
Therapeutic Potential of Tricyclo-DNA antisense oligonucleotides
Abstract
Oligonucleotide therapeutics hold great promise for the treatment of various diseases and the antisense field is constantly gaining interest due to the development of more potent and nuclease resistant chemistries. Despite a rather low success rate with only three antisense drugs being clinically approved, the frontiers of AON therapeutic applications have increased over the past three decades and continue to expand thanks to a steady increase in understanding the mechanisms of action of these molecules, progress in chemical modification and delivery.
In this review, we will examine the recent advances obtained with the tricyclo-DNA chemistry which displays unique pharmacological properties and unprecedented uptake in many tissues after systemic administration. We will review their specific properties and their therapeutic applications mainly for neuromuscular disorders, including exon-skipping for Duchenne muscular dystrophy and exon-inclusion for spinal muscular atrophy, but also aberrant splicing correction for Pompe disease. Finally, we will discuss their advantages and potential limitations, with a focus on the need for careful toxicological screen early in the process of AON drug development.
INTRODUCTION
The development of the antisense oligonucleotide (AON)-based approach started with the work of Zamecnik and Stephenson in 1978 when oligonucleotides were used as tools to downregulate the expression of specific genes [1]. The earliest strategies aimed to modulate gene expression through inhibition of protein translation via steric hindrance of translation initiation sites within the 5′ untranslated region of the targeted gene. AONs were originally unmodified synthetic DNA complementary to the targeted mRNA but it rapidly became evident that chemical modifications to protect AONs from nuclease degradation were necessary. Since the natural phosphodiester internucleotide linkage is highly sensitive to nucleases, it was the obvious site for chemical modification. Various backbones have been introduced over the years but it is interesting to note that the very first one, the phosphorothiate (PS) linkage is still the most widely used backbone modification in therapeutic AONs. The other prime site for chemical modification is the 2′-position in the sugar moiety and it has been widely used in the antisense field. This includes modifications such as 2′-O-methyl (2′OMe), 2′-O-methoxyethyl (2′O-MOE), 2′-fluorinated (2′F) and 2′-O-aminopropyl analogs. Incorporation of these 2′-modified sugars considerably increases binding affinity and pharmacokinetics of AONs over their 2′-deoxy counterparts. Similar characteristics are found in oligonucleotides with backbones based on Phosphorodiamidate morpholino oligomers (PMO), peptide nucleic acid (PNA), locked nucleic acid (LNA), phosphoramidate and methyl-phophonate derivatives (Fig. 1). These advances in the development of antisense chemistries have led to numerous studies investigating the therapeutic potential of antisense technology and clinical applications (for review see [2]). AONs offer therapeutic options in several ways, including the classical targeted gene knockdown, achieved through the recruitment of RNAse H1 to degrade mRNA at sites of DNA/RNA hybridization upon AON binding. For this purpose however, most AONs containing chemical modifications need to be designed as “gapmers” with a DNA core (or “gap”) to induce RNAse H activity flanked by nuclease resistant-modified ends to improve drug stability and tissue distribution. Fully modified AONs on the other hand can be used for other applications which have rapidly gained attention over the last two decades, amongst others the antisense mediated elimination of mRNA toxicity or the manipulation of alternative splicing where the antisense molecules are also called splice switching oligonucleotides (SSO). In this context, oligonucleotides can be used to modulate the ratio of splicing variants or correct splicing defects, by inducing exon-skipping or exon-inclusion, which opened far reaching implications in the treatment of a variety of diseases. The field has progressed very quickly over the last few years and promising results have been achieved, leading to several clinical evaluations.
Notably, most of these advances have been made for neuromuscular disorders (NMDs) such as Duchenne muscular dystrophy (DMD) using exon-skipping, Spinal muscular atrophy (SMA) using exon re-inclusion or aberrant splicing correction for pompe disease. In this review, we focus on the recent pre-clinical developments using the tricyclo-DNA chemistry which displays unique pharmacological properties and unprecedented uptake in many tissues after systemic administration. We will examine their therapeutic potential in the context of NMDs such as DMD, SMA and the Pompe disease and discuss the promises and challenges of this chemistry.
PHYSICOCHEMICAL PROPERTIES OF TC-DNA
Tricyclo-(tc)-DNA has been designed in the late nineties as a conformationally constrained DNA analogue (Fig. 1) [3, 4]. It is thus a second generation analogue of bicyclo-(bc)-DNA which has been introduced as the first member of the class of conformationally constrained DNA analogues in 1993 [5]. The basic concept behind conformational restriction consists in reducing the torsional flexibility of the sugar phosphate backbone within the repeating nucleotide units, thereby structurally preorganizing the single strand for duplex formation. This reduces the entropy of duplex formation and leads thus to more stable duplexes.
Fully modified tc-DNA containing all four natural nucleobases reveal increases in thermal stability of duplexes with complementary DNA by ca 1.2°C/mod and with complementary RNA of ca 2.4°C/mod [6]. The fidelity of Watson-Crick base-pair formation is slightly increased as compared to natural DNA. As expected the driving force for the increased thermodynamic stability of duplexes is a more favourable entropy term as determined from Tm vs concentration variation experiments. Tc-DNA residues show the highest stabilization if used as fully modified oligonucleotides. Single tc-DNA units do not mix very well with the DNA backbone and even less with the RNA backbone, due to energetic penalties arising from structural dissimilarities at the backbone junction sites [7]. However, tc-DNA can efficiently be used as gapmers as we will see below.
Structurally, tc-DNA is rather an RNA than a DNA-analogue as it prefers an overall A-like duplex conformation with complementary DNA and RNA. This has been demonstrated by molecular modelling and by CD-spectroscopy and could very recently be confirmed by a high resolution NMR structure of a fully modified tc-DNA/RNA duplex (unpublished data).
