Exon 32 Skipping of Dysferlin Rescues Membrane Repair in Patients’ Cells
Abstract
Dysferlinopathies are a family of disabling muscular dystrophies with LGMD2B and Miyoshi myopathy as the main phenotypes. They are associated with molecular defects in DYSF, which encodes dysferlin, a key player in sarcolemmal homeostasis. Previous investigations have suggested that exon skipping may be a promising therapy for a subset of patients with dysferlinopathies. Such an approach aims to rescue functional proteins when targeting modular proteins and specific tissues.
We sought to evaluate the dysferlin functional recovery following exon 32 skipping in the cells of affected patients. Exon skipping efficacy was characterized at several levels by use of in vitro myotube formation assays and quantitative membrane repair and recovery tests. Data obtained from these assessments confirmed that dysferlin function is rescued by quasi-dysferlin expression in treated patient cells, supporting the case for a therapeutic antisense-based trial in a subset of dysferlin-deficient patients.
INTRODUCTION
Mutations in DYSF (MIM# 603009, 2p13, GenBank NM_003494.2) [1, 2], the gene encoding dysferlin, cause a variety of muscular dystrophies collectively referred to as dysferlinopathies, the most significant of which are LGMD2B (LGMD2B; MIM# 253601) and Miyoshi Myopathy (MM; MIM# 254130) [1, 2].
At the onset of disease, usually in the second decade of life, proximal muscles in LGMD2B and distal muscles in MM are the principal muscle groups to be affected. In the early stages of disease, serum levels of creatine kinase are elevated and the histology appears clearly dystrophic with numerous inflammatory foci. In both phenotypes, most patients present with generalized muscle weakness after several years of disease progression, though in some cases, the early presentation is a proximo-distal muscle weakness [3]. Nevertheless, scapular winging is uncommon, and cardiac and respiratory complications do not typically develop amid the typical progression of these dysferlinopathies.
Dysferlin interacts with different muscle proteins involved in at least two pathways: membrane repair and myoblast/myotube membrane fusion. The process of membrane repair is now better understood since many actors have been identified at each of the different stages in the process. Among these participating factors, we have found specific types of lipids, sarcolemmal proteins like dysferlin as well as MG53 and annexins, and also muscle ubiquitous protein like calpain 3 [4–12]. The myoblast/myotube fusion, a key process for muscle formation during development and regeneration, is also poorly described but it appears to be associated with a substantial cytoskeletal rearrangement following the recruitment of a large protein network including dysferlin [10, 13–15]. The abnormalities observed in patients carrying function-modifying mutations in DYSF demonstrate the crucial role of dysferlin for muscle physiology andfunctionality.
As for some muscular dystrophies, several therapeutic strategies are currently being evaluated for dysferlinopathies, including gene therapy approaches [6, 16–19]. Based on the promising results of clinical trials for Duchenne Muscular Dystrophy (DMD) using Antisense Oligonucleotide (AON) approaches for exon 51 skipping of the dystrophin pre-mRNA [20–23] (Sarepta therapeutics and BioMarin, Inc., unpublished data), we recently evaluated the feasibility of a similar strategy for application in dysferlinopathies. In contrast to DMD where exon skipping strategies are intended to restore the reading frame (for patients presenting out-of-frame deletions), in dysferlinopathies the aim is to bypass the mutation in exon 32 without altering the reading frame and function. A prior report had described a mildly affected patient with a mutation causing in-frame skipping of exon 32 at one allele (caused by a lariat branch point mutation) together with a null-allele [24]. We considered these data to constitute a “natural” proof of principle than an exon 32-skipping approach could be therapeutic in dysferlinopathies. In a previous study, we were able to demonstrate its efficient skipping in patient cells carrying mutations in this specific exon, and thus demonstrated the feasibility of exon skipping targeting DYSF [25]. In the present work, we sought to characterize the functional recovery following exon 32 skipping in patient cells with the aim of translating our results to future clinical applications. The efficiency of the exon skipping was assessed by immunoblotting and immunochemistry. In addition, based on the role of dysferlin in myotubes, several functional tests were developed to quantify functional recovery. Our results demonstrate for the first time the rescue of dysferlin functions by a quasi-dysferlin generated by exon skipping in patient cells.
MATERIALS AND METHODS
Ethics statement
The relevant sample was provided, anonymously, by Myobank (Myology Institute) affiliated at EuroBioBank (www.eurobiobank.org). This affiliation certifies that the biopsy was obtained in accordance with the ethical standards laid down in the Declaration of Helsinki and the directive 2004/23/EC of the European Parliament.
Patients
Patient biopsies were obtained from a patient affected with Miyoshi myopathy: Patient 1 NM_0003494.3: c.[3477C>A]+[5979dupA]; p.[Tyr1159 *](stop codon in exon 32)+[Ala1993_Glu1994insArg;Ser1995 *] (stop codon in exon 53); and a patientaffected with LGMD2B: Patient 2: NM_0003498.3: c.[342+1G>A] (intron 4, splicing defect)+[3516_3517delTT]; p.[Ser1173 *] (stop codon in exon 32). Mutational data are described using the nomenclature of the Human Genome Variation Society (www.hgvs.org/mutnomen).
Cell cultures
Myoblasts were expanded in Skeletal Muscle Cell Growth Medium (Promocell) adjusted at 20% FBS final, supplemented with 100 μg/ml of gentamycin (Sigma-Aldrich). At confluence, the medium was changed for a differentiation medium based on the Skeletal Muscle Cell Differentiation medium (Promocell) and supplemented with 100 μg/ml of gentamicin, 10 μg/ml of doxycycline (Sigma-Aldrich) and 100 μg/ml of apotransferrin (Sigma-Aldrich). When needed, cells have been differentiated into myotubes, maintained in cell culture supports coated usingcollagen I (BD Biosciences).
