
Current Translational Research and Murine Models For Duchenne Muscular Dystrophy
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked genetic disorder characterized by progressive muscle degeneration. Mutations in the DMD gene result in the absence of dystrophin, a protein required for muscle strength and stability. Currently, there is no cure for DMD. Since murine models are relatively easy to genetically manipulate, cost effective, and easily reproducible due to their short generation time, they have helped to elucidate the pathobiology of dystrophin deficiency and to assess therapies for treating DMD. Recently, several murine models have been developed by our group and others to be more representative of the human DMD mutation types and phenotypes. For instance, mdx mice on a DBA/2 genetic background, developed by Fukada et al., have lower regenerative capacity and exhibit very severe phenotype. Cmah-deficient mdx mice display an accelerated disease onset and severe cardiac phenotype due to differences in glycosylation between humans and mice. Other novel murine models include mdx52, which harbors a deletion mutation in exon 52, a hot spot region in humans, and dystrophin/utrophin double-deficient (dko), which displays a severe dystrophic phenotype due the absence of utrophin, a dystrophin homolog. This paper reviews the pathological manifestations and recent therapeutic developments in murine models of DMD such as standard mdx (C57BL/10), mdx on C57BL/6 background (C57BL/6-mdx), mdx52, dystrophin/utrophin double-deficient (dko), mdxβgeo, Dmd-null, humanized DMD (hDMD), mdx on DBA/2 background (DBA/2-mdx), Cmah-mdx, and mdx/mTRKO murine models.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common and fatal form of muscular dystrophies with an incidence of 1 in 5,000 boys [1, 2]. It is characterized by progressive muscle wasting and degeneration [3]. Mutations in the DMD gene result in the absence of a protein, dystrophin in the sarcolemma [3]. The DMD gene, the largest known gene in humans, consists of 79 exons and a 14 kb long dystrophin mRNA [4]. Dystrophin has four domains: N-terminal domain, 24 spectrin-like rod-shaped domain, cysteine rich domain and C-terminal domain [5]. The N-terminal domain of dystrophin binds to actin, and the cysteine rich and C-terminal domains of dystrophin bind to dystrophin-glycoprotein complex (DGC), a multimeric protein complex found at the plasma membrane (sarcolemma) of muscle fibers (aka myofibers) [5, 6]. Along with DGC, dystrophin crucially links the actin cytoskeleton of the sarcolemma to the extracellular basement membrane, as illustrated in Figure 1 [5, 7]. In the presence of dystrophin, DGC maintains muscle membrane integrity by serving as a signalling center, and a shock absorber to reduce contraction-induced damage [7]. Mutations in many protein components of DGC (such as dystrophin, sarcoglycans or dystroglycans) lead to various forms of muscular dystrophy and murine models with various dystrophic phenotypes, partly because certain components of DGC are more crucial in function than others [7].
In the absence of dystrophin, almost all components of DGC is either lost or mislocalized, the DGC is rendered dysfunctional and, the sarcolemma is highly susceptible to damage during muscle contraction [8]. Normal skeletal muscles regenerate following injury via satellite cells, which are resident muscle stem cells found beneath the basement membrane of myofibers [9, 10]. However, since dystrophic skeletal muscles undergo rapid degeneration followed by regeneration, these chronic cycles of degeneration and regeneration progressively lead to exhaustion of satellite cell pools [9, 11]. As regeneration slows down and can no longer keep up with rapid degeneration, damaged myofibers are replaced with adipose and fibrotic tissues instead of new muscle tissue [9, 11]. The exhausted regenerative capacity along with chronic inflammation exacerbates the dystrophic phenotype.
The clinical onset and diagnosis of DMD occur between 3– 5 years of age. During this period, the affected children display walking difficulties, and elevated creatine kinase levels [3, 12, 13]. Dystrophic muscles of DMD patients display muscle necrosis, invasion of inflammatory cells, impaired regeneration due to exhausted satellite cell pools, and progressive fibrosis and adiposis [6]. As the disease progresses, the affected individuals are wheelchair bound at around 11 years, require ventilation support and, death ensues due to respiratory or cardiac failure between ages 20 to late 30 [1, 14, 15].
Although there is no cure for DMD right now, the current treatment for DMD has increased the lifespan of patients by 7 years since the 1980s [15]. Current treatments of DMD include steroids, surgery and assisted ventilation. Steroids, such as prednisone and deflazacort, are administered at daily doses of 0.75mg/kg and 0.9mg/kg respectively to prolong ambulation in children with DMD [16– 20]. Continuing steroid treatment into adulthood (after the loss of ambulation) aims to achieve the benefits of the treatment (respiratory muscle strength and delay in scoliosis) with fewer side-effects (weight gain and bone fragility), via an alternative dosing regimens (e.g. alternate day, high-dose weekend, or a 10-day “on” cycling with 10 or 20 days “off”) [20]. Surgery can be considered to correct for lower limb contractures (joint, ankle and knee contractures) and scoliosis [21]. Assisted ventilation has increased the lifespan of DMD patients by 10 years or more [22]. Non-invasive ventilation forces air into the lungs and is used to assist coughing, nocturnal hypoventilation and later during daytime hypoventilation [21]. Non-invasive ventilation is usually preferred over tracheostomy as it ensures a better quality of life while prolonging survival [21, 23, 24].
Interestingly, dystrophin deficiency observed in Becker muscular dystrophy (BMD) patients show varying clinical symptoms, wherein many display a much milder phenotype than DMD patients, and some even display an asymptomatic phenotype [25– 27]. The reading frame theory, which is well substantiated, explains that milder phenotypes observed in BMD are caused by in-frame mutations in the DMD gene. These in-frame mutations maintain the reading frame and result in the formation of truncated, internally deleted dystrophin protein. The reading frame theory explains the difference in phenotypes between DMD and BMD patients in 92% of cases [27]. However, in the remaining 8% of the cases, patients display severe phenotypes with in-frame deletions, duplications, and/or due to epigenetic and environmental factors [28].
Here, we will discuss the developments in therapeutic approaches and these include: Exon skipping, gene replacement therapy, stem cell therapy, utrophin up-regulation and read-through therapy using pharmacological agents. Table 1 provides a brief description of therapeutic approaches of DMD. Subsequently, we will focus specifically on murine models: The merits and caveats of each model and their applications in preclinical research. The mouse models discussed here are the standard mdx (with C57BL/10 background), mdx on C57BL/6 background (C57BL/6-mdx), mdx52, dystrophin/utrophin double-deficient (dko), mdxβgeo, Dmd-null, humanized DMD (hDMD), mdx on DBA/2 background (DBA/2-mdx), Cmah-mdx, and mdx/mTRKO murine models.
Therapeutic approaches
Exon Skipping: Many consider exon skipping using antisense oligonucleotide (AONs) as one of the most promising therapeutic approaches [29– 32]. This approach focuses on restoring the reading frame of dystrophin mRNA using AONs [33– 35]. The quasi-dystrophin produced after exon skipping must be partially functional as it allows for milder phenotypes, similar to those seen in BMD patients [36– 38]. However, exon-skipping is not without limitations: Dystrophin restoration induced by phosphorodiamidate morpholino oligomer (PMO or morpholino) AON exon-skipping lasts for only up to 8 weeks in dystrophic dogs and repeated AON administration is required to sustain its therapeutic effects, and issues with low exon skipping efficiency [39]. To overcome these limitations, developments in exon skipping include multiple exon skipping and, the use of various AON delivery systems to improve efficiency [40]. Using a cocktail of AONs allows for multiple (as opposed to single) exon splicing, thereby potentially increasing the applicability of the treatment to 90% (instead of 60% ) of DMD patients [29, 41]. Skipping exon 45– 55 can potentially treat 63% of DMD patients with deletion mutation [36]. Tricyclo-DNA (tcDNA), a new class of AON higher dystrophin levels in diaphragm (50% ) and heart (40% ) and, 3– 4 fold higher skipping than 2’-O-methyl phosphorothioate (2′OMePS) and PMO at equimolar dosing regimens in mdx treated mice [42]. Moreover, new generation morpholinos such as octa-guanidine conjugated vivo-morpholinos (vPMOs) and peptide-linked PMOs (PPMOs), have a cell-penetration moiety and more effective AON chemistries than unmodified morpholinos [43]. Thereby, they are more efficiently delivered into various tissues and have a higher efficacy of dystrophin rescue [43]. Drisapersen, a 2’OMePS exon-skipping drug (ClinicalTrials.gov: NCT01254019), was unsuccessful at Phase III clinical trial as it did not yield statistically significant improvements in the 6 minute walking distance test (6MWT) compared to placebo [44, 45]. According to post-trail ad hoc analysis, drisapersen failure may be due to variation in patients’ age (large number of older participants), disease severity and standards of care among different countries [46]. Limitations in 6MWT arise when differences in age and height (which affects stride length) of patients’ are observed. According to Goemans et al., pooled analysis of two phase II trials suggested that drisapersen can slow down the disease when treated at younger ages and for an extended time [46, 47]. Currently, drisapersen continues to be developed by BioMarin. While 2’OMePS have ribose rings, a negative charge and are structurally similar to RNA, morpholinos are more stable, less toxic and have reduced off-target effects due to their 6-membered ring (lack of similarity to RNA) and neutral charge [48, 49]. Another clinical trial led by Sarepta Therapeutics is investigating the efficacy and safety of a PMO exon-skipping drug called eteplirsen, in advanced stage DMD patients who can undergo exon 51 skipping (ClinicalTrials.gov: NCT02286947) [50].
Gene replacement: This therapy aims to restore dystrophin expression by replacing the mutant DMD gene with a synthetic substitute using recombinant adeno-associated virus (AAV) vectors [51– 57]. AAV is non-pathogenic, and infects non-dividing cells [33, 58]. However, the AAV vector cannot carry the whole DMD gene due to its small packaging size [33, 59]. In order to accommodate for the small packaging size of the vector, less essential regions of the DMD gene are removed to form micro-dystrophin, a truncated but functional form of dystrophin [56, 59– 63]. Interest in AAV therapy arose from its transduction ability in quiescent satellite cells, persistent expression of delivered transgenes and non-pathogenicity [56, 64– 67]. While AAV vectors display low immunogenicity than other vectors, the host’s humoral and cellular immune responses remain a major concern [68]. Dystrophin epitopes from rare ‘revertant’ (truncated dystrophin-positive) fibers (RFs) could sensitize autoreactive T cells and mount an immune response against the transgene product [69]. However, the potential for an immune response can be reduced by intramuscular administration, doses ranging from 2E11 vg/kg to 1.8E12 vg/kg, pre-screening against vector specific neutralizing antibodies and by administering immunosuppressants [54, 70]. A Phase I clinical trial was recently conducted using AAV2.5 vectors (rAAV2.5-CMV-minidystrophin; ClinicalTrials.gov number: NCT00428935]. Each of the two-dose (2.0E10 vg/kg and 1.0E11 vg/kg) cohort studies of three subjects were administered in the biceps of six DMD patients and was found to be safe and well tolerated [67, 71]. Currently, a Phase I clinical trial involves AAV1 vectors (rAAV1.CMV.huFS344; ClinicalTrials.gov number: NCT02354781) which is administered in quadriceps, tibialis anterior gluteal muscles to six DMD patients at a total dose of 2.4E12 vg/kg [72].
Stem cell therapy: Satellite cells are muscle stem cells that allow for muscle regeneration after injury and are located between the sarcolemma and basal lamina of myofibers [73, 74]. Dystrophic muscles undergo continuous cycles of degeneration and regeneration in the dystrophic muscles eventually reduces the ability of resident satellite cells to regenerate injured muscle [73]. This leads to the loss of muscle mass and compensatory insertion of fibrofatty tissue [73]. A limitation of gene replacement and exon skipping therapies is that the stage of the disease determines the effectiveness of the treatment because fibrofatty tissue replaces muscle cells with the progression of the disease [33, 75]. Ideally, stem cell therapy can overcome this hurdle by allowing for increased muscle regenerative capacity in dystrophic muscles [33, 76]. However, the transplantation of satellite cells show limited migration and self-renewal capacity. Stem cell types such as mesoangioblasts and CD133 + cells are able to enter and self-renew satellite cell niches, contribute to muscle regeneration and, unlike satellite cells and myoblasts, they can be delivered systemically [75, 77]. Mesoangioblasts are blood vessel-associated stem cells, which can pass through the walls of blood vessels and differentiate into myofibers [78]. CD133 + cells are human-derived and can differentiate into muscle stem cells [79]. Other developments include, human induced pluripotent stem cells (iPSCs), which are derived by reprograming adult somatic cells into a pluripotent state, and are similar to embryonic stem cells in morphology and gene expression [75, 80, 81]. The advantage of this therapy includes the production of large numbers of myogenic progenitors, the lack of ethical issues that surrounded embryonic stem cells, and the potential to devise patient-specific iPSCs, ideally preventing a host’s immune response [33]. Another kind of stem cells are mesenchymal cells, which are multipotent and can give rise to many tissues including skeletal and cardiac [82]. Aside from their regenerative properties and ability to be delivered systemically, mesenchymal stem cells are most advantageous for their anti-inflammatory properties [82]. Yet, stem cell therapy comes with challenges such as immune and inflammatory reactions, poor survival and limited migration of injected cells [83– 87].
