
The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies
Abstract
Over sixty years ago John Walton and Frederick Nattrass defined limb girdle muscular dystrophy (LGMD) as a separate entity from the X-linked dystrophinopathies such as Duchenne and Becker muscular dystrophies. LGMD is a highly heterogeneous group of very rare neuromuscular disorders whose common factor is their autosomal inheritance. Sixty years later, with the development of increasingly advanced molecular genetic investigations, a more precise classification and understanding of the pathogenesis is possible.
To date, over 30 distinct subtypes of LGMD have been identified, most of them inherited in an autosomal recessive fashion. There are significant differences in the frequency of subtypes of LGMD between different ethnic populations, providing evidence of founder mutations. Clinically there is phenotypic heterogeneity between subtypes of LGMD with varying severity and age of onset of symptoms. The first natural history studies into subtypes of LGMD are in process, but large scale longitudinal data have been lacking due to the rare nature of these diseases. Following natural history data collection, the next challenge is to develop more effective, disease specific treatments. Current management is focussed on symptomatic and supportive treatments. Advances in the application of new omics technologies and the generation of large-scale biomedical data will help to better understand disease mechanisms in LGMD and should ultimately help to accelerate the development of novel and more effective therapeutic approaches.
INTRODUCTION
Limb girdle muscular dystrophy (LGMD) is an umbrella term given to a group of rare, highly heterogeneous, autosomal neuromuscular disorders. The term LGMD was first used in a seminal paper by John Walton and Frederick Nattrass in 1954 [1]. The authors identified LGMD as a separate entity from the more common X-linked Duchenne and Becker muscular dystrophies (DMD/BMD). Between subtypes of LGMD, there are few common features, with the exception of autosomal inheritance. Other similarities in LGMD, most of which are still relevant, were described as illustrated in this quote from the original paper:
“In our opinion, these cases should be regarded as a distinct clinical and genetic group and we suggest they should be called limb-girdle muscular dystrophy. Leaving aside certain clinical characteristics which may be added later, we feel that the cardinal features of this type are (a) onset usually late in the first or in the second or third decade but sometimes in middle age, (b) commencement of muscular weakness in either the shoulder or pelvic girdle, (c) transmission usually via an autosomal recessive gene, (d) a relatively slow course which nevertheless leads to severe disablement and often death before the normal age.” [1]
A number of the cases identified in the paper by Walton and Nattrass continued to be treated in Newcastle and were subsequently identified as LGMD2A and LGMD2I. There are now over thirty subtypes of LGMD identified. LGMD may present at any age and in the severe forms can show an overlap with congenital muscular dystrophies. Heterogeneity is seen between subtypes in presenting complaint, most but not all, experience proximal muscle weakness or myalgia. Distal muscle weakness and life threatening arrhythmias may on the other hand be the first symptom, which illustrates the wide variation between LGMD subtypes.
Over the last sixty years, molecular genetic investigations have advanced, allowing precise classification and improvements in our understanding of the pathogenesis of LGMD subtypes. Other scientific advances have increased our knowledge of the structure and function of various parts of the sarcolemma and myocyte function. In 1995 a consensus was achieved on the categorisation of subtypes of LGMD [2].
Classification and epidemiology
All form of LGMD are inherited autosomally and the classification is alphanumeric with assignation of number ‘1’ or ‘2’ depending on whether they are inherited dominantly or recessively. A letter is added in order of discovery [2]. To date over 50 genetic loci have been identified [3]. Dominantly inherited LGMD is less common and reported to be only 5–10% of all LGMD [4, 5].
Overall prevalence of LGMD worldwide has been estimated to be 1 in 14,500–45,000 [4, 6]. A study by Norwood et al suggested that within the population of Northern England, LGMD accounts for 6.2% of all neuromuscular patients [6]. There is a wide variation in prevalence of LGMD subtypes in different ethnicities, suggesting potential founder mutations [7]. In the case of many of the newly discovered LGMD subtypes, mutations have only been described in a handful of families worldwide [8, 9]. Reportedly the most common LGMD subtype is LGMD2A (26.5–30% ), then LGMD2I (19% ) although this varies widely between populations [6, 10]. Because of the autosomal mode of inheritance LGMD is supposed to affect both genders equally, although Magnetic resonance imaging (MRI) studies have shown that phenotypic differences may exist in the severity of affected muscles [11]. and in LGMD2L males seem to be more frequently affected [12].