Identification of target sequences for dysferlin exon 32 skipping
Bioinformatics analyses, to find targetable sequences within exon 32 of the DYSF gene and its surrounding intronic sequences, were realized using www.umd.be/HSF/ and http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process5home[tools/ESE3/esefinder.cgi?process5home] [25–27].
AONs and transfection
The antisense oligonucleotides (AONs) used are as follows: AON B 5′-GCGUAGAUGGUAGCGGUUCCC-3′; AON D 5′-ACCUACCAGAAAAAGAGUCCUU-3′. AON were synthesized by Eurogentec and contain 2′-O-methyl RNA and full-length phosphorothioate backbones (Figure S1-A). Myoblast cultures were transfected using oligofectamine (Life Technologies) according to the manufacturer’s instructions. Each AON was transfected one, two or three times at 800 nM every two days starting at day 0 of differentiation. Mock experiments were performed using the transfection reagent (oligofectamine) without AON. Control conditions were tested using a non-specific AON: 5′- GGAUCCCGCUCAGGAGUGCUG-3′. All experiments were conducted 48 hours after last transfection (day 6 of differentiation).
RNA extraction
RNAs were extracted from cultured cells using PureLink™RNA Mini Kit (Life technologies) and RNA was purified using DNAse Free Kit (Life technologies). Five hundred ng of RNA was reverse transcribed into cDNA using High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Saint Aubin, France) primed by random hexamers according to the manufacturer’s instructions. PCR amplifications were performed with 1 μL of RT-product using primer pairs flanking the targeted exons (Forward: 5′-CCAGGAGCCAGAGATTCG-3′Reverse: 5′- TGCTTTCAATGTCCTCCTTG-3′) for 35 cycles. Dysferlin RNA exon skipping was analyzed by classical RT-PCR for AON study. The products of PCR were separated by electrophoresis in a 1.5% agarose gel stained with 5% ethidium bromide.
Immunoblot
Detection of dysferlin was performed using the following protocol with primary anti-dysferlin antibodies (NCL-Hamlet 2, Cliniscience) diluted 1:300 or (NCL-Hamlet, Abcam) diluted 1:200 on untreated and treated patient cell protein extracts. Immunoblots were done using a whole protein extract sample prepared in lysis buffer (100 mM Tris buffer at pH 6.8, 4% SDS, 10% glycerol and 1% 2-mercaptoethanol). A 1% reducing agent (Life Technologies) was added and the samples were boiled for ten minutes. 20 μg of sample was loaded onto each lane of a 3–8% NuPage SDS–PAGE (Life Technologies). Proteins were then separated under electrophoresis at 80 volts at room temperature. Proteins were finally transferred onto nitrocellulose membranes (at 260 mA for 2 h at 4°C). Membranes were incubated with blocking buffer (1% non-fat milk powder in TBS-T) for one hour at room temperature. Primary antibodies were then diluted in blocking buffer and incubated with the membranes at room temperature for 1 h, with constant agitation. After washing in TBS-T, membranes were then incubated with the relevant secondary antibodies, which were diluted 1:10,000 in blocking buffer, for one hour at room temperature. The membranes were finally washed in TBS-T and developed using Pico Chemiluminescent Substrate (Pierce). GAPDH (Millipore) was detected using a dilution of 1:10,000.
Immunofluorescence
Cells were grown on Lab-TEK II™(Fisher Scientific) according to the previously explained protocol. Throughout the procedure, cells were kept at room temperature. After treatment, cells were fixed with 4% paraformaldehyde for 10 minutes and then washed in PBS for 10 minutes. Cells were then incubated for 10 minutes with a permeabilization solution (200 μL of PBS 1X +0.5% triton X-100 + protease inhibitors cocktail) (Roche). From there, cells were exposed to a blocking buffer (PBS+ 1% BSA + protease inhibitors cocktail) for 30 minutes. The primary antibody was applied in blocking buffer for 3 hours at room temperature, followed by one wash in PBS for 10 minutes and 1 hour of contact with the secondary antibody in blocking buffer. After a wash in PBS for 10 minutes, cells were fixed using a 4% paraformaldehyde solution in PBS for 10 minutes, then washed again. Finally, coverslips were mounted with Vectashield-Dapi 25 ng/mL, and kept at 4°C until pictures were taken. Dysferlin was detected using NCL-Hamlet at a dilution of 1:200. Caveolin-3 (BD Biosciences) was detected using a dilution of 1:1,000, and desmin (Fischer Scientific) using a dilution of 1:100. Scale bars are indicated on each picture.
Observation was performed using a Zeiss apotome microscope, and images were processed withAxioVision software and/or ImageJ software.
Membrane wounding assay
To induce damage, an area of the sarcolemma of the myotubes was irradiated at full power for 1 second with a two-photon laser-scanning microscope. The Multi-Photon apparatus consisted of a mode-locked Titanium-Sapphire laser system tuned to a 1035 nm excitation with 100 fs pulses at 76 MHz. The microscope was an inverted LEICA SP3. Images were captured for 3 minutes after the irradiation at 7-second intervals. Images were analyzed with ImageJ software.
Osmotic shock assay
Prior to osmotic shock, myotubes were incubated with 10 μM calcein-AM and 25 μg/ml DAPI (Life Technologies) for 20 minutes, followed by washing out the cells with PBS. Hypo-osmotic shock was performed by incubating cells with a differentiation medium diluted appropriately in deionized water [1:9 dilution (complete differentiation medium: H2O) approximating 30 mOsm hypo-osmotic shocks]]. Wide-field epifluorescence microscopy was performed using a 10X 0.3 NA objective and an EMCCD camera (Hamamatsu Photonics) in a Zeiss Axiovert 200 inverted microscope. Cell fluorescence was followed for 450 seconds immediately after medium change to hypo-osmotic medium. Acquisitions were performed with a time interval of 3 seconds. Images were analyzed with ImageJ software. Myotubes were selected as cells containing two or more nuclei as indicated by DAPI staining. Scale bars are indicated on each picture.
