227, 228]. In patient fibroblasts and Epg5–/– MEFs, autophagic flux restriction was related to altered maturation of autophagic vesicles (at the fusion or degradation steps) and processing of endo/lysosomal compartments (Fig. 3). Whether these defects contribute to pathology in muscle and non-muscle tissues from Vici patients, and whether they can be rescued by modulating autophagy induction or expressing potentially compensating proteins, remains to be investigated.
Inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia – IBMPFD is an autosomal dominant multi-systemic disorder caused by mutations in the VCP gene, encoding a type II member of the AAA+-ATPase family, p97/VCP (Valosin Containing Protein), located in the endoplasmic reticulum of all cells. Most IBMPFD patients suffer from muscle weakness and characteristic alterations, including rimmed vacuoles (RV) and tubulofilamentous depositions are observed in muscle cells. Accumulation of p62 and ubiquitinated proteins points to autophagy defects in IBMPFD [229, 230].By functioning as an ubiquitin-selective segregase, VCP is involved in protein processing, recycling and degradation [231]. VCP deficiency impairsprotein quality control by altering the unfolded protein response (UPR) and endoplasmic reticulum-associated protein degradation (ERAD) [232] (Fig. 3). VCP mutants may also limit the clearance of the resulting protein aggregates by altering the formation of aggresomes and their delivery to the autophagy machinery [233]. Moreover, several reports suggest that VCP plays a role in endosome and autophagic vesicle trafficking: Autophagosomes accumulate in muscle from IBMPFD patients and VCP-deficient mice, as well as in primary human myoblasts [229, 234–237]. Ju et al. further established in vitro that VCP deficiency impairs autophagosome maturation and fusion, hence leading to non-degradative vesicles [238] (Fig. 3). Nonetheless, a recent paper indicated that, although immature, autophagic vesicles are acidified and contain lysosomal enzymatic activity, suggesting a defect in the ultimate stages of their maturation [239].
Interestingly, autophagy and endo-lysosomal trafficking defects in IBMPFD seem to impair mTORC1 activation and thereby worsen muscle alterations by promoting autophagy induction. Hence, whilst rapamycin treatment aggravated muscle weakness and atrophy of VCPR155H-transgenic mice, electroporation of an active form of Rheb or Akt/PKB in mutant muscle increased fibre size [240]. Alternative strategies aiming at normalizing autophagic flux in muscle, such as exercise or a lipid-enriched diet, ameliorated the phenotype of VCPR155H-KI homozygous and heterozygous mice [241–243]. However, even if changes in LC3 and p62 levels were reported in the treated mice, further analyses are required to establish whether those strategies impact on the autophagic flux, and whether they do so by limiting autophagy induction or by releasing the degradation steps. This may be essential to identify optimal therapeutic targets and strategies for IBMPFD.
Autosomal-Dominant Centronuclear Myopathy – Dominant mutations in the DNM2 gene, encoding the GTPase dynamin 2, cause AD-CNM which are histologically recognized by the prominent internalization of myonuclei, the presence of sarcoplasmic radial strands and a predominance of type I fibres [244, 245]. AD-CNM is not included in AVM, and no hallmark of autophagy blockade is present in muscle biopsies from patients. However, as DNM2 is involved in intracellular membrane trafficking, endocytosis and cytoskeleton modulation, its deficiency may impact on autophagy and on the endo/lysosomal system. Data pointing in this direction were obtained in KI (p.R465W) mutant mice, a recognized model for the disease [246, 247]. Early lethality of homozygous mutant mice was related to defects in global body metabolism consistent with defective autophagic flux in metabolic organs during the neonatal period [246]. A slight increase in LC3II and p62 levels was detected in liver, but not in muscle, from homozygous neonates. Accumulation of p62 and non-degradative autophagosomes was also found in DNM2-deficient MEFs [246]. The presence of abnormal autophagosome maturation was supported by reduced acidification of autophagic vesicles, which may involve impaired fusion between autophagosome and endo/lysosomal compartments and/or defective lysosomal biogenesis (Fig. 3). Accumulation of LC3II and of autophagic vesicles in muscle from starved, 6 month-old heterozygous mice further pointed to altered autophagosome maturation in diseased muscle. However, the autophagic flux also seems compromised at the induction steps in mutant MEFs, with reduced LC3II accumulation observed upon bafilomycin treatment [246]. Hence, DNM2 deficiency may lead to combined defects in autophagosome formation and maturation. Since there was no drastic accumulation of autophagy vacuoles in diseased muscle, one may assume that reduced induction may predominate in DNM2-deficient muscle. This would be consistent with the clinical and histological similarity to XLMTM, related to impaired autophagy induction (see above).
Muscle pathologies related to impaired degradation steps
X-linked myopathy with excessive autophagy – XMEA specifically affects skeletal muscle and ranges from severe neonatal to slowly progressive late-onset forms; it was only recently linked to mutations in the VMA21 gene, which encodes a major chaperone of the lysosomal proton pump V-ATPase [248–250]. Muscle histology revealed fibre splitting with internalized capillaries, complement C5b-9 deposition and AVSF, as described for Danon diseased muscle [251] (Fig. 3). While there is currently no mouse model for the disease, analyses conducted in fibroblasts and lymphoblasts from XMEA patients have brought information regarding potential pathomechanisms. Ramachadran et al. established that VMA21 deficiency restricts assembly and activity of the V-ATPase, thereby leading to increased lysosomal pH, altered lysosomal enzymatic activity and strongly reduced long-lived protein degradation [248]. Co-localization of autophagosome and lysosome markers suggests an effective fusion of these vesicles but a defect in autophagy degradation steps (Fig. 3). Hence, (macro)autophagy, but also microautophagy and CMA may be impaired in XMEA. Based on the increased expression of some autophagy genes (e.g. Beclin1, LC3), the authors further suggested that autophagy blockade may lead to mTORC1 inhibition by reducing the release of amino acids, and thereby to increased autophagy induction (Fig. 3). This may aggravate autolysosome proliferation and enlargement [248]. Hence, mTORC1 dysregulation as well as potential activation of TFEB, which may explain the observed autophagic vesicle extrusion at the plasma membrane still need to be documented in XMEA muscle cells. Lastly, development of a mouse model for the pathology is needed to compare autophagy impairment in skeletal muscle vs unaffected tissues (including cardiac muscle [252]) and to determine the consequences of modulating autophagy, mTORC1 and/or TFEB on the muscle phenotype.
Muscle diseases with autophagy perturbation related to abnormal protein aggregation
Myofibrillar myopathies – MFM are rare neuromuscular disorders characterized by muscle weakness, related to focal myofibril disintegration and caused by mutations in genes encoding structural proteins (e.g. desmin or filamin C) or chaperones (BAG3, αB-crystallin) found in the Z disk region [253]. Protein aggregates and RV constitute a hallmark of MFM and point to autophagy perturbation [254, 255] (Fig. 3). This is supported by the presence of autophagy-related proteins (e.g. p62, LC3) in inclusions, revealed by proteomic analysis following laser-microdissection [256]. Whether accumulation of autophagy-related proteins reflects an adaptive induction of autophagy in response to protein aggregates or is rather a consequence of autophagy impairment, remains unclear. Of note, some mutations in structural proteins favour the aggregation of their mutated form, in the absence of an autophagy defect [257]. In cardiomyocytes, aggregate-prone mutated forms of desmin or αB-crystallin target autophagy induction in the early stages of the disease, but probably insufficiently to prevent their accumulation over a prolonged time period [258, 259]. Interestingly, BAG3 and αB-crystallin function as chaperones for structural proteins, and either prevent their aggregation or favour their degradation. In particular, BAG3 is involved in CASA, which is induced upon mechanical tension during muscle contraction and permits to degrade damaged contractile components, such as filamin C [67]. BAG3-deficient mice develop a lethal myopathy marked by myofibrillar degeneration and myofibre apoptosis [260]. Abnormal aggregation of mutated BAG3P209L also reproduced myofibrillar disintegration in zebrafish, likely by trapping wild-type BAG3 [261]. In these conditions, aggregates may perturb the endo/lysosomal function as well as the maturation of the autophagosome, thereby leading to a vicious cycle where increased induction and impaired autophagic degradation accelerate aggregate and vacuole formation. If so, therapeutic interventions aiming at inducing autophagy should be applied in the early stages of the disease, before aggregates accumulate. Beneficial effects of such strategies have been obtained in desmin-related cardiomyopathy but remain to be tested in skeletal muscle [257, 262–264].
Similar mechanisms may underlie Oculopharyngeal Muscular Dystrophy (OPMD) with the expanded, mutated form of PABPN1 (Poly(A) Binding Protein Nuclear 1) being prone to aggregate in myonuclei. Whether dysregulated expression of autophagy-related proteins detected in OPMD reflects an induction and/or a blockade of autophagy remains to be established [160, 265, 266]. As for MFM related to BAG3 or αB-crystallin deficiency, muscle alterations caused by dominant mutations in DNAJB6 (associated to Limb-girdle muscular dystrophy 1D/E), encoding another co-chaperone likely involved in CASA, may imply excessive protein aggregation, compensatory autophagy induction and/or perturbation of the autophagic flux, leading to Z-disk degradation, debris accumulation and fibre vacuolization [267–269]. Mutations in the SIL1 gene, which encodes a co-chaperone for BiP, a central quality controller for ER proteins, lead to a degenerative, vacuolar myopathy (as part of the Marinesco-Sjögren syndrome). Pathogenesis may also include induction of inefficient autophagy, in response to nuclear/ER stress and misfolded protein accumulation [270].
Muscle disease with abnormal autophagy features but no molecular mechanisms established
Sporadic Inclusion Body Myositis (sIBM) is an in-flammatory, degenerative muscle disorder clinically marked by atrophy and weakness of distal muscle. Histologically, sIBM is characterized by lymphocytic infiltration, RV and intracellular amyloid deposition [271]. Although the pathomechanisms remain unclear, accumulation of autophagic vacuoles, ubiquitinated protein aggregates and abnormal mitochondria led to the hypothesis that autophagy impairment contributes to the disease [272, 273]. Consistently, reduced activity of lysosomal enzymes, as well as increased levels of p62 and LC3II were found in sIBM muscle biopsies and in human myofibres subjected to sIBM-mimicking culture conditions [274, 275]. Reduced GSK3-dependent phosphorylation of Nbr1 may promote the aggregation of ubiquitinated proteins, despite its increased expression in sIBM biopsies [276, 277]. Further, altered CMA leading to α-synuclein accumulation and reduced mitophagy due to lysosomal dysfunction may contribute to mitochondrial abnormalities detected in diseased muscle [271, 278]. Notwithstanding, elevated expression of some autophagy- and CMA-related components was reported in sIBM muscle biopsies, which may reflect increased induction of the process [278, 279]. Although therapeutic strategies potentiating lysosomal activity (e.g. sodium phenylbutyrate) are proposed to limit myofibre vacuolization and toxic product accumulation, further experiments overcoming the limits of the current in vivo and in vitro disease models are needed to assess the involvement of autophagy in sIBM onset and progression [280].
