
Adult Onset Leigh Syndrome in the Intensive Care Setting: A Novel Presentation of a C12orf65 Related Mitochondrial Disease
Abstract
Background:
Mitochondrial disease can present at any age, with dysfunction in almost any tissue making diagnosis a challenge. It can result from inherited or sporadic mutations in either the mitochondrial or the nuclear genome, many of which affect intraorganellar gene expression. The estimated prevalence of 1/4300 indicates these to be amongst the commonest inherited neuromuscular disorders, emphasising the importance of recognition of the diagnostic clinical features.
Objective:
Despite major advances in our understanding of the molecular basis of mitochondrial diseases, accurate and early diagnoses are critically dependent on the fastidious clinical and biochemical characterisation of patients. Here we describe a patient harbouring a previously reported homozygous mutation in C12orf65, a mitochondrial protein of unknown function, which does not adhere to the proposed distinct genotype-phenotype relationship.
Methods:
We performed clinical, biochemical and molecular analysis including whole exome sequencing on patient samples and cell lines.
Results:
We report an extremely rare case of an adult presenting with Leigh-like disease, in intensive care, in the 5th decade of life, harbouring a recessively inherited mutation previously reported in children. A global reduction in intra-mitochondrial protein synthesis was observed despite normal or elevated levels of mt-RNA, leading to an isolated complex IV deficiency.
Conclusions:
All the reported C12orf65 mutations have shown an autosomal recessive pattern of inheritance. Mitochondrial disease causing mutations inherited in this manner are usually of early onset and associated with a severe, often fatal clinical phenotype. Presentations in adulthood are usually less severe. This patient’s late adulthood presentation is in sharp contrast emphasising the clinical variability that is characteristic of mitochondrial disease and illustrates why making a definitive diagnosis remains a formidable challenge.
ABBREVIATIONS
Mt | Mitochondrial |
COX | Cytochrome c oxidase, CIV |
H&E | Hematoxylin and eosin |
MRI | Magnetic resonance imaging |
MRS | Magnetic resonance spectroscopy |
BN-PAGE | Blue native polyacrylamide gel electrophoresis |
RC | Respiratory chain |
OXPHOS | Oxidative phosphorylation |
LONP | Mitochondrial Lon Protease Homolog |
CLPX | Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit |
INTRODUCTION
The mitochondrial genome (mtDNA) encodes 13 open reading frames in addition to the tRNAs and rRNAs required for intramitochondrial protein synthesis [1]. The remaining components that comprise or facilitate assemble the mitoribosome are encoded by the nucleus. Mutations in any of these genes that affect expression of mtDNA can potentially cause mitochondrial (mt) disease [2–5], a heterogeneous group of disorders characterized by compromised oxidative phosphorylation. The application of whole exome sequencing to patients with well-characterised mitochondrial disease has revealed new nuclear disease genes to be correlated with pathways where their involvement had not previously been known, or had been predicted but not confirmed [6]. The C12orf65 gene falls into the latter category; it is a member of the mitochondrial release factor [7] family, some but not all members of which have been shown to cleave the terminal tRNA from the nascent peptide in the mitoribosome, allowing the newly made protein to escape the translation machinery.
Despite its predicted role in mt-translation, this was not confirmed until the first report of a patient presenting with a pathogenic mutation in C12orf65 [OMIM 613541] leading to decreased mt-protein synthesis [8]. Subsequently a number of patients with C12orf65 mutations have been reported presenting with similar symptoms of mitochondrial disease [8–14] (Table 1). The predicted consequence of the mutations is that truncated versions of C12orf65 protein would be synthesised, where the length of the protein depended on the precise position of the stop codon. A clinical review of patients with C12orf65 mutations has proposed that distinct genotype-phenotype relationships can be distinguished in this patient group, despite the broad spectrum of symptoms [9]. These authors suggest that although the degree of protein truncation dictates the severity, an ‘obligatory clinical triad’ of optic atrophy, peripheral neuropathy and spastic paraparesis is characteristic for all patients.
Here, we describe a late presentation of LS and respiratory failure in a patient with long standing schizoaffective disorder, harbouring a previously reported C12orf65 mutation where the severity of the clinical presentation does not correlate with the length of the truncated C12orf65 protein. This mutation predicts one of the shortest aberrant C12orf65 proteins, and although the symptoms of the minimal obligatory triad were present, the patient only presented to the intensive care unit as an adult in the 5th decade of life. We have characterised respiratory capacity in skeletal muscle and fibroblasts, assessed de novo mitochondrial translation in patient fibroblasts and interrogated the steady state levels of respiratory chain proteins, confirming an isolated mitochondrial complex IV deficiency.
All the reported C12orf65 mutations have shown an autosomal recessive pattern of inheritance. Mitochondrial disease causing mutations inherited in this manner are usually of early onset and associated with a severe, often fatal clinical phenotype. The case presented here is in contrast to this pattern emphasising the importance of recognising the breadth of diagnostic clinical features, especially in light of the estimated prevalence of 1:4300, which indicates that these are amongst the most common inherited neuromusculardisorders [5].
MATERIALS AND METHODS
Case report
The male patient was a term birth, to healthy, non-consanguineous parents, and reached developmental milestones appropriately. At 2 years he underwent a surgical procedure for hypospadias and experienced perioperative respiratory arrest. He was felt to have suffered mild hypoxic brain damage, as evidenced by mild dysarthria, mild fixed limb spasticity and cognitive developmental delay but progressed through normal schooling and remained independent. No bulbar dysfunction or regression was ever documented. He underwent a tendon lengthening procedure at age 14 years with no untoward sequelae. He was diagnosed with schizoaffective disorder in his second decade of life requiring multiple hospital admissions but remained relatively asymptomatic in his lateryears.