Fully modified tc-DNA in its phosphate form is highly stable against nuclease activity in murine and human serum and does not elicit RNaseH activity [8]. The latter fact can easily be explained by the preference of tc-DNA for an RNA like structure. Even tc-DNA gapmers with 5 nucleotide tc-DNA wings and a core of 8 natural DNA nucleotides in the center of the sequence are still highly stable in human serum. As expected, such gapmers are RNaseH competent due to the natural DNA window [9].
As common for the 2′-O-alkyl-RNAs or LNAs, tc-DNA can also be prepared with a phosphorothioate backbone (tc-PS-DNA). Besides negative effects like the occurrence of diastereomeric mixtures of oligonucleotides within the same sequence scheme, there are also general advantages such as a significant increase in nuclease stability and cellular uptake and transport. In contrast to most other phosphodiester based oligonucleotide chemistries tc-DNA is already highly nuclease stable in its phosphate form [8, 9]. Hence, there is no need to use tc-PS-DNA just for increasing nuclease resistance. However, it turns out that tc-PS-DNA shows increased plasma transport, cellular uptake and tissue distribution over normal tc-DNA which makes it the candidate of choice for therapeutic applications [10].
Over the years the potential for antisense applications of tc-DNA either in the phosphate (tc-DNA) or thiophosphate (tc-PS-DNA) form has been evaluated in various systems and formats. Outside therapeutic applications of tc-PS-DNA in the field of neuromuscular disorders, which is outlined in more detail below, tc-DNA has been used for splice correction in a cellular assay with HeLa cells stably expressing the human β-globin gene containing two different mutations in intron 2. These mutations activate a cryptic splice site, leading to the inclusion of an aberrant exon. In a comparative study with a tc-DNA 17-mer, masking the cryptic splice site, it appeared that tc-DNA was able to restore correct splicing by a factor of up to 100-fold more efficiently compared to 2′-OMe-PS-RNA [8]. In another set of experiments fully modified tc-DNA has been directed to the cyclophilin A gene in order to induce exon skipping. Again, in a comparative cellular assay, it was shown that a tc-DNA 11-mer showed significant exon skipping while fully modified LNA of the same length was virtually inactive [11]. In another comparative assay, tc-DNA was tested for its capacity to act as a steric blocker AON of human immunodeficiency virus type 1 tat-dependent trans-activation as a means to inhibit HIV viral replication. Here it was found that a fully modified tc-AON showed the same inhibitory activity as an LNA/2′OMe-RNA mixmer of the same length. Corresponding tc-DNA/2′OMe-RNA mixmers were significantly less active demonstrating again that tc-DNA does not mix well with 2′O-Me-RNA (see also above) [12]. In a final benchmark experiment tc-PS-DNA gapmers of lengths between 14 and 20 nt were tested for their capability to downregulate scavenger receptor B1 mRNA in mice. It turned out that the tc-DNA gapmers outperformed corresponding 2′-MOE-RNA gapmers in activity not only in the liver but also in other non-hepatic tissues such as kidney, heart, lung, diaphragm and skeletal muscles [10].
THERAPEUTIC POTENTIAL FOR NMDS
Exon skipping therapy for DMD
As mentioned above, fully modified AONs are particularly useful to manipulate alternative splicing and splice modulation therapy, where the antisense molecules, also called SSOs can be used to correct splicing defects either by inducing exon-skipping, exon-reinclusion or masking aberrant splice splices, is a promising therapy for numerous genetic diseases. One of the most studied examples till date is for DMD where the AONs aim at skipping one or several exons to restore the reading frame (Fig. 2A).
DMD is a lethal X-linked progressive muscle-wasting disease caused by mutations, typically large deletions in the DMD gene, the largest gene in the human genome [13]. Most mutations, including deletions (approximately 65%), duplications, point mutations or other small gene rearrangements disrupt the open reading frame, leading to aberrant translation and, therefore, to the absence of the essential muscle protein dystrophin. Interestingly, the allelic disease Becker muscular dystrophy (BMD), which results in a much milder phenotype, is mainly caused by mutations maintaining the open reading frame and allowing the production of a partially deleted but functional dystrophin [14]. Antisense-mediated exon-skipping strategies for DMD aim to remove the mutated exon alone or together with additional exons to restore the reading frame and consequently induce the expression of “BMD-like” shortened forms of dystrophin retaining crucial functions. Although the exon-skipping approach appears to be applicable to a large proportion of patients (possibly up to approximately 83% of all DMD patients [15]), one should keep in mind that this will not offer a definite cure but an improvement towards a BMD-like phenotype depending on the functionality of the restored dystrophin.
The principle of the exon-skipping therapy for DMD has first been demonstrated by Pramono et al. in 1996 in lymphoblastoid cells and by Dunckley et al. in 1998 in cultured mouse cells in vitro [16, 17]. Since then, numerous in vivo studies have provided pre-clinical evidence for the therapeutic potential of an antisense strategy for DMD in several animal models. In particular, the mdx mouse model, which harbors a nonsense mutation in exon 23, has been used extensively to test efficacy of the AON approach using various oligonucleotide chemistries such as 2′OMe [18], phosphorodiamidate morpholino oligomers (PMO) [19–21], LNA or PNA [22, 23].
Recently, we have demonstrated the therapeutic potential of tc-DNA in this mouse model following systemic treatment for 12 weeks [24]. The tc-DNA-AON was stable and detected in all tested skeletal muscles as well as the heart and brain after intravenous injections. Quantitative PCR revealed effective skipping of exon 23 to levels 5-6-fold higher than that achieved with 2′OMe and PMO AONs, which are the two chemistries currently in trial for DMD. Importantly, this translated into a greater rescue of dystrophin protein levels, particularly in the diaphragm and heart, where levels reached 50% and 40% respectively, compared to wild-type mice. Notably, exon 23 skipping and dystrophin protein expression were seen in the central nervous system (CNS) of only those animals treated with tc-DNA-AON.