GNE myopathy (also reported as hIBM2 or Distal Myopathies with Rimmed Vacuoles) is an autosomal recessive disorder caused by mutations in the GNE gene and predominantly affecting distal muscle. The gene encodes an enzyme essential for sialic acid production; its deficiency may lead to glucoconjugate hypo-sialylation, which may impact on several cell signalling pathways [281]. RV, cytoplasmic and nuclear inclusions, and abnormal mitochondria accumulate in muscle biopsies from patients. A knock-in mouse model expressing a mutated form (D176V) of the enzyme reproduced the muscle phenotype observed in GNE myopathy [282]. In this model, accumulation of Aβ peptide and of its protein precursor (AβPP) preceded RV formation, suggesting that autophagy defects are secondary events in the disease. Accordingly, perturbation in hypo-sialylated protein folding and trafficking may cause ER stress and target autophagy induction, thereby contributing to muscle degeneration [283, 284]. Sialic acid supplementation alleviated muscle alterations in mutant mice [285] and is currently being tested in clinical trials [281]. Other distal myopathies (DM), such as Welander DM (caused by deficiency in TIA1 mRNA binding protein), Vocal Cord and Pharyngeal DM (linked to mutations in the nuclear matrix gene MATR3) or Tibial Muscular Dystrophy, caused by mutations in TTN (encoding Titin), are marked by RV and may involve autophagy perturbation similar to GNE myopathy [286, 287].
For several additional myopathies, such as dysferlinopathies or the rare megaconial type congenital muscular dystrophy [289, 290], autophagy perturbation has been suggested but additional investigations are needed to assess whether these perturbations are part of the pathomechanism leading to muscle alterations and how they can be linked to the genetic origin of the disease.
CONCLUSION
Abnormal autophagic flux has now been suggested to be involved in the pathogenesis of numerous muscle disorders, in which it may significantly contribute to muscle atrophy. However, many of the studies lack a comprehensive picture of the state of the flux in the diseased muscle and thus conclusions on the pathological significance of autophagy impairment often seem premature. In particular, efforts should be made to clarify whether the flux is enhanced in cases where autophagosomes or autophagy markers accumulate. The use of animal models, combined with in vitro analyses of human muscle cells, is required before a conclusion can be made on the pathogenic role of autophagy in a disease. To this end, a comprehensive analysis of the autophagic flux in muscle remains to be conducted, if possible, at different stages of the disease, in the cases of MDC1A, DM1, XLMTM, laminopathies, GSDIIb, AD-CNM and GNE myopathy. Another drawback is the need to elucidate whether a given autophagy defect constitutes a primary pathogenic event or rather a consequence (or adaptive response) to primary pathogenic muscle alterations. Autophagy perturbations in AVM are primarily or secondarily related to lysosome dysfunction and have an established link to the genetic cause (with the exception of sIBM). By contrast, it is delicate, although essential, to decipher the molecular mechanisms leading to signalling dysregulation and abnormal autophagy induction in the case of diseases caused by deficiency in components of the extracellular matrix (e.g. COLVI; laminin) or of the cytoskeleton (e.g. A-type lamins). Once autophagy dysfunction is established, one needs to consider the efficacy of therapeutic approaches aiming at normalizing the flux, both on muscle phenotype and the restoration of the autophagy flux. Along these lines, further investigations to identify signalling pathways at play in autophagy control, or those which may be dysregulated in diseases, will help to develop specific strategies for individual disorders. A major challenge will be to avoid excessive correction of the flux, which may result in another extreme with too much (or alternatively insufficient) protein and organelle disposal. Therapeutic options should aim at balancing the flux, and therefore, the long-term side effects on muscle mass and force should always be considered. Although out of the scope of this review, we would like to emphasize that other pathological conditions associated with skeletal muscle atrophy involve autophagy perturbation. Examples include neurodegenerative disorders, such as spinobulbar muscular atrophy, in which autophagy induction may contribute to muscle wasting [291–293]. Excessive autophagy seems also to contribute to cachexia-induced muscle atrophy [98], while a decline in the autophagic capacity may contribute to age-related muscle alterations [14, 108]. Further understanding of the consequences of autophagy defects in neuromuscular disorders will help to better elucidate the mechanisms at play in these conditions and help to guide us toward therapeutic strategies to counteract the loss of muscle homeostasis.
conflict of interest
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We are very grateful to Dr. Norma Romero and Emmanuelle Lacène (Paris) for providing pictures of muscle biopsies, to Dr. Valérie Allamand (Paris) for her constructive comments on the manuscript, and to Dr. Daniel Jacob Ham for correcting the manuscript. We also thank Eugénie Robin for her help on illustration. P.C. is funded by grants from the Swiss National Science Foundation. The work in the laboratory of M.A.R. is supported by the Swiss Foundation for Research on Muscle Disease, AFM-Telethon and the Swiss National Science Foundation. We apologize to all colleagues whose work was not cited due to space limitation.
REFERENCES
[1] | Guridi M , Tintignac LA , Lin S , Kupr B , Castets P , Ruegg MA . Activation of mTORC1 in skeletal muscle regulates whole-body metabolism through FGF21. Sci Signal. (2015) ;8: (402):ra113 |
[2] | Kim KH , Jeong YT , Oh H , Kim SH , Cho JM , Kim YN , et al . Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. (2013) ;19: (1):83–92. |
[3] | Schreiber A , Peter M . Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochim Biophys Acta. (1843) )1 163–81. |
[4] | Carmignac V , Quere R , Durbeej M . Proteasome inhibition improves the muscle of laminin alpha2 chain-deficient mice. Hum Mol Genet. (2011) ;20: (3):541–52. |
[5] | Bonuccelli G , Sotgia F , Schubert W , Park DS , Frank PG , Woodman SE , et al . Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am J Pathol. (2003) ;163: (4):1663–75. |
[6] | Bonuccelli G , Sotgia F , Capozza F , Gazzerro E , Minetti C , Lisanti MP . Localized treatment with a novel FDA-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx mice. Cell Cycle. (2007) ;6: (10):1242–8. |
[7] | Bodine SC , Baehr LM . Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab.E. (2014) ;307: (6):469–84. |
[8] | Murrow L , Debnath J . Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu Rev Pathol. (2013) ;8: , 105–37. |
[9] | Nixon RA . The role of autophagy in neurodegenerative disease. Nat Med. (2013) ;19: (8):983–97. |
[10] | Cherra SJ , Chu CT . Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. (2008) ;3: (3):309–23. |
[11] | White E , Mehnert JM , Chan CS . Autophagy, Metabolism, and Cancer. Clin Cancer Res. (2015) ;21: (22):5037–46. |
[12] | Deretic V , Saitoh T , Akira S . Autophagy in infection, inflammation and immunity. Nat Rev Immunol. (2013) ;13: (10):722–37. |
[13] | Choi AM , Ryter SW , Levine B . Autophagy in human health and disease. N Engl J Med. (2013) ;368: (19):1845–6. |
[14] | Merlini L , Bonaldo P , Marzetti E . Editorial: Pathophysiological Mechanisms of Sarcopenia in Aging and in Muscular Dystrophy: A Translational Approach. Front Aging Neurosci. (2015) ;7: , 153. |
[15] | Zhang H , Baehrecke EH . Eaten alive: Novel insights into autophagy from multicellular model systems. Trends Cell Biol. (2015) ;25: (7):376–87. |
[16] | Klionsky DJ , Abdelmohsen K , Abe A , Abedin MJ , Abeliovich H , Acevedo Arozena A , et al Guidelines for the use and interpretation of assays for monitoring autophagy 3rd edition Autophagy. (2016) ;12: (1):1–222. |
[17] | He C , Klionsky DJ . Regulation mechanisms and signalling pathways of autophagy. (2009) ;43: , 67–93. |
[18] | Noda NN , Inagaki F . Mechanisms of Autophagy. Annu Rev Biophys. (2015) ;44: , 101–22. |
[19] | Lum JJ , DeBerardinis RJ , Thompson CB . Autophagy in metazoans: Cell survival in the land of plenty. Nat Rev Mol Cell Biol. (2005) ;6: (6):439–48. |
[20] | Kaur J , Debnath J . Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. (2015) ;16: (8):461–72. |
[21] | Parzych KR , Klionsky DJ . An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal. (2014) ;20: (3):460–73. |
[22] | Dice JF . Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. (1990) ;15: (8):305–9. |
[23] | Bandyopadhyay U , Kaushik S , Varticovski L , Cuervo AM . The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. (2008) ;28: (18):5747–63. |
[24] | Cuervo AM , Dice JF . A receptor for the selective uptake and degradation of proteins by lysosomes. Science. (1996) ;273: (5274):501–3. |
[25] | Cuervo AM . Chaperone-mediated autophagy: Dice’s ‘wild’ idea about lysosomal selectivity. Nat Rev Mol Cell Biol. (2011) ;12: (8):535–41. |
[26] | Zoncu R , Bar-Peled L , Efeyan A , Wang S , Sancak Y , Sabatini DM . mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 334. (2011) ;334: (6056):678–83. |
[27] | Sancak Y , Bar-Peled L , Zoncu R , Markhard AL , Nada S , Sabatini DM . Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. (2010) ;141: (2):290–303. |
[28] | Bar-Peled L , Schweitzer LD , Zoncu R , Sabatini DM . Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. (2012) ;150: (6):1196–208. |
[29] | Dibble CC , Manning BD . Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. (2013) ;15: (6):555–64. |
[30] | Jung CH , Ro SH , Cao J , Otto NM , Kim DH . mTOR regulation of autophagy. FEBS Lett. (2010) ;584: (7):1287–95. |
[31] | Chan EY . mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. (2009) ;2: (84):pe51. |
[32] | Hosokawa N , Hara T , Kaizuka T , Kishi C , Takamura A , Miura Y , et al . Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. (2009) ;20: (7):1981–91. |
[33] | Egan D , Kim J , Shaw RJ , Guan KL . The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. (2011) ;7: (6):643–4. |
[34] | Kim J , Kundu M , Viollet B , Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) ;13: (2):132–41. |
[35] | Egan DF , Shackelford DB , Mihaylova MM , Gelino S , Kohnz RA , Mair W , et al . Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. (2011) ;331: (6061):456–61. |
[36] | Lee JW , Park S , Takahashi Y , Wang HG . The association of AMPK with ULK1 regulates autophagy. PLoS One. (2010) ;5: (11):e15394. |
[37] | Zhao M , Klionsky DJ . AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. (2011) ;13: (2):119–20. |
[38] | Shang L , Chen S , Du F , Li S , Zhao L , Wang X . Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. (2011) ;108: (12):4788–93. |
[39] | Nazio F , Strappazzon F , Antonioli M , Bielli P , Cianfanelli V , Bordi M , et al . mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. (2013) ;15: (4):406–16. |