In his fifth decade of life, six months prior to his acute admission, he was noted to have deteriorated physically reporting shortness of breath on exertion and worsening mobility requiring two sticks to mobilise with the need for a wheelchair for longer distances. His dysarthria had also progressively worsened.
At 45-years-old this man presented to his acute care hospital following a fall at home. On admission he was noted to be drowsy, with a reduced Glasgow coma scale of 13 out of 15, low grade pyrexia of 38°C, tachypnoea (respiratory rate 21), tachycardia (100 bpm) and hypoxic (69% on room air). An arterial blood gas showed a pH of 7.20, pCO2 14.7kPa, pO2 9.3kPa and bicarbonate of 37.8 mmol/L consistent with type II respiratory failure. A chest X-ray showed patchy consolidation of the left base, white cell count was 12.2 and CRP less than 1. He was initially commenced on non-invasive ventilation but continued rapid deterioration warranted assisted ventilation. CT head was normal. On transfer to critical care he was treated for a presumed pneumonia, he responded well and was extubated after 48 hours, requiring intermittent non-invasive ventilation only for elevated pCO2. He then developed massive hematemesis and rapidly became haemodynamically unstable (systolic BP 90 mm Hg, pulse rate; 130 bpm) necessitating transfusion. He required urgent laparotomy and repair of a perforated duodenal ulcer but following the anaesthetic had a protracted hospital stay. He remained ventilator dependent via a tracheostomy related to gross disturbance of central respiratory drive compounded by diaphragmatic weakness and respiratory muscle weakness. Bulbar function remained profoundly poor, necessitating the placement of a percutaneous endoscopic gastrostomy feeding tube.
Histochemical and biochemical analyses
Standard histological (H&E) and histochemical (COX/SDH) analyses of skeletal muscle biopsies were performed on fresh-frozen skeletal muscle sections (10 μm) [15]. Mitochondrial respiratory chain complex activities were determined in skeletal muscle and fibroblast homogenates and expressed relative to the activity of the matrix marker enzyme, citrate synthase [16].
Molecular genetics
Total DNA was extracted from muscle; mtDNA rearrangements investigated by long-range PCR [17] and direct sequencing of the entire mtDNA genome was undertaken as previously described [18]. Alignment and variant calling were performed using SeqScape software v2.6 (Life Technologies, Warrington, UK) using the revised Cambridge reference sequence for human mtDNA (GenBank accession: NC_012920.1) [19]. Whole exome sequencing and bioinformatics was performed as previously described [13]. Briefly, genomic DNA was fragmented and enriched by Illumina TruSeqtrademark 62Mb exome capture and sequenced (Illumina HiSeq 2000, 100 bp paired-end reads). Bioinformatics pipeline included alignment to the human reference genome (UCSC hg19) and variant detection (Varscan v2.2, Dindel v1.01) [20]. On-target variant filtering excluded minor allele frequencies (greater >0.01) in several databases and 343 unrelated in-house controls as previously shown. ANNOVAR was used to define homozygous/compound heterozygous variants and protein altering and/or putative ‘disease causing’ mutations [21]. Candidate genes were ranked by previous association with disease phenotypes [22]. The C12orf65 variant was confirmed by Sanger sequencing (primers; Forward 5′-TCATCTAACCCAGGTCCTCAG-3′; Reverse 5′-AGGCTGTGGAACCCTTGG-3′; GenBank accession: NM_152269.4) [18].
Cell culture
Cells were routinely cultured [23] in DMEM supplemented with 10% FCS, 2 mM L-glutamine, 1x NEAA, 1 mM pyruvate, 50 μg/ml uridine, with either 4500 mg/L glucose or 10 mM galactose, at 37°C, in a humidified 5% CO2 atmosphere. Transfection of HEK293 cells with siRNAs was performed with 30nM final concentration as previously described [7]. Control non-targetting (NT) and custom synthesised against C12orf65 (sense 5′-GGG AGA AGC UGA CGU UGU U dTdT) siRNAs were from Eurogentec.
BN-PAGE, SDS-PAGE and western blotting
For BN PAGE, mitochondrial lysates solubilised with dodecylmaltoside (3.2 g/g protein) from patient and control fibroblasts (25 μg per lane) were separated on 4.5–16% gels as described in [24]. For SDS-PAGE, mitochondrial lysates were prepared from patient and control fibroblasts and 12.5 μg per lane separated on 12% gels as described in [23].
Proteins were transferred to PVDF membranes (GE Healthcare) for immunoblotting. Primary antibodies used were; Complex I - NDUFB8, NDUFA9; Complex II - SDHA (70 kDa flavoprotein subunit); Complex III - UQCRC2 (Core 2); Complex IV - COX4, COX2; Porin; MRPL3, CLPX, Complex V (α-subunit) (all Abcam). ICT1, MRPS26, MRPS27, MRPL45 (PTG Labs); HSP60 (BD Biosciences); LonP (Sigma Life Science). Appropriate HRP-conjugated immunoglobulin secondary antibodies (Dako, Glostrup, Denmark) were used and detected with ECL-Prime (GE Healthcare) as per the manufacturer’s instructions.
Metabolic labelling of mitochondrially encoded proteins
This was essentially as described in Chomyn [25]. Patient and control fibroblasts were metabolically labelled ([35S]-methionine/cysteine) for 1 hour with emetine inhibition of cytosolic translation. Total cell lysates (50 μg) were separated by 15% SDS-PAGE. Signals from newly synthesised mt-proteins were detected from dried gels via PhosphoImaging and ImageQuant software (GE Healthcare). Gels were Coomassie blue stained to confirm equal loading.
Northern analysis
Total RNA from control and patient fibroblasts or HEK293 cells was extracted using Trizol (Life Technologies). Aliquots (4 μg) were electrophoresed through 1.2% denaturing agarose, and transferred to Genescreen Plus. Probes were generated using random hexamers on PCR generated templates corresponding to the internal regions of the genes of interest, as previously described [26].