Restoration of dystrophin expression significantly improved the mdx mouse phenotype. The specific force of tibialis anterior muscles was essentially normalized and maintained up to 80% following eccentric contractions (used to measure the structural integrity of muscle fibres). A significant improvement in respiratory function was also noted, to a greater extent than that seen with 2′OMe or PMO AON treatment. In addition, echocardiography revealed that the tc-DNA-AON significantly improved ventricular ejection fraction and shortening fraction. For the first time, we could also demonstrate that tc-DNA-AON also had beneficial effects in the CNS. Tonic immobility (freezing) resulting from a restraint-induced fear response - a highly reproducible behavioral phenotype of mdx mice that is controlled by central mechanisms – was the same in tc-DNA-AON-treated mdx and WT mice, whereas the responses of the 2′OMe- and PMO AON-treated mdx mice did not differ from untreated mdx mice. Intravenous tc-DNA-AON treatment for 20 weeks was similarly effective in a more severe mouse model of DMD, lacking both utrophin and dystrophin (dko mouse model). It partially rescued dystrophin protein expression in all affected tissues, leading to significant phenotypic improvements.
This study demonstrated for the first time some of the unique properties of the tc-DNA-AON such as their ability to efficiently target the cardiac muscle, but also their capacity to cross the blood brain barrier and restore certain behavioural aspects linked to the absence of dystrophin in the CNS.
Using DMD mouse models, we were able to evaluate a broad biodistribution across all tissues and calculated a half life of the tc-DNA AONs of approximately 45 days in skeletal muscles. The long lasting activity of tc-DNA lead to satisfactory levels of dystrophin upon regimens using lower doses designed to balance a possible toxicity due to chronic exposure to high levels of AONs. When such an approach was carried out in mdx mice (4 weeks at 200 mg/kg/wk followed by 8 weeks at 50 mg/kg/wk), high levels of exon skipping and dystrophin restoration were detected and were comparable to that found in mice treated at 200 mg/kg over 12 weeks [24]. Interestingly, when comparing low dose treatment regimen of AONs in mdx mice, we were able to detect significant levels of exon skipping in mice treated with as low as 20 mg/kg of tc-DNA whereas similar dosing regimen using 2′OMe or PMO AONs hardly induced any detectable exon skipping (unpublished data).
It is well accepted in the field that AON uptake is facilitated in dystrophic muscles and an enhanced absorption of AON from the blood circulation of up to 8 fold as compared to wild type muscles was previously reported for 2′OMe for example [25]. This is believed to be caused by the myogenic degeneration and regeneration processes in the mdx mice, associated with a higher permeability of the myofiber membranes and/or vascular system. Interestingly, we found that tc-DNA biodistribution in wild type mice was not so different and levels of exon skipping detected after 4 intravenous injections were only 50% of the levels detected in mdx mice (unpublished data).
Exon inclusion strategy for spinal muscular atrophy (SMA)
As well as modulating splicing to remove exons from the mature transcript, tc-DNA can be used to promote exon-inclusion and a typical example of this strategy is considered as one of the most promising therapeutic approach for SMA. SMA is caused by mutations in the SMN1 (Survival Motor Neuron 1) gene. The SMN locus presents a telomeric copy of SMN1 which encodes the SMN protein, and centromeric copies of SMN2 which produce very few SMN. SMN2 differs from SMN1 by only a C to T transition in the coding sequence of exon 7 (not affecting the protein sequence), which however results in exclusion of exon 7 in about 90% of mature transcripts [26, 27]. Therefore SMN2 gene expression leads only to approximately 10% of functional SMN protein and the copy number of SMN2 thus correlates with the severity of the disease. As an example, severe forms (type I) carry 2 copies while type III SMA patients carry three or four SMN2 copies. Exon 7 inclusion has been shown as a promising therapeutic strategy for SMA by using AONs (Fig. 2B) and in particular 2′O-MethoxyEthyl (2′MOE) and PMO AONs. However, despite advances in antisense oligonucleotides chemistry and design, the delivery of AONs to the central nervous system remains a challenge, and intracerebroventricular (ICV) injections are required in mice to induce exon 7 inclusion in brain [28, 29], although best pre-clinical results in mice were generally obtained when ICV injections were coupled with IP injection, confirming the peripheral role of SMN [30]. This is where the unique properties of tc-DNA, being able to cross the blood brain barrier become particularly interesting allowing us to evaluate the contribution of central and peripheral restoration of SMN following systemic delivery. Tc-DNA AONs annealing to the TSL (terminal stem loop) or the downstream ISS (intronic splicing silencer) improving the strength of the 5′donor splice site were shown to restore SMN expression and activity in SMA cells (Robin et al.; manuscript in preparation). Furthermore, subcutaneous treatment of SMA type III mice with a 15mer tc-DNA targeting the downstream ISS sequence induced an efficient exon 7 inclusion in all tissues including the CNS, leading to an improvement in type III SMA mice phenotype, by preventing tail, ear and toe necrosis. This type III SMA mouse model, which is considered mild compared to the severe type I mouse model, survives and breeds normally [31], allowing a later start of treatment (7 days of age for example with tc-DNA) compared to day 1 for the severe model. Detection of exon 7 inclusion in brain and spinal cord following tc-DNA subcutaneous treatment starting at 7 days of age therefore confirmed the specific properties of tc-DNA AONs and their ability to cross the blood brain barrier at least at low levels (Robin et al.; manuscript in preparation).