[40] | Lamb CA , Yoshimori T , Tooze SA . The autophagosome: Origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. (2013) ;14: (12):759–74. |
[41] | Hamasaki M , Furuta N , Matsuda A , Nezu A , Yamamoto A , Fujita N , et al . Autophagosomes form at ER-mitochondria contact sites. Nature. 495. (2013) )389–93. |
[42] | Bernard A , Klionsky DJ . Autophagosome formation: Tracing the source. Dev Cell. (2013) ;25: (2):116–7. |
[43] | Ravikumar B , Moreau K , Jahreiss L , Puri C , Rubinsztein DC . Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. (2010) ;12: (8):747–57. |
[44] | Yla-Anttila P , Vihinen H , Jokitalo E , Eskelinen EL . 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. (2009) ;5: (8):1180–5. |
[45] | Hayashi-Nishino M , Fujita N , Noda T , Yamaguchi A , Yoshimori T , Yamamoto A . A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. (2009) ;11: (12):1433–7. |
[46] | Hailey DW , Rambold AS , Satpute-Krishnan P , Mitra K , Sougrat R , Kim PK , et al . Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. (2010) ;141: (4):656–67. |
[47] | Rubinsztein DC , Shpilka T , Elazar Z . Mechanisms of autophagosome biogenesis. Curr Biol.R. (2012) ;22: (1):29–34. |
[48] | Matsunaga K , Saitoh T , Tabata K , Omori H , Satoh T , Kurotori N , et al . Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. (2009) ;11: (4):385–96. |
[49] | Pattingre S , Tassa A , Qu X , Garuti R , Liang XH , Mizushima N , et al . Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. (2005) ;122: (6):927–39. |
[50] | Furuya N , Yu J , Byfield M , Pattingre S , Levine B . The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. (2005) ;1: (1):46–52. |
[51] | Di Bartolomeo S , Corazzari M , Nazio F , Oliverio S , Lisi G , Antonioli M , et al . The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. (2010) ;191: (1):155–68. |
[52] | Maejima Y , Kyoi S , Zhai P , Liu T , Li H , Ivessa A , et al . Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. (2013) ;19: (11):1478–88. |
[53] | Fimia GM , Stoykova A , Romagnoli A , Giunta L , Di Bartolomeo S , Nardacci R , et al . Ambra1 regulates autophagy and development of the nervous system. Nature. (2007) ;447: (7148):1121–5. |
[54] | Geng J , Klionsky DJ . The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Re. (2008) ;9: (9):859–64. |
[55] | Ichimura Y , Kirisako T , Takao T , Satomi Y , Shimonishi Y , Ishihara N , et al . A ubiquitin-like system mediates protein lipidation. Nature. 408. (2000) )488–92. |
[56] | Xie Z , Nair U , Klionsky DJ . Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. (2008) ;19: (8):3290–8. |
[57] | Hu YB , Dammer EB , Ren RJ , Wang G . The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl Neurodegener. (2015) ;4: , 18. |
[58] | Amaya C , Fader CM , Colombo MI . Autophagy and proteins involved in vesicular trafficking. FEBS Lett. (2015) ;589: (22):3343–53. |
[59] | Monastyrska I , Rieter E , Klionsky DJ , Reggiori F . Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc. (2009) ;84: (3):431–48. |
[60] | Saftig P , Klumperman J . Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat Rev Mol Cell Biol. (2009) ;10: (9):623–35. |
[61] | Hyttinen JM , Niittykoski M , Salminen A , Kaarniranta K . Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim Biophys Acta. (2013) ;1833: 3 503–10. |
[62] | Yu L , McPhee CK , Zheng L , Mardones GA , Rong Y , Peng J , et al . Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. (2010) ;465: (7300):942–6. |
[63] | Rogov V , Dotsch V , Johansen T , Kirkin V . Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. MolCell. (2014) ;53: (2):167–78. |
[64] | Stolz A , Ernst A , Dikic I . Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. (2014) ;16: (6):495–501. |
[65] | Kirkin V , Lamark T , Sou YS , Bjorkoy G , Nunn JL , Bruun JA , et al . A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. (2009) ;33: (4):505–16. |
[66] | Arndt V , Dick N , Tawo R , Dreiseidler M , Wenzel D , Hesse M , et al . Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. (2010) ;20: (2):143–8. |
[67] | Ulbricht A , Eppler FJ , Tapia VE , van der Ven PF , Hampe N , Hersch N , et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. (2013) ;23: (5):430–5. |
[68] | Komatsu M , Waguri S , Koike M , Sou YS , Ueno T , Hara T , et al . Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. (2007) ;131: (6):1149–63. |
[69] | Lamark T , Johansen T . Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. (2012) ;736905. |
[70] | Kirkin V , McEwan DG , Novak I , Dikic I . A role for ubiquitin in selective autophagy. Mol Cell. (2009) ;34: (3):259–69. |
[71] | Pandey UB , Nie Z , Batlevi Y , McCray BA , Ritson GP , Nedelsky NB , et al . HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. (2007) ;447: (7146)859–63. |
[72] | Boyault C , Gilquin B , Zhang Y , Rybin V , Garman E , Meyer-Klaucke W , et al . HDAC6-p97/VCP controlled polyubiquitin chain turnover. Embo J. (2006) ;25: (14):3357–66. |
[73] | Klionsky DJ , Abdalla FC , Abeliovich H , Abraham RT , Acevedo-Arozena A , Adeli K , et al . Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. (2012) ;8: (4):445–544. |
[74] | Nascimbeni AC , Fanin M , Masiero E , Angelini C , Sandri M . The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ 2012. |
[75] | Nascimbeni AC , Fanin M , Masiero E , Angelini C , Sandri M . Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy. (2012) ;8: (11):1697–700. |
[76] | Castets P , Lin S , Rion N , Di Fulvio S , Romanino K , Guridi M , et al . Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. (2013) ;17: (5):731–44. |
[77] | Klionsky DJ , Cuervo AM , Seglen PO . Methods for monitoring autophagy from yeast to human. Autophagy. (2007) ;3: (3):181–206. |
[78] | Ju JS , Varadhachary AS , Miller SE , Weihl CC . Quantitation of autophagic flux in mature skeletal muscle. Autophagy. (2010) ;6: (7):929–35. |
[79] | Klionsky DJ , Abeliovich H , Agostinis P , Agrawal DK , Aliev G , Askew DS , et al . Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. (2008) ;4: (2):151–75. |
[80] | Zhang J , Wang J , Ng S , Lin Q , Shen HM . Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy. (2014) ;10: (5):901–12. |
[81] | Pellegrini C , Zulian A , Gualandi F , Manzati E , Merlini L , Michelini ME , et al . Melanocytes–a novel tool to study mitochondrial dysfunction in Duchenne muscular dystrophy. J Cell Physiol. (2013) ;228: (6):1323–31. |
[82] | Zulian A , Tagliavini F , Rizzo E , Pellegrini C , Sardone F , Zini N , et al . Melanocytes from Patients Affected by Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy have Dysfunctional Mitochondria That Can be Rescued with Cyclophilin Inhibitors. Front Aging Neurosci. (2014) ;6: , 324. |
[83] | He C , Bassik MC , Moresi V , Sun K , Wei Y , Zou Z , et al . Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 481. (2012) )511–5. |
[84] | Jamart C , Benoit N , Raymackers JM , Kim HJ , Kim CK , Francaux M . Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur J Appl Physiol. (2012) ;112: (8):3173–7. |
[85] | Jamart C , Francaux M , Millet GY , Deldicque L , Frere D , Feasson L . Modulation of autophagy and ubiquitin-proteasome pathways during ultra-endurance running. J Appl Physiol. (2012) ;112: (9):1529–37. |
[86] | Luo L , Lu AM , Wang Y , Hong A , Chen Y , Hu J , et al . Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signalling in aged rats. Exp Gerontol. (2013) ;48: (4):427–36. |
[87] | Grumati P , Coletto L , Schiavinato A , Castagnaro S , Bertaggia E , Sandri M , et al . Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy. (2011) ;7: (12):1415–23. |
[88] | Tassa A , Roux MP , Attaix D , Bechet DM . Class III phosphoinositide 3-kinase–Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J. (2003) ;376: (Pt 3):577–86. |
[89] | Mizushima N , Yamamoto A , Matsui M , Yoshimori T , Ohsumi Y . In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. (2004) ;15: (3):1101–11. |
[90] | Schiaffino S , Hanzlikova V . Studies on the effect of denervation in developing muscle. II. The lysosomal system. J Ultrastruct Res. (1972) ;39: (1):1–14. |
[91] | O’Leary MF , Hood DA . Denervation-induced oxidative stress and autophagy signalling in muscle. Autophagy. (2009) ;5: (2):230–1. |
[92] | Paul PK , Gupta SK , Bhatnagar S , Panguluri SK , Darnay BG , Choi Y , et al . Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. (2010) ;191: (7):1395–411. |
[93] | Vainshtein A , Desjardins EM , Armani A , Sandri M , Hood DA . PGC-1alpha modulates denervation-induced mitophagy in skeletal muscle. Skelet Muscle. (2015) ;5: , 9. |
[94] | Quy PN , Kuma A , Pierre P , Mizushima N . Proteasome-dependent activation of mammalian target of rapamycin complex 1 (mTORC1) is essential for autophagy suppression and muscle remodeling following denervation. J Biol Chem. (2013) ;288: (2):1125–34. |
[95] | Deval C , Mordier S , Obled C , Bechet D , Combaret L , Attaix D , et al . Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J. (2001) ;360: (Pt 1):143–50. |
[96] | Mofarrahi M , Sigala I , Guo Y , Godin R , Davis EC , Petrof B , et al . Autophagy and skeletal muscles in sepsis. PLoS One.e. (2012) ;7: (10):47265. |
[97] | Penna F , Costamagna D , Pin F , Camperi A , Fanzani A , Chiarpotto EM , et al . Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol. (2013) ;182: (4):1367–78. |
[98] | Sandri M . Protein breakdown in cancer cachexia. Semin Cell Dev Biol (2015) ;54: (2016):11–19. |
[99] | Brocca L , Cannavino J , Coletto L , Biolo G , Sandri M , Bottinelli R , et al . The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J Physiol. (2012) ;590: (Pt 20):5211–30. |
[100] | Qiu J , Tsien C , Thapalaya S , Narayanan A , Weihl CC , Ching JK , et al . Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am J Physiol Endocrinol Metab. (2012) ;303: (8):E983–93. |
[101] | Masiero E , Agatea L , Mammucari C , Blaauw B , Loro E , Komatsu M , et al . Autophagy is required to maintain muscle mass. Cell Metab. (2009) ;10: (6):507–15. |
[102] | Nemazanyy I , Blaauw B , Paolini C , Caillaud C , Protasi F , Mueller A , et al . Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med. (2013) ;5: (6):870–90. |
[103] | Dalakas MC . Toxic and drug-induced myopathies. J Neurol Neurosurg Psychiatry. (2009) ;80: (8):832–8. |
[104] | Kuncl RW , Duncan G , Watson D , Alderson K , Rogawski MA , Peper M . Colchicine myopathy and neuropathy. N Engl J Med. (1987) ;316: (25):1562–8. |
[105] | Ching JK , Ju JS , Pittman SK , Margeta M , Weihl CC . Increased autophagy accelerates colchicine-induced muscle toxicity. Autophagy. (2013) ;9: (12):2115–25. |
[106] | Wohlgemuth SE , Seo AY , Marzetti E , Lees HA , Leeuwenburgh C . Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp Gerontol. (2010) ;45: (2):138–48. |
[107] | Wenz T , Rossi SG , Rotundo RL , Spiegelman BM , Moraes CT . Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A. (2009) ;106: (48):20405–10. |
[108] | Garcia-Prat L , Martinez-Vicente M , Perdiguero E , Ortet L , Rodriguez-Ubreva J , Rebollo E , et al . Autophagy maintains stemness by preventing senescence. Nature. (2016) ;529: (7584):37–42. |
[109] | Youle RJ , Narendra DP . Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) ;12: (1):9–14. |