RESULTS
Clinical findings
Neurophysiological evaluation was consistent with an axonal neuropathy. On examination the patient had bilateral ptosis, horizontal gaze paresis and bilateral optic atrophy. He had mild facial muscle weakness, palatal myoclonus, a reduced gag reflex and dysarthria. There was severe distal muscle wasting of upper and lower limbs. Upper and lower limb reflexes were pathologically brisk, with the exception of ankle and plantar reflexes, which were absent. Initial neuropathy screen including serum protein electrophoresis, copper, caeruloplasmin, acylcarnitines, carnitine, phytanic acid, very long chain fatty acids, α-fetoprotein and urinary organic acids were normal. Serum lactate was 3.7 mmol/L (normal range <2.0 mmol/l).
MRI of the head showed extensive swelling and abnormal high T2-weighted signal changes centrally within the medulla and dorsal pons. Additional small foci of restricted diffusion were seen within the brainstem and the cerebellar hemispheres (Fig. 1). Brain MRS showed a lactate peak, with reduced NAA and raised choline in a voxel centred over the brain stem. Combined these radiological findings were consistent with a diagnosis of Leigh syndrome.
Characterisation of respiratory capacity in skeletal muscle and fibroblasts
A diagnostic muscle biopsy was performed to formally investigate a likely mitochondrial disorder. Although the muscle appeared morphologically normal, histochemical analysis of oxidative enzymes including succinate dehydrogenase (SDH) and cytochrome c oxidase (COX) revealed a generalised decrease in COX activity throughout the biopsy sample, confirmed by the sequential COX-SDH assay (Fig. 2A). Spectrophotometric assays of mitochondrial respiratory chain enzyme activities revealed a severe (approximately 20% of control values) and isolated COX (CIV) deficiency in the muscle of our patient (Fig. 2B). The identical biochemical defect was also observed, to a lesser degree, in the patient’s cultured fibroblasts (approximately 50% of control) (Fig. 2C). The activities of all other RC complexes were normal, confirming an isolated CIV deficiency.
Cell morphology (Fig. 2D) and growth rate of fibroblasts derived from the patient were comparable to control cells on glucose. However, these failed to proliferate on galactose (Fig. 2E, F), a substrate that forces cells to utilise oxidative phosphorylation rather than glycolysis for ATP production. This observation confirmed that the cells express a mitochondrial respiratory chain defect, consistent with the decrease in complex IV activity determined spectrophotometrically.
Identification of pathogenic C12orf65 mutation
Diagnostic molecular genetic studies excluded mtDNA depletion, mtDNA rearrangements and mtDNA point mutations as a cause of the respiratory chain defect in muscle. Whole exome capture and sequencing identified a homozygous exonic deletion (c.210delA, p.(Gly72Alafs *13) frameshift mutation (Fig. 3A), predicted to encode a truncated version of the protein due to the introduction of a premature stop codon (Fig. 3B). This was identical to the mutation described in the first report of C12orf65 patients [8]. Subsequent C12orf65 reports also identified truncating mutations [8–13]. Although premature stop codons often trigger non-sense mediated decay (NMD) to degrade transcripts and so prevent synthesis of aberrant polypeptides, control levels of C12orf65 mRNA were reported ([8, 12], and data not shown) and one study detected a truncated C12orf65 protein [11] suggesting that aberrant C12orf65 transcripts identified thus far escape normal NMD.
As with our patient’s mutation, a number of the reported mutations predict not only a truncated protein but also the loss of the highly conserved GGQ motif, a determining feature of release factors. This critical feature is essential for triggering peptidyl-tRNA hydrolysis, which cleaves the ester bond between the P-site tRNA and the nascent polypeptide chain [27, 28]. Since loss of this motif or the whole protein may affect synthesis of all mitochondrially encoded polypeptides, intraorganellar translation was analysed by metabolic labelling.
C12orf65 mutation disrupts expression of respiratory complexes
De novo mitochondrial translation was assessed in patient fibroblasts and indicated a global decrease in protein synthesis (Fig. 4A), as seen in previously reported patients. This reduction in mt-protein synthesis is not a consequence of a reduction in mt-RNA levels, which are modestly elevated (Fig. 4B), again as seen in other C12orf65 patients. When cells are artificially depleted of C12orf65 protein using siRNA, the elevation in mt-RNA levels is more striking (Fig. 4C). A mitoribosome defect is unlikely to be responsible in this patient, as no global differences were observed in the steady state levels of mitoribosomal protein levels (Fig. 4D).
To identify if the synthesised polypeptides are stable, steady state levels of RC proteins were assessed by western blotting following SDS-PAGE. This confirmed decreased levels of complex IV (COX2 expression), consistent with our biochemical assays but also revealed reduced levels of CI and modest changes in CIII subunits (Fig. 4E). To determine if synthesised polypeptides were incorporated into fully assembled oxidative phosphorylation complexes, western blots of mitochondrial fractions separated by 1D BN-PAGE were performed. A reduction in the amount of fully assembled complexes I, IV and V was observed in the patient compared to controls, together with a minor complex III defect, visible in the I+III supercomplex (Fig. 4F). These data are consistent with C12orf65 playing a ubiquitous role in mt-translation but with a more severe effect on CIV activity. This translation defect caused by loss of C12orf65 does not appear to trigger an unfolded protein response as no increase was seen in relevant proteases (Fig. 4G, LONP and CLPX).
DISCUSSION
Here we present the comprehensive clinical, genetic, biochemical, and molecular characterisation of an adult patient harbouring a mutation in the C12orf65 gene. Although 27 patients with recessively inherited C12orf65 mutations have been documented in the literature (Table 1), this individual had an atypical presentation, highlighting how different the aspects of their clinical and/ or molecular features can be.