Aberrant splicing correction for pompe disease
Another mutation affecting normal splicing of mRNA is found in the GAA gene encoding the acid alpha-glucosidase enzyme (GAA), causing Pompe disease or glycogenosis type II (GSD-II), an inherited recessive metabolic myopathy (incidence: 1/40,000). Hundreds of GAA mutations have been identified, but few of them are more common, particularly the [c.-32 -13T>G] mutation [32, 33], which is found in >40% of alleles in adult Caucasian patients and 90% of patients included in the French database [34]. Most severe splice mutations (e.g. totally disabling the splicing reactions) are those that typically affect the consensus donor (GU) or acceptor (AG) splice sites at the 5′ and 3′ ends of introns, thus impairing transesterification reactions. Mutations which affect cis-acting regulatory elements (silencers or enhancers) and other RNA features influencing splicing are usually less severe given that resulting inhibition is often incomplete and some normal transcripts can still be synthesised. The T to G transition provoked by the [c.-32-13T>G] mutation in the GAA gene is one of those, which strongly decreases but not completely abolish the normal inclusion of the second exon into the GAA mRNA during the maturation of the nascent pre-mRNA transcript. The consequence of such mis-splicing is that ensuing Δ2-GAA mRNAs (more than 90% of the GAA transcripts resulting from the mutated gene) lack their start codon and thus cannot be translated into a protein [35]. The [c.–32-13T>G] mutation is located at the 3′ end of the intron 1, within the polypyrimidine (Py) tract, among the branch site (in humans the branch consensus is yUnAy) and the AG splice site, which allows the recruitment of the dimeric U2 auxiliary factor protein or U2AF, identified as a major site of splicing regulation. Therefore, a common explanation for the effect of the [c.-32 -13T>G] mutation is that the T to G transition weakens the strength of the Py tract, which could not any more efficiently recruit U2AF [36]. Nonetheless, the fact that the Py consensus varies somewhat in length and sequence for every intron does not support such an assumption. Instead, the T>G transition might alter the correct removal of intron 1 by modifying the secondary structure of the pre-mRNA at the level of its 3′ acceptor splice site, thus making it ineffective. This is supported by the analysis of the intron-1/exon-2 boundary with the RNAfold software from the ViennaRNA Web Services, which shows major differences for the predicted secondary structures in normal and mutated sequences. In both cases, MFE (minimum free energy) predicted structures show that this particular region of the pre-mRNA could likely form a hairpin, although such a structure is dramatically reinforced in the mutant (Fig. 3A). As a result, the high base-pairing probability of the nucleotides involved in the secondary structure of the mutant pre-mRNA makes almost inaccessible some of the crucial consensus sequences at the 3′ end of intron-1, such as the acceptor splice site (SA1), the pyrimidine tract for U2AF recruitment and the branch site. To summarize, it appears that the T>G transition might alter the correct removal of intron-1 by adding force to a cryptic hairpin self-annihilating SA1.
According to these assumptions, one can propose a method for rescuing a full length GAA mRNA in patient cells with the [c.-32 -13T>G] mutation by using antisense oligonucleotides designed to anneal the 5′ region of exon-2 involved in the putative hairpin described above, hence allowing to set free SA1 and its key upstream cis-elements (Py & branch site) without masking downstream ESE sites (Fig. 3B). Such a rationale has been established ex vivo on skin fibroblasts derived from a compound heterozygote patient harbouring the [c.–32 -13T>G] mutation, by using tc-DNA-AONs targeting the putative hairpin and therefore rescuing the expression of a functional protein (Avril et al;, manuscript in preparation). Considering the very properties of the tc-DNA oligonucleotides, which are efficiently distributed into all skeletal muscles, in particular respiratory muscles, the heart and the CNS, one could anticipate that a splice-switching strategy might likely provide a suitable treatment option for patients with late-onset Pompe disease for whom current treatments are quite ineffective. Interestingly, a recent study using the antisense technology, but targeting the muscle specific glycogen synthase, and not the GAA gene directly, demonstrated encouraging results and could also represent a viable therapeutic strategy for Pompe disease after further development [37].
Specific properties/advantages of tc-DNA
Through these various applications, it appears that the tc-DNA chemistry may represent a new avenue for the synthesis of novel splice-switching drugs for many neuromuscular disorders. The very mechanisms responsible for the enhanced activity of tc-DNA are not yet fully understood. We have previously described using nanoparticle tracking analysis (NTA), Size-Exclusion Chromatography with Multi-Angle Static Light Scattering (SEC-MALS) and dynamic light scattering (DLS) that in contrast to the corresponding 2′OMe and PMO oligomers, tc-DNA AONs spontaneously form defined nanoparticles ranging from 40–100 nm [24, 38]. This propensity to spontaneously self-assemble into nanoparticles could imitates features of transfection reagents and nanoparticle delivery systems [39] and hence could potentially improve cellular uptake compared to 2′OMe and PMO oligomers. While it is not known whether self-assembly of tc-DNA is maintained in vivo and therefore responsible for increased potency, we have demonstrated that this process is maintained in physiological conditions and in the presence of serum proteins [38]. The involvement of class A scavenger receptors (SCARAs) together with other polyI sensitive scavenger receptors (SR) has also been shown in gymnosis conditions in vitro [38]. It is feasible to speculate that the superior activity of tc-DNA is related to its propensity to self-assemble into nanoparticles mediating better SR interaction and subsequent gymnotic uptake, which would fit with the particle-wrapping model for receptor mediated uptake of nanoparticles [39, 40]. The mechanism of spontaneous tc-DNA self-assembly is not well understood. The three additional C atoms increase the hydrophobicity of the molecule and this might impart secondary amphipathic properties on the structure enabling self-assembly via mechanisms resembling the self assembly of ampipathic peptides [41]. Alternatively, the extra rings in the tc-DNA might facilitate stacking and generation of structures resembling poly-G aggregates that were seen to enhance the uptake of AONs and binding to SR [42].
We could also hypothesize that tc-DNA as nanoparticles or free AONs might bind naturally to appropriate, not yet identified, carriers in the bloodstream allowing them to cross endothelial barriers more efficiently. Structural and modeling studies are underway to unravel the mechanism of tc-DNA self assembly and their protein binding profile.