[110] | Hanna RA , Quinsay MN , Orogo AM , Giang K , Rikka S , Gustafsson AB . Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. (2012) ;287: (23):19094–104. |
[111] | Novak I , Kirkin V , McEwan DG , Zhang J , Wild P , Rozenknop A , et al . Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Re. (2010) ;11: (1):45–51. |
[112] | Okatsu K , Saisho K , Shimanuki M , Nakada K , Shitara H , Sou YS , et al . p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. (2010) ;15: (8):887–900. |
[113] | Okatsu K , Uno M , Koyano F , Go E , Kimura M , Oka T , et al . A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem. (2013) ;288: (51):36372–84. |
[114] | Sarraf SA , Raman M , Guarani-Pereira V , Sowa ME , Huttlin EL , Gygi SP , et al . Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. (2013) ;496: (7445):372–6. |
[115] | Ziviani E , Tao RN , Whitworth AJ . Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. (2010) ;107: (11):5018–23. |
[116] | Romanello V , Sandri M . Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Re. (2010) ;12: (6):433–9. |
[117] | Romanello V , Guadagnin E , Gomes L , Roder I , Sandri C , Petersen Y , et al . Mitochondrial fission and remodelling contributes to muscle atrophy. Embo J. (2010) ;29: (10):1774–85. |
[118] | Grumati P , Coletto L , Sabatelli P , Cescon M , Angelin A , Bertaggia E , et al . Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. (2010) ;16: (11):1313–20. |
[119] | Katsetos CD , Koutzaki S , Melvin JJ . Mitochondrial dysfunction in neuromuscular disorders. Semin Pediatr Neurol. (2013) ;20: (3):202–15. |
[120] | Mammucari C , Milan G , Romanello V , Masiero E , Rudolf R , Del Piccolo P , et al . FoxO3 controls autophagy in skeletal muscle in vivo . Cell Metab. (2007) ;6: (6):458–71. |
[121] | Yamazaki Y , Kamei Y , Sugita S , Akaike F , Kanai S , Miura S , et al . The cathepsin L gene is a direct target of FOXO1 in skeletal muscle. Biochem J. (2010) ;427: (1):171–8. |
[122] | Zhao J , Brault JJ , Schild A , Cao P , Sandri M , Schiaffino S , et al . FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. (2007) ;6: (6):472–83. |
[123] | Sanchez AM , Csibi A , Raibon A , Cornille K , Gay S , Bernardi H , et al . AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. (2012) ;113: (2):695–710. |
[124] | Stitt TN , Drujan D , Clarke BA , Panaro F , Timofeyva Y , Kline WO , et al . The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. (2004) ;14: (3):395–403. |
[125] | Sanchez AM , Candau RB , Bernardi H . FoxO transcription factors: Their roles in the maintenance of skeletal muscle homeostasis. Cellular and molecular life sciences: CMLS. (2014) ;71: (9):1657–71. |
[126] | Sardiello M , Palmieri M , di Ronza A , Medina DL , Valenza M , Gennarino VA , et al . A gene network regulating lysosomal biogenesis and function. Science. (2009) ;325: (5939):473–7. |
[127] | Settembre C , Di Malta C , Polito VA , Garcia Arencibia M , Vetrini F , Erdin S , et al . TFEB links autophagy to lysosomal biogenesis. Science. (2011) ;332: (6036)1429–33. |
[128] | Settembre C , Zoncu R , Medina DL , Vetrini F , Erdin S , Huynh T , et al . A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. (2012) ;31: (5):1095–108. |
[129] | Roczniak-Ferguson A , Petit CS , Froehlich F , Qian S , Ky J , Angarola B , et al . The transcription factor TFEB links mTORC1 signalling to transcriptional control of lysosome homeostasis. Sci Signal. (2012) ;5: (228):ra42. |
[130] | Pena-Llopis S , Vega-Rubin-de-Celis S , Schwartz JC , Wolff NC , Tran TA , Zou L , et al . Regulation of TFEB and V-ATPases by mTORC1. Embo J. (2011) ;30: (16):3242–58. |
[131] | Moresi V , Carrer M , Grueter CE , Rifki OF , Shelton JM , Richardson JA , et al . Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci U S A. (2012) ;109: (5):1649–54. |
[132] | Sandri M . Autophagy in skeletal muscle. FEBS Lett. (2010) ;584: (7):1411–6. |
[133] | Mordier S , Deval C , Bechet D , Tassa A , Ferrara M . Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signalling pathway. J Biol Chem. (2000) ;275: (38):29900–6. |
[134] | Castets P , Ruegg MA . MTORC1 determines autophagy through ULK1 regulation in skeletal muscle. Autophagy. (2013) ;9: (9):1435–7. |
[135] | Yamada E , Bastie CC , Koga H , Wang Y , Cuervo AM , Pessin JE . Mouse skeletal muscle fiber-type specific macroautophagy and muscle wasting is regulated by a Fyn/STAT3/Vps34 signalling pathway. Cell Re. (2012) ;1: (5):557–69. |
[136] | Mizushima N . Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. (2009) ;452: , 13–23. |
[137] | Gawlik KI , Durbeej M . Skeletal muscle laminin and MDC1A: Pathogenesis and treatment strategies. Skelet Muscle. (2011) ;1: (1):9. |
[138] | Carmignac V , Svensson M , Körner Z , Elowsson L , Matsumura C , Gawlik KI , et al . Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. (2011) ;20: (24):4891–902. |
[139] | Wu YT , Tan HL , Shui G , Bauvy C , Huang Q , Wenk MR , et al . Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. (2010) ;285: (14):10850–61. |
[140] | Lin YC , Kuo HC , Wang JS , Lin WW . Regulation of inflammatory response by 3-methyladenine involves the coordinative actions on Akt and glycogen synthase kinase 3beta rather than autophagy. J Immunol. (2012) ;189: (8):4154–64. |
[141] | Klein AF , Gasnier E , Furling D . Gain of RNA function in pathological cases: Focus on myotonic dystrophy. Biochimie. (2011) ;93: (11):2006–12. |
[142] | Sicot G , Gourdon G , Gomes-Pereira M . Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: New findings and future challenges. Hum Mol Genet. (2011) ;20: (R2):R116–23. |
[143] | Beffy P , Del Carratore R , Masini M , Furling D , Puymirat J , Masiello P , et al . Altered signal transduction pathways and induction of autophagy in human myotonic dystrophy type 1 myoblasts. Int J Biochem Cell Biol. (2010) ;42: (12):1973–83. |
[144] | Loro E , Rinaldi F , Malena A , Masiero E , Novelli G , Angelini C , et al . Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. (2010) ;17: (8):1315–24. |
[145] | Bargiela A , Cerro-Herreros E , Fernandez-Costa JM , Vilchez JJ , Llamusi B , Artero R . Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis Model Mech. (2015) ;8: (7):679–90. |
[146] | Ueda H , Shimokawa M , Yamamoto M , Kameda N , Mizusawa H , Baba T , et al . Decreased expression of myotonic dystrophy protein kinase and disorganization of sarcoplasmic reticulum in skeletal muscle of myotonic dystrophy. J Neurol Sci. (1999) ;162: (1):38–50. |
[147] | Swash M , Fox KP . Abnormal intrafusal muscle fibres in myotonic dystrophy: A study using serial sections. J Neurol Neurosurg Psychiatry. (1975) ;38: (1):91–9. |
[148] | Ludatscher RM , Kerner H , Amikam S , Gellei B . Myotonia dystrophica with heart involvement: An electron microscopic study of skeletal, cardiac, and smooth muscle. J Clin Pathol. (1978) ;31: (11):1057–64. |
[149] | Dunne PW , Ma L , Casey DL , Harati Y , Epstein HF . Localization of myotonic dystrophy protein kinase in skeletal muscle and its alteration with disease. Cell Motil Cytoskeleton. (1996) ;33: (1):52–63. |
[150] | Guiraud-Dogan C , Huguet A , Gomes-Pereira M , Brisson E , Bassez G , Junien C , et al . DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim Biophys Acta. (2007) ;1772: (11-12):1183–91. |
[151] | Savkur RS , Philips AV , Cooper TA . Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. (2001) ;29: (1):40–7. |
[152] | Llagostera E , Catalucci D , Marti L , Liesa M , Camps M , Ciaraldi TP , et al . Role of myotonic dystrophy protein kinase (DMPK) in glucose homeostasis and muscle insulin action. PLoS One. (2007) ;2: (11):e1134. |
[153] | Denis JA , Gauthier M , Rachdi L , Aubert S , Giraud-Triboult K , Poydenot P , et al . mTOR-dependent proliferation defect in human ES-derived neural stem cells affected by myotonic dystrophy type 1. J Cell Sci. (2013) ;126: (Pt 8):1763–72. |
[154] | Salisbury E , Sakai K , Schoser B , Huichalaf C , Schneider-Gold C , Nguyen H , et al . Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res. (2008) ;314: (11-12):2266–78. |
[155] | Li X , Zhang W , Lv H , Wang ZX , Yuan Y . [Activities of Akt pathway and their correlation with pathological changes in myotonic dystrophy]. Beijing Da Xue Xue Bao. (2010) ;42: (5):526–9. |
[156] | Yadava RS , Foff EP , Yu Q , Gladman JT , Kim YK , Bhatt KS , et al . TWEAK/Fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum Mol Genet. (2015) ;24: (7):2035–48. |
[157] | Allamand V , Brinas L , Richard P , Stojkovic T , Quijano-Roy S , Bonne G . ColVI myopathies: Where do we stand, where do we go? Skelet Muscle ((2011) ;1: , 30. |
[158] | Ramanoudjame L , Rocancourt C , Laine J , Klein A , Joassard L , Gartioux C , et al . Two novel COLVI long chains in zebrafish that are essential for muscle development. Hum Mol Genet. (2015) ;24: (23):6624–39. |
[159] | Chrisam M , Pirozzi M , Castagnaro S , Blaauw B , Polishchuck R , Cecconi F , et al . Reactivation of autophagy by spermidine ameliorates the myopathic defects of collagen VI-null mice. Autophagy. (2015) ;11: (12):2142–52. |
[160] | Merlini L , Nishino I . 201st ENMC International Workshop: Autophagy in muscular dystrophies–translational approach, 1-3 November Bussum, The Netherlands. Neuromuscul Disord. (2014) ;24: (6):546–61. |
[161] | Flanigan KM . Duchenne and Becker muscular dystrophies. Neurol Clin. (2014) ;32: (3):671–88, viii. |
[162] | DiMario JX , Uzman A , Strohman RC . Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev Biol. (1991) ;148: (1):314–21. |
[163] | Anderson JE , Bressler BH , Ovalle WK . Functional regeneration in the hindlimb skeletal muscle of the mdx mouse. J Muscle Res Cell Motil. (1988) ;9: (6):499–515. |
[164] | Petrof BJ , Shrager JB , Stedman HH , Kelly AM , Sweeney HL . Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. (1993) ;90: (8):3710–4. |
[165] | Momma K , Noguchi S , Malicdan MC , Hayashi YK , Minami N , Kamakura K , et al . Rimmed vacuoles in Becker muscular dystrophy have similar features with inclusion myopathies. PLoS One. (2012) ;7: (12):e52002. |
[166] | De Palma C , Morisi F , Cheli S , Pambianco S , Cappello V , Vezzoli M , et al . Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. (2012) ;3: , e418. |
[167] | Spitali P , Grumati P , Hiller M , Chrisam M , Aartsma-Rus A , Bonaldo P . Autophagy is Impaired in the Tibialis Anterior of Dystrophin Null Mice. PLoS Curr. (2013) )5. |
[168] | Pauly M , Daussin F , Burelle Y , Li T , Godin R , Fauconnier J , et al . AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol. (2012) ;181: (2):583–92. |
[169] | Pal R , Palmieri M , Loehr JA , Li S , Abo-Zahrah R , Monroe TO , et al . Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat Commun. (2014) ;5: , 4425. |
[170] | Hindi SM , Sato S , Choi Y , Kumar A . Distinct roles of TRAF6 at early and late stages of muscle pathology in the mdx model of Duchenne muscular dystrophy. Hum Mol Genet. (2014) ;23: (6):1492–505. |
[171] | Peter AK , Crosbie RH . Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp Cell Res. (2006) ;312: (13):2580–91. |
[172] | Dogra C , Changotra H , Wergedal JE , Kumar A . Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signalling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J Cell Physiol. (2006) ;208: (3):575–85. |