LGMD subtypes are often grouped according to which protein is affected and many are allelic with other conditions.
Pathogenesis
LGMD is caused by multiple genes encoding for proteins within the sarcolemma, cytosol or nucleus of the myocyte (see Table 1 and Fig. 1). The lack of a common aetiology explains some of the differences in severity of phenotypes. The heterogeneous nature of LGMD makes it difficult to define a common pathway of myocyte damage. Possible mechanisms include membrane instability [33], errors in formation of a functional dystroglycan complex [60], and defects in muscle repair mechanisms [32, 61]. It is likely that the majority of the LGMD subtypes eventually develop membrane instability, similar to dystrophinopathies, which ultimately leads to muscle fibre degeneration. High levels of intracellular calcium have been implicated in myocytic damage [62], although the exact mechanism of calcium ion influx is not well described. It has been suggested that the influx of calcium ions activates proteolysis, eventually leading to apoptosis/necrosis of the myocytes [63]. Damage of muscle fibres leads to the release of inflammatory cytokines and consecutively neutrophils and macrophages are dispatched to degrade cellular debris [64]. Muscle satellite cells (undifferentiated myocyte progenitor cells) replace the damaged or necrotic tissue [65]. Eventually muscular repair mechanisms and satellite cell populations are overwhelmed and deposition of fibrotic (collagen) tissue and adipose tissue occurs [66].
Natural history of LGMD
A thorough understanding of the progression of LGMD subtypes is dependent on the completion of longitudinal natural history studies. Natural history studies are essential in defining disease specific outcome measures and in developing care standards. To date, small scale cross-sectional studies and case series provide most of the information we have on the natural history of LGMD. The rarity of LGMD makes recruitment to larger longitudinal studies challenging. Longitudinal natural history studies have been completed on LGMD2I [67]. and a large cohort of LGMD2B are currently being assessed in an international longitudinal study, the results of which are not yet published. LGMD1B and 2A cohorts have been described, although no longitudinal natural history studies have been accomplished.
Patients with LGMD display predominantly symmetrical, proximal muscle weakness, although asymmetry and distal involvement can be seen in LGMD2B and 2L. Typical features and complications of LGMD may include respiratory compromise, cardiomyopathy, cardiac arrhythmias, scapular winging, calf hypertrophy, rigid spine and limb contractures, but almost all of the known LGMD subtypes show clinical heterogeneity and assuming a diagnosis based on “classical” phenotypes may be misleading. Less common associated features may aid to the diagnosis, as for example rippling muscle disease in LGMD1C [68]. Serum creatinine kinase (CK) levels are almost always raised in LGMD2, although the degree to which the CK is raised depends on the subtype, with LGMD2B and 2L having exceptionally high serum CK activities. Autosomal dominantly inherited LGMD commonly have a lower serum CK level than recessively inherited forms, except for LGMD1C, which can present with CK levels >2000 IU/l.
A brief summary of the features of the more common and distinctive subtypes of LGMD is listed below.
LGMD1A (myotilinopathy)
LGMD 1A is allelic with myotilin-associated myofibrillar myopathy and most patients with MYOT mutations have been described with the latter phenotype. The onset of weakness occurs typically during adult life and patients show normal or mildly elevated serum CK levels [69]. Other features can include respiratory failure, cardiomyopathy and dysarthric speech [69]. Over the past years several imaging studies using muscle MRI have been performed, showing a selective pattern of involvement. Of the hamstring muscles the semimembranosus muscle is most severely affected whereas the adjacent semitendinosus muscle is often well preserved [70]. Most patients with LGMD1A show the characteristic changes of a myofibrillar myopathy in their muscle biopsy.
