
Neo-epitope Peptides as Biomarkers of Disease Progression for Muscular Dystrophies and Other Myopathies
Abstract
For several decades, serological biomarkers of neuromuscular diseases as dystrophies, myopathies and myositis have been limited to routine clinical biochemistry panels. Gauging the pathological progression is a prerequisite for proper treatment and therefore identifying accessible, easy to monitor biomarkers that can predict the disease progression would be an important advancement. Most muscle diseases involve accelerated muscle fiber degradation, inflammation, fatty tissue substitution and/or fibrosis. All these pathological traits have been shown to give rise to serological peptide biomarkers in other tissues, underlining the potential application of existing biomarkers of such traits in muscle disorders. A significant quantity of tissue is involved in these pathological mechanisms alongside with qualitative changes in protein turnover in myofibrillar, extra-cellular matrix and immunological cell protein fractions accompanied by alterations in body fluids. We propose that protein and peptides can leak out of the afflicted muscles and can be of use in diagnosis, prediction of pathology trajectory and treatment efficacy. Proteolytic cleavage systems are especially modulated during a range of muscle pathologies, thereby giving rise to peptides that are differentially released during disease manifestation. Therefore, we believe that pathology-specific post-translational modifications like cleavages can give rise to neoepitope peptides that may represent a promising class of peptides for discovery of biomarkers pertaining to neuromuscular diseases.
BACKGROUND
Loss of muscle mass and function compromises health through impaired metabolic homeostasis and quality of life via hindered freedom of motion. Besides being an issue in the form of cachexia and sarcopenia, it is additionally important in a range of neuromuscular diseases, namely muscular dystrophies, congenital myopathies and myositis. These are a diverse group of pathologies affecting more than 2 million people in Europe alone that are characterized by loss of muscle mass and function, frequently accompanied by fibrosis and/or inflammation possibly as a secondary phenomenon (further detail on epidemiology: http://neuromuscular.wustl.edu/).
These diseases are mostly chronic and manifest at varying ages, from infancy to adulthood. Most are progressive and the severe ones are associated with significantly shortened lifespan, while others cause lifelong disability [1].
While the various forms of myositis are idiopathic, mainly inflammatory conditions, muscular dystrophies and congenital myopathies are genetic disorders that affect the structural integrity or critical signalling cascades controlling the muscle fibres [2].
Duchenne’s muscular dystrophy is the most common childhood dystrophy, where the absence of a critical subsarcolemmal protein, dystrophin, initiates a pathophysiological cascade eventually leading to leakage, disruption and death of the muscle fibres, generating sustained regeneration cycles [3]. The exposure of intracellular proteins to the extracellular environment, may elicit immunological reactions and thereby contribute to chronic inflammation which leads to abnormal regeneration and endomysial fibrosis [3, 4].
Considering the size of the muscle mass, the quantitative and qualitative changes in protein turnover associated with these pathologies and the degree of leakiness in healthy and diseased muscle fibres, muscle pathologies can result in release of pathology-specific peptides that can be used as serological or urinary biomarkers [5, 6].
DISEASE DESCRIPTION
The term muscular dystrophy covers more than 30 inherited diseases, associated with skeletal muscle degeneration and causing progressive weakness. Dystrophies manifest a range of effects including, fatty tissue invasion and muscle wasting through an abnormal dynamic between necrosis and regeneration, thereby affecting the metabolic fitness, fibrosis and/or inflammation [7, 8]. Since it is unfeasible to describe all of the diseases in a single review, the focus will be only on the most frequent ones and on their similar characteristics.
Duchenne and Becker muscular dystrophies (DMD and BMD) are two similar pathologies that constitute the most common causes of pathological muscle loss, aside from Sarcopenia and cachexia. Both are characterized by progressive fibrosis[4, 9, 10], inflammation, and displacement of muscle by fat or connective tissue [11]. This is more pronounced in DMD, usually leading to death in the third to fourth decade of life, while the milder and more variable BMD result in a life expectancy shortened by 0–20 years.
Limb-Girdle Muscular Dystrophy (LGMD) is a group of heterogeneous muscle dystrophies. Although they are clinically quite diverse, most manifestations are also characterized by degenerating myofibers, and later on infiltration of T-cells and macrophages, leading to a secondary inflammation [12, 13].
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant inherited neuromuscular disorder ranking third in muscular dystrophy frequency [2, 14–16]. This muscular dystrophy predominantly affects face muscles, upper arms and the scapula and leads to premature disability and loss of independence despite not reducing life expectancy severely. Part of the disease consists of perivascular inflammation with observed fiber necrosis, phagocytosis and increased tissue regeneration [17].
Inflammatory myopathies or myositis cause muscle atrophy and weakness through inflammation processes [18]. Furthermore, myositis can lead to pulmonary fibrosis, respiratory failure and death [19].
Shared pathological traits
Despite being different diseases with different etiologies, dystrophies and myositis share several traits that provide us with a common ground in which to search for biomarkers. Overlapping and non-overlapping traits between the individual myopathies are of great interest from a monitoring perspective, as they permit stratification. Shared traits for the myopathies are the presence of abnormal rates of turnover of muscle fiber components, inflammation or fibrosis, resulting in the production of peptide biomarkers [20–22] (Table 1). Although varying in manifestation and extent, monitoring these traits in each disease is useful, both for clinical assessment as well as prognosis of the disease course or treatment efficacy. Based upon experiences from inflammatory and degenerative pathologies in other tissues, we believe that detecting and assessing the presence or levels of these indicators in serum is a viable method to diagnose or determine the extent of the disease.
Inflammation
One of the traits of dystrophies is impaired structural integrity of muscle fibres leading to leakage of their contents, provoking an immune response. Increased expression of major histocompatibility complex type II is shown in both dystrophies and myositis and a consistent T lymphocyte invasion is seen in both human and animal models of muscular dystrophy. Likewise, it has been shown that genes coding for cytokines which induce apoptosis and inflammation (TNF-α, TGF-β, NF-κB and IL-6) are over-expressed in DMD patients. [3, 23, 24]. Fragments of MMP-degraded collagen I and III have in these pathologies been shown to characterize the inflammatory phenotype and in the case of osteoarthritis even separate patient with predominant inflammatory (synovitis) and mechanical osteochondral defects [25, 26]. Correspondingly, it has been shown in mouse and dog muscular dystrophy models that the dystrophic phenotype is associated with increases of MMP expression and that inhibition of MMP expression or activity actively ameliorates the dystrophic phenotype [27, 28].
Inflammation and fibrosis are sometimes interconnected as the proliferation of inflammatory cells leads to overexpression of cytokines that stimulate collagen accretion, i.e. TGF-β, and push native cells towards fibrogenic phenotypes (TNF, TFG-β1), contributing to scar tissue formation [29]. In the myocardium, this fibrosis leads to cardiac dysfunction which a prominent contributor to mortality in DMD patients [30].
Fibrosis
Fibrosis has been consistently reported to be present in various dystrophies after repeated regeneration cycles. Particularly with dystrophies, development of significant intramuscular fibrosis and replacement of functional muscle tissue with collagenous tissue is a problem. The satellite cells contributing to the formation of new muscle fibers, gradually lose their myogenic lineage and switch towards a fibrogenic phenotype, becoming myofibroblasts or fibroblasts. This leads to accumulation of interstitial collagens (types 1 through 6) andproteoglycans, which in turn results in changes in quality and quantity of extra-cellular matrix tissue (ECM)[4, 31]. The arrangement and distribution of the extracellular matrix around the contractile fibers is complex and differentially affected by musclediseases. Collagen I dominates the perimysium while type III collagen is found both in endomysium and epimysium, further associating with collagen V [32]. Thus, the normally well-ordered intramuscular connective tissue expands by taking the place of degraded muscle fibers with possible alterations in the organization of collagens (isoform ratios or crosslinking) [33]. Although describing the most aggressive development seen in DMD, the initial events in this process manifest in most dystrophies. Furthermore, dystrophic fibrosis has been shown to be associated with increased expression of gelatinases and collagenases MMP-1, -2, and -9 [34].