[173] | Barton ER , Morris L , Musaro A , Rosenthal N , Sweeney HL . Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. (2002) ;157: (1):137–48. |
[174] | Eghtesad S , Jhunjhunwala S , Little SR , Clemens PR . Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol Med. (2011) ;17: (9-10):917–24. |
[175] | Mouisel E , Vignaud A , Hourde C , Butler-Browne G , Ferry A . Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signalling in skeletal muscles of venerable (18-24-month-old) dystrophic mdx mice. Muscle Nerve. (2010) ;41: (6):809–18. |
[176] | von Maltzahn J , Renaud JM , Parise G , Rudnicki MA . Wnt7a treatment ameliorates muscular dystrophy. Proc Natl Acad Sci U S A. (2012) ;109: (50):20614–9. |
[177] | Gurpur PB , Liu J , Burkin DJ , Kaufman SJ . Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol. (2009) ;174: (3):999–1008. |
[178] | Boppart MD , Burkin DJ , Kaufman SJ . Activation of AKT signalling promotes cell growth and survival in alpha7beta1 integrin-mediated alleviation of muscular dystrophy. Biochim Biophys Acta. (1812) )4 439–46. |
[179] | Burkin DJ , Wallace GQ , Nicol KJ , Kaufman DJ , Kaufman SJ . Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. (2001) ;152: (6):1207–18. |
[180] | Gregorevic P , Plant DR , Lynch GS . Administration of insulin-like growth factor-I improves fatigue resistance of skeletal muscles from dystrophic mdx mice. Muscle Nerve. (2004) ;30: (3):295–304. |
[181] | Shavlakadze T , White J , Hoh JF , Rosenthal N , Grounds MD . Targeted expression of insulin-like growth factor-I reduces early myofiber necrosis in dystrophic mdx mice. Mol Ther. (2004) ;10: (5):829–43. |
[182] | Risson V , Mazelin L , Roceri M , Sanchez H , Moncollin V , Corneloup C , et al . Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol. (2009) ;187: (6):859–74. |
[183] | Kim MH , Kay DI , Rudra RT , Chen BM , Hsu N , Izumiya Y , et al . Myogenic Akt signalling attenuates muscular degeneration, promotes myofiber regeneration and improves muscle function in dystrophin-deficient mdx mice. Hum Mol Genet. (2011) ;20: (7):1324–38. |
[184] | Bibee KP , Cheng YJ , Ching JK , Marsh JN , Li AJ , Keeling RM , et al . Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. Faseb J. (2014) ;28: (5):2047–61. |
[185] | Whitehead NP , Kim MJ , Bible KL , Adams ME , Froehner SC . A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc Natl Acad Sci U S A. (2015) ;112: (41):12864–9. |
[186] | De Palma C , Perrotta C , Pellegrino P , Clementi E , Cervia D . Skeletal muscle homeostasis in duchenne muscular dystrophy: Modulating autophagy as a promising therapeutic strategy. Front Aging Neurosci. (2014) ;6: , 188. |
[187] | Fetalvero KM , Yu Y , Goetschkes M , Liang G , Valdez RA , Gould T , et al . Defective autophagy and mTORC1 signalling in myotubularin null mice. Mol Cell Biol. (2013) ;33: (1):98–110. |
[188] | Shelton GD , Rider BE , Child G , Tzannes S , Guo LT , Moghadaszadeh B , et al . X-linked myotubular myopathy in Rottweiler dogs is caused by a missense mutation in Exon 11 of the MTM1 gene. Skelet Muscle. (2015) ;5: (1):1. |
[189] | Al-Qusairi L , Prokic I , Amoasii L , Kretz C , Messaddeq N , Mandel JL , et al . Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. Faseb J. (2013) ;27: (8):3384–94. |
[190] | Buj-Bello A , Laugel V , Messaddeq N , Zahreddine H , Laporte J , Pellissier JF , et al . The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad SciU S A. (2002) ;99: (23):15060–5. |
[191] | Joubert R , Vignaud A , Le M , Moal C , Messaddeq N , Buj-Bello A . Site-specific Mtm1 mutagenesis by an AAV-Cre vector reveals that myotubularin is essential in adult muscle. Hum Mol Genet. (2013) ;22: (9):1856–66. |
[192] | Vergne I , Roberts E , Elmaoued RA , Tosch V , Delgado MA , Proikas-Cezanne T , et al . Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. (2009) ;28: (15):2244–58. |
[193] | Tosch V , Rohde HM , Tronchere H , Zanoteli E , Monroy N , Kretz C , et al . A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet. (2006) ;15: (21):3098–106. |
[194] | Hnia K , Kretz C , Amoasii L , Bohm J , Liu X , Messaddeq N , et al . Primary T-tubule and autophagy defects in the phosphoinositide phosphatase Jumpy/MTMR14 knockout mice muscle. Adv Biol Regul. (2012) ;52: (1):98–107. |
[195] | Dowling JJ , Low SE , Busta AS , Feldman EL . Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet. (2010) ;19: (13):2668–81. |
[196] | Azibani F , Muchir A , Vignier N , Bonne G , Bertrand AT . Striated muscle laminopathies. Semin Cell Dev Biol. (2014) ;29: , 107–15. |
[197] | Park YE , Hayashi YK , Bonne G , Arimura T , Noguchi S , Nonaka I , et al . Autophagic degradation of nuclear components in mammalian cells. Autophagy. (2009) ;5: (6):795–804. |
[198] | Ramos FJ , Chen SC , Garelick MG , Dai DF , Liao CY , Schreiber KH , et al . Rapamycin reverses elevated mTORC1 signalling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med.144ra. (2012) ;4: (144):03. |
[199] | Choi JC , Muchir A , Wu W , Iwata S , Homma S , Morrow JP , et al . Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. (2012) ;4: (144):144ra02. |
[200] | Greenberg SA , Pinkus JL , Amato AA . Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve. (2006) ;34: (4):406–16. |
[201] | Malicdan MC , Nishino I . Autophagy in lysosomal myopathies. Brain Pathol. (2012) ;22: (1):82–8. |
[202] | Nishino I , Fu J , Tanji K , Yamada T , Shimojo S , Koori T , et al . Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. (2000) ;406: (6798)906–10. |
[203] | Tanaka Y , Guhde G , Suter A , Eskelinen EL , Hartmann D , Lullmann-Rauch R , et al . Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. (2000) 406: (6798)902–6. |
[204] | Eskelinen EL . Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. (2006) ;27: (5-6):495–502. |
[205] | Eskelinen EL , Schmidt CK , Neu S , Willenborg M , Fuertes G , Salvador N , et al . Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell. (2004) ;15: (7):3132–45. |
[206] | Beertsen W , Willenborg M , Everts V , Zirogianni A , Podschun R , Schroder B , et al . Impaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J Immunol. (2008) ;180: (1):475–82. |
[207] | Huynh KK , Eskelinen EL , Scott CC , Malevanets A , Saftig P , Grinstein S . LAMP proteins are required for fusion of lysosomes with phagosomes. Embo J. (2007) ;26: (2):313–24. |
[208] | Eskelinen EL , Illert AL , Tanaka Y , Schwarzmann G , Blanz J , Von Figura K , et al . Role of LAMP-2 in lysosome biogenesis and autophagy. Mol Biol Cell. (2002) ;13: (9):3355–68. |
[209] | Rothaug M , Stroobants S , Schweizer M , Peters J , Zunke F , Allerding M , et al . LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol Commun. (2015) ;3: , 6. |
[210] | Malicdan MC , Noguchi S , Nonaka I , Saftig P , Nishino I . Lysosomal myopathies: An excessive build-up in autophagosomes is too much to handle. NeuromusculDisord. (2008) ;18: (7):521–9. |
[211] | Lim JA , Li L , Raben N . Pompe disease: From pathophysiology to therapy and back again. Front Aging Neurosci. (2014) ;6: , 177. |
[212] | Raben N , Nagaraju K , Lee E , Kessler P , Byrne B , Lee L , et al . Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. (1998) ;273: (30):19086–92. |
[213] | Takikita S , Myerowitz R , Zaal K , Raben N , Plotz PH . Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches. Mol Genet Metab. (2009) ;96: (4):208–17. |
[214] | Raben N , Hill V , Shea L , Takikita S , Baum R , Mizushima N , et al . Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. (2008) ;17: (24):3897–908. |
[215] | Raben N , Takikita S , Pittis MG , Bembi B , Marie SK , Roberts A , et al . Deconstructing Pompe disease by analyzing single muscle fibers: To see a world in a grain of sand. Autophagy. (2007) ;3: (6):546–52. |
[216] | Spampanato C , Feeney E , Li L , Cardone M , Lim JA , Annunziata F , et al . Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. (2013) ;5: (5):691–706. |
[217] | Fukuda T , Ewan L , Bauer M , Mattaliano RJ , Zaal K , Ralston E , et al . Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. (2006) ;59: (4):700–8. |
[218] | Douillard-Guilloux G , Raben N , Takikita S , Batista L , Caillaud C , Richard E . Modulation of glycogen synthesis by RNA interference: Towards a new therapeutic approach for glycogenosis type II. Hum Mol Genet. (2008) ;17: (24):3876–86. |
[219] | Douillard-Guilloux G , Raben N , Takikita S , Ferry A , Vignaud A , Guillet-Deniau I , et al . Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease. Hum Mol Genet. (2010) ;19: (4):684–96. |
[220] | Raben N , Schreiner C , Baum R , Takikita S , Xu S , Xie T , et al . Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder–murine Pompe disease. Autophagy. (2010) ;6: (8):1078–89. |
[221] | Lim JA , Li L , Kakhlon O , Myerowitz R , Raben N . Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy. (2015) ;11: (2):385–402. |
[222] | Shea L , Raben N . Autophagy in skeletal muscle: Implications for Pompe disease. Int J Clin Pharmacol Ther. (2009) ;47: (Suppl 1):S42–7. |
[223] | Fukuda T , Ahearn M , Roberts A , Mattaliano RJ , Zaal K , Ralston E , et al . Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther. (2006) ;14: (6):831–9. |
[224] | Medina DL , Fraldi A , Bouche V , Annunziata F , Mansueto G , Spampanato C , et al . Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. (2011) ;21: (3):421–30. |
[225] | Martina JA , Diab HI , Lishu L , Jeong AL , Patange S , Raben N , et al . The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. (2014) ;7: (309):ra9. |
[226] | Takikita S , Schreiner C , Baum R , Xie T , Ralston E , Plotz PH , et al . Fiber type conversion by PGC-1α activates lysosomal and autophagosomal biogenesis in both unaffected and Pompe skeletal muscle. PLoS One. (2010) ;5: (12):e15239. |
[227] | Cullup T , Kho AL , Dionisi-Vici C , Brandmeier B , Smith F , Urry Z , et al . Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. (2013) ;45: (1):83–7. |
[228] | Zhao H , Zhao YG , Wang X , Xu L , Miao L , Feng D , et al . Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. (2013) ;200: (6):731–41. |
[229] | Nalbandian A , Donkervoort S , Dec E , Badadani M , Katheria V , Rana P , et al . The multiple faces of valosin-containing protein-associated diseases: Inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci. (2011) ;45: (3):522–31. |
[230] | Broccolini A , Mirabella M . Hereditary inclusion-body myopathies. Biochim Biophys Acta. (1852) )4 644–50. |
[231] | Yamanaka K , Sasagawa Y , Ogura T . Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta. (2012) ;1823: (1):130–7. |
[232] | Chou TF , Brown SJ , Minond D , Nordin BE , Li K , Jones AC , et al . Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A. (2011) ;108: (12):4834–9. |
[233] | Ju JS , Miller SE , Hanson PI , Weihl CC . Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem. (2008) ;283: (44):30289–99. |
[234] | Nalbandian A , Llewellyn KJ , Badadani M , Yin HZ , Nguyen C , Katheria V , et al . A progressive translational mouse model of human valosin-containing protein disease: The VCP(R155H/+) mouse. Muscle Nerve. (2013) ;47: (2):260–70. |