Statistical analysis
Individual means were compared using the parametric Student’s t-test or with the non-parametric Mann-Whitney test. The powers of the tests were strictly superior at 75% . Differences were considered to be statistically significant if p < 0.01 and the size of the effect was >20% .
RESULTS
As reported previously, we were able to efficiently bypass exon 32-containing mutations in DYSF by use of AONs (Wein et al, 2010). We thus evaluated the efficacy of the AON-induced splicing event on muscle cells from a control individual. These cells were transfected, using oligofectamine, with two AONs (B and D) (Figure S1A). In mock conditions, the transfection reagent (oligofectamine) without AON was used. RT-PCRs were done using a forward primer at exon 31 and a reverse primer at exon 33. We were able to demonstrate exon 32 skipping by the presence of a 215 bp transcript corresponding to an mRNA deleted of the 78 nucleotides of exon 32. In this experiment both AONs were found to be efficient and were used in all subsequent experiments (Figure S1B). As dysferlin is supposed to be expressed during myotube formation, we chose to perform our AON treatment during the differentiation process. We performed single or multiple treatments (every two days) on cells from Patients 1 and 2, starting at cellular confluence until 6 days of differentiation, when myotubes are present. The presence of a 215 bp band indicated that exon 32 was successfully skipped by both AONs, and exon-exon boundaries were verified (Fig. 1A, Figure S1B-D).
We then determined that the quasi-dysferlin protein generated following the exon 32 skipping is stable in treated patient cells. The predicted molecular weight of the quasi-dysferlin is close to the full-length isoform, 238 kDa and 241 kDa respectively. In protein extracts from cells treated with AONs B and D, we observed the expression of the quasi-dysferlin (Fig. 1B, Figure S2A). This protein is observed as early as 48 hours after the start of the treatment.
We evaluated the gain of function brought about by the expression of the quasi-dysferlin using different tests. Quasi-dysferlin is present and localized at the plasma membrane of myotubes only in treated patients’ cells. Co-localization with caveolin 3 labeling confirmed the membrane targeting of quasi-dysferlin (Fig. 2).
We decided to verify if quasi-dysferlin could restore the myotubes’ membrane repair capacities, which is the main function described for dysferlin [4].
We performed membrane wounding/repair assays both on muscle cells obtained from patient 1 as well as controls. To analyze this function we set up a laser wounding assay on in vitro culture. The extent of the lesion on myotubes was quantified in the presence of FM 1–43 dye and either with or without calcium as described previously [4, 6, 17]. In this assay, we observed that myotubes treated with AON successfully repaired membrane lesions in the presence of calcium, as seen by the low incorporation of FM1–43, whereas untreated patient myotubes exhibited a substantial level of incorporation (Fig. 3A). Quantitative analysis confirmed that both AON treatments restored the ability of patient cells to repair their membranes to a level which was similar to that observed in control myotubes (Fig. 3B).
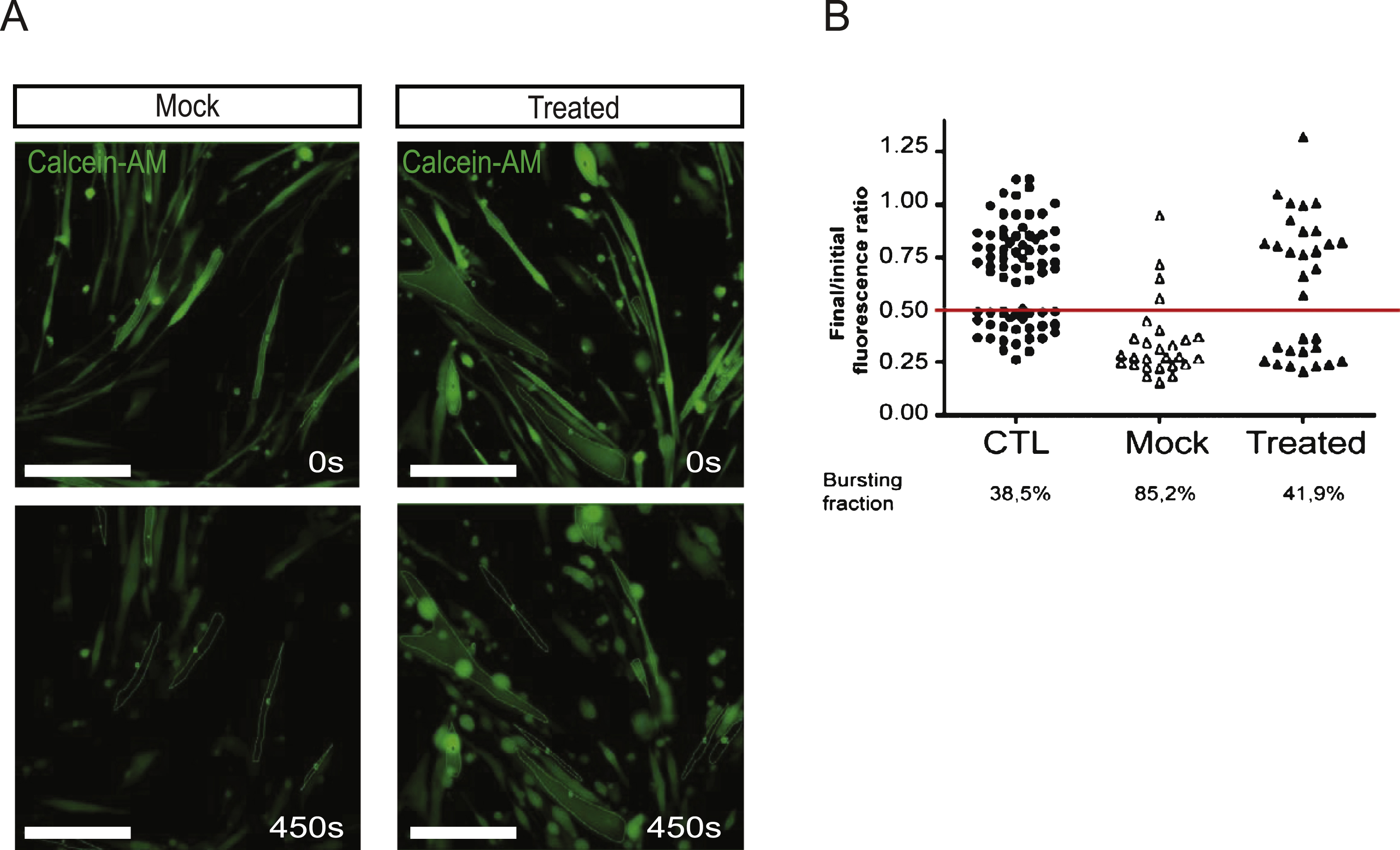
In order to further investigate the function of the quasi-dysferlin, we carried out an osmotic shock assay adapted from a previous study [28]. Hypo-osmotic shock rapidly induces cell swelling which increases the membrane tension, and therefore applies a mechanical stress at the cell surface. Patient and control myotubes were subjected to a 30 mOsm hypo-osmotic shock for 7.5 min. To monitor the integrity of the plasma membrane, we followed the intracellular accumulation of calcein-AM, a cell-permeable non-fluorescent molecule that becomes fluorescent inside intact cells and leaks out of the cells if the plasma membrane is ruptured (Fig. 4A). We could restrict our analysis to fully differentiated myotubes by selecting cells with polynucleated syncytia as revealed by DAPI staining of the nucleus (data not shown). The level of fluorescence is maintained around its initial value in myotubes whose plasma membrane remains intact during osmotic shock. In contrast, cell bursting events lead, in all observed cases, to a ratio signal value that is below 0.5 and therefore represents the bursting threshold. Mock-transfected patient myotubes showed a high rate of bursting events (85.2% ) compared to control cells (36.5% ), in agreement with the known defect of membrane repair observed in these patients (Fig. 4B). One single AON transfection in patient cells was sufficient to increase membrane integrity since the measured level of bursting was close to that of control myotubes (41.9% ) (Fig. 4B). These results show that restoration of dysferlin by AON treatment protects patient cells from mechanical stress. The total numberof nuclei per cells in these experiments was also calculated and represented as a graph in Figure S2B.