Muscle protein turnover
It is obvious from a macroscopic perspective of the muscle that the loss of functional muscle cells is associated with a change in muscle protein turnover. Stable isotope studies have shown that baselineprotein synthesis does not appear to be impaired in dystrophic patients [35]. However, considering that 2–400 grams of muscle is turned over a day in healthy adults, a small percentile deficit in the synthesis and degradation ratio can easily amount to several kilos of muscle per year. Such a difference is adequate to explain the observed muscle loss with even the most aggressive dystrophies, but it would be almost impossible to detect using stable isotope methodology.
In dystrophies and myositis, proteases from the MMP, calpain, caspase and cathepsin groups are known to contribute to muscle catabolism. Upregulation of MMP-7 and cathepsins H and L were demonstrated in DMD patients [36] along with a significant upregulation of MMP-2 expression [37] while progressive increase of MMP-9 levels was more recently noted in serum from DMD patients [38]. Findings were supported by experiments held on mdx mice, demonstrating increased MMP activity and involvement in pathogenesis [39]. Moreover, various cathepsin mRNA isoforms were upregulated in atrophying muscle of rats [40, 41]. Calpains (CAPN) are calcium-dependent proteases that have been linked to muscle degradation and necrosis [42] but mutations on the CAPN3 gene which is exclusively expressed in skeletal muscle leads to LGMD [43], possibly because of a protective role of the CAPN3 protease activity in exercise induced stress [44]. Lastly, caspase activity is related to muscle atrophy in various muscle wasting models involving myonuclear apoptosis [45].
BIOMARKERS
BIPED (Burden of disease, Investigative, prognostic, efficacy and diagnostic) Classification
A biomarker is defined as a characteristic that can be objectively measured and evaluated as “an indicator of normal biological processes, pathogenic processes or pharmacological responses to therapeutic or other health care interventions” [46]. Biomarkers can be qualitative or quantitative (e.g. histological assessment vs. Bone mineral density) and are commonly classified as “dry” or “wet”. The latter is everything biochemical and the former is everything that is not, including imaging techniques (such as MR or PET), questionnaires, clinical descriptions, etc.
Biomarkers can serve different purposes and thus have different inherent uses and restrictions.Diagnosis, description of the disease severity/progression or the impact of a potential treatment are some of the areas where biomarkers can be utilized. A suggested classification of different markers, the BIPED criteria, was given by the Osteoarthritis Biomarkers Network, providing practical requirements and recommendations for each category [47]. This nomenclature can be applied to muscle pathologies. In the BIPED criteria, there are five categories of markers: Burden of disease, Investigative, prognostic, efficacy and diagnostic which constitute the acronym of the proposed nomenclature (BIPED). For each of these classifications, requirements with respect to sensitivity, specificity and robustness are provided (Table 2). Different categories are not mutually exclusive and candidate biomarkers can belong to more than one category. Biomarker classification facilitates the selection of surrogate endpoints in a clinical study setting. Proper selection of endpoints is vital in clinical cohorts, to expedite and enhance the process by either deselecting non-responding individuals earlier in the study or promote better initial selection of candidates, e.g. fast progressors or otherwise at-risk populations. Avoiding inclusion or prolonged involvement of unsuitable patients in the trials can mitigate unnecessary time and fund consummation, accelerating the trial process [48].
Existing wet and dry biomarkers used in myopathies
Gene testing is available for a number of dystrophies, but despite the marked progression in the identification of specific mutations that can be verified by molecular methods [49], not all genes implicated have yet been discovered, limiting the sensitivity of genetic diagnostics [50, 51]. Muscle biopsies are often employed to identify the specificity of the pathology, the extent of degeneration, fatty tissue replacement or to be used in immunohistochemical assays. While diagnosis is thus adequately covered, assessing burden of disease and making qualified prognoses for disease trajectory is hampered by the severe lack of specific, robust or practical biomarkers.
When characterizing dystrophic patients, both the current and projected quantity and quality of the muscle is of interest. A range of biophysical techniques as air displacement plethysmography (ADP), biological impedance analysis/spectroscopy (BIA/BIS), Magnetic resonance imaging (MRI), Computed Tomopgraphy (CT), Dual X-ray Absorptiometry DXA) can all be used to assess muscle volume, while MRI and microCT can even provide information about fibrosis [52, 53]. However, availability, cost, radiation exposure, accuracy, validation and to some extent culture amongst clinicians have limited the use of all of these tools in routine clinical practice [7, 54–58]. Practically, muscle function or quality is assessed through functional testing Isometric grip strength, sitting and rising from a chair or six minutes walking tests are common examples of the functional tests used in clinical studies and settings [59].
Most dystrophies and myopathies cause muscle weakness, increased incidence of muscle fiber degeneration and regeneration and increase levels of Creatine Kinase (CK) levels in serum. Presence of CK and myoglobin (Mb) in serum are indicators of general muscle injury or degradation, but neither are capable of indicating the source or the extent.
Creatine Kinase is so far the main biomarker for assessing disease activity (“burden of disease”) and has been extensively used as a marker of sarcolemma disruption and membrane leakage during the past 50 years. Aberrant CK levels are obligatory for diagnosis of some dystrophies as in BMD [60]. As a biomarker though, CK is far from ideal, as it varies strongly between and within patients [11]. In DMD/BMD, CK is increased up to 200-fold but the levels lower with age, fibrosis progression and loss of muscle mass [11, 61]. Asymptomatic individuals with abnormal dystrophin genes but normal CK levels are also known to exist [62]. In Limb-girdle muscular dystrophy (LGMD), elevation of CK is not observed in general but can be very high in the autosomal recessive forms that tend to have an earlier onset as well. Levels of CK can also be increased in Facioscapulohumeral dystrophy (FSHD) albeit rarely, as with congenital myopathies and myositis [61].
Therefore, CK cannot provide viable information on the extent of the damage and biopsies cannot be repeated over time to facilitate sufficient monitoring over the course of a treatment. Imaging techniques (DXA, MRI and CT) are rarely used in monitoring of muscle diseases as the information they provide is considered of limited clinical utility, compared to the expense and possible radiation exposure associated with their use, while strength and endurance tests (sitting/rising, walking test) suffers from high variability and risk of intra-rater differences [6].
More importantly, little or no robust protein based biomarkers are available for prognosis of the disease or early indicators of response to a treatment. In clinical trials, it is of paramount importance to have biomarkers that can detect treatment response at an early stage. The ability to exclude patients that show no progression or better yet select better-suitedcandidates for cohorts is a key factor for successful clinical studies. It has to be noted that there are studies that have demonstrated the utility of some serum biomarkers as potential candidates for prognosis, stratification or response to treatment [5, 63–67].
Neoepitope biomarkers
One possible type of biomarkers showing promise in this regard is neoepitope peptide biomarkers. These are biomarkers in which pathology-specific post-translational modifications to distinct proteins generate disease-specific epitopes. Peptides whose production in vivo is pathology-related may therefore be used as biomarkers. In this respect, neoepitope peptides produced through pathology-specific proteolytic cleavage are particularly interesting, because proteolytic activity is well known to be modulated in a large number of pathologies [3, 68–70]. As the sarcolemma becomes permeable, smaller peptides produced upon proteolysis can leak into the circulation more easily than do intact proteins, making them measurable in serum and plasma or even urine [71].
Existing neo-epitope biomarkers
An example of this is the carboxy-terminal collagen crosslinked biomarker of collagen type I (CTX-I) cleaved by Cathepsin K, that has become one of the primary biomarkers of bone turnover [72, 73]. As collagen type I is the predominant matrix protein in bone and Cathepsin K is almost exclusively expressed by osteoclasts, the abundance of the fragment is tightly associated with total osteoclast activity, thus related to the process of bone loss/turnover. This feature enables the CTX-I assay to detect changes in bone loss much earlier than e.g. DXA. CTX-I has been successful at prognosing changes in the bone turnover [74].