A further crucial feature of the tc-DNA chemistry is that it displays higher RNA binding properties than 2′OMe and PMO [43], thus permitting use of AONs of decreased length. Importantly, this offers the advantage of reducing the mass of synthetic nucleotides administered while keeping biological effect, which could reduce toxicity induced by accumulation of AONs. In a recent study, we compared the effect and therapeutic potential of a 15-nt and a 13-nt tc-DNA in the mdx mouse model (Relizani et al., manuscript in preparation) and demonstrated that systemic delivery of 13nt-tcDNA allows restoration of dystrophin in skeletal muscles at similar levels to a 15nt. Remarkably in the heart, the 13nt-tcDNA induces significantly higher levels of exon skipping and dystrophin restoration than its 15nt counterpart, suggesting a potential size advantage for uptake in the cardiac muscle. However, these unique uptake properties are not entirely due to the small size of tc-DNA since treatment with a 20nt tc-DNA also leads to efficient uptake in the cardiac muscle (unpublished data).
While using small size AONs offers an advantage in terms of cost of production, decreasing the sequence length makes it increasingly more challenging to identify a unique target site. Furthermore, increased binding energy has the capacity to not only increase potency against the intended target but also to “off-targets” and the so-called hybridization-mediated off-targets effects (OTEs) may become a more prevalent concern. However because splicing modulation is highly dependent on target sequence position in the intended RNA target, effects on unintended off-target RNAs containing perfect match are extremely unlikely with splice switching AONs as opposed to gapmers AONs recruiting RNAse H [44, 45]. OTEs have not been noted with great abundance in preclinical or clinical studies to date, probably because not all off-target binding actually leads to functional effects. Not all sites on mRNA are accessible to an ASO, nor are all off-target mRNAs in tissues that receive pharmacologic concentrations of an ASO as they are differentially and temporally expressed in different tissues.
One of the unique opportunities of AONs-based therapeutics is that putative off-target interactions can be predicted using sequence alignment algorithms and pragmatic genomic screening strategies are in place in most companies developing AON-based drugs to delineate potential OTEs. The Oligonucleotide safety Working group (OSWG) recommends assessment of OTEs for AONs during drug discovery and development, both computationally and experimentally [46] and such studies are currently on-going for tc-DNA.
While these recommendations and screening are necessary, OTEs are generally not the main concern in the antisense field. Instead, the toxicities noted have been overwhelmingly associated with those expected by class. Each class of AON from first to third generation has stereotypic toxicity profiles and these nuances are important for the toxicologic pathologist to be aware of [44]. We have recently evaluated the toxicological profile of tc-DNA AONs in DMD mouse model and shown that high dose tc-DNA treatment (200 mg/kg/wk for 12 weeks) was well-tolerated in all mice. Histopathological findings in tc-DNA-treated animals were generally limited to minimal glomerular changes and few cell necrosis in proximal tubules (Relizani et al., manuscript in preparation). Serum biochemistry analysis following 12 weeks of treatment did not reveal any significant increase in transaminases or bilirubin, and only a slight variation in serum creatinine and urinary kidney toxicity biomarkers levels could be detected, which is typical of the PS-AONs accumulation in kidneys. Similarly, no toxic effects were observed at any dosage using SMA mouse models during the course of our experiments (with respect to clinical observations, organ weights, macroscopic observations at necropsy). These results suggest an encouraging safety profile for tc-DNA although one should remain cautious until full toxicological studies are completed since we cannot exclude a possible acute toxicity which would be sequence specific as it has previously been reported with the LNAs, inducing unexpected, sequence specific hepatotoxicity [47].
CONCLUSION
Within the last few years the prospect of successful AON-based splicing therapy for neuromuscular disorders has moved a step closer, particularly for DMD and SMA. Many of these promising therapies have now entered clinical trials and encouraging results have been obtained in most cases, while also showing the limitations of currently used chemistries. The large number of planned and on-going trials using the antisense technology reflects the optimism of these approaches to regulate gene expression and modulate splicing. Much of this optimism can be attributed to the development of novel oligonucleotides modifications, such as the tc-DNA described in this review or the use of cell penetrating peptide which also demonstrated extremely promising results. Recent preclinical work in animal models using these new classes of AONs, which improve tissue uptake, target binding affinity as well as resistance to nuclease degradation, suggest that solutions to the current challenges of systemic and tissue targeted delivery are close at hand. While we focused this review on DMD, SMA and Pompe disease, it should be noted that many other NMDs could benefit from the progresses described here and several preclinical studies have actually demonstrated the therapeutic potential of AONs in other pathologies as reviewed extensively by Van Roon-Mom and Aartsma-Rus [48] and Disterer and colleagues [49].
The very properties of tc-DNA described in this review make them particularly attractive as AONs drugs for such genetic diseases, and clinical evaluation of tc-DNA for the treatment of DMD is currently being planned. However, the promise of tc-DNA still crucially depends on how well it will be tolerated in humans.
With continued success, we will hopefully see a gradual implementation of novel modifications which will improve potency of these drugs over the coming years and that will increasingly extend lifespan and improve the quality of life for patients with neuromuscular diseases.
COMPETING FINANCIAL INTERESTS
Christian Leumann and Luis Garcia are co-funders of Synthena, which produces tricyclo-DNA oligomers.
ACKNOWLEDGMENTS
The authors have financial support from the Agence nationale de la recherche (ANR – Chair of Excellence HandiMedEx), the Association Monegasque contre les myopathies (AMM) and the Duchenne Parent project France (DPPF).