DISCUSSION
This study demonstrates the possibility of restoring functionality in dysferlin-deficient human muscle cells by skipping of an exon bearing a deleterious mutation. The functional normalization was obtained using AONs targeting exon 32. Both AONs induced the splicing out of exon 32 and the expression of a deleted isoform of dysferlin in two different patient cell lines. To evaluate the functionality of this isoform, we employed a variety of assays that measure the myoblast/myotube fusion process as well as the membrane repair ability.
In the present study we have improved the skipping efficiency obtained in our previous work [25], by designing the new AON D targeting the splice donor site of exon 32. The skipping efficiency seems to be quite high, given that almost all the transcripts produced seem to be skipped. This efficiency seems greater than what has been obtained in DMD using a similar AON chemistry [29]. The difference observed here can be explained by several parameters as they were previously discussed [30].
This skipped mRNA was stable enough to produce a quasi-dysferlin deleted of 26-residues, preserving the frame and located at the beginning of the C2D domain. When considering our approach, it can be observed that a discrepancy exists between the high level of exon skipping and the relatively low level of protein expression. This could possibly be attributed to a decrease in the stability between the full-length dysferlin and the quasi-dysferlin deleted of exon 32; this specific issue should be addressed in future experiments. Nevertheless, this low level of quasi-dysferlin expression is sufficient to rescue the membrane repair capacity of treated patient cells, confirming that pivotal aspects of dysferlin function are rescued upon bypassing the mutation in exon 32. Cells in affected patients are more susceptible to membrane mechanical constraints or at least membrane disorganization. We have shown here that they are more sensitive to a localized lesion or to an increase of membrane tension induced by osmotic pressure. One could hypothesize that this is due to the abnormal recruitment of membrane vesicles at the sarcolemma in the absence of dysferlin. Vesicle recruitment involves various proteins, among them caveolin 3. However we did not detect any difference in caveolin 3 localization after AON treatment. Since we have demonstrated that AON treated patient cells were resistant to both osmotic pressure and sarcolemma lesions, we can hypothesize that this is due to the proper recruitment of membrane vesicles at the sites of injury.
It has been proposed that dysferlin could induce membrane blebbing on cells consequent to a hypo-osmotic shock; these blebs probably result from the rapid membrane surface increase and variations of membrane tension [5, 9, 31]. Absence of dysferlin could impair this mechanism. Based on these observations, we set up a test to follow and quantify the resistance of myotubes to increased membrane tension induced by hypo-osmotic shock in defined time bands. To resist the membrane tension, cells augment the surface of their sarcolemma by the recruitment of additional membrane from reservoirs. These membrane reservoirs could be either vesicles or large invaginations of sarcolemma that could be reorganized by the protein complex involving dysferlin [5, 7, 9–11, 28, 32]. Altogether, this hypothesis can explain how dysferlin-positive myotubes accommodate acute mechanical stresses and are in accordance with previous published data from other groups [4, 8, 31, 33].
Our results establish a proof of concept for exon skipping as a therapeutic perspective for dysferlinopathies. Another group has also demonstrated that a mutant dysferlin pseudoexon could be skipped [34]. Such an approach has already been successfully applied in the case of DMD. After the encouraging results obtained in two independent phase II/III clinical trials for the skipping of dystrophin exon 51 [20–22, 35], BioMarin is moving forward with market authorization for applications involving the skipping of this exon using Drisapersen (Food and Drug Administration approval has been given and European Medicines Agency approval is still under review). Meanwhile, Sarepta is currently extending a phase II/III trial (NTC02255552) using Eteplirsen. In addition, phase II clinical trials are ongoing for exons 44, 45 and 53 using 2’-O-methyl chemistry and for exon 53 using PMO chemistry [36].