[235] | Nalbandian A , Llewellyn KJ , Kitazawa M , Yin HZ , Badadani M , Khanlou N , et al . The homozygote VCP(R(1)(5)(5)H/R(1)(5)(5)H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS One.e. (2012) ;7: (9):46308. |
[236] | Weihl CC , Miller SE , Hanson PI , Pestronk A . Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet. (2007) ;16: (8):919–28. |
[237] | Vesa J , Su H , Watts GD , Krause S , Walter MC , Martin B , et al . Valosin containing protein associated inclusion body myopathy: Abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul Disord. (2009) ;19: (11):766–72. |
[238] | Ju JS , Fuentealba RA , Miller SE , Jackson E , Piwnica-Worms D , Baloh RH , et al . Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. (2009) ;187: (6):875–88. |
[239] | Tresse E , Salomons FA , Vesa J , Bott LC , Kimonis V , Yao TP , et al . VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. (2010) ;6: (2):217–27. |
[240] | Ching JK , Elizabeth SV , Ju JS , Lusk C , Pittman SK , Weihl CC . mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet. (2013) ;22: (6):1167–79. |
[241] | Nalbandian A , Nguyen C , Katheria V , Llewellyn KJ , Badadani M , Caiozzo V , et al . Exercise training reverses skeletal muscle atrophy in an experimental model of VCP disease. PLoS One. (2013) ;8: (10):e76187. |
[242] | Llewellyn KJ , Nalbandian A , Jung KM , Nguyen C , Avanesian A , Mozaffar T , et al . Lipid-enriched diet rescues lethality and slows down progression in a murine model of VCP-associated disease. Hum Mol Genet. (2014) ;23: (5):1333–44. |
[243] | Llewellyn KJ , Walker N , Nguyen C , Tan B , BenMohamed L , Kimonis VE , et al . A Fine Balance of Dietary Lipids Improves Pathology of a Murine Model of VCP-Associated Multisystem Proteinopathy. PLoS One. (2015) ;10: (7):e0131995. |
[244] | Durieux AC , Prudhon B , Guicheney P , Bitoun M . Dynamin 2 and human diseases. J Mol Med (Berl). (2010) ;88: (4):339–50. |
[245] | Jungbluth H , Gautel M . Pathogenic mechanisms in centronuclear myopathies. Front Aging Neurosci. (2014) ;6: , 339. |
[246] | Durieux AC , Vassilopoulos S , Laine J , Fraysse B , Brinas L , Prudhon B , et al . A centronuclear myopathy–dynamin 2 mutation impairs autophagy in mice. Traffic. (2012) ;13: (6):869–79. |
[247] | Durieux AC , Vignaud A , Prudhon B , Viou MT , Beuvin M , Vassilopoulos S , et al . A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet. (2010) ;19: (24):4820–36. |
[248] | Ramachandran N , Munteanu I , Wang P , Ruggieri A , Rilstone JJ , Israelian N , et al . VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. (2013) ;125: (3):439–57. |
[249] | Crockett CD , Ruggieri A , Gujrati M , Zallek CM , Ramachandran N , Minassian BA , et al . Late adult-onset of X-linked myopathy with excessive autophagy. Muscle Nerve. (2014) ;50: (1):138–44. |
[250] | Ruggieri A , Ramachandran N , Wang P , Haan E , Kneebone C , Manavis J , et al . Non-coding VMA21 deletions cause X-linked myopathy with excessive autophagy. Neuromuscul Disord. (2015) ;25: (3):207–11. |
[251] | Dowling JJ , Moore SA , Kalimo H , Minassian BA . X-linked myopathy with excessive autophagy: A failure of self-eating. Acta Neuropathol. (2015) ;129: (3):383–90. |
[252] | Saraste A , Koskenvuo JW , Airaksinen J , Ramachandran N , Munteanu I , Udd B , et al . No cardiomyopathy in X-linked myopathy with excessive autophagy. Neuromuscul Disord. (2015) ;25: (6):485–7. |
[253] | Selcen D . Myofibrillar myopathies. Neuromuscul Disord. (2011) ;21: (3):161–71. |
[254] | Weihl CC , Iyadurai S , Baloh RH , Pittman SK , Schmidt RE , Lopate G , et al . Autophagic vacuolar pathology in desminopathies. Neuromuscul Disord. (2015) ;25: (3):199–206. |
[255] | Claeys KG , Fardeau M . Myofibrillar myopathies. Handb Clin Neurol. (2013) ;113: , 1337–42. |
[256] | Kley RA , Serdaroglu-Oflazer P , Leber Y , Odgerel Z , van der Ven PF , Olive M , et al . Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain. (2012) ;135: (Pt 9):2642–60. |
[257] | Cabet E , Batonnet-Pichon S , Delort F , Gausseres B , Vicart P , Lilienbaum A . Antioxidant Treatment and Induction of Autophagy Cooperate to Reduce Desmin Aggregation in a Cellular Model of Desminopathy. PLoS One. (2015) ;10: (9):e0137009. |
[258] | Tannous P , Zhu H , Johnstone JL , Shelton JM , Rajasekaran NS , Benjamin IJ , et al . Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad SciU S A. (2008) ;105: (28):9745–50. |
[259] | Zheng Q , Su H , Ranek MJ , Wang X . Autophagy and p62 in cardiac proteinopathy. Circ Res. (2011) ;109: (3):296–308. |
[260] | Homma S , Iwasaki M , Shelton GD , Engvall E , Reed JC , Takayama S . BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. (2006) ;169: (3):761–73. |
[261] | Ruparelia AA , Oorschot V , Vaz R , Ramm G , Bryson-Richardson RJ . Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. (2014) ;128: (6):821–33. |
[262] | Maloyan A , Sayegh J , Osinska H , Chua BH , Robbins J . Manipulation of death pathways in desmin-related cardiomyopathy. Circ Res. (2010) ;106: (9):1524–32. |
[263] | Bhuiyan MS , Pattison JS , Osinska H , James J , Gulick J , McLendon PM , et al . Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. (2013) ;123: (12):5284–97. |
[264] | Pattison JS , Osinska H , Robbins J . Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. (2011) ;109: (2):151–60. |
[265] | Anvar SY , t Hoen PA , Venema A , van der Sluijs B , van Engelen B , Snoeck M , et al . Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients. Skelet Muscle. (2011) ;1: (1):15. |
[266] | Raz Y , Raz V . Oculopharyngeal muscular dystrophy as a paradigm for muscle aging. Front Aging Neurosci. (2014) ;6: , 317. |
[267] | Bengoechea R , Pittman SK , Tuck EP , True HL , Weihl CC . Myofibrillar disruption and RNA-binding protein aggregation in a mouse model of limb-girdle muscular dystrophy 1D. Hum Mol Genet. (2015) ;24: (23):6588–602. |
[268] | Sarparanta J , Jonson PH , Golzio C , Sandell S , Luque H , Screen M , et al . Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. (2012) ;44: (4):450–5, S1-2. |
[269] | Harms MB , Sommerville RB , Allred P , Bell S , Ma D , Cooper P , et al . Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol. (2012) ;71: (3):407–16. |
[270] | Roos A , Buchkremer S , Kollipara L , Labisch T , Gatz C , Zitzelsberger M , et al . Myopathy in Marinesco-Sjogren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathol. (2014) ;127: (5):761–77. |
[271] | Askanas V , Engel WK , Nogalska A . Sporadic inclusion-body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim Biophys Acta. (1852) )4 633–43. |
[272] | Lünemann JD , Schmidt J , Schmid D , Barthel K , Wrede A , Dalakas MC , et al . Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. (2007) ;61: (5):476–83. |
[273] | Lünemann JD , Schmidt J , Dalakas MC , Münz C . Macroautophagy as a pathomechanism in sporadic inclusion body myositis. Autophagy. (2007) ;3: (4):384–6. |
[274] | Nogalska A , D’Agostino C , Terracciano C , Engel WK , Askanas V . Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol. (2010) ;177: (3):1377–87. |
[275] | Nogalska A , Terracciano C , D’Agostino C , King Engel W , Askanas V . p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. (2009) ;118: (3):407–13. |
[276] | Nicot AS , Lo Verso F , Ratti F , Pilot-Storck F , Streichenberger N , Sandri M , et al . Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy. (2014) ;10: (6):1036–53. |
[277] | D’Agostino C , Nogalska A , Cacciottolo M , Engel WK , Askanas V . Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol. (2011) ;122: (5):627–36. |
[278] | Cacciottolo M , Nogalska A , D’Agostino C , Engel WK , Askanas V . Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol Appl Neurobiol. (2013) ;39: (7):750–61. |
[279] | Kumamoto T , Ueyama H , Tsumura H , Toyoshima I , Tsuda T . Expression of lysosome-related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol. (2004) ;107: (1):59–65. |
[280] | Nogalska A , D’Agostino C , Engel WK , Askanas V . Sodium phenylbutyrate reverses lysosomal dysfunction and decreases amyloid-beta42 in an in vitro-model of inclusion-body myositis. Neurobiol Dis. (2014) ;65: , 93–101. |
[281] | Nishino I , Carrillo-Carrasco N , Argov Z . GNE myopathy: Current update and future therapy. J Neurol Neurosurg Psychiatry. (2015) ;86: (4):385–92. |
[282] | Malicdan MC , Noguchi S , Nonaka I , Hayashi YK , Nishino I . A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. (2007) ;16: (22):2669–82. |
[283] | Li H , Chen Q , Liu F , Zhang X , Li W , Liu S , et al . Unfolded protein response and activated degradative pathways regulation in GNE myopathy. PLoS One. (2013) ;8: (3):e58116. |
[284] | Kumamoto T , Ito T , Horinouchi H , Ueyama H , Toyoshima I , Tsuda T . Increased lysosome-related proteins in the skeletal muscles of distal myopathy with rimmed vacuoles. Muscle Nerve. (2000) ;23: (11):1686–93. |
[285] | Yonekawa T , Malicdan MC , Cho A , Hayashi YK , Nonaka I , Mine T , et al . Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain. (2014) ;137: (Pt 10):2670–9. |
[286] | Screen M , Raheem O , Holmlund-Hampf J , Jonson PH , Huovinen S , Hackman P , et al . Gene expression profiling in tibial muscular dystrophy reveals unfolded protein response and altered autophagy. PLoS One. (2014) ;9: (3):e90819. |
[287] | Udd B . Distal myopathies. Curr Neurol Neurosci Rep. (2014) ;14: (3):434. |
[288] | Fanin M , Nascimbeni AC , Angelini C . Muscle atrophy, ubiquitin-proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve. (2014) ;50: (3):340–7. |
[289] | Mitsuhashi S , Hatakeyama H , Karahashi M , Koumura T , Nonaka I , Hayashi YK , et al . Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum Mol Genet. (2011) ;20: (19):3841–51. |
[290] | Mitsuhashi S , Ohkuma A , Talim B , Karahashi M , Koumura T , Aoyama C , et al . A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am J Hum Genet. (2011) ;88: (6):845–51. |
[291] | Rusmini P , Crippa V , Cristofani R , Rinaldi C , Cicardi ME , Galbiati M , et al . The Role of the Protein Quality Control System in SBMA. J Mol Neurosci. (2015) . |
[292] | Chua JP , Reddy SL , Merry DE , Adachi H , Katsuno M , Sobue G , et al . Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet. (2014) ;23: (5):1376–86. |
[293] | Yu Z , Wang AM , Adachi H , Katsuno M , Sobue G , Yue Z , et al . Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. (2011) ;7: (10):e1002321. |
Figures and Tables
Fig.1
Overview of mechanisms and proteins involved in autophagy in mammals. In (macro)autophagy, initiation leads to the formation of a phagophore, which engulfs large cytoplasmic parts and expands to give rise to autophagosomes. Autophagy induction depends on the balance of several regulatory pathways converging on the Ulk1 complex (A). Autophagy ensures selective degradation of proteins and organelles, mediated by different autophagy cargo receptors (p62, Nbr1) and chaperone/co-chaperone proteins (Hsp, BAG3) (B). After fusion with the lysosomes and/or endosomes, degradation of the autolysosomal content by lysosomal enzymes permits recycling of metabolites. Of note, lysosomes are also involved in degradation associated with microautophagy and chaperone-mediated autophagy (CMA) (C). Red lines represent inhibition; green arrows show activation. Bcln1, Beclin1; MVB, multivesicular bodies; Ub, ubiquitin.