Utrophin upregulating is another viable therapy because utrophin is a protein very similar to dystrophin with 80% amino acid sequence homology and takes the functional role of dystrophin during foetal muscle development [88]. The advantage of induced utrophin expression is that it could potentially prevent an immune response against dystrophin [89]. A drug called Biglycan, is a proteoglycan found endogenously in mice and humans, which stabilizes the muscle membrane by recruiting utrophin to the sarcolemma [90]. SMT C1100 is another oral drug that upregulates utrophin and reduces muscular dystrophy in mdx mice [91]. However, phase 1a clinical trial showed low plasma levels of SMT C1100 and, a phase 1b clinical trial (which was recently completed) tested the safety and tolerability of SMT C1100 at higher doses (however, the results are not yet published) (ClinicalTrials.gov number: NCT02056808) [91]. SMT022357 is a second generation drug with better metabolic and physiochemical profile than SMT C1100 [92]. It shows increased utrophin expression in cardiac, respiratory, and skeletal muscles in mdx mice and decreases necrosis and fibrosis [92]. Utrophin upregulation cannot completely restore muscle function to normal, possibly due to its inability to bind to neuronal nitric oxide synthase (nNOS) and/or due its structural differences to dystrophin [93]. Nevertheless, utrophin upregulation improves muscle function and reduces muscular dystrophy, and is applicable to all patients regardless of their mutation type [93].
Read through therapy involves suppression of nonsense mutations in DMD patients [94– 96]. Gentamicin, an antibiotic allows for read through of premature termination codon (PTC) mutations, i.e. nonsense mutation, by replacing a stop codon with a new amino acid to continue translation [95, 97, 98]. However, it is not used clinically in DMD patients due to serious dose limiting toxicities including a hearing loss. PTC124 (also known as Ataluren) is a drug that appears more potent than gentamicin in restoring dystrophin expression although there exist some controversies regarding its read through ability [99]. Ataluren is currently being investigated in a phase III trial for its efficacy during a 6 minute walk test in DMD patients with nonsense mutations (ClinicalTrials.gov number: NCT01826487) [99, 100]. Generally, the applicability of read through therapies is limited to around 10 – 15% of DMD cases [101].
Endonuclease-based gene repair: Nuclease-mediated genome editing creates site-specific double stranded breaks in DNA [102, 103]. This cellular DNA repair mechanisms, such as homologous recombination (HR) or non-homologous end joining (NHEJ) mechanisms, result in insertions or deletions at break points that may lead to wild-type sequence correction [104]. The four engineered endonucleases recently developed include meganucleases, zinc-finger nucleases, transcription activator-like effector nucleases (TALEN) and, clustered regularly interspaced short palindromic repeat/Cas9 (CRISPR/Cas9) [102, 104– 106]. This therapy is able to restore the normal reading frame of the dystrophin gene, delete a nonsense codon and knockout a gene [103]. This therapy recently emerged in DMD studies, allowing permanent gene correction (by precise modifications at the target locus), and hence, overcomes the hurdle of transient mRNA correction (which calls for continuous drug administration) associated in AON-exon skipping and pharmacological read through therapies [106]. The advantage of this therapy is that it creates precise modifications at the target locus, and hence, yields a specific protein product with predictable functionality [105].
Murine Models of DMD
To name a few, among the many different animal models of DMD, are zebrafish, dog and pig models. Homozygous sap mutant zebrafish have a nonsense mutation at the N-terminal domain of sapje (sap) locus (an orthologue of DMD locus), resulting in the loss of dystrophin, muscle degeneration and, extensive fibrosis and inflammation [107]. The zebrafish model is useful for screening small-molecule drugs and visualizing molecular processes in vivo as the embryos and larvae are translucent [107]. However, these non-mammalian zebrafish models are phylogenetically far apart from humans. The commonly studied, Golden Retriever muscular dystrophy (GRMD) dog model harbours a mutation in intron 6, leading to a premature stop codon in exon 8, and are more similar to DMD patients in disease severity than mouse models [108– 110]. Beagle-based canine X-linked muscular dystrophy (CXMD) dogs are crossed to GRMD to contain the same mutation but are smaller and easier to handle than GRMD [111]. However, dogs with identical mutations can show large differences in dystrophic phenotype, which can blur end points and confound data interpretation [112– 115]. Pigs are more similar in anatomy, physiology, and genetics to humans than dogs and mice, but the newly developed pig models are not yet used in preclinical studies. Transgenic pig with a mutation in DMD exon 52 show symptoms similar to DMD patients, such as, elevated serum creatine levels, lack of functional dystrophin, and progressive fibrosis [111, 116, 117]. However, it also displays upregulation of utrophin (dystrophin homologue) as observed in mouse models [116, 117]. While the spontaneous substitution of arginine to tryptophan, in exon 41 results in dystrophinopathy, the affected pigs display a BMD-like (and not a DMD) phenotype [118, 119].
Murine models are often used to lay the groundwork for DMD studies including the pathogenesis of DMD and, the efficacy and toxicity of therapeutics [6]. However, murine models also have limitations such as lack of host immune responses to therapeutic agents (e.g.: Vector capsids) and, small size (compromising the ability to produce and deliver scaled-up amount of vectors to large volumes of muscles) [120]. Nevertheless, murine models are valuable animal models for research as they can be bred and genetically engineered with relative ease, and they are less expensive than other large animal models such as dogs and pigs. Many mouse models such as hDMD, Cmah-mdx, mdx/mTRKO and DBA/2 background have been recently developed. Table 2 provides a brief summary of the dystrophic features of murine models discussed in this review paper.
Mdx on C57BL/10 background
Features of mdx mice: M dx, a commonly used classic mouse model, harbors a spontaneous point mutation at exon 23 of the Dmd gene, leading to the loss of dystrophin. M dx arose from an inbred strain of C57BL/10. M dx pathogenesis involves increase in creatine kinase levels, muscle degeneration, variation of fiber size, and centrally nucleated fibers (CNFs) indicative of muscle regeneration [6, 121]. While young mdx mice display mild cardiomyopathy, older mdx mice (especially female mice between ages 20 to 22 months) show severe dilated cardiomyopathy, frequent premature ventricular contractions, and cardiac fibrosis [122, 123]. Mdx has a much milder phenotype and normal lifespan compared to DMD patients: It does not exhibit impaired regeneration, accumulation of fibrofatty tissue, reduced myofiber number, except for in the diaphragm [124, 125]. The mild phenotype of mdx mice can be explained by (1) high regenerative capacity: The satellite cell pools of C57BL/10 were able to renew themselves even after 50 cycles of severe degeneration-regeneration (2) upregulation of utrophin, a dystrophin homologue, throughout their lifespan (unlike DMD patients), attenuating the effects of dystrophin deficiency [126].
Involvement in therapeutic approaches: The mdx (C57BL/10 background, C57BL/10-mdx) mouse is the most widely used model of DMD [127, 128]. In an effort to reduce the mild dystrophic phenotype of mdx mice, high dose irradiation of mdx muscles were employed to block muscle regeneration [129, 130]. For instance, one study irradiated hind limb muscles of mdx mice which prevented the expansion of revertant fibers (RFs), and showed that RF expansion depends on muscle regeneration [131]. Another study genetically labelled (LacZ reporter) myofibers which were then transplanted in irradiated hindlimb muscles of mdx mice, resulting in self renewal of satellite stem cell pools [132]. Mdx mice on various immunodeficient backgrounds, such as mdx-null and recombinase-activating gene (Rag)2-/γ chain-/C5- mice (which is required for V(D) rearrangement), were created to evaluate gene and cell therapies, without the compounding effects of an immune response [120]. Meng et al. reported that the efficiency of transplanting human muscle stem cells (pericytes and CD133 + cells) into mouse muscles depends on the environment and the mouse strain [133]. They reported that there were more myofibers and satellite cells of donor origin in (Rag)2-/γ chain-/C5- mice than mdx-nude mice and, that cryoinjured muscles provided a more permissive environment for transplantation than irradiated muscles [133]. Mdx mice have also been used in developing pharmacological treatments of DMD, such as VBP15. VBP15, a synthetic corticosteroid oral drug, inhibits NF-κB and doesn’t lead to side effects associated with currently used steroids (e.g. prednisolone) since it doesn’t stimulate glucocorticoid-responsive element (GRE) transactivation [134]. Mdx mice treated with VBP15 (15 mg/kg) showed increase force in extensor digitorum longus (EDL) muscles by 12% and 16% in the two preclinical trials, while prednisolone showed no increase in force [134]. For maximal force exerted by forelimb muscles of mdx mice, VBP15 showed increase in force while prednisolone showed a decrease compared to non-treated mdx mice likely because the mdx mice treated with prednisolone displayed growth retardation [134]. Aside for improvements in muscle strength, VBP15 (15 mg/kg) treated mice showed a 38% reduction in inflammatory foci compared to non-treated [134]. VBP15 in currently undergoing a randomized, double-blinded and placebo-controlled phase 1 clinical trial in healthy adults, to evaluate the safety of VBP15 after a single dose and after 14 daily doses of VBP15 (ClinicalTrials.gov Identifier: NCT02415439). Arginine pyruvate is another pharmacological drug and was shown to protect mdx mice against cardiac hypertrophy by 25% , ventricular dilation by 20% , and kyphosis by 94% [128].
Mdx mice on C57BL/6 background
Features of C57BL/6-mdx mice: M dx on C57BL/6 background (C57BL/6-mdx) is a novel murine model that is valuable in comparative studies, involving the use of mouse models such as mdx52. The C57BL/10 genetic background of mdx mice poses as a barrier to analyze and compare the phenotype of other mouse models such as mdx52 (which possesses a C57BL/6 genetic background). C57BL/10 inbred strain is akin to and shares a common origin with C57BL/6 but differs in allelic variants at H9, Igh2 and Lv loci [136]. C57BL/6 genetic background was employed in mdx2cv, mdx3cv, mdx4cv and mdx5cv, which were created by treating the mice with chemical mutagens (ethylnitrosourea (ENU)) (see Table 2) [137] Mdx2cv and mdx3cv mice both harbor a point mutation at the splice acceptor site in intron 42 and in intron 65, respectively. Mdx4cv mice harbor a nonsense mutation in exon 53. A point mutation in mdx5cv mice causes a new splice site in exon 10 [67]. The different mutation locations in these mdx strains relative to the seven different promoters in the Dmd gene leads to a wide array of dystrophin isoforms and hence, these mutants might be useful in studies involving dystrophin function and expression [67]. Aside from being useful in comparative studies involving mouse models with similar genetic background, C57BL/6-mdx mice cannot recapitulate the DMD phenotype any better than mdx mice.
Involvement in therapeutic approaches: There are not many therapeutic studies that involve the use of C57BL/6-mdx mice. Wang et al. reported that induced pluripotent stem cells (iPSCs) from muscle fibroblasts of 14 month C57BL/6-mdx mice (14m-MuF-iPSCs), showed lower proliferation and reprogramming activity than younger C57BL/6-mdx mice [135]. They also showed that the inhibition of TGF-β and BMP signalling stabilized the 14m-MuF-iPSCs, which differentiated into skeletal muscles as efficiently as iPSCs from younger C57BL/6-mdx mice [135]. Fukada et al. report that C57BL/6 strain has the best self-renewal capacity among four inbred strains of mdx mice: C57BL/6, DBA/2, BALB/c, and C3H/HeN [138]. C57BL/6-mdx mice are observed to have a significantly higher count of RFs than mdx52 at all age groups (2, 6, 12 and 18 months) examined [139]. Since the background of these murine model were identical, the results suggest that age, the type and the location of the mutation in the Dmd gene influences the expression and expansion of RFs in skeletal muscles [139].