Dysferlin exon 32 encodes the N-terminal portion of the C2D domain, therefore deletion of exon 32 would most likely lead to loss of function of the entire domain without major consequences [24, 37]. The C2D domain is composed of the exons 31, 32, 33 and 34 whose deletion would maintain the open reading frame of dysferlin mRNA. In addition to dysferlin exon 32, the skipping of exon 34 has also been reported from control cells [38]. It would therefore be feasible to attempt multiple exon skipping so as to bypass the entire C2D domain of dysferlin using the previously validated AONs. These methods of multiple exon skipping have already been used with success in DMD treatment for the simultaneous skipping of at least 6 consecutive exons [39–51]. Exon skipping could also be used to remove pseudoexons. Recently, a mutation creating a pseudoexon between exons 44 and 45 of DYSF has been identified [34, 46]. Exon skipping using an AON targeting this pseudoexon restored a normal mRNA and increased dysferlin expression, opening the way for such an approach in dysferlinopathies [34].
Although some improvements may be necessary to bring about more routine use of AON-based therapies, rapid advances being made for DMD will certainly apply to other rare muscle disorders, and will allow a rapid translation of proof of principle into clinical applications in the future. At least a subset of 20 patients (with 6 different truncating mutations) should thus benefit from antisense-induced exon 32 skipping.
CONFLICT OF INTEREST STATEMENT
MB, MK, LG and NL have a patent for dysferlin exon 32 skipping (#US20120208865).
ACKNOWLEDGMENTS
We would like to acknowledge Danielle Depetris for technical assistance and J. Wes Ulm for proofreading. This work was supported by the Association Française contre les Myopathies (AFM), Institut National de la Santéet de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), UniversitéPierre et Marie Curie (UPMC), PPMD, MyoAge (EC 7th FP, contract 223576 to GBB) and the Jain Foundation. N.W. and F.B. have received PhD fellowship grants from the AFM and Fondation pour la Recherche Médicale respectively. YM has a fellowship grant from the European Community’s Seventh Framework Program (FP7/2007-2013/NEUROMICS) under grant agreement n°2012-305121.
Appendices
The supplementary table and figure are available in the electronic version of this article: http://dx.doi.org/10.3233/JND-150109.
REFERENCES
1 | Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M(1998) A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2BNat Genet20: 13742Epub 1998/09/10. |
2 | Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C(1998) Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophyNat Genet20: 13136Epub 1998/09/10 |
3 | Nguyen K, Bassez G, Krahn M, Bernard R, Laforet P, Labelle V, etal (2007) Phenotypic study in 40 patients with dysferlin gene mutations:High frequency of atypical phenotypesArch Neurol64: 811761182Epub 2007/08/19. |
4 | Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R(2003) Defective membrane repair in dysferlin-deficient muscular dystrophyNature. 423168172Epub 2003/05/09. |
5 | Defour A, Van der Meulen JH, Bhat R, Bigot A, Bashir R, Nagaraju K(2014) Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretionCell Death Dis5: e1306Epub 2014/06/27. |
6 | Krahn M, Wein N, Bartoli M, Lostal W, Courrier S, Bourg-Alibert N(2010) A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathySci Transl Med2: 5050ra69Epub 2010/09/24. |
7 | Lek A, Evesson FJ, Lemckert FA, Redpath GM, Lueders AK, Turnbull L(2013) Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repairJ Neurosci33: 1250855094Epub 2013/03/22. |
8 | Marg A, Schoewel V, Timmel T, Schulze A, Shah C, Daumke O(2012) Sarcolemmal repair is a slow process and includes EHD2Traffic9999: 9999 |
9 | McDade JR, Archambeau A, Michele DE(2014) Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repairFaseb J28: 836603670Epub 2014/05/03. |
10 | Posey ADJr, Pytel P, Gardikiotes K, Demonbreun AR, Rainey M, George M(2011) Endocytic recycling proteins EHD1 and EHD2 interact with fer-1-like-5 (Fer1L5) and mediate myoblast fusionJ Biol Chem286: 973797388Epub 2010/12/24. |
11 | Redpath GM, Woolger N, Piper AK, Lemckert FA, Lek A, Greer PA(2014) Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repairMol Biol Cell25: 1930373048Epub 2014/08/22. |
12 | Roche JA, Lovering RM, Roche R, Ru LW, Reed PW, Bloch RJ(2010) Extensive mononuclear infiltration and myogenesis characterize recovery of dysferlin-null skeletal muscle from contraction-induced injuriesAm J Physiol Cell Physiol.C298: 2298312Epub 2009/11/20. |
13 | de Luna N, Gallardo E, Soriano M, Dominguez-Perles R, de la Torre C, Rojas-Garcia R(2006) Absence of dysferlin alters myogenin expression and delays human muscle differentiation “in vitro”J Biol Chem281: 251709217098Epub 2006/04/13. |
14 | Demonbreun AR, Fahrenbach JP, Deveaux K, Earley JU, Pytel P, McNally EM(2011) Impaired muscle growth and response to insulin-like growth factor 1 in dysferlin-mediated muscular dystrophyHum Mol Genet20: 4779789Epub 2010/12/04. |
15 | Balasubramanian A, Kawahara G, Gupta VA, Rozkalne A, Beauvais A, Kunkel LM(2014) Fam65b is important for formation of the HDAC6-dysferlin protein complex during myogenic cell differentiationFaseb J28: 7295569Epub 2014/04/02. |
16 | Grose WE, Clark KR, Griffin D, Malik V, Shontz KM, Montgomery CL(2012) Homologous recombination mediates functional recovery of dysferlin deficiency following AAV5 gene transferPLoS One7: 6e39233Epub 2012/06/22. |
17 | Lostal W, Bartoli M, Bourg N, Roudaut C, Bentaib A, Miyake K(2010) Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transferHum Mol Genet19: 101897907Epub 2010/02/16. |
18 | Philippi S, Lorain S, Beley C, Peccate C, Precigout G, Spuler SDysferlin rescue by spliceosome-mediated pre-mRNA trans-splicing targeting introns harbouring weakly defined 3’ splice sitesHum Mol Genet2015Epub 2015/04/24. |