On a similar note, recent research has shown that fragments of collagen produced through cleavage of MMP (Matrix metalloproteinase) or ADAMTS (A Disintegrin And Metalloproteinase) can function as biomarkers in conditions of aberrant ECM turnover [75]. The rationale is that altered abundance or activity of several MMP, ADAMTS, calpain and caspase proteases have been shown to be involved in protein turnover changes in many connective tissue pathologies. A range of proteolytic peptides derived from collagens 1 to 6 has shown to work as biomarkers of ankylosing spondylitis [76], OA [77], RA [78], kidney fibrosis [79], lung fibrosis [75] and liver fibrosis [80]. Also, some of these collagen peptide biomarkers as well as proteolytic fragments ofC-reactive protein (CRP), namely MMP-cleaved CRP (CRPM), have shown to be biomarkers of the tissue inflammation associated with arthritic conditions and the response to anti-inflammatory treatment in these conditions [25, 81]. Because of the shared molecular origins of these markers and the proteases involved in the previously described muscle pathologies there is a high likelihood that several of these biomarker can find use in neuromuscular disorders (Table 3).
Clinical experience and potential in muscle pathologies
An interesting neo-epitope describing fragmentation of the C-terminus of agrin by neurotrypsin in the neuromuscular junction has been recently presented [82]. The agrin fragment correlates with neurogenic sarcopenia. This marker aims at assessing the underlying condition at the neuromuscular junction, which correlates with the decline of muscle mass in males.
In clinical immobilization studies, changes of MMP-generated collagen VI product levels were found to be associated with muscle regrowth following immobilization [83]. Other MMPs and collagen combinations studied for liver or kidney fibrosis, including biomarkers as collagen I fragment generated by MMP -2, -9 and -13 (C1M) [84] or the MMP-9 proteolytically revealed neo-epitope of type III collagen, (C3M) [85] can be used as a biomarker relating to muscle protein turnover. Also, an immunoassay using antibodies against the collagen type III propeptide before the interaction with N-terminal proteases has been also proposed in assessing collagen III formation in liver fibrosis models [86].
Based on the progressive nature of fibrosis as the disease escalates (e.g. DMD, BMD), markers predicting fibrosis propensity by measuring the continuous synthesis/degradation processes of collagen in other tissues can be evaluated on their potential against muscle diseases.
As it has been shown that intracellular proteins such as creatine kinase and myoglobin can leak into the extracellular compartment and thus work as biomarkers of muscle disruption, it seems likely that other proteins or protein fragments from the muscle can enter the circulation. An MMP-degraded fragment of titin has been shown to be detectable in serum from healthy young men [87]. This indicates that metabolic byproducts from intracellular protein degradation can enter the circulation. Generated peptides can be possibly used as biomarkers of the myofibrillar proteins turnover in the muscle and thus provide information about ongoing muscle catabolism or anabolism. Indeed, serum Troponin I has been shown to be increased in dystrophic patients [88]. These intracellular proteins, particularly from the myofibrlliar fraction as they display the greatest degree of tissue specificity, show promise as biomarkers of muscle turnover.
POTENTIAL FOR NEOEPITOPE BIOMARKERS IN EXISTING AND EMERGING THERAPIES
As previously explained, myopathologies are either congenital or occult/idiopathic and in general there are currently no FDA-approved disease-modifying therapies. In most cases, treatment is symptomatic and aims at maintaining the self ambulatory ability for patients [60, 62]. Current scientific efforts are being directed towards several different treatment modalities, with the dominant approaches being based on anabolic/anticatabolic, anti-fibrotics or anti-inflammatory therapies, underlining the need for biomarkers monitoring these individual aspects of the diseases. The current pipeline for drugs targeting few of the most common causes of muscle wasting can be seen in Fig. 1.
While routine clinical biochemistry biomarkers provide measures of inflammation, e.g. IL-6, TNF-alpha or C-reactive protein, they are less well-suited for characterization of muscle fibrosis and catabolism. Since existing biomarkers have proven inadequate to detect subtle changes in the pathology, assessing treatment effects becomes challenging. A panel of varying peptide biomarkers of both formation and degradation could be of value in characterizing the fibrotic aspects of inflammatory myopathies and dystrophies and thereby contribute to the clinical characterization of subjects. Neoepitope biomarkers have demonstrated potential in describing these processes.
Antiinflammatory
In myositis, inflammation is part of the cause of the pathology, whereas in congenital dystrophies, it is a consequence of the chronic muscle disruption. In both cases, this contributes to symptoms and declined functionality, making it a relevant pathological mechanism to treat. Glucocorticoids have been shown to slow loss of locomotor function in DMD or even provide temporary improvements and is recommended as first-line treatment in both myositis and dystrophies [89–92] Biological anti-inflammatory drugs like inhibitors of TNF-alpha, IL-2, IL-6 or type I interferon have already shown promise in this regard in both inflammatory myopathies and dystrophies [93–97].
Likewise, in inflammatory myopathies, disproportionate activation of inflammatory pathways and increased proteolytic rates have been shown to be associated with overexpression of MMPs, localized in the muscle fibers [68, 98]. Muscle and tendonare weightbearing connective tissues with considerable expression and turnover of collagens, and inflammation in muscle pathologies is associated with increases in MMP activity. Thus, proteolyticfragments generated by proteases induced by inflammation-driven proteases, such as MMPs, appear to be good biomarker candidates. It has to be noted that some common medical interventions for those diseases (e.g. corticosteroids) could create a masking effect when trying to use collagen biomarkers some compounds induce cartilage resorption, tendon degradation or modify protein expression [99, 100]. Although it would create an extra burden in detecting significant differences in patient’s biomarker levels it could still be viable as an indicator within a larger biomarker panel.
Antifibrotic
Fibrosis is by definition the consequence of abnormal ECM turnover, resulting in excessive deposition of ECM proteins, especially collagens I, III and VI [101]. It has to be noted that the distribution of collagen isoforms in the various layers of the extracellular matrix of skeletal muscle tissues and the arrangement of the connective tissue associated with contractile fibers is extremely complex and differentially affected in muscle diseases [102]. Nevertheless, fragments of these constituents or their corresponding propeptides detected in circulation have been shown to reflect the extent of liver fibrosis and ECM remodeling in rats [84, 86, 103] and even demonstrate response to antifibrotic treatment [104, 105].
This has also been demonstrated as the case for myopathies and dystrophies, where endo- and perimysial fibrosis in the form of accumulated coallagen I, III, IV and fibronectin predominates [106, 107]. In DMD, endomysial fibrosis has been one of the strongest predictors of motor deterioration [31]. Therefore, early identification of loss in muscle strength or mass can be achieved by following closely the extent of fibrosis in the muscle tissue. Inhibiting the fibrotic processes may contribute to improved muscular function and this is the purpose of several current and previous pharmacologicaltrials.
Myostatin inhibitors and several putative antifibrotic treatment routes are being looked into. Previous preclinical and in vitro studies havedocumented that myostatin inhibitors downregulates markers of fibrosis, but this has not been shown conclusively in clinical models yet [108, 109].
The most advanced in terms of clinical trials is HT-100, a delayed-release Halofuginone preparation. It exerts its effect through inhibition of fibrosis and inflammation, in part through blocking of TGF-beta signaling [110], while it also directly inhibits collagen type I synthesis. At the time of writing it has initiated Phase II trials (by Akashi, NCT02525302) for DMD treatment.
Assays directed at monitoring changes in the aforementioned pathologic manifestations have been successful in providing information for developing therapies during antifibrotic clinical trials. Biomarker characterization in a Halo/Akashi trial of Halofuginone trial helped reveal fibrosis collagen turnover, incidentally through use of neoepitope collagen peptide fibrosis biomarkers, i.e. PINP and C3M [110] and can be applied in future clinical cohorts.
Anabolic
As loss of muscle tissue and associated muscle function is a trait of most myopathies, therapeutic approaches aimed at restoring functional muscle mass are under examination, with particular focus on pharmacological interventions. While the efficacy of exercise is questionable, anabolic drugs have shown some promise, although none of these have reached FDA approval yet.
Essentially, there are only two classes of drugs that display consistent myoanabolic properties and those are anabolic androgenic s and inhibitors of the endogenous cytokine myostatin. Inhibition of myostatin (also known as GDF-8) has been shown to result in muscle hypertrophy and myostatin inhibitor drugs are at the time of writing in clinical trials for myositis (Novartis’ bimagrumab in phase III, NCT01423110) and Duchenne (PF-06252616 for Pfizer, phase II, NCT02310763).