Mdx52 mice
Features of mdx52: M dx52 mice, developed in 1997 by Araki and colleagues, contain a deletion of exon 52 of the Dmd gene, resulting in the absence of full-length dystrophin [140]. These mice exhibit muscle necrosis, regeneration and hypertrophy, and more importantly, lacks the expression of two of the four shorter dystrophin isoforms, Dp140 and Dp260 (Fig. 3) [140]. Since the mouse models of that time (except for mdx3cv) expressed all dystrophin isoforms, mdx52 was developed to study how deficiency in these isoforms influences the disease phenotype. While mdx52 mice display skeletal muscle pathology similar to mdx mice, the location of its deletion mutation, advantageously corresponds, to the hot spot region (exons 45– 55) of mutations in DMD patients. Approximately 70% of DMD deletion mutations are located in this central region [141, 142]. Additionally, absence of Dp260 isoform in mdx52 mice causes abnormal electroretinograms (ERG) similar to DMD and BMD patients, who lack Dp260 due to mutations in exon 44– 53 [143, 144]. Figure 2A shows that mdx52 mice have lower RF expansion (low RFs numbers within a single cluster) than age-matched mdx mice (which amounts to a 58% lower RF expansion at 12 months of age as reported by Echigoya et al., 2013) [139]. Hence, it is thought to be a better mouse model at evaluating dystrophin restoring therapies because naturally existing RF might prevent accurate assessment of a therapeutic efficacy.
Involvement in Therapeutic Approaches: Exon 51 skipping is the most common target for single exon skipping therapies and is applicable to 13% of all DMD patients [34, 38]. Skipping exon 51 using PMOs restored bodywide expression of in-frame dystrophin (20% – 30% of normal levels) in mdx52 mice along with improved muscle function [145]. Exon 51 skipping induced by intramuscular PMO injection in mdx52 mice was recently shown to have the highest percentage of dystrophin positive fibers at 5 weeks of age, when muscle regeneration was very active [146]. PMO uptake into muscle cells of mdx52 seems effective during myogenic differentiation to myotube formation; specifically PMO and 2’OMePS were most efficiently delivered in dystrophic muscles at early stages of C2C12 myotube formation [146].
Multiple exon skipping of exons 45– 55 in whole body skeletal muscles using vPMOs restored dystrophin expression up to 15% and ameliorated skeletal muscle pathology in mdx52 mice [145, 147]. This multiple exon skipping therapy is theoretically applicable to 63% of DMD patients with out-of-frame deletion mutations [34, 38]. In addition, this specific mutation is associated with exceptionally mild BMD patients or asymptomatic individuals [148, 149]. M dx52 is a valuable model for evaluating exon skipping therapies as its deletion mutation is associated with the hot spot region of the human D MD gene.
Dko mice
Features of dko mice: Dko is a double deficient mouse model that lacks dystrophin and utrophin [150]. Dko was developed to reflect the absence of utrophin protein observed in adult DMD patients, and thereby devise a more severe phenotype than mdx mouse model. Dystrophic features of dko mutants include severe and progressive muscle wasting, weight loss after weaning, abnormal breathing rhythms, early onset of joint contractures and kyphosis leading to slack posture and premature death between 4 to 20 weeks [150, 151]. Although respiratory failure appears to be the primary cause of death in dko mutants, cardiomyopathy and swallowing difficulties due to weak tongue muscles might be contributing factors [150, 151]. However, since dko mice die prematurely (mostly around 10 weeks), they are hard to generate and maintain [152]. Dko mice have more severe dystrophic phenotype than mdx because they lack compensatory utrophin expression that is present in mdx mice [150, 151]. Recent studies suggest that as little as 5% dystrophin expression levels can extend the lifespan of dko mice [153, 154]. Clinical symptoms such as waddling gait, kyphosis and short life span observed in dko mice are similar to those observed in DMD patients [150, 151]. Dko mice also express higher levels of immunoproteasome than mdx and display severe atrophy [155]. Mdx (C57BL/10 background) and utrophin-deficient (C57BL/6 background) mice were crossed multiple times to obtain dko mice with hybrid genetic background [150]. It might be more useful to mate C57BL/6-mdx with utrophin-deficient mice to rule out differences in genetic background.
Involvement in Therapeutic Approaches: D ko mutants have been used in gene therapies testing, such as exon skipping, and gene replacement using virus vectors. PPMO targeting exon 23 restored dystrophin expression in almost all skeletal muscles and restored expression of dystrophin associated protein such as glycosylated dystroglycan and neuronal nitric synthase in all age groups of dko mutants [156]. It was found that early treatment of PPMO (i.e. during 20– 29 days of age) restored dystrophin expression in almost all skeletal muscles of dko mice and resulted in delayed disease progression, prevented severe kyphosis and eye infection, and increased life span of dko mutants [156]. However, treatment of PPMO at an advanced stage of the disease had little effect on dko mice even in the presence of high levels of dystrophin [156]. The likely reasons for this finding in later stage are severe loss of muscle fibres and its replacement by fibrotic tissue, along with severe kyphosis [156]. Utrophin upregulation therapy is advantageous in immune response evasion against dystrophin. D ko mutants were also used to test the efficacy of utrophin minigene delivery using adenovirus vectors [157]. Utrophin minigene was found in nearly 95% of muscle fibers 30 days after injection along with a significant reduction in necrosis and an 85% reduction of centrally nucleated fibers (likely due reduced degeneration) was observed in TA muscles compared to non-treated dko mice [157]. Recently, small nuclear RNAs (U7snRNA) along with AONs were packaged into AAV vector (scAAV9-U7ex23) and intravenously injected into dko mice [158]. This approach of using small nuclear RNA in antisense mediated-exon skipping therapy was employed to overcome hurdles such as, low efficacy in cardiac muscles, poor uptake and rapid clearance of the drug [158]. Treated dko mice displayed increased dystrophin levels (among 45% to 95% ) in all muscles including cardiac muscle, improved muscle function, and increased lifespan (50.2 weeks compared to 10.2 weeks in non-treated dko mice) [158].
Mdxβgeo
Features of mdxβgeo: M dxβgeo contains an insertion of a gene trap vector (ROSAβgeo) in exon 63 of the Dmd gene, resulting in the loss of cysteine rich and C-terminal domains (as illustrated in Figure 3A) [159]. This mouse model was developed by Wertz & Fuchtbauer in 1998 [159]. And unlike the spontaneous and ethylnitrosourea (ENU)-induced mutant mice of that time, mdxβgeo had all isoforms mutated and could detect the Dmd gene expression early in embryogenesis and in adult organs (such as the brain, liver, eye, pancreas and lung) by staining for β-galactosidase (LacZ reporter) [159]. Mdxβgeo mice display a loss of dystrophin isoforms (including Dp71), abnormally dilated esophagus, cardiac hypertrophy, and other typical dystrophic features such as muscle degeneration, cellular infiltration, and regenerated fibers with centrally located nuclei [159]. Full-length dystrophin was absent in skeletal muscles, however, trace amounts of PCR product reflecting wild-type mRNA was detected in the brain [159]. Krasowska et al. used mdxβgeo and inhibitory synaptic markers (such as neuroligin2 and vesicular GABA transporter) to show that cognitive impairments in DMD patients might be due to aberrant clustering of receptors at inhibitory synapses in the hippocampus [160].
Dmd-null
Features of Dmd-null mice: Dmd-null mice contain a deletion of the entire D md gene on mouse chromosome X using a Cre-loxP recombination technique [161]. Dmd-null mice were developed to prevent the expression of all dystrophin isoforms (Fig. 3B illustrates dystrophin isoforms) [161]. While mdxβgeo may express dystrophin isoforms, Dmd-null mice can express neither revertant fibers nor any of the isoforms as its alternative splicing (exon skipping) ability is lost due to the deletion of the entire gene [162]. Dmd-null mice display muscle hypertrophy, behavioural abnormality, infertility and other dystrophic features similar to mdx mice [161]. These mice are useful in transgenic studies that investigate the function of dystrophin isoforms [161].
hDMD mice
Features of hDMD: Humanized DMD mouse model (B6.DBA2.129-hDMDtg/tg) has been engineered to carry the complete human DMD gene in chromosome 5 of the mouse genome (wild type) [163, 164]. This is not a disease model as it allows for the expression of full-length human dystrophin protein as well as intrinsic murine dystrophin. ‘t Hoen and colleagues designed the humanized DMD model (hDMD) to assess the efficacy and safety of human specific AONs in vivo for sequence specific therapies such as exon-skipping [163]. The hDMD mouse model might provide further insight into gene regulation, genomic stability, and frequency of mutations and recombination in the DMD gene [163]. The hDMD mouse model might potentially be engineered in future to carry mutations in the human DMD gene in a dystrophin-deficient, mdx background [163].
Involvement in Therapeutic Approaches: The hDMD murine model is advantageous to test sequence specific therapies such as exon skipping. Optimization of human specific AONs could only be previously conducted in vitro. hDMD mice are very useful as it allows for preclinical testing and optimization of human specific AONs in vivo [165, 166]. Goyenvalle et al. employed the hDMD mouse model to evaluate the in vivo efficacy of 11 different U7 small-nuclear RNA in the splicing of exon 45– 55 [167]. Their constructs, which were packaged in an AAV vector, could achieve an efficient multi-exon skipping of at least 3 exons in the DMD gene [167]. On the other hand, crossing hDMD mice with mdx or dko mouse models rescued the dystrophic phenotype as human dystrophin compensated for the lack of dystrophin in the mice [164]. Histological results showed normal fiber size, absence of CNFs and lack of fibrosis [164]. Ongoing experiments aim to induce deletions in the human DMD gene of the hDMD/mdx mouse. This would have great value in preclinical in vivo studies of muscle function, dystrophin expression and the overall success of a particular AON treatment.
Mdx on DBA/2 background
Features of DBA/2-mdx mice: M dx on DBA/2 background (DBA/2, DBA/2-mdx) has a more severe dystrophic phenotype than mdx (C57BL/10 background) and shares more histopathological features with DMD patients. Fukada and colleagues developed the DBA/2-mdx mouse model which is available in Jackson laboratory and Central Institute for Experimental Animals (CIEA) Japan. The DBA/2 inbred strain is considered a challenging breeder and possesses many mutated genes: They are highly susceptible to hearing loss (Cdh23ahl), eye abnormalities reflective of glaucoma (GpnmbR150X and Tyrp1isa), extremely intolerant to alcohol and morphine (Klrd1DBA/2J) [168, 169]. Unlike C57BL/6 strain, DBA/2 strain is susceptible to audiogenic seizures and resistant atherosclerotic aortic lesions [170– 172]. Moreover, DBA/2 mice also display shorter life spans, more pronounced weight loss with age (sarcopenia) and significantly lower self-renewal efficiency of satellite cells than that of C57BL/6 [138]. Unlike mdx mice, mdx on a DBA/2 background show reduced muscle mass, increased fibrosis, and fatty tissue accumulation and reduced regeneration potential of satellite cells, resulting in prominent muscle weakness [138]. Figure 2B shows that DBA/2-mdx mice show a lower percentage of CNFs than mdx and mdx52 mice at 2 months (a 33% reduction of CNFs was shown from unpublished data). The self-renewal ability of satellite cells might explain the difference in phenotypes between mdx and DBA/2-mdx mice [61, 92].
Involvement in therapeutic approaches: DBA/2-mdx is a very new murine model and hence, there are not many therapeutic studies involving its use. Imatinib, a tyrosine kinase inhibitor, blocks the expression of PDGFRα (tyrosine kinase receptors) in skeletal muscle mesenchymal progenitors and reduces fibrosis in DBA/2-mdx mice [173]. Additionally, the therapeutic dose of imatinib does not influence the proliferation of myoblasts in vitro and its use may be promising for stem cell therapies [173].