The precise role of C12orf65 has not yet been fully elucidated, however, studies depleting cells of this protein together with analysis of patient cell lines carrying mutations clearly indicates a role in mt-protein synthesis. This is consistent with the evolutionary conservation of the GGQ domain that determines its ribosome dependency and designation as a translation release factor [7]. Deleterious mutations that disrupt the GGQ motif are predicted to result in a more severe phenotype, whereas downstream C-terminal mutations have been reported with more favourable phenotypic presentations that typically lack cognitive impairment [9]. The patient described herein, harbours a mutation in the C12orf65 gene that would, therefore, predict both a severe and early-onset presentation. This prediction is reinforced by the same mutation having originally been reported in children with impairment of mitochondrial translation and severe decreases in activities of complexes I, IV and V [8]. In distinction to these expectations this patient presented in adulthood (aged 45 years) with milder symptoms and an isolated complex IV defect. Although the ‘obligatory clinical triad’ of symptoms was present there were other distinctive features in this patient’s presentation.
The patient suffered from shortness of breath and associated respiratory muscle weakness, consistent with a long term deficiency in respiratory chain (RC) enzyme activities. To date, only 3 published cases of C12orf65-related mitochondrial disease have reported respiratory failure; but age of onset varies from 4 to 14 years. This unusual cause of failure to wean from a respirator in a man in his fifth decade of life was, therefore, likely precipitated by a marked catabolic state, resulting from compromised respiratory chain activity. Spectrophotometric analysis of respiratory chain complexes indicated a severe and isolated complex IV deficiency, consistent with the growth defect seen in patient fibroblasts when challenged to respire on galactose. Intriguingly however, our molecular investigations of individual mt-RNA species, proteins and OXPHOS complex formation revealed a complex regulation of gene expression. Elevation of the mt-mRNAs encoding OXPHOS components did not correlate with elevated protein levels, nor did modest decreases in protein levels of specific complex I and V components compromise complex activities. Indeed, biochemically we were able to find only an isolated complex IV deficiency. This lack of complex I dysfunction also marked this phenotype as different from the biochemical defects reported in published cases.
Of the mitochondrial disease patients for whom there is no current molecular diagnosis, approximately 30% exhibit defects in mitochondrial translation [6]. Incorporating whole exome sequencing into diagnostic algorithms is facilitating molecular genetic diagnoses but it is clear that identifying a pathogenic mutation in a gene encoding a relevant mitochondrial protein is only the first step towards understanding the molecular basis of mitochondrial disease. For example, mitochondrial aminoacyl tRNA synthetases (mt-aaRS) are responsible for adding the appropriate amino acid to the cognate mt-tRNA. The lack of a correctly charged mt-tRNA would be predicted to impair all intra-mitochondrial translation irrespective of which of mt-tRNA was in short supply. However, recessively-inherited defects in different aminoacyl mt-tRNA synthetases lead to very different clinical phenotypes exhibiting marked tissue specificity [29]. Similarly, different mt-tRNA mutations would be predicted to affect overall intra-mitochondrial translation but the degree to which each gene product is synthesised cannot be so easily correlated with either the particular mutant mt-tRNA or the relevant mt-aaRS. There are clearly other factors that play a part in modifying the severity of the defects and further research is required to dissect what these are and how they contribute to the clinicalphenotype.
Defective C12orf65-induced RC deficiency may be responsible for a retrograde signal that causes the increase in mitochondrial RNA seen in all patient samples where mt-RNA levels were analysed (ibid and [8]), and in cell lines following siRNA mediated depletion of C12orf65. This may potentially reflect a compensatory response directed at overcoming the OXPHOS defect, however the defective C12orf65 prevents any subsequent increase in protein synthesis. The cellular ATP requirements in this patient were partially sustained by glycolysis, however under conditions of higher energy demands the consequences of the C12orf65 defect manifested as respiratory failure with defective OXPHOS.
Of the identified C12orf65 mutations this individual harbours a previously reported frameshift mutation that eliminates the GGQ domain and prematurely truncates the protein ten amino acids beyond this domain, generating one of the shortest reported C12orf65 proteins (Fig. 2B, [13]). In distinction to the published clinical reports of C12orf65 mutations, which are consistent with the shortest proteins correlating with the more severe symptoms (summarised in [13]), this individual only presented in his fifth decade of life with a milder and more restricted panoply of symptoms. Mitochondrial disease was until recently classified as a rare disorder [30, 31]. However, advances in our understanding of the genetics, biochemistry, and clinical presentations of these conditions have resulted in increasingly effective diagnoses. This improved recognition of mitochondrial disease is confirmed in a recent study indicating an estimated prevalence of 1:4300 making these some of the most common inherited neuromuscular disorders [5].
In summary, we demonstrate for the first time, an example where the severity of the clinical phenotype is independent of the position of the truncating mutation in the C12orf65 gene. Hence, cases such as the one presented here, reinforce the complexity of the clinical presentations of mitochondrial disease, where variable combinations of symptoms, with different degrees of severity, can manifest at any age.
CONFLICT OF INTEREST STATEMENT FOR ALL AUTHORS
The authors have no conflict of interest to report.
COMPETING FINANCIAL INTEREST
The authors declare no competing financial interests.