19 | Sondergaard PC, Griffin DA, Pozsgai ER, Johnson RW, Grose WE, Heller KN(2015) AAV. Dysferlin overlap vectors restore function in dysferlinopathy animal modelsAnnals of Clinical and Translational Neurology2: 3256270Epub 2015/03/31. |
20 | Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K(2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation studyLancet378: 9791595605 |
21 | van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M(2007) Local dystrophin restoration with antisense oligonucleotide PRO051N Engl J Med357: 2626772686Epub 2007/12/28. |
22 | Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G(2014) Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 studyLancet Neurol13: 10987996Epub 2014/09/12. |
23 | Flanigan KM, Voit T, Rosales XQ, Servais L, Kraus JE, Wardell C(2014) Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: Results of a double-blind randomized clinical trialNeuromuscul Disord24: 11624Epub 2013/12/11. |
24 | Sinnreich M, Therrien C, Karpati G(2006) Lariat branch point mutation in the dysferlin gene with mild limb-girdle muscular dystrophyNeurology66: 711141116Epub 2006/04/12. |
25 | Wein N, Avril A, Bartoli M, Beley C, Chaouch S, Laforet P(2010) Efficient bypass of mutations in dysferlin deficient patient cells by antisense-induced exon skippingHum Mutat31: 2136142Epub 2009/12/03. |
26 | Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR(2003) ESEfinder: A web resource to identify exonic splicing enhancersNucleic Acids Res31: 1335683571Epub 2003/06/26. |
27 | Desmet FO, Beroud C(2012) Bioinformatics and mutations leading to exon skippingMethods Mol Biol867: 1735Epub 2012/03/29. |
28 | Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D(2011) Cells respond to mechanical stress by rapid disassembly of caveolaeCell144: 3402413Epub 2011/02/08. |
29 | Aartsma-Rus A, Kaman WE, Bremmer-Bout M, Janson AA, den Dunnen JT, van Ommen GJ(2004) Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cellsGene Ther11: 1813911398Epub 2004/07/02. |
30 | Aartsma-Rus A(2012) Overview on DMD exon skippingMethods Mol Biol867: 97116Epub 2012/03/29. |
31 | Wang B, Yang Z, Brisson BK, Feng H, Zhang Z, Welch EM(2010) Membrane blebbing as an assessment of functionalrescue of dysferlin-deficient human myotubes via nonsense suppressionJ Appl Physiol109: 3901905Epub 2010/06/19. |
32 | Oulhen N, Onorato TM, Ramos I, Wessel GM(2014) Dysferlin is essential for endocytosis in the sea star oocyteDev Biol388: 194102Epub 2013/12/26. |
33 | Kerr JP, Ziman AP, Mueller AL, Muriel JM, Kleinhans-Welte E, Gumerson JD(2013) Dysferlin stabilizes stress-induced Ca2+signaling in the transverse tubule membraneProc Natl Acad Sci U S A110: 512083120836Epub 2013/12/05. |
34 | Dominov JA, Uyan O, Sapp PC, McKenna-Yasek D, Nallamilli BR, Hegde M(2014) A novel dysferlin mutant pseudoexon bypassed with antisense oligonucleotidesAnnals of clinical and translational neurology1: 9703720Epub 2014/12/11. |
35 | Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N(2011) Systemic administration of PRO051 in Duchenne’s muscular dystrophyN Engl J Med364: 1615131522Epub 2011/03/25. |
36 | Arechavala-Gomeza V, Anthony K, Morgan J, Muntoni F(2012) Antisense oligonucleotide-mediated exon skipping for duchenne muscular dystrophy: Progress and challengesCurr Gene Ther12: 3152160Epub 2012/04/27. |
37 | Azakir BA, Di Fulvio S, Salomon S, Brockhoff M, Therrien C, Sinnreich M(2012) Modular dispensability of dysferlin’s C2 domainsreveals rational design for mini-dysferlin moleculesJ Biol ChemEpub 2012/06/28. |
38 | Aartsma-Rus A, Singh KH, Fokkema IF, Ginjaar IB, van Ommen GJ, den Dunnen JT(2010) Therapeutic exon skipping for dysferlinopathies? Eur J Hum Genet18: 8889894Epub 2010/02/11. |
39 | Aartsma-Rus A, Janson AA, Kaman WE, Bremmer-Bout M, van Ommen GJ, den Dunnen JT(2004) Antisense-induced multiexon skipping for Duchenne muscular dystrophy makes more senseAm J Hum Genet74: 18392Epub 2003/12/19. |
40 | Goyenvalle A, Wright J, Babbs A, Wilkins V, Garcia L, Davies KE(2012) Engineering multiple U7snRNA constructs toinduce single and multiexon-skipping for Duchenne muscular dystrophyMol Ther20: 612121221Epub 2012/02/23. |
41 | Echigoya Y, Yokota T(2014) Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotidesNucleic Acid Therapeutics24: 15768Epub 2014/01/02. |
42 | Adkin CF, Meloni PL, Fletcher S, Adams AM, Muntoni F, Wong B(2012) Multiple exon skipping strategies to by-pass dystrophin mutationsNeuromuscul Disord.22: 4297305Epub 2011/12/21. |
43 | Beroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Humbertclaude V, Monnier N(2007) Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophyHum Mutat28: 2196202Epub 2006/10/17. |
44 | Aartsma-Rus A, Kaman WE, Weij R, den Dunnen JT, van Ommen GJ, van Deutekom JC(2006) Exploring the frontiers of therapeutic exon skipping for Duchenne muscular dystrophy by double targeting within one or multiple exonsMol Ther14: 3401407Epub 2006/06/07. |
45 | Gualandi F, Trabanelli C, Rimessi P, Calzolari E, Toffolatti L, Patarnello T(2003) Multiple exon skipping and RNA circularisation contribute to the severe phenotypic expression of exon 5 dystrophin deletionJ Med Genet40: 8e100Epub 2003/08/16. |
46 | Kergourlay V, Rai G, Blandin G, Salgado D, Beroud C, Levy N(2014) Identification of splicing defects caused by mutations in the dysferlin geneHum Mutat35: 1215321541Epub 2014/10/15. |
Figures and Tables
Fig.1
AON B efficiently skipped dysferlin exon 32 in human patient cells. (A) Efficiency of exon skipping using AON B was assessed by RT-PCR on cells from patient 2. PCR was used to amplify the region between exons 31 and 33 (293 bp with or 215 bp without exon 32). RT::negative control of reverse transcription. Mock: negative control of transfection. Scrambled: nonspecific AON sequence. AON B: single transfection of AON B. CTL: positive control, cells were transfected with dysferlin full-length (native dysferlin). MM: molecular marker. (B) Western Blot experiment performed with proteins from (AON B or scrambled) treated patient cells or control cells. Hamlet was used to detect dysferlin and GAPDH was used for normalization.