REFERENCES
[1] | Clayton NP , Nelson CA , Weeden T , Taylor KM , Moreland RJ , Scheule RK , et al. Antisense Oligonucleotide-mediated Suppression of Muscle Glycogen Synthase 1 Synthesis as an Approach for Substrate Reduction Therapy of Pompe Disease. Mol Ther Nucleic Acids. (2014) ;3: :e206. Epub 2014/10/29. |
[2] | Zamecnik PC , Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. (1978) ;75: (1):280–4. Epub 2015/08/19. |
[3] | McClorey G , Wood MJ. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr Opin Pharmacol. (2015) ;24: , 52–8. |
[4] | Steffens R , Leumann CJ. Synthesis and Thermodynamic and Biophysical Properties of Tricyclo-DNA. J Am Chem Soc. (1999) ;121: (14):3249–55. |
[5] | Steffens R , Leumann CJ. Tricyclo-DNA: A phosphodiester-backbone based DNA analog exhibiting strong complementary base-pairing properties. J Am Chem Soc. (1997) ;119: (47):11548–9. |
[6] | Tarköy M , Leumann C. Synthesis and Pairing Properties of Decanucleotides from (3′S,5′R)-2′-Deoxy-3′, 5′-ethanoβ-D-ribofuranosyladenine and -thymine. Angew Chem Int Ed. (1993) ;32: (10):1432–4. |
[7] | Renneberg D , Leumann CJ. Watson-Crick base-pairing properties of tricyclo-DNA. J Am Chem Soc. (2002) ;124: , 5993–6002. |
[8] | Ittig D , Gerber AB , Leumann CJ. Position-dependent effects on stability in tricyclo-DNA modified oligonucleotide duplexes. Nucleic Acids Res. (2011) ;39: (1):373–80. |
[9] | Renneberg D , Schümperli D , Leumann CJ. Biological and antisense properties of tricyclo-DNA. Nucleic Acids Res. (2002) ;30: , 2751–7. |
[10] | Ittig D , Renneberg D , Vonlanthen D , Luisier S , Leumann CJ. Oligonucleotide analogues: From supramolecular principles to biological properties. In: Hocec M, editor. XIII Symposium on the chemistry of nucleic acid components: Spindleruv. Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Prague: Collection Symposium Series. (2005) ;21–6. |
[11] | Murray S , Ittig D , Koller E , Berdeja A , Chappell A , Prakash TP , et al. TricycloDNA-modified oligo-2′-deoxyribonucleotides reduce scavenger receptor B1 mRNA in hepatic and extra-hepatic tissues—a comparative study of oligonucleotide length, design and chemistry. Nucleic Acids Res. (2012) ;40: (13):6135–43. |
[12] | Ittig D , Liu S , Renneberg D , Schümperli D , Leumann CJ. Nuclear antisense effects in cyclophilin A pre-mRNA splicing by oligonucleotides: A comparison of tricyclo-DNA with LNA. Nucleic Acids Res. (2004) ;32: , 346–53. |
[13] | Ivanova G , Reigadas S , Ittig D , Arzumanov A , Andreola ML , Leumann C , et al. Tricyclo-DNA Containing Oligonucleotides as Steric Block Inhibitors of HIV-1 Tat-Dependent Trans Activation and HIV-1 Infectivity. Oligonucleotides. (2007) ;17: (1):54–65. |
[14] | Muntoni F , Torelli S , Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. (2003) ;2: (12):731–40. |
[15] | Monaco AP , Bertelson CJ , Liechti-Gallati S , Moser H , Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. (1988) ;2: (1):90–5. |
[16] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: (3):293–9. |
[17] | Pramono ZA , Takeshima Y , Alimsardjono H , Ishii A , Takeda S , Matsuo M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem Biophys Res Commun. (1996) ;226: (2):445–9. Epub 1996/09/13. |
[18] | Dunckley MG , Manoharan M , Villiet P , Eperon IC , Dickson G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum Mol Genet. (1998) ;7: (7):1083–90. |
[19] | Lu QL , Mann CJ , Lou F , Bou-Gharios G , Morris GE , Xue SA , et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. (2003) ;9: (8):1009–14. |
[20] | Fletcher S , Honeyman K , Fall AM , Harding PL , Johnsen RD , Wilton SD. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med. (2006) ;8: (2):207–16. |
[21] | Fletcher S , Honeyman K , Fall AM , Harding PL , Johnsen RD , Steinhaus JP , et al. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol Ther. (2007) ;15: (9):1587–92. |
[22] | Lu QL , Rabinowitz A , Chen YC , Yokota T , Yin H , Alter J , et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci U S A. (2005) ;102: (1):198–203. |
[23] | Ivanova GD , Arzumanov A , Abes R , Yin H , Wood MJ , Lebleu B , et al. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. (2008) ;36: (20):6418–28. |
[24] | Yin H , Lu Q , Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther. (2008) ;16: (1):38–45. |
[25] | Goyenvalle A , Griffith G , Babbs A , Andaloussi SE , Ezzat K , Avril A , et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med. (2015) . Epub 2015/02/03. |
[26] | Heemskerk H , de Winter C , van Kuik P , Heuvelmans N , Sabatelli P , Rimessi P , et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol Ther. (2010) ;18: (6):1210–7.Epub 2010/04/22. |
[27] | Lorson CL , Hahnen E , Androphy EJ , Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. (1999) ;96: (11):6307–77. Epub 1999/05/26. |
[28] | Monani UR , Lorson CL , Parsons DW , Prior TW , Androphy EJ , Burghes AH , et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. (1999) ;8: (7):1177–83. Epub 1999/06/17. |
[29] | Hua Y , Sahashi K , Hung G , Rigo F , Passini MA , Bennett CF , et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. (2010) ;24: (15):1634–44. |