ACKNOWLEDGMENTS
This work was supported by the Wellcome Trust [096919Z/11/Z and 074454/Z/04/Z to ZCL, RNL, PFC, RWT]; the UK National Institute for Health Research Biomedical Research Centre for Ageing and Age-related Diseases award to Newcastle upon Tyne Hospitals NHS Foundation Trust [GSG] and the UK NHS Specialized Services and Newcastle upon Tyne Hospitals NHS Foundation Trust that support the ‘Rare Mitochondrial Disorders of Adults and Children’ Diagnostic Service [http://www.newcastle-mitochondria.com/]; National Institute for Health Research HCS Doctoral Fellowship [(NIHR-HCS-D12-03-04) to CLA]. The sponsors had no role in the design of study or interpretation of results. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Ethical approval - Local study approval was granted (NRES Committee North East - Newcastle & North Tyneside 1). All clinical investigations were evaluated according to the Declaration of Helsinki. The authors thank Tran Hornig-Do, Gavin Falkous and Francesco Bruni for technical advice.
REFERENCES
1 | Anderson S (1981) Sequence and organization of the human mitochondrial genome Nature 290: 457 465 |
2 | Pearce S, Nezich CL, Spinazzola A (2013) Mitochondrial diseases: Translation matters Molecular and cellular neurosciences 55: 1 12 |
3 | Greaves LC, Reeve AK, Taylor RW, Turnbull DM (2012) Mitochondrial DNA and disease The Journal of Pathology 226: 2 274 286 |
4 | Falk MJ (2015) Mitochondrial Disease Sequence Data Resource (MSeqDR): A global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities Molecular Genetics and Metabolism 114: 3 388 396 |
5 | Gorman GS (2015) Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease Annals of Neurology 77: 5 753 759 |
6 | Taylor RW (2014) Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies JAMA 312: 1 68 77 |
7 | Richter R, Rorbach J, Pajak A, Smith PM, Wessels HJ, Huynen MA, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM (2010) A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome EMBO J 29: 6 1116 1125 |
8 | Antonicka H (2010) Mutations in C12orf65 in Patients with Encephalomyopathy and a mitochondrial translation defect Am J Hum Genet 87: 1 115 122 |
9 | Spiegel R (2014) Delineation of C12orf65-related phenotypes: A genotype-phenotype relationshiEuropean Journal of Human Genetics: EJHG 22: 8 1019 1025 |
10 | Buchert R (2013) Mutations in the mitochondrial gene C12ORF65 lead to syndromic autosomal recessive intellectual disability and show genotype phenotype correlation European Journal of Medical Genetics 56: 11 599 602 |
11 | Shimazaki H (2012) A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55) Journal of Medical Genetics 49: 12 777 784 |
12 | Tucci A (2014) Novel C12orf65 mutations in patients with axonal neuropathy and optic atrophy Journal of Neurology, Neurosurgery, and Psychiatry 85: 5 486 492 |
13 | Pyle A (2014) Behr’s Syndrome is Typically Associated with Disturbed Mitochondrial Translation and Mutations in the C12orf65 Gene Journal of Neuromuscular Diseases 1: 55 63 |
14 | Imagawa E (2015) Homozygous V116* mutation in C12orf65 results in Leigh syndrome Journal of Neurology, Neurosurgery, and Psychiatry 10.1136/jnnp-2014-310084 |
15 | Old SL, Johnson M (1989) Methods of microphotometric assay of succinate dehydrogenase and cytochrome c oxidase activities for use on human skeletal muscle Histochem J 21: 545 555 |
16 | Kirby DM, Thorburn DR, Turnbull DM, Taylor RW (2007) Biochemical assays of respiratory chain complex activity Methods in Cell Biology 80: 93 119 |
17 | Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, Taylor RW, Turnbull DM, Taivassalo T (2008) Resistance training in patients with single, large-scale deletions of mitochondrial DNA Brain: A Journal of Neurology 131: Pt 11 2832 2840 |
18 | Tuppen HA (2010) The M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families Brain: A Journal of Neurology 133: 10 2952 2963 |
19 | Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA Nature Genetics 23: 2 147 |
20 | Horvath R (2012) A new phenotype of brain iron accumulation with dystonia, optic atrophy, and peripheral neuropathy Movement disorders: Official Journal of the Movement Disorder Society 27: 6 789 793 |
21 | Wang K, Li M, Hakonarson H (2010) ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data Nucleic Acids Research 38: 16 e164 |
22 | Lieber DS (2013) Targeted exome sequencing of suspected mitochondrial disorders Neurology. 80: 19 1762 1770 |
23 | Serre V, Rozanska A, Beinat M, Chretien D, Boddaert N, Munnich A, Rotig A, Chrzanowska-Lightowlers ZM (1832) Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency Biochimica et Biophysica Acta 8 1304 1312 |
24 | Hornig-Do HT (2014) Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations EMBO Molecular Medicine 6: 2 183 193 |
25 | Chomyn A (1996) In vivo labeling and analysis of human mitochondrial translation products Methods Enzymol 264: 197 211 |
26 | Chrzanowska-Lightowlers ZM, Preiss T, Lightowlers RN (1994) Inhibition of mitochondrial protein synthesis promotes increased stability of nuclear-encoded respiratory gene transcripts J Biol Chem 269: 44 27322 27328 |
27 | Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis RNA 5: 8 1014 1020 |
28 | Seit-Nebi A, Frolova L, Justesen J, Kisselev L (2001) Class-1 translation termination factors: Invariant GGQ minidomain is essential for release activity and ribosome binding but not for stop codon recognition Nucleic Acids Res 29: 19 3982 3987 |
29 | Konovalova S, Tyynismaa H (2013) Mitochondrial aminoacyl-tRNA synthetases in human disease Molecular Genetics and Metabolism 108: 4 206 211 |
30 | Hurko O (2013) Drug development for rare mitochondrial disorders Neurotherapeutics: The Journal of the American Society for Experimental Neuro Therapeutics 10: 2 286 306 |
31 | Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nature reviews Genetics 6: 5 389 402 |
32 | Heidary G, Calderwood L, Cox GF, Robson CD, Teot LA, Mullon J, Anselm I (2014) Optic atrophy and a Leigh-like syndrome due to mutations in the C12orf65 gene: Report of a novel mutation and review of the literature Journal of Neuro-Ophthalmology: The Official Journal of the North American Neuro-Ophthalmology Society 34: 1 39 43 |
Figures and Tables
Fig.1
MRI revealed abnormalities consistent with mitochondrial disease. Cranial MRI was performed showing swelling and abnormal high T2-weighted signal changes centrally within the dorsal pons (A, red arrow) and medulla (B, C red arrows). Other small foci of restricted diffusion were detected and also seen within the cerebellar hemispheres (A, yellow arrow).