Several soluble biomarkers have already been shown to follow the degradation and formation processes in muscle remodeling [86]. Loss of muscle strength and reconstitution of muscle mass was described during an immobilization/remobilization study by measuring collagen VI turnover [83]. It has also been shown that circulating collagen III fragments measured in human plasma can define formation of muscle mass [86]. However, there is a scarcity of biomarkers of change in muscle mass, with stable isotope-based measurement of protein synthesis (and/or degradation) being the currently only viable method.
As discussed previously, myofibrillar proteins and protein fragments, such as the previously described MMP-cleaved Titin fragment, can enter the circulation and may show promise as biomarkers of muscle protein turnover and by extension, catabolism or anabolism. However, these peptide biomarkers have yet to show promise in terms of being biomarkers of anabolism, or catabolism and by extension anabolic treatment response.
FUTURE BIOMARKER DEVELOPMENT
While serological biomarkers of muscle mass would be of utility in characterization of myopathic or dystrophic patients in clinical care, biomarkers indicative of myoanablism or catabolism would be of great benefit in identifying at-risk populations in many different clinical scenarios and treatment efficacy in anabolic drug trials. Discovering peptide biomarkers of myoanabolism should be a priority to the biomarker research niche.
Suggested parent proteins for peptide fragment biomarkers could be proteins from the Dystrophin-associated protein complex (DAPC), endo- or perimysial ECM, the sarcolemma or the muscle contractile apparatus itself, i.e. sarcomeric proteins (Fig. 2).
If peptides exist that have multiple pathology-specific PTMs, this allows for higher specificity of the individual biomarker and possibly for further stratification capabilities. Therefore, identification of peptides from relevant parent proteins with secondary pathology-specific PTMs is important as this can give rise to more specific biomarkers. Relevant PTMs in this context could be increased oxidative nitrosylation or carbonylation, citrullination, crosslinking or cleavage mediated by proteases upregulated as part of a disease’s pathology, e.g. over expression of proteases. As these PTMs are related to the pathological process, (e.g. defective ECM remodeling), peptides containing them may translate into biomarkers that respond earlier to pathology changes in contrast to biomarkers that are related to the outcome of the process (e.g. imaging techniques or histopathology in the case of fibrosis). Indeed, this has been shown to work for other connective tissue pathologies like osteoporosis, rheumatoid arthritis, osteoarthritis and several types of organ fibrosis [111]. Hence, due to the inherent qualities of neoepitopes, there is potential in using them as prognostic biomarkers as well as in monitoring disease progression.
CONCLUSION
Dystrophies and inflammatory myopathies are serious diseases with limited treatment options. Despite the differences in the pathologies of thediseases, a common thread can be identified through muscle fiber degeneration, inflammation and/orfibrosis. Several routes for developing pharmacological interventions against one or all of these traits are being explored.
Neoepitope peptide biomarkers hold great potential in this respect as, they have been successful in characterizing localized pathological protein turnover, fibrosis and inflammation in a range of other diseases. With both dystrophies and myositis, more muscle fibres are being degraded and built up at any one time point, than in the healthy condition. This situation is dissimilar to the normal steady state turnover of muscle proteins and we hypothesize that this difference produces characteristic peptides derived from the muscle proteins that are measures of diseases activity.
Treatments are mainly targeted on dealing with the inflammation or fibrosis either directly(anti-inflammatory, anti-fibrotic) or indirectly (exon skipping). Suggested biomarkers (C1M, PINP etc.) have shown capability in describing and monitoring the extent of these clinical features. We believe that by using neoepitope biomarkers, improvedcharacterization of these traits could strengthen or speed up ongoing clinical trial efforts as they can contribute to disease progression monitoring, treatment efficacy and stratification/selection for participating patients. Assessment of a pharmaceutical entity’s potential will be much easier for both the industry as well as the medical personnel, leading to higher quality submissions to the regulatory agencies.
CONFLICT OF INTERESTS
The authors are involved in protein based biomarker research but are otherwise impartial to the technologies and approaches discussed.
REFERENCES
[1] | Mercuri E , Muntoni F . Muscular dystrophies. Lancet. (2013) ;381: :845–860. |
[2] | Reed P , Porter NC , Strong J , Pumplin DW , Corse AM , Luther PW , et al. Sarcolemmal reorganization in facioscapulohumeral muscular dystrophy. Ann Neurol. (2006) ;59: :289–297. |
[3] | Shin J , Tajrishi MM , Ogura Y , Kumar A . Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol. (2013) ;45: :2266–2279. |
[4] | Klingler W , Jurkat-Rott K , Lehmann-Horn F , Schleip R . The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. (2012) ;31: :184–195. |
[5] | Rouillon J , Poupiot J , Zocevic A , Amor F , Léger T , Garcia C , et al. Serum proteomic profiling reveals fragments of MYOM3 as potential biomarkers for monitoring the outcome of therapeutic interventions in muscular dystrophies. Hum Mol Genet. (2015) ;24: :4916–4932. |
[6] | Scharf G , Heineke J . Finding good biomarkers for sarcopenia. J Cachexia Sarcopenia Muscle. (2012) ;3: :145–148. |
[7] | Nedergaard A , Karsdal MA , Sun S , Henriksen K . Serological muscle loss biomarkers: An overview of current concepts and future possibilities. J Cachexia Sarcopenia Muscle. (2013) ;4: :1–17. |
[8] | Clark KA , McElhinny AS , Beckerle MC , Gregorio CC . Striated muscle cytoarchitecture: An intricate web of form and function. Annu Rev Cell Dev Biol. (2002) ;18: :637–706. |
[9] | Brown KJ , Marathi R , Fiorillo AA , Ciccimaro EF , Sharma S , Rowlands DS , et al. Accurate quantitation of dystrophin protein in human skeletal muscle using mass spectrometry. J Bioanal Biomed. (2012) ;Suppl 7: :1–16. |
[10] | Oak SA , Zhou YW , Harry W , Jarrett HW . Mechanisms of Signal Transduction: Skeletal Muscle Signaling Pathway through the Dystrophin Glycoprotein Complex and Skeletal Muscle Signaling Pathway through the Dystrophin Glycoprotein Complex and Rac1 *. J Biol chem. (2003) ;78: :39287. doi: 10.1074/jbc.M305551200 |
[11] | Zatz M , Rapaport D , Vainzof M , Passos-Bueno MR , Bortolini ER , Pavanello RDC , et al. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J Neurol Sci. (1991) ;102: :190–196. |
[12] | Hauerslev S , Sveen ML , Vissing J , Krag TO . Protein turnover and cellular stress in mildly and severely affected muscles from patients with limb girdle muscular dystrophy type 2I. PLoS One. (2013) ;8: :e66929. |
[13] | Laval SH , Bushby KMD . Limb-girdle muscular dystro-phies–from genetics to molecular pathology. Neuropathol Appl Neurobiol. (2004) ;30: :91–105. |
[14] | Kekou K , Fryssira H , Sophocleous C , Mavrou A , Manta P , Metaxotou C . Facioscapulohumeral muscular dystrophy molecular testing using a non radioactive protocol. Mol Cell Probes. (2005) ;9: :422–424. |