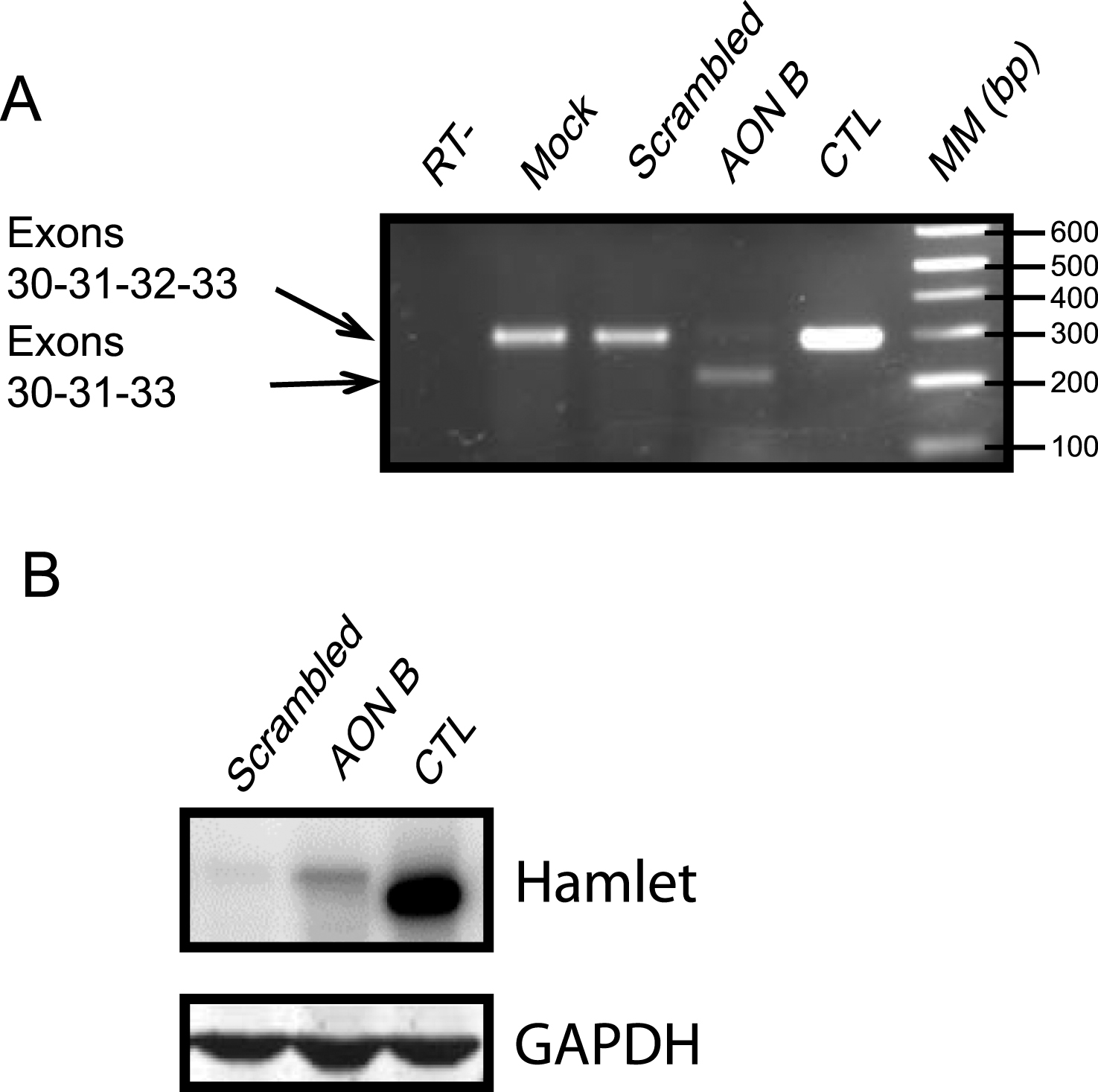
Fig.2
Exon 32-skipped dysferlin is correctly localized. Caveolin 3 labeling: maturation of myotubes and production of dysferlin were evidenced by Hamlet 1 labeling (Bars = 10 μm, arrow pointed to colocalized signal). DAPI was used as a nucleus marker. All images were captured by an apotome microscope.