[30] | Hua Y , Sahashi K , Rigo F , Hung G , Horev G , Bennett CF , et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. (2011) ;478: (7367):123–6. Epub 2011/10/08. |
[31] | Osman EY , Miller MR , Robbins KL , Lombardi AM , Atkinson AK , Brehm AJ , et al. Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum Mol Genet. (2014) ;23: (18):4832–45. |
[32] | Hsieh-Li HM , Chang JG , Jong YJ , Wu MH , Wang NM , Tsai CH , et al. A mouse model for spinal muscular atrophy. Nat Genet. (2000) ;24: (1):66–70. |
[33] | Kroos MA , Pomponio RJ , Hagemans ML , Keulemans JL , Phipps M , DeRiso M , et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology. (2007) ;68: (2):110–5. Epub 2007/01/11. |
[34] | Zampieri S , Buratti E , Dominissini S , Montalvo AL , Pittis MG , Bembi B , et al. Splicing mutations in glycogen-storage disease type II: Evaluation of the full spectrum of mutations and their relation to patients’ phenotypes. Eur J Hum Genet. (2010) ;19: (4):422–31. Epub 2010/12/24. |
[35] | Laforet P , Laloui K , Granger B , Hamroun D , Taouagh N , Hogrel JY , et al. The French Pompe registry. Baseline characteristics of a cohort of 126 patients with adult Pompe disease. Revue neurologique. (2013) ;169: (8-9):595–602. Epub 2013/09/07. |
[36] | Raben N , Nichols RC , Martiniuk F , Plotz PH. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum Mol Genet. (1996) ;5: (7):995–1000. Epub 1996/07/01. |
[37] | Dardis A , Zanin I , Zampieri S , Stuani C , Pianta A , Romanello M , et al. Functional characterization of the common c-32-13T>G mutation of GAA gene: Identification of potential therapeutic agents. Nucleic Acids Res. (2014) ;42: (2):1291–302. Epub 2013/10/24. |
[38] | Ezzat K , Aoki Y , Koo T , McClorey G , Benner L , Coenen-Stass A , et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano letters. (2015) ;15: (7):4367–73. Epub 2015/06/05. |
[39] | Nel AE , Madler L , Velegol D , Xia T , Hoek EM , Somasundaran P , et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. (2009) ;8: (7):543–57. Epub 2009/06/16. |
[40] | Gao H , Shi W , Freund LB. Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci U S A. (2005) ;102: (27):9469–74. Epub 2005/06/24. |
[41] | Pujals S , Fernandez-Carneado J , Lopez-Iglesias C , Kogan MJ , Giralt E. Mechanistic aspects of CPP-mediated intracellular drug delivery: Relevance of CPP self-assembly. Biochim Biophys Acta. (2006) ;1758: (3):264–79. Epub 2006/03/21. |
[42] | Wu CC , Castro JE , Motta M , Cottam HB , Kyburz D , Kipps TJ , et al. Selection of oligonucleotide aptamers with enhanced uptake and activation of human leukemia B cells. Hum Gene Ther. (2003) ;14: (9):849–60. Epub 2003/06/28. |
[43] | Renneberg D , Bouliong E , Reber U , Schumperli D , Leumann CJ. Antisense properties of tricyclo-DNA. Nucleic Acids Res. (2002) ;30: (13):2751–7. Epub 2002/06/28. |
[44] | Frazier K. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologist’s perspective. Toxicol Pathol. (2015) ;43: (1):78–89. Epub 2014/11/12. |
[45] | Kamola PJ , Kitson JD , Turner G , Maratou K , Eriksson S , Panjwani A , et al. In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. (2015) ;43: (18):8638–50. Epub 2015/09/05. |
[46] | Lindow M , Vornlocher HP , Riley D , Kornbrust DJ , Burchard J , Whiteley LO , et al. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat Biotechnol. (2012) ;30: (10):920–23. Epub 2012/10/12. |
[47] | Swayze EE , Siwkowski AM , Wancewicz EV , Migawa MT , Wyrzykiewicz TK , Hung G , et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. (2006) ;35: (2):687–700. Epub 2006/12/22. |
[48] | van Roon-Mom WM , Aartsma-Rus A. Overview on applications of antisense-mediated exon skipping. Methods Mol Biol. (2012) ;867: , 79–96. Epub 2012/03/29. |
[49] | Disterer P , Kryczka A , Liu Y , Badi YE , Wong JJ , Owen JS , et al. Development of therapeutic splice-switching oligonucleotides. Hum Gene Ther. (2014) ;25: (7):587–98. Epub 2014/05/16. |
Figures and Tables
Fig.1
Chemical structures of a selection of oligonucleotide analogues that have been tested as antisense agents (2′OMe: 2′OMethyl, 2′MOE: 2′OMethoxyethyl, LNA: Locked nucleic acid, PMO: Phosphorodiamidate morpholino oligomer, PNA: Peptide nucleic acid, bc-DNA: Bicyclo-DNA, tc-DNA: Tricyclo-DNA).

Fig.2
Antisense oligonucleotides mediated splice correction for various neuromuscular diseases. A - Exon skipping rationale for DMD. Patients with DMD carry mutations disrupting the open-reading frame of the dystrophin pre-mRNA. In this example, exon 50 is deleted, creating an out-of-frame mRNA and leading to the synthesis of a truncated non-functional or unstable dystrophin. The tc-DNA AON directed against exon 51 can induce effective skipping of exon 51 and restore the open reading frame, therefore generating an internally deleted but partly functional dystrophin. B – Exon inclusion rationale for SMA. Exon 7 of SMN2 gene is spliced out in 90% of the mature mRNA leading to only 10% of functional SMN protein. Tc-DNA AON can mask the ISS inducing inclusion of exon 7 and leading to a functional SMN protein.