[15] | Hengstman GJ , van Venrooij WJ , Vencovsky J , Moutsopoulos HM , van Engelen BG . The relative prevalence of dermatomyositis and polymyositis in Europe exhibits a latitudinal gradient. Ann Rheum Dis. (2000) ;59: :141–142. |
[16] | Clapp J , Mitchell LM , Bolland DJ , Fantes J , Corcoran AE , Scotting PJ , et al. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am J Hum Genet. (2007) ;81: :264–279. |
[17] | Flanigan KM , Harper SQ . Facioscapulohumeral Dystrophy. In: Muscle Disease: Pathology and Genetics, Second edition. (2013) , pp 288–297. |
[18] | Selva O’Callaghan A , Trallero Araguás E . Inflammatory myopathies. Dermatomyositis, polymyositis, and inclusion body myositis. Reumatol Clin. (2008) ;4: :197–206. |
[19] | Hengstman GJD , De Bleecker JL , Feist E , Vissing J , Denton CP , Manoussakis MN , et al. Open-label trial of anti-TNF-alpha in dermato- and polymyositis treated concomitantly with methotrexate. Eur Neurol. (2008) ;59: :159–163. |
[20] | Ayoglu B , Chaouch A , Lochmüller H , Politano L , Bertini E , Spitali P , et al. Affinity proteomics within rare diseases: A BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol Med. (2014) ;6: :1–19. |
[21] | Scotton C , Passarelli C , Neri M , Ferlini A . Biomarkers in rare neuromuscular diseases. Exp Cell Res. (2013) ;1–6. |
[22] | Burch PM , Pogoryelova O , Goldstein R , Bennett D , Guglieri M , Straub V , et al. Muscle-derived proteins as serum biomarkers for monitoring disease progression in three forms of muscular dystrophy. J Neuromuscul Dis. (2015) ;2: :241–255. |
[23] | Chen Y-W , Nagaraju K , Bakay M , McIntyre O , Rawat R , Shi R , et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. (2005) ;65: :826–834. |
[24] | Haslett JN , Sanoudou D , Kho AT , Bennett RR , Greenberg SA , Kohane IS , et al. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci U S A. (2002) ;99: :15000–15005. |
[25] | Bay-jensen AC , Byrjalsen I , Siebuhr AS . Serological biomarkers of joint tissue turnover predict tocilizumab response at baseline. J Clin Rheumatol. (2014) ;20: :332–335. |
[26] | van Spil WE , DeGroot J , Lems WF , Oostveen JCM , Lafeber FPJG . Serum and urinary biochemical markers for knee and hip-osteoarthritis: A systematic review applying the consensus BIPED criteria. Osteoarthritis Cartilage. (2010) ;18: :605–612. |
[27] | Fukushima K , Nakamura A , Ueda H , Yuasa K , Yoshida K , Takeda S , et al. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (CXMDJ). BMC Musculoskelet Disord. (2007) ;8: :54. |
[28] | Li H , Mittal A , Makonchuk DY , Bhatnagar S , Kumar A . Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. (2009) ;18: :2584–2598. |
[29] | Gosselin LE , Williams JE , Personius K , Farkas GA . A comparison of factors associated with collagen metabolism in different skeletal muscles from dystrophic (mdx) mice: Impact of pirfenidone. Muscle Nerve. (2007) ;35: :208–216. |
[30] | Tandon A , Villa CR , Hor KN , Jefferies JL , Gao Z , Towbin JA , et al. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in duchenne muscular dystrophy. J Am Heart Assoc. (2015) ;4: . doi: 10.1161/JAHA.114.001338 |
[31] | Desguerre I , Mayer M , Leturcq F , Barbet J-P , Gherardi RK , Christov C . Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol. (2009) ;68: :762–773. |
[32] | Gillies AR , Lieber RL . Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve. (2011) ;44: :318–331. |
[33] | Haus JM , Carrithers JA , Trappe SW , Trappe TA . Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol. (2007) ;47306: :2068–2076. |
[34] | Chen X , Li Y . Role of matrix metalloproteinases in skeletal muscle. Cell Adhes Migr. (2009) ;3: :337–341. |
[35] | Rennie M , Edwards R , Griggs R , Matthews S , Read M , Matthews D , et al. Total protein-turnover and muscle protein-synthesis by stable isotope techniques in duchenne muscular-dystrophy. Eur J Clin Invest. (1982) ;12: :32–34. |
[36] | Sohar I , Laszlo A , Gaal K , Mechler F . Cysteine and metalloproteinase activities in serum of Duchenne muscular dystrophic genotypes. Biol Chem Hoppe Seyler. (1988) ;369: (Suppl):277–279. |
[37] | von Moers A , Zwirner A , Reinhold A , Brückmann O , van Landeghem F , Stoltenburg-Didinger G , et al. Increased mRNA expression of tissue inhibitors of metalloproteinase-1 and -2 in Duchenne muscular dystrophy. Acta Neuropathol. (2005) ;109: :285–293. |
[38] | Nadarajah VD , van Putten M , Chaouch A , Garrood P , Straub V , Lochmüller H , et al. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in Duchenne muscular dystrophy (DMD). Neuromuscul Disord. (2011) ;21: :569–578. |
[39] | Kumar A , Bhatnagar S , Kumar A . Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am J Pathol. (2010) ;177: :248–260. |
[40] | Stevenson EJ , Giresi PG , Koncarevic A , Kandarian SC . Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J Physiol. (2003) ;551: :33–48. |
[41] | Taillandier D , Aurousseau E , Meynial-Denis D , Bechet D , Ferrara M , Cottin P , et al. Coordinate activation of lysosomal, Ca 2+-activated and ATP-ubiquitin-dependent proteinases in the unweighted rat soleus muscle. Biochem J. (1996) ;316: (Pt 1):65–72. |
[42] | Goll DE , Thompson VF , Li H , Wei W , Cong J . The Calpain System. Physiol Rev. (2003) ;1990: :731–801. |
[43] | Hauerslev S , Sveen M-L , Duno M , Angelini C , Vissing J , Krag TO . Calpain 3 is important for muscle regeneration: Evidence from patients with limb girdle muscular dystrophies. BMC Musculoskelet Disord. (2012) ;13: :43. |
[44] | Ojima K , Kawabata Y , Nakao H , Nakao K , Doi N , Kitamura F , et al. Dynamic distribution of muscle-specific calpain in mice has a key role in physical-stress adaptation and is impaired in muscular dystrophy. J Clin Invest. (2010) ;120: :2672–2683. |
[45] | Jackman RW , Kandarian SC . The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol.C. (2004) ;287: :834–843. |
[46] | Aronson JK . Biomarkers and surrogate endpoints. Br J Clin Pharmacol. (2005) ;59: :491–494. |
[47] | Bauer DC , Hunter DJ , Abramson SB , Attur M , Corr M , Felson D , et al. Classification of osteoarthritis biomarkers: A proposed approach. Osteoarthritis Cartilage. (2006) ;14: :723–727. |
[48] | FDA report. Challenge and opportunity on the critical path to new medical products. US Dep Heal Hum Serv Food drug Adm. (2004) . |
[49] | Trollet C , Athanasopoulos T , Popplewell L , Malerba A , Dickson G . Gene therapy for muscular dystrophy: Current progress and future prospects. Expert Opin Biol Ther. (2009) ;9: (7):849–866. |
[50] | Schwartz M , Hertz JM , Sveen ML , Vissing J . LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology. (2005) ;64: :1635–1637. |
[51] | Bonne G , Leturcq F , Ben YR . Emery-Dreifuss Muscular Dystrophy. Gene Rev. (2004) . http://www.ncbi.nlm.nih.gov/books/NBK1436/. (accessed 22 Apr. 2015). |
[52] | Gong QY , Phoenix J , Kemp GJ , García-Fiñana M , Frostick SP , Brodie DA , et al. Estimation of body composition in muscular dystrophy by MRI and stereology. J Magn Reson Imaging. (2000) ;12: :467–475. |
[53] | Kampschulte M , Schneider CR , Litzlbauer H , Tscholl C , Schneider D , Zeiner C , et al. Quantitative 3D micro-CT imaging of human lung tissue quantitative 3-D-mikro-CT-bildgebung von humanem lungengewebe. Exp Radiol. (2013) ;869–876. |
[54] | Damilakis J , Adams JE , Guglielmi G , Link TM . Radiation exposure in X-ray-based imaging techniques used in osteoporosis. Eur Radiol. (2010) ;20: :2707–2714. |