Cmah-mdx mice
Features of Cmah-mdx: Cmah-mdx mice, developed by Chandrasekharan and colleagues, harbor two mutations: A deletion mutation in the C mah gene (Cmahtm1Avrk) and a nonsense mutation in exon 23 of the Dmd gene (Dmdmdx) [174]. The CMAH gene is required for the expression of N-acetylneuraminic acid (Neu5Ac), a type of sialic acid that is incorporated in glycan structures such as glycoproteins and glycolipids [175, 176]. Mice lacking only the Cmah gene display impairments in humoral immune function, coordination, hearing and wound healing [177, 178]. While the Cmah gene is expressed in mice, it is naturally inactive in humans [179]. Knocking-out the Cmah allele eliminates Neu5Ac in all cells of the mdx mice and humanizes the glycan structures in mice [178, 180]. Chandrasekharan et al. reports that changing the sialylation in mdx mice, brought about by the Cmah gene deletion, enhances the disease severity in the mice [174]. In contrast to mdx mice, Cmah-mdx mice showed increased mortality, loss of ambulation, and increased cardiac and skeletal impairment at an earlier age and/or to a greater extent [174]. At 11 months of age, nearly 50% of the Cmah mice died [174]. In comparison to mdx, Cmah-mdx mice at 8 months showed a 70% reduction in constant speed (5 rpm) rotarod test (loss of ambulation), and a reduction in peak force by 88% and 66% for diaphragm and cardiac trabeculae, respectively [174]. Cmah-mdx mice also had increased fibrosis in the quadriceps at 6 weeks of age, increased regions of necrosis in the heart at 3 months of age and, increased fibrosis in the diaphragm relative to mdx mice at 6 months of age [174]. Chandrasekharan et al. discussed two mechanisms that leads to the accelerated and more severe dystrophic phenotypes in Cmah-mdx mice: 1) diminished function of dystrophin-glycoprotein complex including reduced binding of extracellular matrix proteins to α−dystroglycan and reduced utrophin upregulation, 2) increased activation of complement (C5b-9) driven by increased expression of antibodies specific to dietary Neu5Gc, a foreign glycan in Cmah-deficient mice [174]. Currently, there are no published therapeutic approaches involving Cmah-mdx mice, a mouse model recently developed in 2010.
mdx/mTRKO
Features of mdx/mTRKO mice: mdx/mTRKO was generated by crossing mdx4cv mice with mice containing deletion in the RNA component TERC (mTR) of telomerase [181]. Telomerase is an enzyme that maintains the length of telomeres, which are DNA repeats that protect chromosomes from aberrant recombination, fusion and degradation [181]. Mdx/mTRKO was developed, as many studies showed that DMD patients progressively loose muscle regenerative capacity with age and, that telomere shortening increases with age in DMD patients and correlates with reduced regeneration [181]. Unlike mdx mice, mdx/mTRKO (with dystrophin deficiency and telomerase dysfunction) show a more severe dystrophic phenotype as seen in humans: impaired self-renewal capacity of stem cells, muscle wasting, accumulation of fibrosis and calcium deposits, increased creatine kinase levels, kyphosis, dilated cardiomyopathy, heart failure and shortened lifespan of around 12 months [181]. Mourkioti et al. suggest that dystrophin deficiency coupled with oxidative stress and metabolic demands of cardiac muscles leads to accelerated telomere shortening and progressive cardiomyopathy [182].
CONCLUSIONS
Although murine models differ in some respects to the clinical manifestations of DMD in humans, they are still valuable for basic and cost effective investigations involving pathogenesis, and in preclinical trials. Developments in murine models of DMD are essential for overcoming limitations of existing murine models such as mdx and for higher success in clinical trials. Modifications to mdx mice are useful for reducing the discrepancies in dystrophic phenotypes between mice and humans. For instance, inducing secondary mutations (e.g. Cmah-deficient mdx mice) that have important cellular effects (e.g. altering the form of glycosylation) or, modifying the genetic background (e.g. DBA/2-mdx mice), leads to increased severity of dystrophic phenotype observed in mdx (C57BL/10 genetic background) mice. Genetic background influences phenotype: DBA/2 inbred strain has a much lower regenerative capacity of satellite cells than C57BL/10 and C57BL/6 inbred strains, and DBA/2 inbred strain is shown to display reduced muscle weight and myofiber numbers than C57BL/6 inbred strain. Mdx52 mice are similar to and have the same genetic background as C57BL/6-mdx mice, but provide an added value, since it carries a deletion mutation corresponding to the hot spot region (exons 45– 55) of the DMD gene. DBA/2-mdx, mdx/mTRKO and dko mouse models provide a more severe dystrophic phenotype than mdx. Mdxβgeo and Dmd-null mice lack dystrophin isoforms (including Dp71) and revertant fiber expression, and hence, may be useful in assessing the efficacy of dystrophin amelioration in preclinical trials. The hDMD mouse model is useful for optimizing human specific sequences of AONs in pre-clinical trials. Overall, developments in murine models greatly help in their contributions to the therapeutic approaches for DMD in preclinical trials.
ACKNOWLEDGMENTS
This work is supported by Muscular Dystrophy Canada, Jesse’s Journey, The Friends of Garrett Cumming Research Fund, HM Toupin Neurological Science Research Fund, Canadian Institutes of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), Canada Foundation for Innovation (CFI), and Women and Children’s Health Research Institute (WCHRI). The project is supported financially through AIHS Summer Studentship Award, and Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowships for Research Abroad.
REFERENCES
[1] | Hoffman EP , Brown RH Jr , Kunkel LM . Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. (1987) ;51: (6):919–28. |
[2] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Annals of neurology. (2012) ;71: (3):304–13. |
[3] | Moser H . Duchenne muscular dystrophy: Pathogenetic aspects and genetic prevention. Human genetics. (1984) ;66: (1):17–40. |
[4] | Koenig M , Hoffman EP , Bertelson CJ , Monaco AP , Feener C , Kunkel LM . Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. (1987) ;50: (3):509–17. |
[5] | Campbell KP . Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell. (1995) ;80: (5):675–9. |
[6] | Nakamura A , Takeda S . Mammalian models of Duchenne Muscular Dystrophy: Pathological characteristics and therapeutic applications. Journal of Biomedicine & Biotechnology. (2011) ;2011: :184393. |
[7] | Whitmore C , Morgan J . What do mouse models of muscular dystrophy tell us about the DAPC and its components? Int J Exp Pathol. (2014) ;95: (6):365–77. |
[8] | Lapidos KA , Kakkar R , McNally EM . The dystrophin glycoprotein complex - Signaling strength and integrity for the sarcolemma. Circulation research. (2004) ;94: (8):1023–31. |
[9] | Wallace GQ , McNally EM . Mechanisms of Muscle Degeneration, Regeneration, and Repair in the Muscular Dystrophies. Annual review of physiology. (2009) ;71: :37–57. |
[10] | Collins CA , Olsen I , Zammit PS , Heslop L , Petrie A , Partridge TA , et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. (2005) ;122: (2):289–301. |
[11] | Rahimov F , Kunkel LM . The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. The Journal of cell biology. (2013) ;201: (4):499–510. |
[12] | Duchenne . The Pathology of Paralysis with Muscular Degeneration (Paralysie Myosclerotique), or Paralysis with Apparent Hypertrophy. British medical journal. (1867) ;2: (363):541–2. |
[13] | Hoffman EP , Kunkel LM . Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron. (1989) ;2: (1):1019–29. |
[14] | Eagle M , Baudouin SV , Chandler C , Giddings DR , Bullock R , Bushby K . Survival in Duchenne muscular dystrophy: Improvements in life expectancy since and the impact of home nocturnal ventilation. Neuromuscular disorders: NMD. (2002) ;12: (10):926–9. |
[15] | Passamano L , Taglia A , Palladino A , Viggiano E , D’Ambrosio P , Scutifero M , et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta myologica: Myopathies and cardiomyopathies: Official journal of the Mediterranean Society of Myology/edited by the Gaetano Conte Academy for the study of striated muscle diseases. (2012) ;31: (2):121–5. |
[16] | Biggar WD , Harris VA , Eliasoph L , Alman B . Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscular disorders: NMD. (2006) ;16: (4):249–55. |
[17] | Houde S , Filiatrault M , Fournier A , Dube J , D’Arcy S , Berube D , et al. Deflazacort use in Duchenne muscular dystrophy: An 8-year follow-up. Pediatric neurology. (2008) ;38: (3):200–6. |
[18] | Griggs RC , Moxley RT 3rd , Mendell JR , Fenichel GM , Brooke MH , Pestronk A , et al. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Archives of neurology. (1991) ;48: (4):383–8. |
[19] | Mendell JR , Moxley RT , Griggs RC , Brooke MH , Fenichel GM , Miller JP , et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. The New England journal of medicine. (1989) ;320: (24):1592–7. |
[20] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: (1):77–93. |
[21] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Implementation of multidisciplinary care. Lancet Neurol. (2010) ;9: (2):177–89. |
[22] | Wollinsky KH , Kutter B , Geiger PM . Long-term ventilation of patients with Duchenne muscular dystrophy: Experiences at the Neuromuscular Centre Ulm. Acta myologica: Myopathies and cardiomyopathies: Official journal of the Mediterranean Society of Myology/edited by the Gaetano Conte Academy for the study of striated muscle diseases. (2012) ;31: (3):170–8. |
[23] | Wagner KR , Lechtzin N , Judge DP . Current treatment of adult Duchenne muscular dystrophy. Biochimica et biophysica acta. (2007) ;1772: (2):229–37. |
[24] | Eagle M , Baudouin SV , Chandler C , Giddings DR , Bullock R , Bushby K . Survival in Duchenne muscular dystrophy: Improvements in life expectancy since and the impact of home nocturnal ventilation. Neuromuscular Disorders. (2002) ;12: (10):926–9. |
[25] | Monaco AP , Kunkel LM . Cloning of the Duchenne/Becker muscular dystrophy locus. Advances in human genetics. (1988) ;17: :61–98. |
[26] | Aartsma-Rus A , Van Deutekom JC , Fokkema IF , Van Ommen GJ , Den Dunnen JT . Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle & nerve. (2006) ;34: (2):135–44. |
[27] | Koenig M , Beggs AH , Moyer M , Scherpf S , Heindrich K , Bettecken T , et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. American journal of human genetics. (1989) ;45: (4):498–506. |
[28] | Beggs AH , Hoffman EP , Snyder JR , Arahata K , Specht L , Shapiro F , et al. Exploring the Molecular-Basis for Variability among Patients with Becker Muscular-Dystrophy - Dystrophin Gene and Protein Studies. American journal of human genetics. (1991) ;49: (1):54–67. |