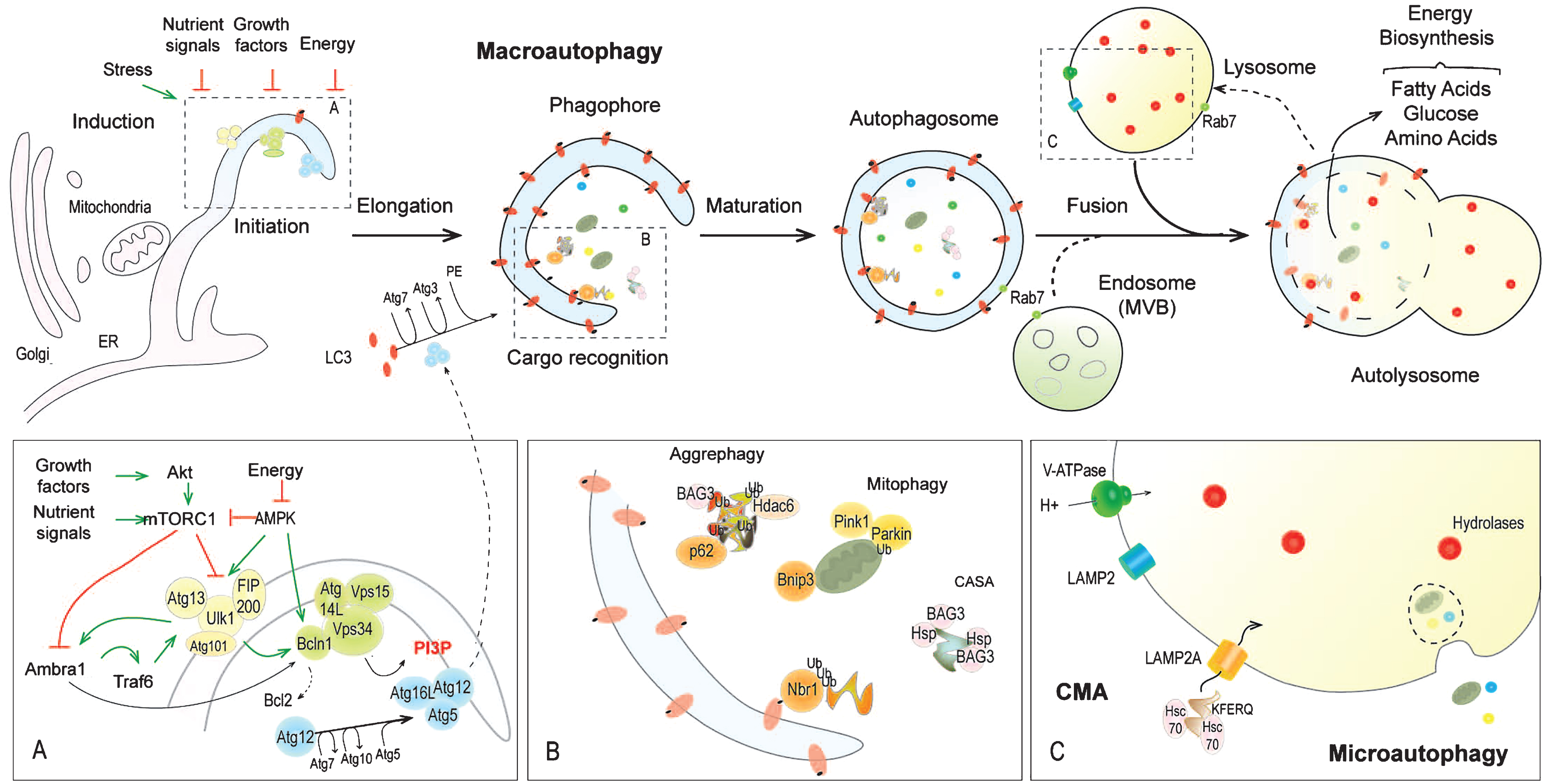
Fig.2
Neuromuscular disorders related to dysregulation of autophagy induction. In control (Ctrl) muscle, autophagy induction is determined by the state of the Ulk1 complex, which is inhibited by Akt/PKB-mTORC1 signalling and activated by the AMPK pathway. The autophagic flux varies depending on nutritive and stress stimuli, which can promote prolonged autophagy induction via FoxO-dependent expression of autophagy genes. In pathological conditions, excessive or insufficient autophagic flux may contribute to muscle damage. In DM1 and MDC1A, Akt/PKB inhibition would promote autophagy induction, while Akt/PKB-mTORC1 activation seems to restrict autophagic flux in XLMTM, COLVI-RM, DMD and laminopathies. Of note, autophagy induction may be enhanced in XLMTM due to abnormal Vps34 activation caused by Mtm1 deficiency. Treatments, which proved some efficacy in animal models, are indicated in green. Red lines represent inhibition; green arrows show activation. In pathological conditions, red and green arrows indicate abnormal inhibition and activation of signalling pathways, respectively. Black and white arrowheads in the hematein & eosin staining of muscle cross-sections indicate fat and degeneration regions, respectively; arrows show vacuolated fibres. The mutated proteins are indicated by an asterisk. COL, collagen; DG, dystroglycan; IR, insulin receptor; IRS, insulin receptor substrate; m, month; NOS, NO synthase; PI3K, phosphoinositide-3 kinase; ROS, reactive oxygen species; SF, splicing factor; SG, sarcoglycan; y, year. Scale bar, 100 μm.