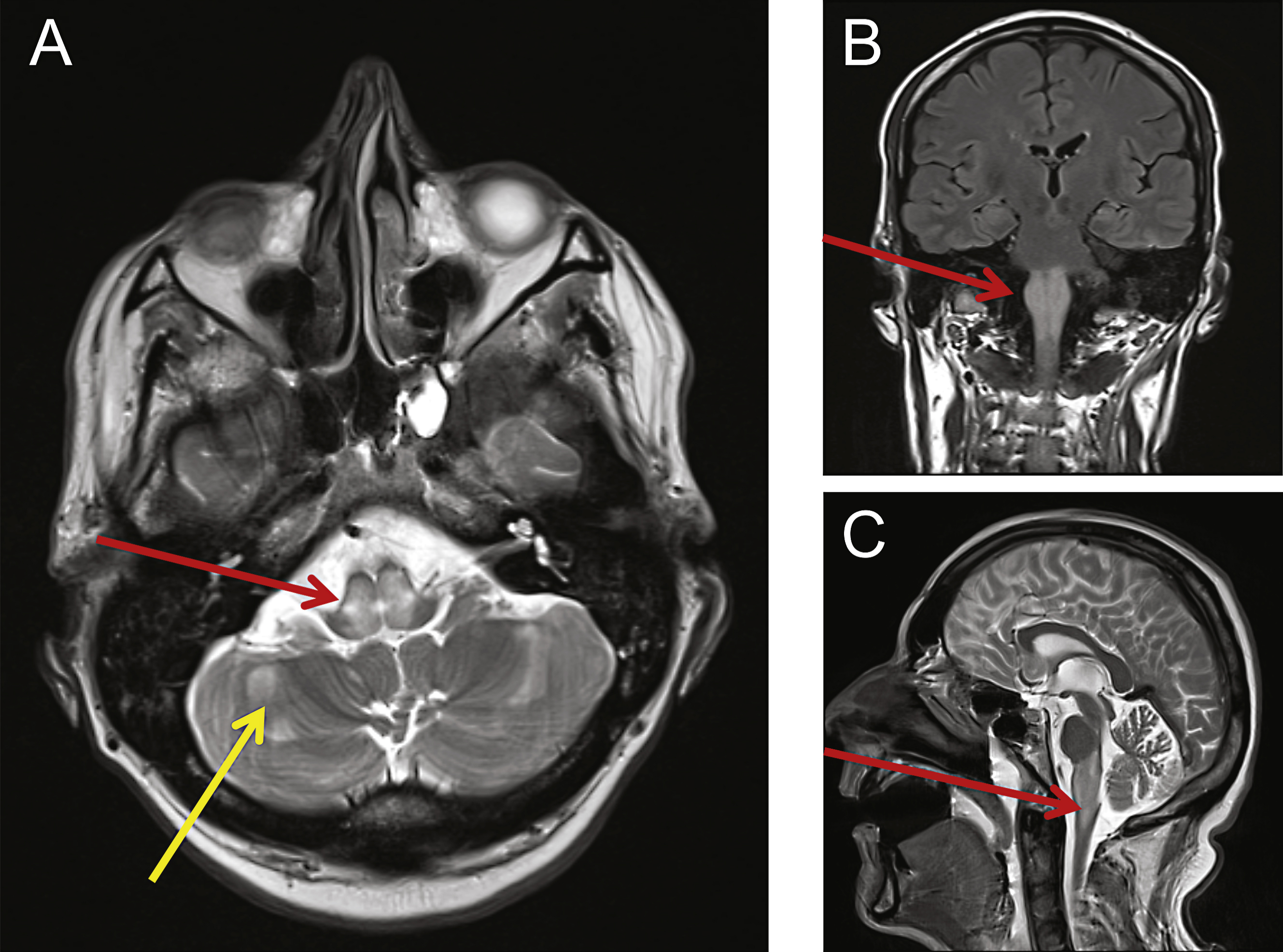
Fig.2
Histochemical, biochemical and cellular analyses indicate mitochondrial dysfunction. A. Serial cryosections of the patient’s muscle biopsy (upper panels) were assessed histologically (H&E) and histochemically for oxidative enzyme activities including succinate dehydrogenase (SDH) and cytochrome c oxidase (COX) and the sequential COX-SDH reaction and compared to controls (lower panels). Respiratory chain complex activities were determined spectrophotometrically in skeletal muscle (B) and fibroblast (C) homogenates and presented as a % of control values. Morphology (D) and growth of patient and control fibroblasts was monitored over 6 days in glucose (E) and galactose (F) n = 3 experimental repeats. Representative differences in density and morphology of the patient and control fibroblasts after 6 days in galactose are presented (D). Scale bar = 50 μm.