[29] | Touznik A , Lee JJ , Yokota T . New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert opinion on biological therapy. (2014) ;14: (6):809–19. |
[30] | Lee EJ , Kim AY , Lee EM , Lee MM , Min CW , Kang KK , et al. Therapeutic effects of exon skipping and losartan on skeletal muscle of mdx mice. Pathology international. (2014) ;64: (8):388–96. |
[31] | Guncay A , Yokota T . Antisense oligonucleotide drugs for Duchenne muscular dystrophy: How far have we come and what does the future hold? Future Med Chem. (2015) ;7: (13):1631–5. |
[32] | Lu QL , Yokota T , Takeda S , Garcia L , Muntoni F , Partridge T . The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy. Mol Ther. (2011) ;19: (1):9–15. |
[33] | Seto JT , Bengtsson NE , Chamberlain JS . Therapy of Genetic Disorders-Novel Therapies for Duchenne Muscular Dystrophy. Current pediatrics reports. (2014) ;2: (2):102–12. |
[34] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human mutation. (2009) ;30: (3):293–9. |
[35] | Hoffman EP , Fischbeck KH , Brown RH , Johnson M , Medori R , Loike JD , et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N Engl J Med. (1988) ;318: (21):1363–8. |
[36] | Beroud C , Tuffery-Giraud S , Matsuo M , Hamroun D , Humbertclaude V , Monnier N , et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Human mutation. (2007) ;28: (2):196–202. |
[37] | Muntoni F , Torelli S , Ferlini A . Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. (2003) ;2: (12):731–40. |
[38] | Aoki Y , Nakamura A , Yokota T , Saito T , Okazawa H , Nagata T , et al. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol Ther. (2010) ;18: (11):1995–2005. |
[39] | Lee JJ , Yokota T . Antisense therapy in neurology. J Pers Med. (2013) ;3: (3):144–76. |
[40] | Yokota T , Nakamura A , Nagata T , Saito T , Kobayashi M , Aoki Y , et al. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic acid therapeutics. (2012) ;22: (5):306–15. |
[41] | Echigoya Y , Yokota T . Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides. Nucleic acid therapeutics. (2014) ;24: (1):57–68. |
[42] | Goyenvalle A , Griffith G , Babbs A , El Andaloussi S , Ezzat K , Avril A , et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nature medicine. (2015) ;21: (3):270. |
[43] | Wu B , Li Y , Morcos PA , Doran TJ , Lu P , Lu QL . Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Molecular therapy: The journal of the American Society of Gene Therapy. (2009) ;17: (5):864–71. |
[44] | GlaxoSmithKline. A Clinical Study to Assess the Efficacy and Safety of GSK2402968 in Subjects With Duchenne Muscular Dystrophy (DMD114044) 2014 [cited 2015 May 04]. Available from: https://clinicaltrials.gov/ct2/show/NCT01254019 |
[45] | Lu Q-l , Cirak S , Partridge T . What Can We Learn From Clinical Trials of Exon Skipping for DMD? Molecular Therapy Nucleic Acids. (2014) ;3: (e152). |
[46] | Aartsma-Rus A , Ferlini A , Goemans N , Pasmooij AM , Wells DJ , Bushby K , et al. Translational and regulatory challenges for exon skipping therapies. Hum Gene Ther. (2014) ;25: (10):885–92. |
[47] | Goemans N , Voit T , McDonald CM , Mercuri E , Wilson R , Wardell C , et al. Pooled analyses of efficacy parameters in patients with Duchenne muscular dystrophy (DIVED): Results from the drisapersen (DRIS) clinical trial programme. Neuromuscular Disorders. (2014) ;24: (9-10):829–30. |
[48] | Douglas AGL , Wood MJA . Splicing therapy for neuromuscular disease. Molecular and Cellular Neuroscience. (2013) ;56: :169–85. |
[49] | Amantana A , Iversen PL . Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Current opinion in pharmacology. (2005) ;5: (5):550–5. |
[50] | Kaye E , Laforet G . Safety Study of Eteplirsen to Treat Advanced Stage Duchenne Muscular Dystrophy 2015 [cited 2015 May 04]. Available from: https://clinicaltrials.gov/ct2/show/NCT02286947. |
[51] | Lostal W , Kodippili K , Yue Y , Duan D . Full-length dystrophin reconstitution with adeno-associated viral vectors. Human gene therapy. (2014) ;25: (6):552–62. |
[52] | Jarmin S , Kymalainen H , Popplewell L , Dickson G . New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert opinion on biological therapy. (2014) ;14: (2):209–30. |
[53] | Wright JF . Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene therapy. (2008) ;15: (11):840–8. |
[54] | Athanasopoulos T , Graham IR , Foster H , Dickson G . Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene therapy. (2004) ;11: :S109-S21. |
[55] | Heller KN , Montgomery CL , Shontz KM , Janssen PML , Clark KR , Mendell JR , et al. AAV Mediated Overexpression of Human alpha 7 Integrin Leads to Histological and Functional Improvement in Dystrophic Mice. Molecular Therapy. (2013) ;21: :S111-S. |
[56] | Koo T , Popplewell L , Athanasopoulos T , Dickson G . Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Human gene therapy. (2014) ;25: (2):98–108. |
[57] | Benchaouir R , Robin V , Goyenvalle A . Gene and splicing therapies for neuromuscular diseases. Frontiers in bioscience. (2015) ;20: :1190–233. |
[58] | Yoshimura M , Sakamoto M , Ikemoto M , Mochizuki Y , Yuasa K , Miyagoe-Suzuki Y , et al. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Molecular therapy: The journal of the American Society of Gene Therapy. (2004) ;10: (5):821–8. |
[59] | Gregorevic P , Allen JM , Minami E , Blankinship MJ , Haraguchi M , Meuse L , et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nature medicine. (2006) ;12: (7):787–9. |
[60] | Sakamoto M , Yuasa K , Yoshimura M , Yokota T , Ikemoto T , Suzuki M , et al. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochemical and biophysical research communications. (2002) ;293: (4):1265–72. |
[61] | Watchko J , O’Day T , Wang B , Zhou LQ , Tang Y , Li J , et al. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Human gene therapy. (2002) ;13: (12):1451–60. |
[62] | Wang B , Li J , Xiao X . Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proceedings of the National Academy of Sciences of the United States of America. (2000) ;97: (25):13714–9. |
[63] | Zhang Y , Yue Y , Li L , Hakim CH , Zhang K , Thomas GD , et al. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Human molecular genetics. (2013) ;22: (18):3720–9. |
[64] | Yuasa K , Yoshimura M , Urasawa N , Ohshima S , Howell JM , Nakamura A , et al. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene therapy. (2007) ;14: (17):1249–60. |
[65] | Howell JM , Lochmuller H , O’Hara A , Fletcher S , Kakulas BA , Massie B , et al. High-level dystrophin expression after adenovirus-mediated dystrophin minigene transfer to skeletal muscle of dystrophic dogs: Prolongation of expression with immunosuppression. Human gene therapy. (1998) ;9: (5):629–34. |
[66] | Ghosh SS , Gopinath P , Ramesh A . Adenoviral vectors - A promising tool for gene therapy. Applied biochemistry and biotechnology. (2006) ;133: (1):9–29. |
[67] | Mendell JR , Campbell K , Rodino-Klapac L , Sahenk Z , Shilling C , Lewis S , et al. Dystrophin immunity in Duchenne’s muscular dystrophy. The New England journal of medicine. (2010) ;363: (15):1429–37. |
[68] | Zaiss AK , Muruve DA . Immune responses to adeno-associated virus vectors. Current gene therapy. (2005) ;5: (3):323–31. |
[69] | Klein CJ , Coovert DD , Bulman DE , Ray PN , Mendell JR , Burghes AH . Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): Evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. American journal of human genetics. (1992) ;50: (5):950–9. |
[70] | Okada T , Takeda S . Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy. Pharmaceuticals. (2013) ;6: (7):813–36. |
[71] | Bowles DE , McPhee SWJ , Li CW , Gray SJ , Samulski JJ , Camp AS , et al. Phase 1 Gene Therapy for Duchenne Muscular Dystrophy Using a Translational Optimized AAV Vector. Molecular Therapy. (2012) ;20: (2):443–55. |
[72] | Rodino-Klapac LR , Janssen PM , Shontz KM , Canan B , Montgomery CL , Griffin D , Heller K , Schmelzer L , Handy C , Clark KR , Sahenk Z , Mendell JR , Kaspar BK . Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Human molecular genetics. (2013) ;22: (24):4929–37. |
[73] | McNally EM , Pytel P . Muscle diseases: The muscular dystrophies. Annual review of pathology. (2007) ;2: :87–109. |
[74] | Peault B , Rudnicki M , Torrente Y , Cossu G , Tremblay JP , Partridge T , et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Molecular therapy: The journal of the American Society of Gene Therapy. (2007) ;15: (5):867–77. |
[75] | Konieczny P , Swiderski K , Chamberlain JS . Gene and cell-mediated therapies for muscular dystrophy. Muscle & nerve. (2013) ;47: (5):649–63. |
[76] | Dumont NA , Wang YX , Rudnicki MA . Intrinsic and extrinsic mechanisms regulating satellite cell function. Development (Cambridge, England). (2015) ;142: (9):1572–81. |
[77] | Meng J , Chun S , Asfahani R , Lochmuller H , Muntoni F , Morgan J . Human skeletal muscle-derived CD133(+) cells form functional satellite cells after intramuscular transplantation in immunodeficient host mice. Molecular therapy: The journal of the American Society of Gene Therapy. (2014) ;22: (5):1008–17. |
[78] | Tedesco FS , Hoshiya H , D’Antona G , Gerli MF , Messina G , Antonini S , et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Science translational medicine. (2011) ;3: (96):96ra78. |
[79] | Torrente Y , Belicchi M , Sampaolesi M , Pisati F , Meregalli M , D’Antona G , et al. Human circulating AC133(+) stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. The Journal of clinical investigation. (2004) ;114: (2):182–95. |
[80] | Tedesco FS , Gerli MF , Perani L , Benedetti S , Ungaro F , Cassano M , et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Science translational medicine. (2012) ;4: (140):140ra89. |
[81] | Park IH , Zhao R , West JA , Yabuuchi A , Huo H , Ince TA , et al . Reprogramming of human somatic cells to pluripotency with defined factors. Nature. (2008) ;451: (7175):141–6. |
[82] | Markert CD , Atala A , Cann JK , Christ G , Furth M , Ambrosio F , et al. Mesenchymal stem cells: Emerging therapy for Duchenne muscular dystrophy. PM & R: The journal of injury, function, and rehabilitation. (2009) ;1: (6):547–59. |
[83] | Meregalli M , Farini A , Parolini D , Maciotta S , Torrente Y . Stem cell therapies to treat muscular dystrophy: Progress to date. BioDrugs: Clinical immunotherapeutics, biopharmaceuticals and gene therapy. (2010) ;24: (4):237–47. |
[84] | Maffioletti SM , Noviello M , English K , Tedesco FS . Stem cell transplantation for muscular dystrophy: The challenge of immune response. BioMed research international. (2014) :964010. |
[85] | Huard J , Cao B , Qu-Petersen Z . Muscle-derived stem cells: Potential for muscle regeneration. Birth defects research Part C, Embryo today: Reviews. (2003) ;69: (3):230–7. |
[86] | Skuk D , Caron N , Goulet M , Roy B , Espinosa F , Tremblay JP . Dynamics of the early immune cellular reactions after myogenic cell transplantation. Cell transplantation. (2002) ;11: (7):671–81. |