[55] | Levine JA , Abboud L , Barry M , Reed JE , Sheedy PF , Jensen MD . Measuring leg muscle and fat mass in humans: Comparison of CT and dual-energy X-ray absorptiometry elderly patients Measuring leg muscle and fat mass in humans: Comparison of CT and dual-energy X-ray absorptiometry. J Appl Physiol. (2014) ;88: :452–456. |
[56] | Visser M , Fuerst T , Lang T , Salamone L , Tamara B . Validity of fan-beam dual-energy X-ray absorptiometry for measuring fat-free mass and leg muscle mass. J Appl Physiol. (2014) ;87: :452–456. |
[57] | Clark BC , Cook SB , Ploutz-Snyder LL . Reliability of techniques to assess human neuromuscular function in vivo . J Electromyogr Kinesiol. (2007) ;17: :90–101. |
[58] | Lee RC , Wang Z , Heymsfield SB . Skeletal muscle mass and aging: Regional and whole-bod measurment methods. Can J Appl Physiol. (2001) ;26: :102–122. |
[59] | McDonald CM , Henricson EK , Abresch RT , Florence JM , Eagle M , Gappmaier E , et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: Longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. (2013) ;48: :343–356. |
[60] | Witting N , Duno M , Vissing J . Deletion of exon 26 of the dystrophin gene is associated with a mild Becker muscular dystrophy phenotype. Acta Myol. (2011) ;30: :182–184. |
[61] | Cardamone M , Darras BT , Ryan MM . Inherited myopathies and muscular dystrophies. Semin Neurol. (2008) ;28: :250–259. |
[62] | Witting N , Duno M , Petri H , Krag T , Bundgaard H , Kober L , et al. Anoctamin 5 muscular dystrophy in Denmark: Prevalence, genotypes, phenotypes, cardiac findings, and muscle protein expression. J Neurol. (2013) ;260: :2084–2093. |
[63] | Jeanson-Leh L , Lameth J , Krimi S , Buisset J , Amor F , Le Guiner C , et al. Serum profiling identifies novel muscle miRNA and cardiomyopathy-related miRNA biomarkers in golden retriever muscular dystrophy dogs and duchenne muscular dystrophy patients. Am J Pathol. (2014) ;184: :2885–2898. |
[64] | Li X , Li Y , Zhao L , Zhang D , Yao X , Zhang H , et al. Circulating muscle-specific miRNAs in duchenne muscular dystrophy patients. Mol Ther Nucleic Acids. (2014) ;3: :e177. |
[65] | Zaharieva IT , Calissano M , Scoto M , Preston M , Cirak S , Feng L , et al. Dystromirs as serum biomarkers for monitoring the disease severity in Duchenne muscular dystrophy. PLoS One. (2013) :8: . doi: 10.1371/journal.pone.0080263 |
[66] | Mizuno H , Nakamura A , Aoki Y , Ito N , Kishi S , Yamamoto K , et al. Identification of muscle-specific MicroRNAs in serum of muscular dystrophy animal models: Promising novel blood-based markers for muscular dystrophy. PLoS One. (2011) ;6: :14–19. |
[67] | Coenen-Stass AML , McClorey G , Manzano R , Betts CA , Blain A , Saleh AF , et al. Identification of novel, therapy-responsive protein biomarkers in a mouse model of Duchenne muscular dystrophy by aptamer-based serum proteomics. Sci ReNature Publishing Group. (2015) ;5: :17014. |
[68] | Ahtikoski AM , Tuominen H , Korpelainen JT , Takala TES , Oikarinen A . Collagen synthesis and degradation in polyneuropathy and myopathies. Muscle Nerve. (2004) ;30: :602–608. |
[69] | Holmberg C , Ghesquière B , Impens F , Gevaert K , Kumar JD , Cash N , et al. Mapping proteolytic processing in the secretome of gastric cancer-associated myofibroblasts reveals activation of MMP-1, MMP-2, and MMP-3. J Proteome Res. (2013) ;12: :3413–2422. |
[70] | Brundel BJJM , Ausma J , Van Gelder IC , Van Der Want JJL , Van Gilst WH , Crijns HJGM , et al. Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc Res. (2002) ;54: :380–389. |
[71] | Allen DG , Whitehead NP . Duchenne muscular dystrophy–what causes the increased membrane permeability in skeletal muscle? Int J Biochem Cell Biol. (2011) ;43: :290–294. |
[72] | Rosen HN , Moses AC , Garber J , Iloputaife ID , Ross DS , Lee SL , et al. Serum CTX: A new marker of bone resorption that shows treatment effect more often than other markers because of low coefficient of variability and large changes with bisphosphonate therapy. Calcif Tissue Int. (2000) ;66: :100–103. |
[73] | Rosenquist C , Fledelius C , Christgau S , Pedersen BJ , Bonde M , Qvist P , et al. Serum CrossLaps One Step ELISA. First application of monoclonal antibodies for measurement in serum of bone-related degradation products from C-terminal telopeptides of type I collagen. Clin Chem. (1998) ;44: :2281–2289. |
[74] | Garnero P , Ferreras M , Karsdal MA , Nicamhlaoibh R , Risteli J , Borel O , et al. The type I collagen fragments ICTP and CTX reveal distinct enzymatic pathways of bone collagen degradation. J Bone Miner Res. (2003) ;18: :859–867. |
[75] | Leeming , Leeming , Sand JM , Nielsen MJ , Genovese , Martinez , et al. Serological investigation of the collagen degradation profile of patients with chronic obstructive pulmonary disease or idiopathic pulmonary fibrosis. Biomark Insights. (2012) ;119. |
[76] | Bay-Jensen A-C , Wichuk S , Byrjalsen I , Karsdal MA , Maksymowych WP . Two novel diagnostic biomarkers of cartilage degradation and connective tissue inflammation are predictive of radiographic disease progression. Osteoarthr Cartil. (2012) ;20: :S90. |
[77] | Bay-Jensen AC , Liu Q , Byrjalsen I , Li Y , Wang J , Pedersen C , et al. Enzyme-linked immunosorbent assay (ELISAs) for metalloproteinase derived type II collagen neoepitope, CIIM-Increased serum CIIM in subjects with severe radiographic osteoarthritis. Clin Biochem. (2011) ;44: :423–429. |
[78] | Bay-Jensen AC , Karsdal MA , Vassiliadis E , Wichuk S , Marcher-Mikkelsen K , Lories R , et al. Circulating citrullinated vimentin fragments reflect disease burden in ankylosing spondylitis and have prognostic capacity for radiographic progression. Arthritis Rheum. (2013) ;65: :972–980. |
[79] | Genovese F , Manresa AA , Leeming DJ , Karsdal MA , Boor P . The extracellular matrix in the kidney: A source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair. (2014) ;7: :4. |
[80] | Vassiliadis E , Oliveira CP , Alvares-da-Silva MR , Zhang C , Carrilho FJ , Stefano JT , et al. Circulating levels of citrullinated and MMP-degraded vimentin (VICM) in liver fibrosis related pathology. Am J Transl Res. (2012) ;4: :403–414. |
[81] | Siebuhr A , Bay-Jensen AC , Leeming DJ , Plat A , Byrjalsen I , Christiansen C , et al. Serological identification of fast progressors of structural damage with rheumatoid arthritis. Arthritis Res Ther. (2013) ;15: :R86. |
[82] | Drey M , Sieber CC , Bauer JM , Uter W , Dahinden P , Fariello RG , et al. C-terminal Agrin Fragment as a potential marker for sarcopenia caused by degeneration of the neuromuscular junction. Exp Gerontol. (2013) ;48: :76–80. |
[83] | Nedergaard A , Sun S , Karsdal MA , Henriksen K , Kjær M , Lou Y , et al. Type VI collagen turnover-related peptides-novel serological biomarkers of muscle mass and anabolic response to loading in young men. J Cachexia Sarcopenia Muscle. (2013) ;4: :267–275. |
[84] | Leeming D , He Y , Veidal S , Nguyen Q , Larsen D , Koizumi M , et al. A novel marker for assessment of liver matrix remodeling: An enzyme-linked immunosorbent assay (ELISA) detecting a MMP generated type I collagen neo-epitope (C1M). Biomarkers. (2011) ;16: :616–628. |
[85] | Barascuk N , Veidal SS , Larsen L , Larsen D V , Larsen MR , Wang J , et al. A novel assay for extracellular matrix remodeling associated with liver fibrosis: An enzyme-linked immunosorbent assay (ELISA) for a MMP-9proteolytically revealed neo-epitope of type III collagen. Clin Biochem. (2010) ;43: :899–904. |