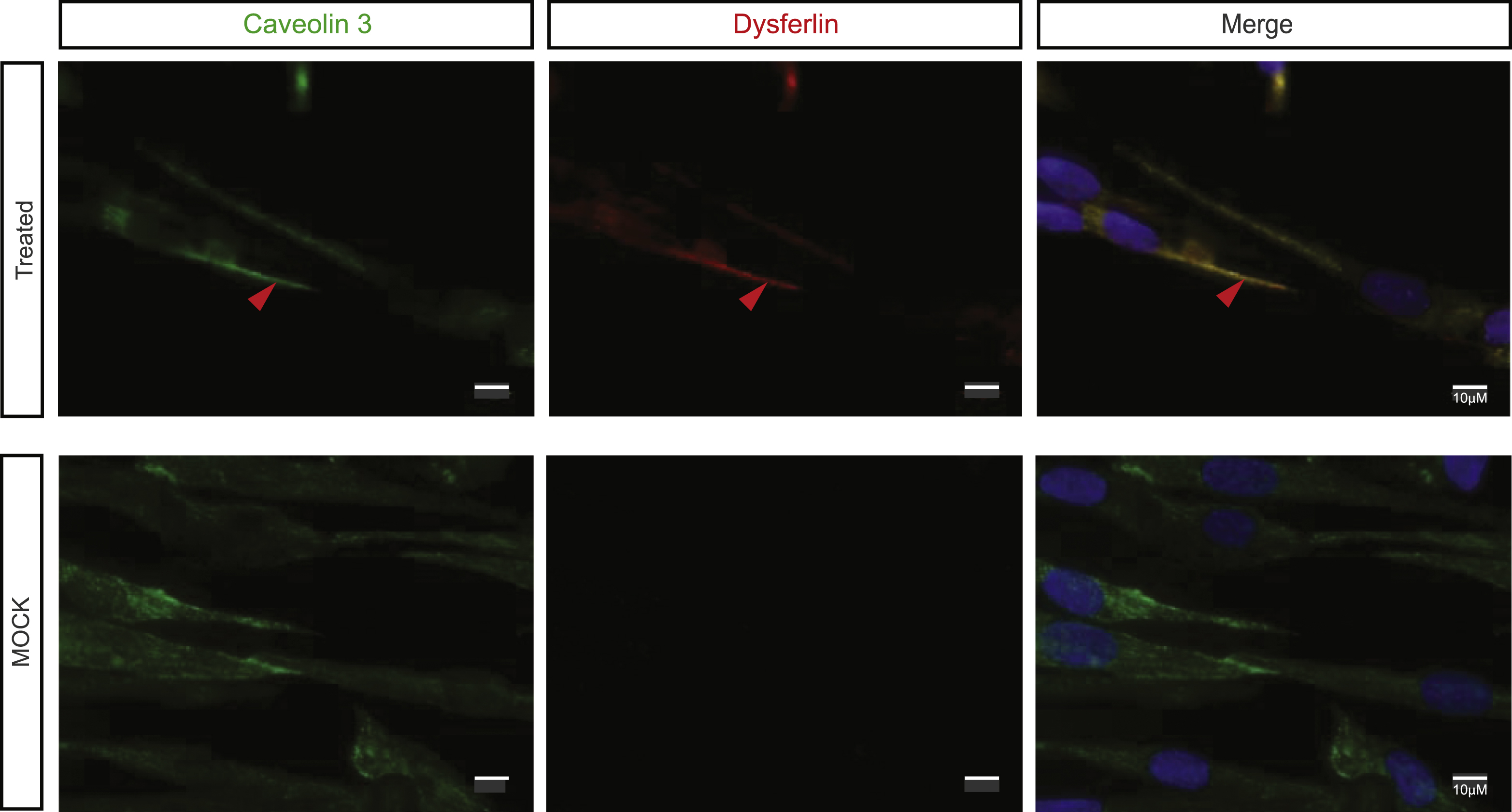
Fig.3
Quasi-dysferlin increases the membrane repair capacity. (A) Membrane repair consequent to a bi-photon laser injury at the sarcolemma was visualized in presence of FM1-43 dye. Mock treated cells along with AON B and AON D treated cells from patient 1 were compared for three minutes at intervals of 7 seconds. Pictures at 0, 1, 2 and 3 minutes are shown. Arrows indicate the site of lesion. The scale (of false colors used) is represented on the left. (B) Box plots represent the rate of change of fluorescence (Δ[fluorescence]/Δt) in the myotubes from control (n = 9), mock-treated patient cells (n = 9), AON B treated patient cells (n = 10) and AON D treated patient cells (n = 9). Boxes extend from the 25th to the 75th percentile values. Minimum and maximum values are indicated by the dots at the ends of the vertical lines. Horizontal bars indicate the median value. * p < 0.01 compared to mock-treated patient cells.
![Quasi-dysferlin increases the membrane repair capacity. (A) Membrane repair consequent to a bi-photon laser injury at the sarcolemma was visualized in presence of FM1-43 dye. Mock treated cells along with AON B and AON D treated cells from patient 1 were compared for three minutes at intervals of 7 seconds. Pictures at 0, 1, 2 and 3 minutes are shown. Arrows indicate the site of lesion. The scale (of false colors used) is represented on the left. (B) Box plots represent the rate of change of fluorescence (Δ[fluorescence]/Δt) in the myotubes from control (n = 9), mock-treated patient cells (n = 9), AON B treated patient cells (n = 10) and AON D treated patient cells (n = 9). Boxes extend from the 25th to the 75th percentile values. Minimum and maximum values are indicated by the dots at the ends of the vertical lines. Horizontal bars indicate the median value. *
p < 0.01 compared to mock-treated patient cells.](https://content.iospress.com:443/media/jnd/2015/2-3/jnd-2-3-jnd150109/jnd-2-3-jnd150109-g003.jpg)
Fig.4
AON-treated patients cells are resistant to hypo-osmotic shock. (A) Membrane rupture in response to hypo-osmotic shock was assessed using calcein-AM to distinguish between intact and burst cells. Low-magnification epifluorescence snapshots of calcein-AM in mock and treated cells at the beginning (t = 0 s, top) and at the end (t = 450 sec, bottom) of the hypo-osmotic shock (Bar = 100 μm). (B) Plot of final/initial fluorescence intensity ratio for control myotubes (WT, n = 74, dark dots), for patient 2 myotubes untreated (MOCK, n = 27, white triangles), treated with AON D (n = 31, dark triangles) during the hypo-osmotic shock. The bursting fraction corresponds to the cell population whose fluorescence ratio is below the selected bursting threshold. The bursting threshold has been set to 0.5 of normalized fluorescence intensity (horizontal red line).