[87] | Skuk D , Roy B , Goulet M , Chapdelaine P , Bouchard JP , Roy R , et al. Dystrophin expression in myofibers of Duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Molecular therapy: The journal of the American Society of Gene Therapy. (2004) ;9: (3):475–82. |
[88] | Love DR , Hill DF , Dickson G , Spurr NK , Byth BC , Marsden RF , et al. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. (1989) ;339: (6219):55–8. |
[89] | Hirst RC , McCullagh KJ , Davies KE . Utrophin upregulation in Duchenne muscular dystrophy. Acta myologica: Myopathies and cardiomyopathies: Official journal of the Mediterranean Society of Myology/edited by the Gaetano Conte Academy for the study of striated muscle diseases. (2005) ;24: (3):209–16. |
[90] | Amenta AR , Yilmaz A , Bogdanovich S , McKechnie BA , Abedi M , Khurana TS , Fallon JR . Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci U S A. (2011) ;108: :762–7. |
[91] | Tinsley JM FR , Storer R , Wilkes F , Potter AC , Squire SE , Powell DS , Cozzoli A , Capogrosso RF , Lambert A , Wilson F , Wren S , De Luca A , Davies KE . Daily treatment with SMT Ca novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PloS one. (2011) ;6: :e19189. |
[92] | Guiraud SS , Squire SE , Edwards B , Chen H , Burns DT , Shah N , Babbs A , Davies SG , Wynne GM , Russell AJ , Elsey D , Wilson FX , Tinsley JM , Davies KE . Second-generation compound for the modulation of utrophin in the therapy of DMD. Human molecular genetics. (2015) ;24: (2412). |
[93] | van Westering TL , Betts CA , Wood MJ . Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules. (2015) ;20: (5):8823–55. |
[94] | Bushby K , Finkel R , Wong B , Barohn R , Campbell C , Comi GP , et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle & nerve. (2014) ;50: (4):477–87. |
[95] | Barton-Davis ER , Cordier L , Shoturma DI , Leland SE , Sweeney HL . Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. The Journal of clinical investigation. (1999) ;104: (4):375–81. |
[96] | Howard M , Frizzell RA , Bedwell DM . Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nature medicine. (1996) ;2: (4):467–9. |
[97] | Wagner KR , Hamed S , Hadley DW , Gropman AL , Burstein AH , Escolar DM , et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Annals of neurology. (2001) ;49: (6):706–11. |
[98] | Manuvakhova M , Keeling K , Bedwell DM . Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. Rna. (2000) ;6: (7):1044–55. |
[99] | Welch EM , Barton ER , Zhuo J , Tomizawa Y , Friesen WJ , Trifillis P , et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. (2007) ;447: (7140):87–91. |
[100] | Spiegel R . Phase 3 Study of Ataluren in Patients With Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD) [cited 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT01826487. |
[101] | Hoffman EP , Bronson A , Levin AA , Takeda S , Yokota T , Baudy AR , et al. Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. The American journal of pathology. (2011) ;179: (1):12–22. |
[102] | Chapdelaine P , Pichavant C , Rousseau J , Paques F , Tremblay JP . Meganucleases can restore the reading frame of a mutated dystrophin. Gene therapy. (2010) ;17: (7):846–58. |
[103] | Rousseau J , Chapdelaine P , Boisvert S , Almeida LP , Corbeil J , Montpetit A , et al. Endonucleases: Tools to correct the dystrophin gene. Journal of Gene Medicine. (2011) ;13: (10):522–37. |
[104] | Long CZ , McAnally JR , Shelton JM , Mireault AA , Bassel-Duby R , Olson EN . Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. (2014) ;345: (6201):1184–8. |
[105] | Ousterout DG , Kabadi AM , Thakore PI , Perez-Pinera P , Brown MT , Majoros WH , et al. Correction of Dystrophin Expression in Cells From Duchenne Muscular Dystrophy Patients Through Genomic Excision of Exon 51 by Zinc Finger Nucleases. Molecular Therapy. (2015) ;23: (3):523–32. |
[106] | Ousterout DG , Perez-Pinera P , Thakore PI , Kabadi AM , Brown MT , Qin XX , et al. Reading Frame Correction by Targeted Genome Editing Restores Dystrophin Expression in Cells From Duchenne Muscular Dystrophy Patients. Molecular Therapy. (2013) ;21: (9):1718–26. |
[107] | Berger J , Currie PD . Zebrafish models flex their muscles to shed light on muscular dystrophies. Disease models & mechanisms. (2012) ;5: (6):726–32. |
[108] | Sharp NJ , Kornegay JN , Van Camp SD , Herbstreith MH , Secore SL , Kettle S , et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. (1992) ;13: (1):115–21. |
[109] | Cooper BJ , Winand NJ , Stedman H , Valentine BA , Hoffman EP , Kunkel LM , et al. The Homolog of the Duchenne Locus Is Defective in X-Linked Muscular-Dystrophy of Dogs. Nature. (1988) ;334: (6178):154–6. |
[110] | Kornegay JN , Cundiff DD , Bogan DJ , Bogan JR , Okamura CS . The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscular disorders: NMD. (2003) ;13: (6):493–500. |
[111] | Yu X , Bao B , Echigoya Y , Yokota T . Dystrophin-deficient large animal models: Translational research and exon skipping. American journal of translational research. (2015) ;7: (8):1314–31. |
[112] | Ambrosio CE , Valadares MC , Zucconi E , Cabral R , Pearson PL , Gaiad TP , et al. Ringo, a Golden Retriever Muscular Dystrophy (GRMD) dog with absent dystrophin but normal strength. Neuromuscular Disorders. (2008) ;18: (11):892–3. |
[113] | Banks GB , Chamberlain JS . The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Current topics in developmental biology. (2008) ;84: :431–53. |
[114] | Schatzberg SJ , Olby NJ , Breen M , Anderson LVB , Langford CF , Dickens HF , et al. Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscular Disorders. (1999) ;9: (5):289–95. |
[115] | Bretag AH . Stem cell treatment of dystrophic dogs. Nature. (2007) ;450: (7173):E23-E. |
[116] | Klymiuk N , Blutke A , Graf A , Krause S , Burkhardt K , Wuensch A , et al. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Human molecular genetics. (2013) ;22: (21):4368–82. |
[117] | Selsby JT , Ross JW , Nonneman D , Hollinger K . Porcine Models of Muscular Dystrophy. Ilar J. (2015) ;56: (1):116–26. |
[118] | Nonneman DJ , Brown-Brandl T , Jones SA , Wiedmann RT , Rohrer GA . A defect in dystrophin causes a novel porcine stress syndrome. BMC genomics. (2012) :13. |
[119] | Hollinger K , Yang CX , Montz RE , Nonneman D , Ross JW , Selsby JT . Dystrophin insufficiency causes selective muscle histopathology and loss of dystrophin-glycoprotein complex assembly in pig skeletal muscle. Faseb Journal. (2014) ;28: (4):1600–9. |
[120] | McGreevy JW , Hakim CH , McIntosh MA , Duan DS . Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Disease models & mechanisms. (2015) ;8: (3):195–213. |
[121] | Bulfield G , Siller WG , Wight PA , Moore KJ . X chromosome-linked muscular dystrophy (mdx) in the mouse. Proceedings of the National Academy of Sciences of the United States of America. (1984) ;81: (4):1189–92. |
[122] | Bostick B , Yue Y , Duan D . Gender influences cardiac function in the mdx model of Duchenne cardiomyopathy. Muscle & nerve. (2010) ;42: (4):600–3. |
[123] | Wehling-Henricks M , Jordan MC , Roos KP , Deng B , Tidball JG . Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Human molecular genetics. (2005) ;14: (14):1921–33. |
[124] | Stedman HH , Sweeney HL , Shrager JB , Maguire HC , Panettieri RA , Petrof B , et al. The Mdx Mouse Diaphragm Reproduces the Degenerative Changes of Duchenne Muscular-Dystrophy. Nature. (1991) ;352: (6335):536–9. |
[125] | Tanabe Y , Esaki K , Nomura T . Skeletal-Muscle Pathology in X-Chromosome-Linked Muscular-Dystrophy (Mdx) Mouse. Acta neuropathologica. (1986) ;69: (1-2):91–5. |
[126] | Grady RM , Teng HB , Nichol MC , Cunningham JC , Wilkinson RS , Sanes JR . Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell. (1997) ;90: (4):729–38. |
[127] | Bulfield G , Siller WG , Wight PAL , Moore KJ . X-Chromosome-Linked Muscular-Dystrophy (Mdx) in the Mouse. P Natl Acad Sci-Biol. (1984) ;81: (4):1189–92. |
[128] | Vianello S , Bouyon S , Benoit E , Sebrie C , Boerio D , Herbin M , et al. Arginine butyrate per os protects mdx mice against cardiomyopathy, kyphosis and changes in axonal excitability. Neurobiology of disease. (2014) ;71: :325–33. |
[129] | Pagel CN , Partridge TA . Covert persistence of mdx mouse myopathy is revealed by acute and chronic effects of irradiation. Journal of the neurological sciences. (1999) ;164: (2):103–16. |
[130] | Hodgetts SI , Grounds MD . Irradiation of dystrophic host tissue prior to myoblast transfer therapy enhances initial (but not long-term) survival of donor myoblasts. Journal of cell science. (2003) ;116: (20):4131–46. |
[131] | Yokota T , Lu QL , Morgan JE , Davies KE , Fisher R , Takeda S , et al. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. Journal of cell science. (2006) ;119: (13):2679–87. |
[132] | Shi XZ , Garry DJ . Muscle stem cells in development, regeneration, and disease. Genes & development. (2006) ;20: (13):1692–708. |
[133] | Meng J , Bencze M , Asfahani R , Muntoni F , Morgan JE . The effect of the muscle environment on the regenerative capacity of human skeletal muscle stem cells. Skeletal muscle. (2015) ;5: :11. |
[134] | Heier CR , Damsker JM , Yu Q , Dillingham BC , Huynh T , Van der Meulen JH , et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO molecular medicine. (2013) ;5: (10):1569–85. |
[135] | Wang B , Miyagoe-Suzuki Y , Yada E , Ito N , Nishiyama T , Nakamura M , et al. Reprogramming efficiency and quality of induced Pluripotent Stem Cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages. PLoS currents. (2011) ;3: :RRN1274. |
[136] | The Jackson Laboratory. C57BL/10J [cited 2015]. Available from: https://www.jax.org/strain/000665. |
[137] | Chapman VM , Miller DR , Armstrong D , Caskey CT . Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc Natl Acad Sci U S A. (1989) ;86: (4):1292–6. |
[138] | Fukada S , Morikawa D , Yamamoto Y , Yoshida T , Sumie N , Yamaguchi M , et al. Genetic background affects properties of satellite cells and mdx phenotypes. The American journal of pathology. (2010) ;176: (5):2414–24. |
[139] | Echigoya Y , Lee J , Rodrigues M , Nagata T , Tanihata J , Nozohourmehrabad A , et al. Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice. PloS one. (2013) ;8: (7):e69194. |
[140] | Araki E , Nakamura K , Nakao K , Kameya S , Kobayashi O , Nonaka I , et al. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochemical and biophysical research communications. (1997) ;238: (2):492–7. |
[141] | Den Dunnen JT , Grootscholten PM , Bakker E , Blonden LA , Ginjaar HB , Wapenaar MC , et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. American journal of human genetics. (1989) ;45: (6):835–47. |
[142] | Nobile C , Marchi J , Nigro V , Roberts RG , Danieli GA . Exon-intron organization of the human dystrophin gene. Genomics. (1997) ;45: (2):421–4. |
[143] | Kameya S , Araki E , Katsuki M , Mizota A , Adachi E , Nakahara K , et al. Dp260 disrupted mice revealed prolonged implicit time of the b-wave in ERG and loss of accumulation of beta-dystroglycan in the outer plexiform layer of the retina. Human molecular genetics. (1997) ;6: (13):2195–203. |