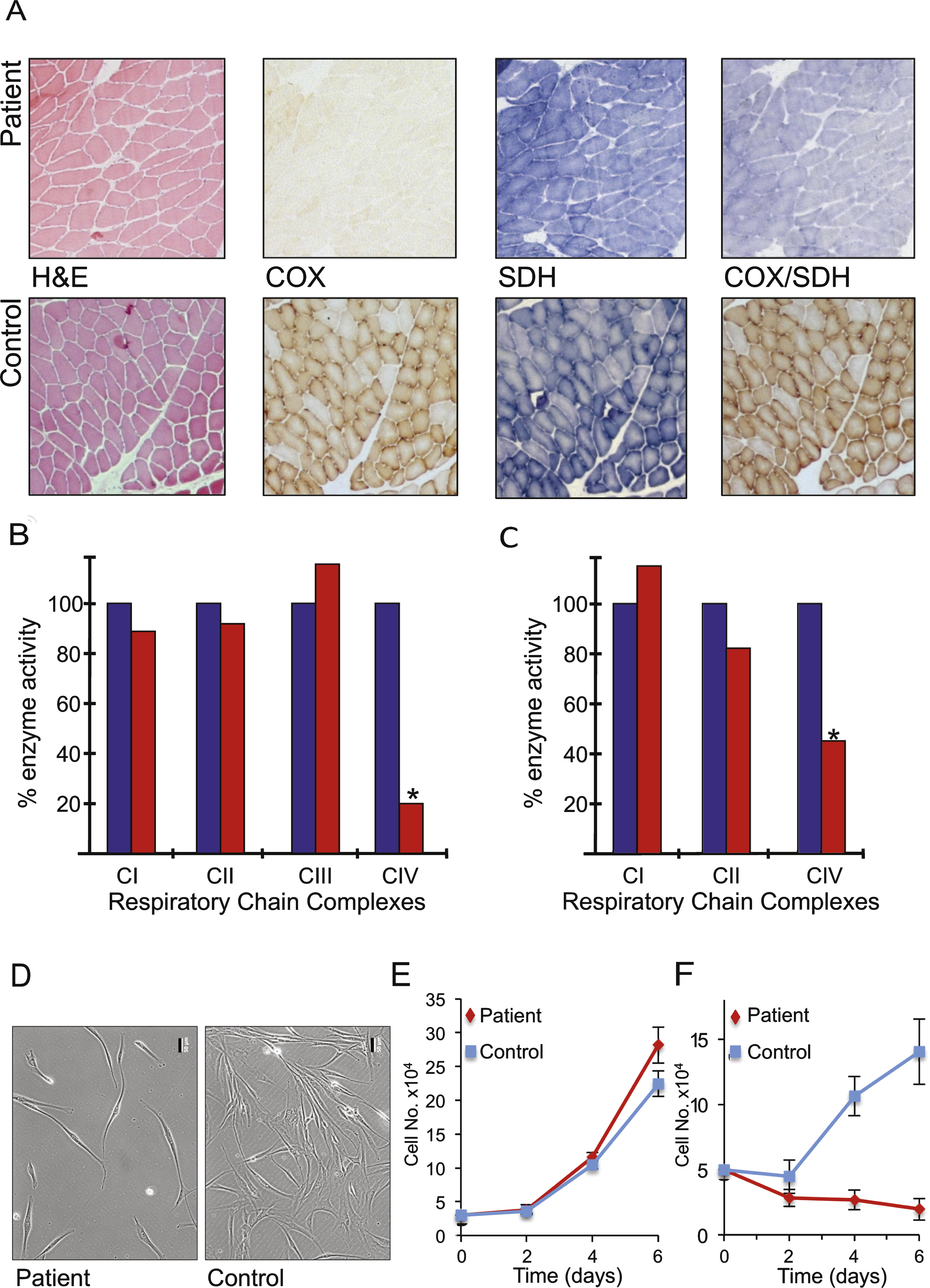
Fig.3
Sanger sequence analysis of C12orf65 patient gene. A. Region of an electropherogram of Sanger sequencing of the C12orf65 gene from the patient (upper panel) and wildtype control (lower panel), aligned to the GenBank accession: NM_152269.4 [19]. The green arrow indicates the position of the adenine nucleotide in control sample that is deleted from the patient’s genome. B. Schematic demonstrating the consequence of the single nucleotide deletion on the peptide sequence. The resultant frameshift introduces amino acid changes and a premature UGA stop codon (grey box) predicted to encode a truncated protein. The alterations to the functional conserved GGQ motif are also boxed in grey.
![Sanger sequence analysis of C12orf65 patient gene. A. Region of an electropherogram of Sanger sequencing of the C12orf65 gene from the patient (upper panel) and wildtype control (lower panel), aligned to the GenBank accession: NM_152269.4 [19]. The green arrow indicates the position of the adenine nucleotide in control sample that is deleted from the patient’s genome. B. Schematic demonstrating the consequence of the single nucleotide deletion on the peptide sequence. The resultant frameshift introduces amino acid changes and a premature UGA stop codon (grey box) predicted to encode a truncated protein. The alterations to the functional conserved GGQ motif are also boxed in grey.](https://content.iospress.com:443/media/jnd/2015/2-4/jnd-2-4-jnd150121/jnd-2-4-jnd150121-g003.jpg)
Fig.4
Gene expression analyses of OXPHOS components and related proteins. A. Intramitochondrial protein synthesis was measured in cell lysates by incorporation of 35S-methionine and cysteine into fibroblasts derived from patient (P) or control (C). The relative positions of the mt-proteins are indicated. Protein loading was confirmed by Coomassie blue (CBB) staining of the gel. B. Northern blot analysis was performed on patient and control RNA (4 μg) to quantify the steady state levels of the RNA species indicated. The cytosolic 18 S rRNA was used as a loading control. C. Similar analyses were performed on human kidney cells (HEK293) following 6 days siRNA mediated depletion of C12orf65 (lanes 1–3), or a non-targetting control siRNA (lanes 4–6) reflecting 3 experimental repeats. The cytosolic 18 S rRNA was used a loading control. Westerns of patient and control mitochondrial lysates (12.5 μg) were separated by 12% SDS:PAGE to detect levels of mitoribosomal proteins (D), respiratory chain proteins (E) or mitochondrial proteases (G) using porin as a loading control. F. Assembly and integrity of the OXPHOS complexes was determined by blue native 4.5–16% gradient PAGE of mitochondrial lysates (25 μg) from patient and control. After transfer, Western blot analysis was used to visualize all OXPHOS complexes with subunit specific antibodies as follows: complex I (NDUFA9), complex III (Core 2) and complex II (SDHA), complex IV (COX4) and complex V (α-subunit).