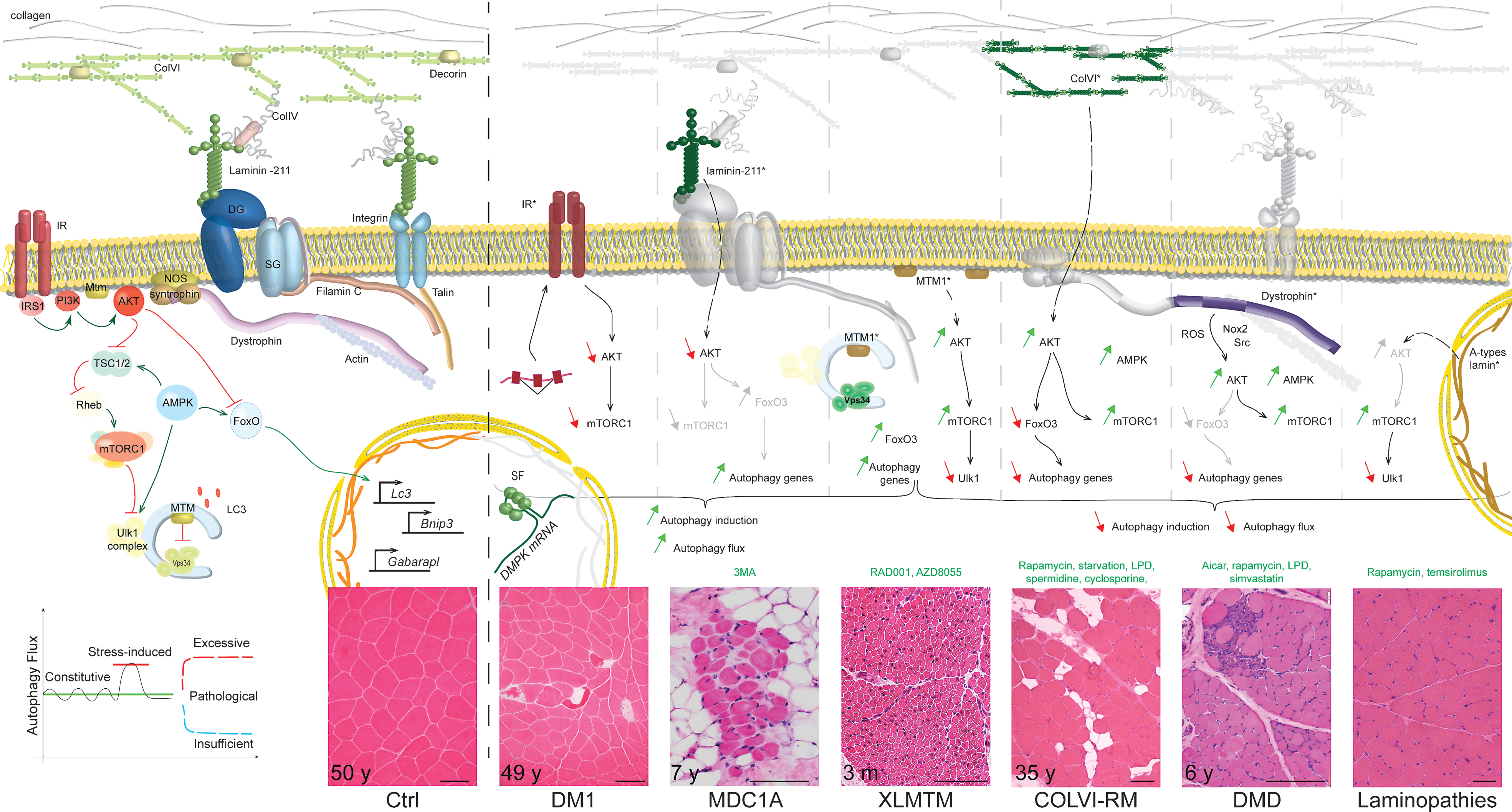
Fig.3
Neuromuscular disorders related to defective autophagosome maturation or lysosomal dysfunction. In myopathies with rimmed vacuoles (RV) (e.g. IBMPFD and MFM), autophagic vesicles would enlarge due to blockade of the maturation and fusion steps, likely caused by the accumulation of protein aggregates. In IBMPFD, VCP deficiency may also directly alter autophagosome maturation. In lysosomal storage disorders (LSD), defective autophagosome maturation/fusion and/or altered degradation steps lead to the formation of autophagic vacuoles with sarcolemmal features (AVSF) or enlarged lysosomes; glycogen massively accumulates in these vesicles in GSDIIb and GSDII diseases. Centronuclear myopathies related to DNM2 (AD-CNM) deficiency may also involve a defect in maturation/fusion steps. Of note, lysosome biogenesis and autophagy induction seem to be perturbed in some of these diseases. Red lines represent inhibition; green arrows show activation. Arrows in the H&E-stained muscle cross-sections indicate vacuoles. The pathogenic protein is indicated by an asterisk. CryAB, α-crystallin B chain; Des, desmin; Flnc, filamin C; M6PR, mannose-6 phosphate receptor; Ub, ubiquitin; y, year. Scale bar, 100 μm.
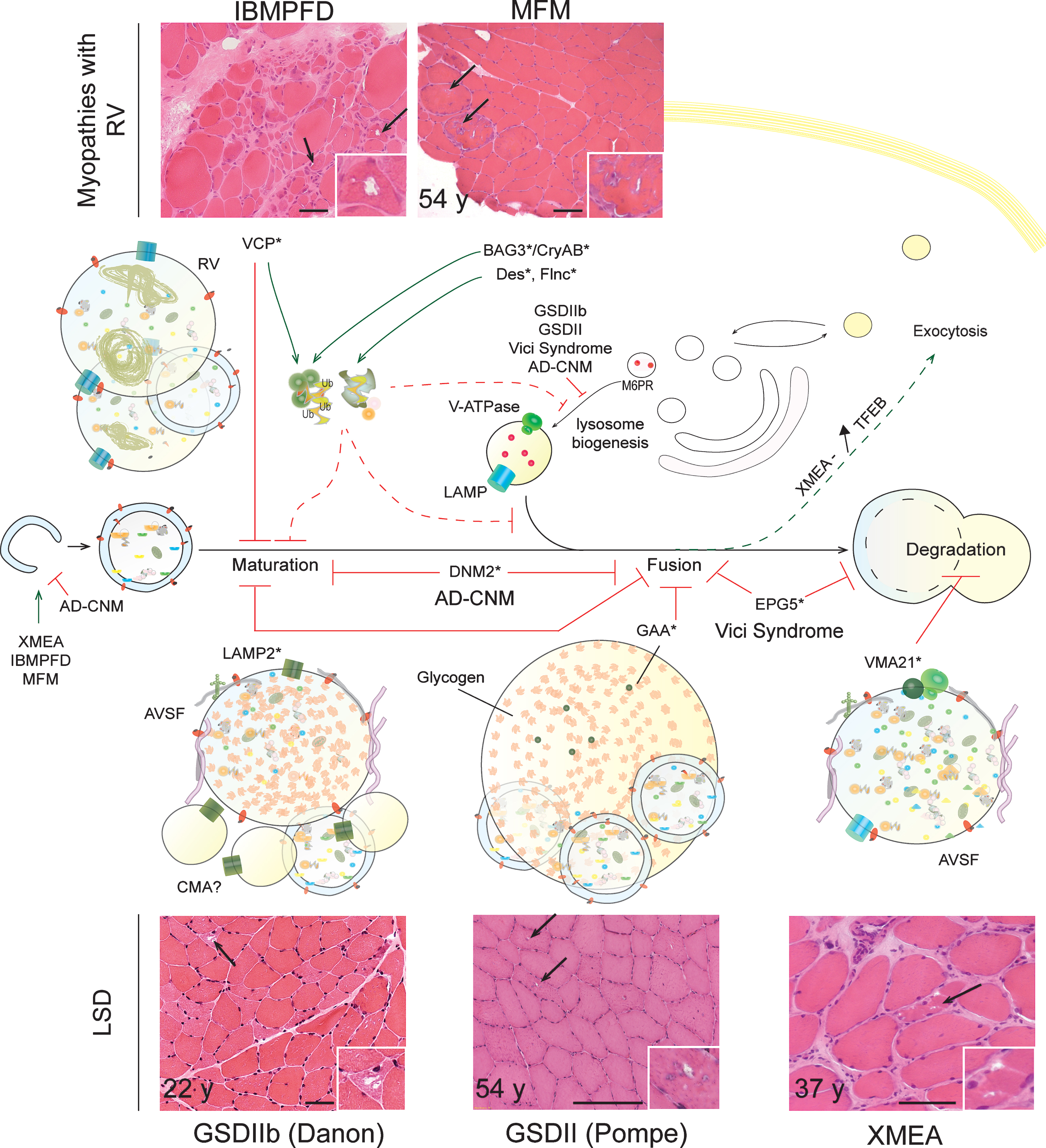
Table 1
Neuromuscular disorders related to autophagy defects – Signs, type of deregulation and possible therapeutic strategies
| Autophagy features | |||||||||
| Disorder | Mutated | Animal | Histology signs | Genes/proteins | Flux in | Regulatory | Treatment | References | |
| gene | model used | muscle | signaling | ||||||
| CMD | MDC1A | LAMA2 | dy 3K /dy 3K mice | Degeneration | ↑ Transcript, ↑LC3II | Increased? | ↓Akt | 3-MA | 137–140 |
| ColVI-RM | COL6 | Col6a1-/- mice | Abnormal organelles (mitochondria and SR), apoptosis, degeneration | ↓Transcript ↓LC3II, (↑p62) | Blocked at induction | ↑Akt, ↓FoxO, ↑mTORC1, ↑AMPK LPD, rapamycin, cyclosporin1, spermidine | LPD, rapamycin, cyclosporin1, spermidine | 87, 118, 157–160 | |
| Dystrophies | Laminopathies | LMNA | Lmna-/- mice | Vacuoles, aggregates | ↑LC3, ↑UlkP757 | Blocked at induction? | ↑mTORC1, ↑Akt | Rapamycin, temsirolimus | 196–199 |
| DMD, BMD | DMD | mdx mice | Abnormal organelles, (aggregates, vacuoles) | ↓Transcript ↓LC3II, (↑p62) | Blocked at induction | ↑Akt, ↑mTORC1, ↑AMPK, ↑Traf6 | LPD, rapamycin, AICAR | 161–186 | |
| OPMD | PABPN1 | – | Aggregates, (RV) | – | – | – | – | 160,265,266 | |
| LGMD1D/E | DNAJB6 | – | Aggregates, vacuoles | – | Impaired CASA? | – | – | 267–269 | |
| DM1 | DMPK | Dmpk-/- mice, | Vacuoles, apoptosis | ↑LC3II | – | ↓Akt/mTORC1 | – | 141–156 | |
| DM1 flies | |||||||||
| CNM | XLMTM | MTM1 | Mtm1-/- mice | AV, glycogen, aggregates, abnormal organelles | ↑ Transcript ↑LC3, ↑p62, ↑UlkP757 | Increased then blocked at maturation/fusion? | ↑Akt, mTORC1 ↑FoxO | RAD001, AZD8055 | 187–195 |
| AD-CNM | DNM2 | Dnm2R465W mice | – | ↑LC3II (St) | Blocked at induction/maturation? | – | – | 244–247 | |
| LSD | GSDIIb | LAMP2 | Lamp2-/- mice | AVSF, glycogen, aggregates | ↑LC3II | Blocked at maturation/fusion? Altered lysosome biogenesis? CMA? | – | – | 102, 201–209 |
| GSDII | GAA | GAA-/- mice | AV, glycogen, autophagic build-up, abnormal organelles | ↑LC3, ↑p62 | Blocked at fusion, altered lysosome biogenesis | – | ERT+autophagy inhibition | 210–226 | |
| Vici synd. | EPG5 | Epg5-/- mice | AV, glycogen, abnormal organelles, degeneration | ↑LC3, ↑p62/Nbr1 | Blocked at fusion/degradation? | ↓Akt, ↑FoxO | – | 227, 228 | |
| XMEA | VMA21 | – | AVSF | (↑ Transcript) ↑LC3II, ↑p62 | Blocked at degradation? Increased induction? | ↓mTORC1, ↑TFEB | – | 248–252 | |
| MyopathywithRV | IBMPFD | VCP | VCPR155H mice | RV, aggregates | ↑LC3II, ↑p62 | Blocked at fusion, increased induction | ↓mTORC1 | Exercise, HFD | 229–243 |
| sIBM | – | – | RV, aggregates, abnormal organelles | ↑LC3II, ↑p62/Nbr1 | Blocked at fusion/degradation? Increased induction? | ↓mTORC1 | – | 271–280 | |
| MFM | BAG3, CRYAB FLNC, ... | Bag3-/- mice, Bag3P209L zebrafish, CryAB R120G mice | Aggregates, RV | ↑LC3, ↑p62 | Blocked? Impaired CASA? Increased induction? | – | Rapamycin (CryABR120G - heart) | 253–264 | |
| GNE myopathy | GNE | – | RV, aggregates, abnormal organelles | – | – | – | – | 281–285 | |
AV, autophagic vacuole; AVSF, AV with sarcolemmal features; CASA, chaperone-assisted selective autophagy; CMA, chaperone-mediated autophagy; CMD, congenital muscular dystrophy; CNM, centronuclear myopathy; ERT, enzyme replacement therapy; HFD, high fat diet; LPD, low protein diet; 3-MA, 3-methyladenine; RV, rimmed vacuoles. Italics indicate elements remaining to be confirmed in skeletal muscle; brackets indicate non predominant pathological features.