[144] | Sigesmund DA , Weleber RG , Pillers DA , Westall CA , Panton CM , Powell BR , et al. Characterization of the ocular phenotype of Duchenne and Becker muscular dystrophy. Ophthalmology. (1994) ;101: (5):856–65. |
[145] | Aoki Y , Yokota T , Nagata T , Nakamura A , Tanihata J , Saito T , et al. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc Natl Acad Sci U S A. (2012) ;109: (34):13763–8. |
[146] | Aoki Y , Nagata T , Yokota T , Nakamura A , Wood MJ , Partridge T , et al. Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-alpha2 chain-null congenital muscular dystrophy mice. Human molecular genetics. (2013) ;22: (24):4914–28. |
[147] | Echigoya Y , Aoki Y , Miskew B , Panesar D , Touznik A , Nagata T , et al. Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45-55 with a cocktail of vivo-morpholinos in mdx52 mice. Molecular therapy Nucleic acids. (2015) ;4: :e225. |
[148] | Ferreiro V , Giliberto F , Muniz GM , Francipane L , Marzese DM , Mampel A , et al. Asymptomatic Becker muscular dystrophy in a family with a multiexon deletion. Muscle & nerve. (2009) ;39: (2):239–43. |
[149] | Aoki Y , Yokota T , Wood MJ . Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. BioMed research international. (2013) :402369. |
[150] | Deconinck AE , Rafael JA , Skinner JA , Brown SC , Potter AC , Metzinger L , et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. (1997) ;90: (4):717–27. |
[151] | Grady RM , Teng H , Nichol MC , Cunningham JC , Wilkinson RS , Sanes JR . Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell. (1997) ;90: (4):729–38. |
[152] | Zhou L , Rafael-Fortney JA , Huang P , Zhao XS , Cheng G , Zhou X , et al. Haploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx mice. Journal of the neurological sciences. (2008) ;264: (1-2):106–11. |
[153] | Li D , Yue Y , Duan D . Marginal level dystrophin expression improves clinical outcome in a strain of dystrophin/utrophin double knockout mice. PloS one. (2010) ;5: (12):e15286. |
[154] | van Putten M , Hulsker M , Young C , Nadarajah VD , Heemskerk H , van der Weerd L , et al. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology. (2013) ;27: (6):2484–95. |
[155] | Chen CN , Graber TG , Bratten WM , Ferrington DA , Thompson LV . Immunoproteasome in animal models of Duchenne muscular dystrophy. Journal of muscle research and cell motility. (2014) ;35: (2):191–201. |
[156] | Wu B , Cloer C , Lu P , Milazi S , Shaban M , Shah SN , et al. Exon skipping restores dystrophin expression, but fails to prevent disease progression in later stage dystrophic dko mice. Gene Ther. (2014) ;21: (9):785–93. |
[157] | Wakefield PM , Tinsley JM , Wood MJ , Gilbert R , Karpati G , Davies KE . Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene therapy. (2000) ;7: (3):201–4. |
[158] | Goyenvalle A , Wright J , Babbs A , Wilkins V , Garcia L , Davies KE . Engineering Multiple U7snRNA Constructs to Induce Single and Multiexon-skipping for Duchenne Muscular Dystrophy. Molecular Therapy. (2012) ;20: (6):1212–21. |
[159] | Wertz KaF , E.M. Dmd(mdx-beta geo): A new allele for the mouse dystrophin gene. Developmental dynamics: An official publication of the American Association of Anatomists. (1988) ;212: :229–41. |
[160] | Krasowska E , Zablocki K , Gorecki DC , Swinny JD . Aberrant Location of Inhibitory Synaptic Marker Proteins in the Hippocampus of Dystrophin-Deficient Mice: Implications for Cognitive Impairment in Duchenne Muscular Dystrophy. PloS one. (2014) ;9: (9). |
[161] | Kudoh H , Ikeda H , Kakitani M , Ueda A , Hayasaka M , Tomizuka K , et al. A new model mouse for Duchenne muscular dystrophy produced by 2. 4 Mb deletion of dystrophin gene using Cre-loxP recombination system. Biochemical and biophysical research communications. (2005) ;328: (2):507–16. |
[162] | Kudoh H , Ikeda H , Kakitani M , Ueda A , Hayasaka M , Tomizuka K , et al. A new model mouse for Duchenne muscular dystrophy produced by 2.4 Mb deletion of dystrophin gene using Cre-loxP recombination system. Biochemical and biophysical research communications. (2005) ;328: (2):507–16. |
[163] | t Hoen PA , de Meijer EJ , Boer JM , Vossen RH , Turk R , Maatman RG , et al. Generation and characterization of transgenic mice with the full-length human DMD gene. The Journal of biological chemistry. (2008) ;283: (9):5899–907. |
[164] | Veltrop M , van der Kaa J , Claassens J , van Vliet L , Verbeek S , Aartsma-Rus A . Generation of embryonic stem cells and mice for duchenne research. PLoS currents. (2013) :5. |
[165] | Wu B , Benrashid E , Lu P , Cloer C , Zillmer A , Shaban M , et al. Targeted skipping of human dystrophin exons in transgenic mouse model systemically for antisense drug development. PloS one. (2011) ;6: (5):e19906. |
[166] | Bremmer-Bout M , Aartsma-Rus A , de Meijer EJ , Kaman WE , Janson AA , Vossen RH , et al. Targeted exon skipping in transgenic hDMD mice: A model for direct preclinical screening of human-specific antisense oligonucleotides. Molecular therapy: The journal of the American Society of Gene Therapy. (2004) ;10: (2):232–40. |
[167] | Goyenvalle A , Wright J , Babbs A , Wilkins V , Garcia L , Davies KE . Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Molecular therapy: The journal of the American Society of Gene Therapy. (2012) ;20: (6):1212–21. |
[168] | Howell GR , Soto I , Ryan M , Graham LC , Smith RS , John SW . Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J Neuroinflammation. (2013) ;10: :76. |
[169] | Williams PA , Howell GR , Barbay JM , Braine CE , Sousa GL , John SW , et al. Retinal ganglion cell dendritic atrophy in DBA/2J glaucoma. PLoS One. (2013) ;8: (8):e72282. |
[170] | The Jackson Laboratory. DBA/2J [cited 2015]. Available from: https://www.jax.org/strain/000671. |
[171] | The Jackson Laboratory. C57BL/6J [cited 2015]. Available from: https://www.jax.org/strain/000664. |
[172] | Nishina PM , Wang J , Toyofuku W , Kuypers FA , Ishida BY , Paigen B . Atherosclerosis and plasma and liver lipids in nine inbred strains of mice. Lipids. (1993) ;28: (7):599–605. |
[173] | Ito T , Ogawa R , Uezumi A , Ohtani T , Watanabe Y , Tsujikawa K , et al. Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-marker expression in muscle mesenchymal progenitors. Neuromuscular disorders: NMD. (2013) ;23: (4):349–56. |
[174] | Chandrasekharan K , Yoon JH , Xu Y , deVries S , Camboni M , Janssen PM , et al. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Science translational medicine. (2010) ;2: (42):42ra54. |
[175] | Kawano T , Koyama S , Takematsu H , Kozutsumi Y , Kawasaki H , Kawashima S , et al. Molecular cloning of cytidine monophospho-N-acetylneuraminic acid hydroxylase. Regulation of species- and tissue-specific expression of N-glycolylneuraminic acid. The Journal of biological chemistry. (1995) ;270: (27):16458–63. |
[176] | Shaw L , Schneckenburger P , Carlsen J , Christiansen K , Schauer R . Mouse liver cytidine-5’-monophosphate-N-acetylneuraminic acid hydroxylase. Catalytic function and regulation. European journal of biochemistry/FEBS. (1992) ;206: (1):269–77. |
[177] | The Jackson Laboratory. B6.129X1-Cmahtm1Avrk/J [cited 2015]. Available from: https://www.jax.org/strain/017588 |
[178] | Hedlund M , Tangvoranuntakul P , Takematsu H , Long JM , Housley GD , Kozutsumi Y , et al. N-glycolylneuraminic acid deficiency in mice: Implications for human biology and evolution. Molecular and cellular biology. (2007) ;27: (12):4340–6. |
[179] | Varki A . Colloquium paper: Uniquely human evolution of sialic acid genetics and biology. Proceedings of the National Academy of Sciences of the United States of America. (2010) ;107: (Suppl 2):8939–46. |
[180] | Chou HH , Takematsu H , Diaz S , Iber J , Nickerson E , Wright KL , et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proceedings of the National Academy of Sciences of the United States of America. (1998) ;95: (20):11751–6. |
[181] | Sacco A , Mourkioti F , Tran R , Choi J , Llewellyn M , Kraft P , et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. (2010) ;143: (7):1059–71. |
[182] | Mourkioti F , Kustan J , Kraft P , Day JW , Zhao MM , Kost-Alimova M , et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nature cell biology. (2013) ;15: (8):895–904. |
[183] | Tanganyika-de Winter CL , Heemskerk H , Karnaoukh TG , van Putten M , de Kimpe SJ , van Deutekom J , et al. Long-term Exon Skipping Studies With 2’-O-Methyl Phosphorothioate Antisense Oligonucleotides in Dystrophic Mouse Models. Molecular therapy Nucleic acids. (2012) ;1: :e44. |
[184] | Scheuerbrandt G . Prosensa’s continued development of exon-51 skipping with drisapersen after the failure of its phase-III clinical trial. |
[185] | Therapeutics S . Safety Study of Eteplirsen to Treat Advanced Stage Duchenne Muscular Dystrophy 2015 [cited 2015 May 04]. Available from: https://clinicaltrials.gov/ct2/show/NCT02286947 |
[186] | Yin H , Price F , Rudnicki MA . Satellite cells and the muscle stem cell niche. Physiological reviews. (2013) ;93: :23–67. |
[187] | University; EOA . Efficacy of Umbilical Cord Mesenchymal Stem Cells in Duchenne Muscular Dystrophy [cited May 04]. Available from: https://clinicaltrials.gov/ct2/show/NCT[clinicaltrials.gov/ct2/show/NCT]. (2014) ). |
[188] | Institute; NBaS. Stem Cell Therapy in Duchenne Muscular Dystrophy 2014 [cited 2015 May 04]. Available from: https://clinicaltrials.gov/ct2/show/NCT02241434. |
[189] | Therapeutics; P. Phase 3 Study of Ataluren in Patients With Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD) 2014 [cited 2015 May 04]. Available from: https://clinicaltrials.gov/ct2/show/NCT01826487. |
Figures and Tables
Fig.1
Dystrophin links actin cytoskeleton to the dystrophin glycoprotein complex. In normal muscles, the N-terminal domain of dystrophin binds to actin. Dystrophin then, subsequently interacts with the components of DGC: It interact with neuronal nitric oxide synthase (nNOS) at the region between exon 42 to exon 45, then, its cysteine rich domain binds to β-dystroglycan, and lastly, its C-terminal domain binds to syntrophin and dystrobrevin.
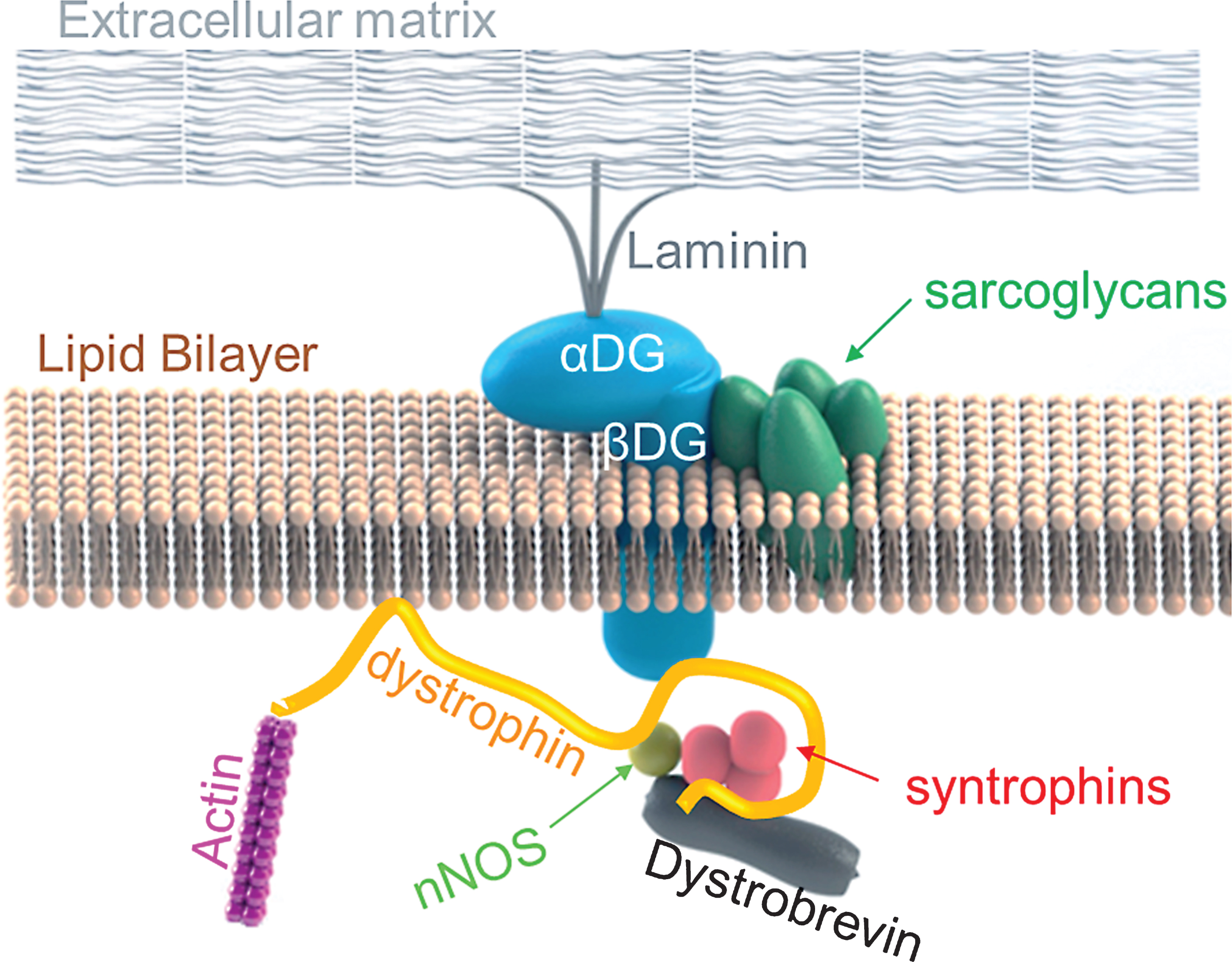
Fig.2
Histology concerning RF expression and CNFs observed in dystrophic mice models of mdx, mdx52 and/or mdx-DBA/2) (A) Mdx52 mice show lower number of RFs in a single cluster than mdx52 mice at 12 months of age. Echigoya et al., 2013 showed that mdx52 has a 58% lower RF expansion than age-matched mdx mice of 12 months. The tibialis anterior (TA) muscles of mdx and mdx52 were immunostained with a rabbit polyclonal antibody against C-terminal domain (position at 3,661– 3,677 amino acids; Abcam, Bristol, UK). Bars = 50μm. (B) Hematoxylin and eosin stained images for TA muscles of mdx, mdx52 and mdx-DBA/2 mice at 2 months of age. Arrows indicate centrally nucleated fibers. Bars = 100μm.