[86] | Nielsen MJ , Nedergaard AF , Sun S , Veidal SS , Larsen L , Zheng Q , et al. The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am J Transl Res. (2013) ;5: :303–315. |
[87] | Sun S , Henriksen K , Karsdal MA , Armbrecht G , Belavý DL , Felsenberg D , et al. Measurement of a MMP-2 degraded Titin fragment in serum reflects changes in muscle turnover induced by atrophy. Exp Gerontol. (2014) ;58: :83–89. |
[88] | Hathout Y , Brody E , Clemens PR , Cripe L , DeLisle RK , Furlong P , et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci. (2015) ;112: :201507719. |
[89] | Manzur AY , Kuntzer T , Pike M , Swan A . Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. (2004) ;CD003725. |
[90] | Micks RH , Mullaney J . Dermatomyositis successfully treated by prednisone. Ir J Med Sci. (1958) ;333–334. |
[91] | van de Vlekkert J , Hoogendijk JE , de Haan RJ , Algra A , van der Tweel I , van der Pol WL , et al. Oral dexamethasone pulse therapy versus daily prednisolone in sub-acute onset myositis, a randomised clinical trial. Neuromuscul Disord. (2010) ;20: :382–389. |
[92] | Ramanan AV , Campbell-Webster N , Ota S , Parker S , Tran D , Tyrrell PN , et al. The effectiveness of treating juvenile dermatomyositis with methotrexate and aggressively tapered corticosteroids. Arthritis Rheum. (2005) ;52: :3570–3578. |
[93] | Aggarwal R , Oddis CV . Therapeutic advances in myositis. Curr Opin Rheumatol. (2012) ;24: :635–641. |
[94] | Okiyama N , Sugihara T , Iwakura Y , Yokozeki H , Miyasaka N , Kohsaka H . Therapeutic effects of interleukin-6 blockade in a murine model of polymyositis that does not require interleukin-17A. Arthritis Rheum. (2009) ;60: :2505–2512. |
[95] | Efthimiou P . Tumor necrosis factor-alpha in inflammatory myopathies: Pathophysiology and therapeutic implications. Semin Arthritis Rheum. (2006) ;36: :168–172. |
[96] | Hengstman GJD , van den Hoogen FHJ , Barrera P , Netea MG , Pieterse A , van de Putte LBA , et al. Successful treatment of dermatomyositis and polymyositis with anti-tumor-necrosis-factor-alpha: Preliminary observations. Eur Neurol. (2003) ;50: :10–15. |
[97] | Mammen AL , Sartorelli V . IL-6 Blockade as a therapeutic approach for duchenne muscular dystrophy. Ebio Medicine. (2015) ;2–3. |
[98] | Evans WJ . Skeletal muscle loss: Cachexia, sarcopenia, and inactivity 1–3. (2010) ;91: :1123–1127. |
[99] | Wei AS , Callaci JJ , Juknelis D , Marra G , Tonino P , Freedman KB , et al. The effect of corticosteroid on collagen expression in injuredrotator cuff tendon. J bone Jt surg Am. (2011) ;88: :1331–1338. |
[100] | Ay M , Kuntzer T , Pike M , Av S . Glucocorticoid corticosteroids for Duchenne muscular dystrophy (Review). Cochrane Collab (2009) ;1 http://www.ncbi.nlm.nih.gov/pubmed/18254031 |
[101] | Gressner AM , Weiskirchen R . Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J Cell Mol Med. (2006) ;10: :76–99. |
[102] | Lieber RL , Ward SR . Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am J Physiol Cell Physiol. (2013) ;305: :C241–C252. |
[103] | Veidal SS , Vassiliadis E , Bay-Jensen A-C , Tougas G , Vainer B , Karsdal MA . Procollagen type I N-terminal propeptide (PINP) is a marker for fibrogenesis in bile duct ligation-induced fibrosis in rats. Fibrogenesis Tissue Repair. (2010) ;3: :5. |
[104] | Schierwagen R , Leeming DJ , Klein S , Granzow M , Nielsen MJ , Sauerbruch T , et al. Serum markers of the extracellular matrix remodeling reflect antifibrotic therapy in bile-duct ligated rats. Front Physiol. (2013) . doi: 10.3389/fphys.2013.00195 |
[105] | Zhou L , Lu H . Targeting fibrosis in Duchenne muscular dystrophy. J Neuropathol Exp Neurol. (2010) ;69: :771–776. |
[106] | Duance VC , Stephens HR , Dunn M , Bailey AJ , Dubowitz V . A role for collagen in the pathogenesis of muscular dystrophy? Nature. (1980) ;284: :470. |
[107] | Foidart M , Foidart JM , Engel WK . Collagen localization in normal and fibrotic human skeletal muscle. Arch Neurol. (1981) ;38: :152–157. |
[108] | Zhao BL , Kollias HD , Wagner KR . Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. (2008) ;283: :19371–19378. |
[109] | Artaza JN , Singh R , Ferrini MG , Braga M , Tsao J , Gonzalez-Cadavid NF . Myostatin promotes a fibrotic phenotypic switch in multipotent C3H 10T1/2 cells without affecting their differentiation into myofibroblasts. J Endocrinol. (2008) ;196: :235–249. |
[110] | Zion O , Genin O , Kawada N , Yoshizato K , Roffe S , Nagler A , et al. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas. (2009) ;38: :427–435. |
[111] | Karsdal MA , Henriksen K , Leeming DJ , Woodworth T , Vassiliadis E , Bay-Jensen A-C . Novel combinations of Post-Translational Modification (PTM) neo-epitopes provide tissue-specific biochemical markers–are they the cause or the consequence of the disease? Clin Biochem. (2010) ;43: :793–804. |
[112] | Cynthia Martin F , Hiller M , Spitali P , Oonk S , Dalebout H , Palmblad M , et al. Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics - Clin Appl. (2014) ;8: :269–278. |
[113] | Norwood FLM , Harling C , Chinnery PF , Eagle M , Bushby K , Straub V . Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain. (2009) ;132: :3175–3186. |
[114] | Van der Kooi AJ , Barth PG , Busch HF , de Haan R , Ginjaar HB , van Essen AJ , et al. The clinical spectrum of limb girdle muscular dystrophy. A survey in The Netherlands. Brain. (1996) ;119: (Pt 5):1471–1480. |
[115] | Cooper D , Upadhhyaya M . Facioscapulohumeral Muscular Dystrophy (FSHD): Clinical Medicine and Molecular Cell Biology. (2004) . |
[116] | Tawil R , van der Maarel SM , Tapscott SJ . Facioscapulohumeral dystrophy: The path to consensus on pathophysiology. Skelet Muscle. (2014) ;4: :12. |
[117] | Choy EHS , Isenberg DA . Treatment of dermatomyositis and polymyositis. Rheumatology (Oxford). (2002) ;41: :7–13. |
[118] | Orphanet. Orphanet Report Series Prevalence of rare diseases: Bibliographic data. Rare Dis. (2014) ;1–29. |
[119] | Bernatsky S , Joseph L , Pineau CA , Bélisle P , Boivin JF , Banerjee D , et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: Age, sex and regional differences. Ann Rheum Dis. (2009) ;68: :1192–1196. |
[120] | Needham M , Corbett A , Day T , Christiansen F , Fabian V , Mastaglia FL . Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci. (2008) ;15: :1350–1353. |
[121] | Böhm J , Vasli N , Malfatti E , Le Gras S , Feger C , Jost B , et al. An integrated diagnosis strategy for congenital myopathies. PLoS One. (2013) ;8: :e67527. |
[122] | McNeil SM , Woulfe J , Ross C , Tarnopolsky MA . Congenital inflammatory myopathy: A demonstrative case and proposed diagnostic classification. Muscle Nerve. (2002) ;259–264. |
[123] | Vassiliadis E , Veidal SS , Barascuk N , Mullick JB , Clausen RE , Larsen L , et al. Measurement of matrix metalloproteinase 9-mediated collagen type III degradation fragment as a marker of skin fibrosis. BMC Dermatol. (2011) ;11: :6. |
[124] | Rouillon J , Zocevic A , Leger T , Garcia C , Camadro J-M , Udd B , et al. Proteomics profiling of urine reveals specific titin fragments as biomarkers of Duchenne muscular dystrophy. Neuromuscul Disord. (2014) ;24: :563–573. |
Figures and Tables
Fig.1
Ongoing clinical trials for DMD/BMD and LGMD diseases. The current stage at the time of writing for the most advanced in each category is illustrated. Indicatively “Exon skipping” drugs is a promising class, in which Drisapersen/PRO051 (Prosensa), Eteplirsen/AVI-4658 (Sarepta) are currently in phase III [65] Ataluren/PTC-124 has received marketing approval in Europe (but not in US) under the name Translarna mda.gov.