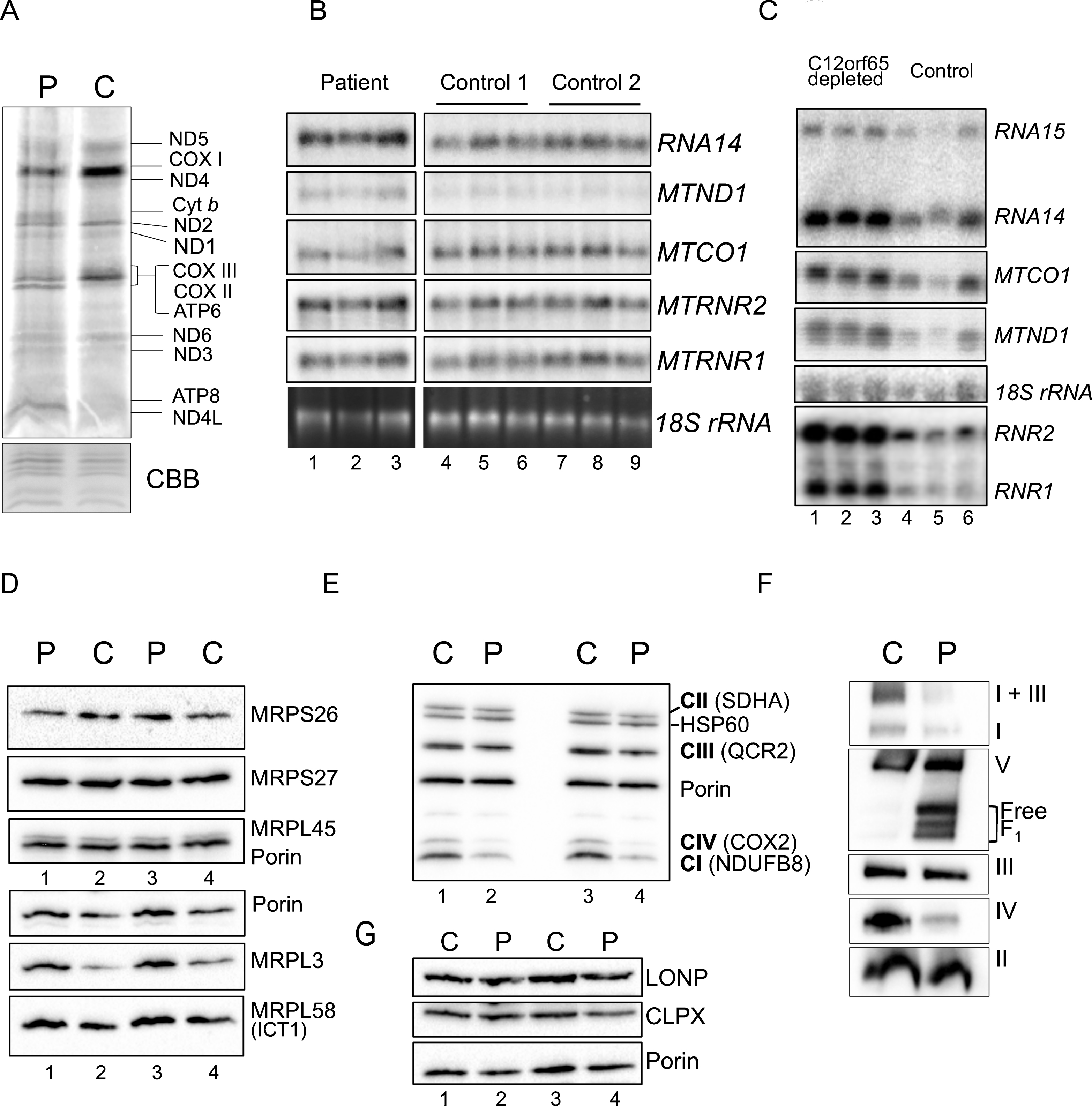
Table 1
Summary of clinical features reported in previously reported patients * harbouring pathogenic mutations in C12orf65
| Clinical Symptoms | Reference |
| Optic atrophy † | [8, 9, 11–14, 32] |
| Pyramidal signs † | [9, 10, 12, 13, 32] |
| Intellectual disability | [8–10, 12–14] |
| Neuropathy (including distal weakness, foot deformity †) | [8, 9, 11–14] |
| Ophthalmoparesis | [8, 10, 13, 32] |
| Facial muscle weakness | [8, 13, 32] |
| Cerebellar involvement (intentional tremor, dysmetria, ataxia) | [8, 13, 32] |
| MRI changes consistent with Leigh syndrome | [8, 13, 32] |
| Dysmorphic facies (hypertelorism, broad nasal bridge) | [10, 13] |
| Bulbar involvement | [8, 32] |
| Respiratory compromise/failure | [8, 32] |
| Macular colloid bodies | [12] |
| Retinitis pigmentosa | [12] |
| Cardiomyopathy | [32] |
| Arthrogryposis | [10] |
| Scoliosis | [12] |
| Paralytic ileus | [8] |
| Urinary stress incontinence | [8] |
| Ovarian failure | [14] |
*Previously reported patients (n = 27), †Clinical triad symptom.