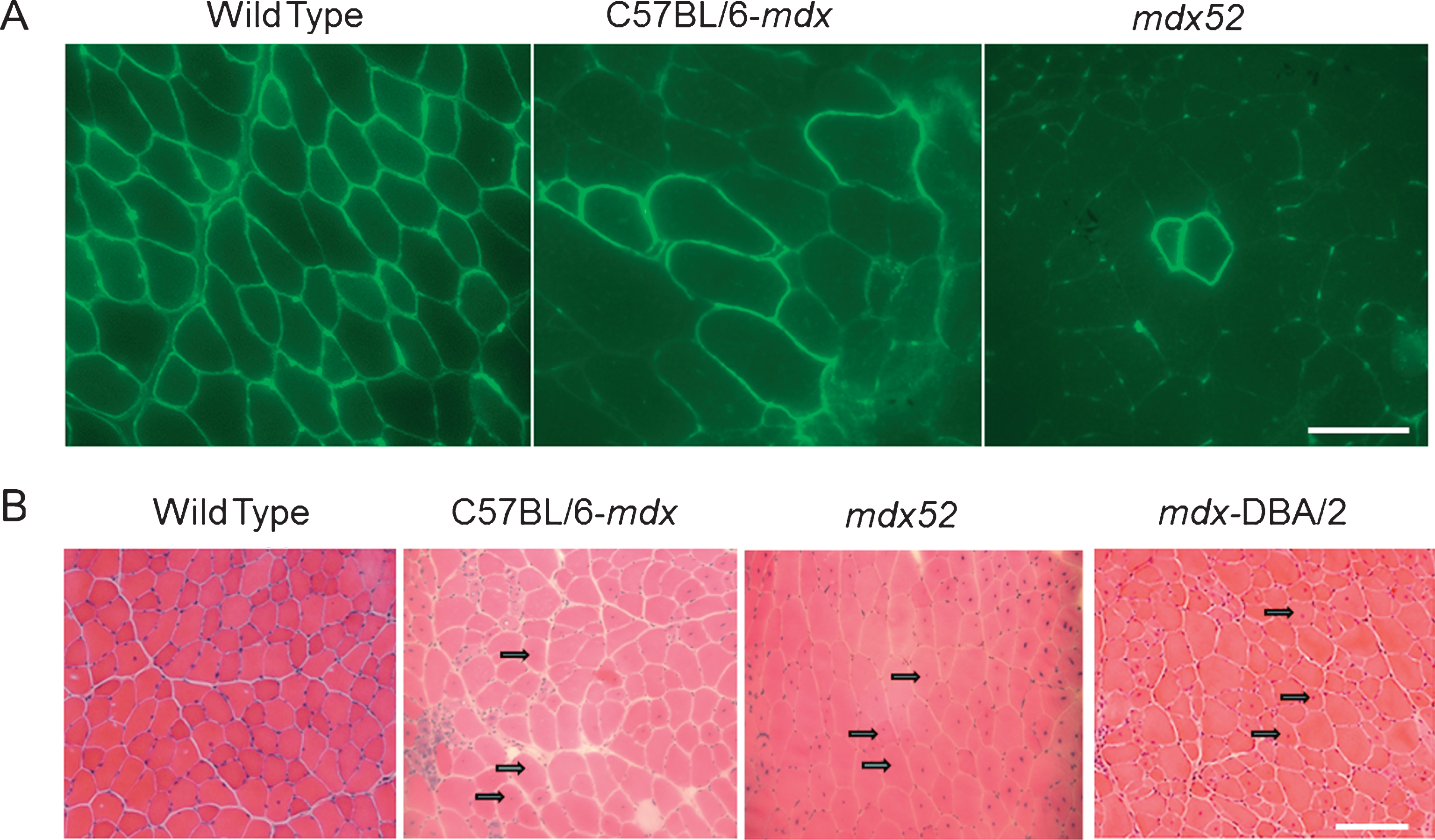
Fig.3
The promoters and isoforms of the dystrophin gene, and the location of mutations in murine models. (A) The location of different promoters (brain (B), muscle (M), Purkinje (P), retinal (R), brain-3 (B3), Schwann cell (S), and general (G)) of the dystrophin gene is displayed alongside with the location of mutations observed in some murine models (and also illustrates the insertion of the ROSAβgeo in 3’ end of exon 63 in mdxβgeo). Yellow rectangles represent exons. (B) The promoters of Dp427 results in “full-length” dystrophin protein (consisting of the N-terminal actin-binding domain, rod domain, WW domain, cysteine rich domain (Cys) and C-terminal domain (CT)). The remaining promoters lead to shortened dystrophin isoforms.
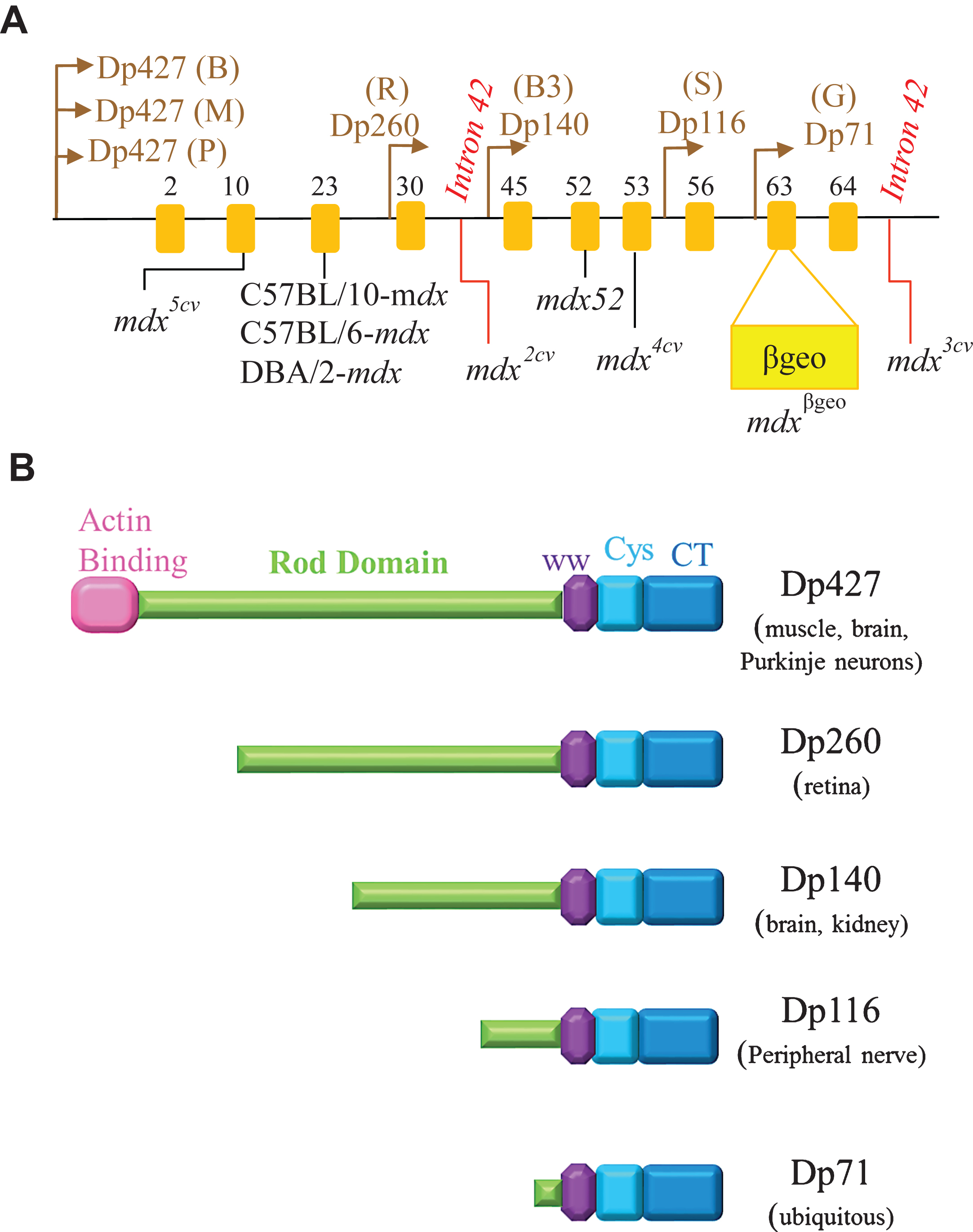
Table 1
Overview of therapeutic approaches and its associated glossary of terms
| Exon skipping therapy | Antisense oligonucleotides are used to splice one or multiple exons in pre-mRNA to restore the reading frame |
| Antisense oligonucleotides (AONs) | Short synthetic nucleic acids that target specific sequences of pre-mRNA, modulating the splicing pattern to allow for in-frame dystrophin mRNA. Some of the AONs developed are 2’-O-methyl phosphorothioate (2’OMePS), phosphorodiamidate morpholino oligomers (PMOs), Vivo-morpholinos (vPMOs) and peptide-linked PMOs (PPMOs). Each of these AONs has different chemistries but the latter two have cell-penetrating moieties. |
| Gene replacement therapy | Provides a substitute for dystrophin in a dystrophin-null background by packaging a truncated form of the dystrophin gene in vectors such as the non-pathogenic recombinant adeno-associated virus (rAAV) vector. |
| Stem cell therapy | Involves stem cell transplantation, proliferation and differentiation into muscle cells and hence, contributes to increased muscle regeneration, preventing muscle wasting and fibrosis. |
| Induced pluripotent stem cells (iPSC) | Adult somatic cells that are genetically reprogrammed into an embryonic stem cell-like pluripotent state and hence, can differentiate into myofibers and increase muscle regeneration capacity. |
| Utrophin upregulation therapy | Aims to increase levels of utrophin, a protein similar to dystrophin, in dystrophic muscles to compensate for the absence of dystrophin. Pharmacological drugs, such as SMT C1100, SMT022357 and Biglycan, are shown to increase utrophin levels. |
| Read-through therapy | Pharmacological agents, such as Ataluren (aka PTC124), are used to replace a premature stop codon (nonsense mutation) with a new amino acid, allowing for continued translation of dystrophin protein. |
| Endonuclease-based gene repair | DNA gene editing technique: Endonucleases used to create site-specific breaks in double-stranded DNA, which initiates DNA repair and gene correction. |
Table 2
Mutation types and phenotypic features of murine models of Duchenne muscular dystrophy
| Murine models | Molecular Mutation | Phenotype | References |
| mdx (C57BL/10 genetic background) | Spontaneous point mutation in exon 23 of the Dmd gene. | Skeletal muscle degeneration-regeneration, necrosis, little fibrosis, utrophin upregulation and, greater regenerative capacity than DMD patients. | (121) |
| mdx (C57BL/6 genetic background) | Spontaneous point mutation in exon 23 of the Dmd gene. | Similar to C57BL/10-mdx, used for comparative studies, greatest regenerative capacity than other inbred strains of mdx. | (135) |
| Mdx2cv | Intron 42 point mutation | C57BL/6 background and the chemically induced mutation creates a new splice acceptor site. | (137) |
| Mdx3cv | Intron 65 point mutation | C57BL/6 background and the chemically induced mutation creates a new splice acceptor site. | (137) |
| Mdx4cv | Nonsense mutation at exon 53 | C57BL/6 background and harbours a chemically induced nonsense mutation. | (137) |
| Mdx5cv | Point mutation at exon 10 of Dmd | C57BL/6 background and the chemically induced mutation causes a new splice site in exon 10. | (137) |
| mdx52 (C57BL/6 genetic background) | Deletion mutation in exon 52 of the Dmd gene | Variation in myofiber size, skeletal muscles are hypertrophic, muscle degeneration-regeneration cycles, necrosis, lower RFs than C57BL/6-mdx | (140) |
| dko | Double deficient of the Dmd and Utr genes | Severe and progressive muscle wasting, weight loss after weaning, abnormal breathing rhythms, early onset of joint contractures, short life span and kyphosis by 20 weeks | (150) |
| mdxβgeo | Insertion of ROSA β-geo gene trap vector in exon 63 | Loss of most dystrophin isoforms (including Dp71), cardiac hypertrophy, abnormally dilated esophagus. (Note: The cysteine rich and C-terminal domains are lost in these mice) | (159) |
| D md -null | Deletion of the entire dystrophin gene | Produced by Cre-loxP technology. Lacks revertant fibers and all dystrophin isoforms. Displays muscle hypertrophy, behavioural abnormality and infertility. | (162) |
| hDMD | Knock-in of the complete human DMD gene in chromosome 5 of mouse genome. | No dystrophic phenotype | (163) |
| mdx (DBA/2 genetic background) | Spontaneous point mutation in exon 23 of the Dmd gene. | Lower muscle mass, greater fibrosis and fatty tissue accumulation, and lower regenerative capacity of satellite cells than C57BL/10-mdx mice. | (138) |
| Cmah-mdx (C57BL/10 genetic background) | Deletion mutation in the C mah gene and spontaneous point mutation in exon 23 of the Dmd gene | Nearly 50% mortality at 11 months of age, loss of ambulation by 8 months, greater fibrosis than mdx (C57BL/10) mice in skeletal muscles like diaphragm and quadriceps, and necrosis in the heart by 3 months | (174) |
| m dx/mTRKO | Exon 23 point mutation and deletion of RNA component TERC (mTR) of telomerase | Severe dystrophic phenotype: Impaired self-renewal capacity, severe muscle wasting, accumulation of fibrosis and calcium deposits, increase creatine kinase levels, kyphosis, dilated cardiomyopathy, heart failure and shortened lifespan (12 months). | (181) |