![Ongoing clinical trials for DMD/BMD and LGMD diseases. The current stage at the time of writing for the most advanced in each category is illustrated. Indicatively “Exon skipping” drugs is a promising class, in which Drisapersen/PRO051 (Prosensa), Eteplirsen/AVI-4658 (Sarepta) are currently in phase III [65] Ataluren/PTC-124 has received marketing approval in Europe (but not in US) under the name Translarna mda.gov.](https://content.iospress.com:443/media/jnd/2016/3-3/jnd-3-3-jnd160150/jnd-3-jnd160150-g001.jpg)
Fig.2
Anatomy of the muscle structure: The extracellular matrix, the sarcolemma and the intracellular domains are illustrated, depicting the relations between the different constituents. Proteins that are affected by mutations or deletions have the respective disease indicated in parentheses.
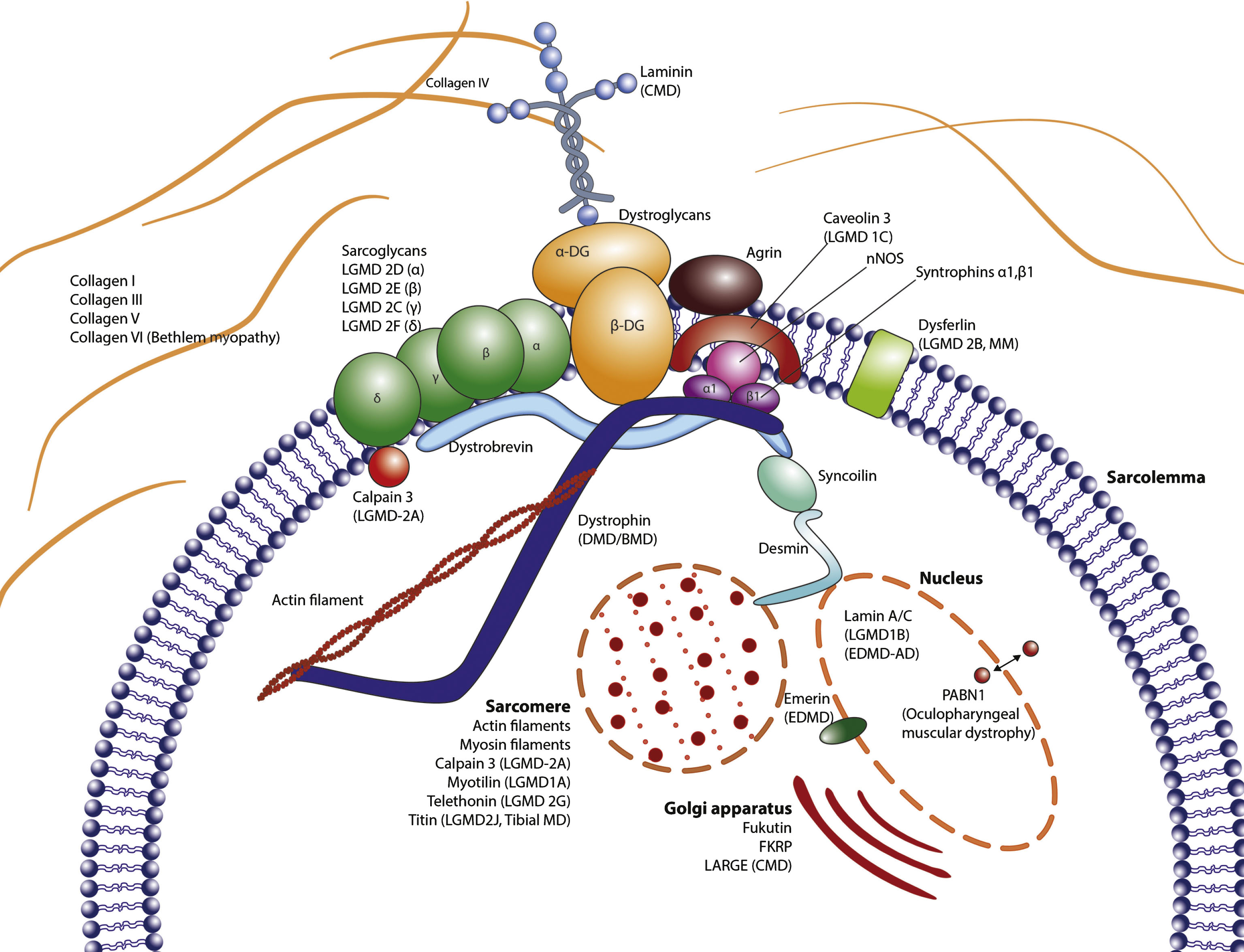
Table 1
Prevalence of Myopathies and Dystrophies; histological and biochemical disease characteristics
| Condition | Biochemical imbalance | Phenotype | Prevalence per 100.000 | References |
| Muscular dystrophies | ||||
| Duchenne Muscular Dystrophy (DMD) | Increased CK, MMP1, MMP2, MMP7, MMP9, fibronectin Cathepsins H and L | Fibrosis, inflammation, muscle wasting, fat substitution | 8–29 (males) | [1, 34, 36, 37, 112] |
| Becker Muscular Dystrophy (BMD) | 7–29 (males) | |||
| Limb-Girdle Muscular Dystrophy (LGMD) | Heterogenous (absence of proteins, dysfunctional interactions etc.) | Atrophy, endomysial fibrosis, inflammation in some cases | 0.8–2.3 | [43, 113, 114] |
| Facioscapulohumeral dystrophy | DUX4 expression in muscle | Fibrosis, perivascular infiltration, endomysial inflammation | 5 | [14, 17, 115, 116] |
| Inflammatory Myopathies | ||||
| Polymyositis (PM) | Pro-/inflammatory markers increased | Acute inflammatory onset (responsive to immunomodulation), muscle fiber atrophy, lung fibrosis | 6.3-7.1 | [117–119] |
| Dermatomyositis (DM) | 6.3 | |||
| Inclusion Body Myositis (IBM) | Chronic inflammation, muscle fiber atrophy, lung fibrosis (rare) | 1.5 (general population) 5.1 (people >50 years ol d) | [120] | |
| Congenital myopathies | Heterogeneous | Central nucleated fibers, Type I muscle fiber prevalence. rare inflammation | 6 (per live births) | [121, 122] |
Table 2
BIPED criteria for muscular dystrophy/myopathy. Adapted description of the BIPED criteria from the initially proposed by the osteoarthritis Network [47]
| Burden of disease | Investigative | Prognostic | Efficacy | Diagnostic | |
| Definition | Biomarker associated with the extent or severity of muscle loss | Biomarker not meeting criteria for another category | Predicts onset or progression | Indicative of treatment efficacy | Differentiates diseased groups from non-diseased |
| Subjects | Must manifest muscular dystrophy/myopathy | NA | With and/or without diagnosed muscular dystrophy/myopathy1 | With muscular dystrophy/myopathy | With and or without muscular dystrophy/myopathy |
| Design | Cross-sectional, case control | NA | Longitudinal | Controlled trial | Cross-sectional or case-control |
| Outcomes | Extent of severity of muscular dystrophy/myopathy | NA | New or worsening muscular dystrophy/myopathy | New or ameliorated muscular dystrophy/myopathy | Muscolar dystrophy vs no muscular dystrophy/myopathy |
| Criteria | Significant association between marker and extent or severity of muscular dystrophy/myopathy | NA | Significant association between marker and onset or progression of muscular dystrophy/myopathy | Significant association between marker and treatment effect | Significant association between marker and muscular dystrophy/myopathy diagnosis |
| Examples | Creatine Kinase, strength tests, biopsy | None or very limited selection | Muscle mass, strength, endurance | DNA test, biopsy histochemistry, imaging |
Table 3
Neoepitope biomarkers with relevance to neuromuscular disorders covering a spectrum of processes connected to the disease
| Type of marker | Related to process | Application in muscle | Refs |
| C1M, C3M | Collagen 1,3 degradation | Inflammation in muscle tissue | [123] |
| PINP, C1M | Collagen I synthesis, ECM remodeling | Fibrosis | [84, 103] |
| CAF | Agrin fragmentation | Functional disintegration in neuromuscular | [82] |
| junction, sarcopenia | |||
| Titin | Muscle metabolism | Muscle turnover | [87, 124] |