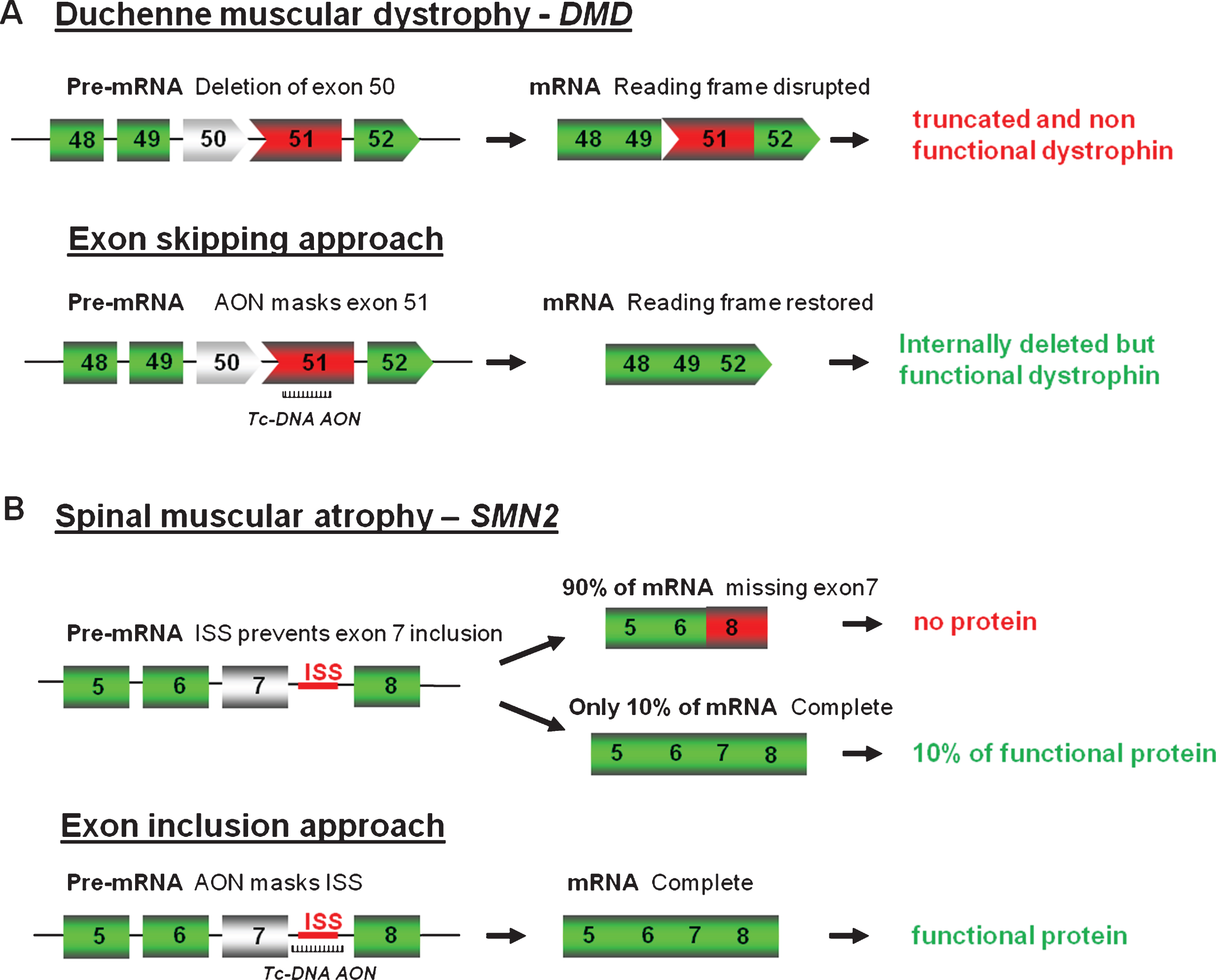
Fig.3
A - ESE finder and MFE (minimal free energy) secondary structure predictions: Notice that the beginning of exon-2 is markedly deprived of ESE (exon splice enhancer) sites binding SRSF (Serine/arginine splicing factors) but shows ESS (exon splice silencer) sites for hnRNP A1 (boxed – yellow contributes to the 5′ UTR; light purple designates coding sequence). The T>G transition might alter the correct removal of intron-1 by modifying the secondary structure of the pre-mRNA at the level of its 3′ acceptor splice site (e.g. generating a locked hairpin), thus making it ineffective. B - Splice switching rationale to rescue the [c.-32-13 T>G] common mutation: MFE analysis of the region spanning the end of intron-1 and the beginning of exon-2 in the presence of a 15-mer tcDNA-AON annealing exon-2 at position [+7 to +22]. The frequency of the new MFE structure is 25% instead of 65%. (SA1: Acceptor splice site 1, U2AF: U2 auxilliary factor, U2snRNP: U2 small nuclear ribonucleoprotein).
![A - ESE finder and MFE (minimal free energy) secondary structure predictions: Notice that the beginning of exon-2 is markedly deprived of ESE (exon splice enhancer) sites binding SRSF (Serine/arginine splicing factors) but shows ESS (exon splice silencer) sites for hnRNP A1 (boxed – yellow contributes to the 5′ UTR; light purple designates coding sequence). The T>G transition might alter the correct removal of intron-1 by modifying the secondary structure of the pre-mRNA at the level of its 3′ acceptor splice site (e.g. generating a locked hairpin), thus making it ineffective. B - Splice switching rationale to rescue the [c.-32-13 T>G] common mutation: MFE analysis of the region spanning the end of intron-1 and the beginning of exon-2 in the presence of a 15-mer tcDNA-AON annealing exon-2 at position [+7 to +22]. The frequency of the new MFE structure is 25% instead of 65%. (SA1: Acceptor splice site 1, U2AF: U2 auxilliary factor, U2snRNP: U2 small nuclear ribonucleoprotein).](https://content.iospress.com:443/media/jnd/2016/3-2/jnd-3-2-jnd160146/jnd-3-jnd160146-g003.jpg)