LGMD1B (laminopathy)
Laminopathy is allelic with several other multisystem disorders (i.e. lipodystrophy, Emery Dreifuss muscular dystrophy) and may present from the first to the fourth decade of life. Features include severe cardiomyopathy and potentially life threatening cardiac arrhythmias [71]. Cardiology follow up is therefore very important. Due to the risk of life threatening arrhythmias, LGMD 1B is an important diagnosis to exclude in patients with an unidentified LGMD following muscle biopsy analysis, which generally shows unspecific findings. The only way to successfully exclude LGMD1B is by genetic testing. Rigid spine and limb contractures are also associated but not universal [71]. Serum CK levels are either normal or mildly elevated, with MRI studies have shown the most affected muscles include adductor magnus, semimembranosus, long head of the biceps femoris, soleus and medial head of the gastrocnemius [72].
LGMD1C (caveolinopathy)
Caveolinopathy comprises 1-2% of all LGMD [73, 74]. It usually presents within the first decade of life. The most common presenting complaint is primarily myalgia, followed by ‘rippling muscles’ and proximal muscle weakness [68]. It may be possible to elicit the ‘Rippling’ of the muscles by percussion with a tendon hammer. Levels of serum CK may be moderate to high; 4–25 times normal levels [75]. Hypertrophic cardiomyopathy and cardiac arrhythmias can occasionally occur in this disease and annual reviews are suggested [76].
LGMD1E, 2R (The desminopathies)
Desmin is a structural protein involved in membrane stabilisation. Most mutations in the DES gene have been associated with myofibrillar myopathy and also cardiomyopathy. LGMD 1E has been described in families in America and Finland [77, 78] with LGMD 2R only reported in a Turkish Family [54]. Patients typically present in adulthood with progressive weakness. The dominant form (LGMD1E) has a strong association with cardiomyopathy [77] with both recessive and dominant forms associated with cardiac arrhythmias [23]. Serum CK levels may be normal or may show a modest increase (up to 4 times normal levels). On muscle MRI patients with desminopathies typically show involvement of the semitendinosus, gracilis and sartorius muscles and muscle biopsies show characteristic features of myofibrillar myopathies with desmin accumulation [79, 80].
LGMD2A (calpainopathy)
Calpainopathy, probably the most common form of LGMD worldwide, typically first presents from 2–40 years [6, 30]. The phenotype is highly variable but studies have estimated that loss of ambulation can occur from 5–39 years [30]. Respiratory compromise is common at later stages, but less severe than in the LGMDs associated with the dystrophin-glycoprotein complex. Patients with LGMD2A don’t show cardiac involvement. Serum CK is typically moderately raised, at 3–20 times normal levels. MRI studies have suggested that the muscles of the shoulder girdle and of the posterior compartments of the legs are most severely affected, with relative sparing of the sartorius, gracilis and vastus medialis muscles [81]. (see ‘Fig. 2’) Muscle biopsies show dystrophic features with sometimes prominent inflammatory infiltrates and loss of calpain 3 expression by Western blotting can suggest the diagnosis, which should be genetically confirmed.
LGMD2B (dysferlinopathy)
Dysferlinopathy is one of the more common forms of LGMD. One Italian study estimated this to be the cause of 18% of all LGMDs in their population [74]. Age of onset is variable (typically second to third decade of life) and progression is often slow [82]. Cardiomyopathy is not generally a feature in LGMD2B. Serum CK activity may be very high; often up to 40 times of normal levels. MRI studies have shown gluteal muscles, tensor fasciae latae, semitendinosus, semimembranosus, biceps femoris and triceps surae to be the most severely affected muscles [83]. Dysferlin-deficiency with more prominent involvement of the lower legs muscles is also known as Miyoshi myopathy. Immunoanalysis of dysferlin in muscle biopsies typically shows a lack of expression by immunohistochemistry and Western blotting.
LGMD2C, 2D, 2E, 2F, (The sarcoglycanopathies)
The sarcoglycanopathies combined account for 12% of all LGMD in an Italian population [84]. The four types (2C, 2D, 2E, 2F) relate to the specfic sarcoglycan protein which is missing, γ, α, β, δ, respectively. Phenotypically, sarcoglycanopathy resembles dystrophinopathy, with severe, progressive proximal muscular weakness [85]. Symptoms may present in early life – typically 4–7 years old – and presentation may occur up to the second decade. Respiratory failure and cardiomyopathy are common features and should be actively screened for. Serum CK activity is generally very high and the diagnosis is typically established by a lack or reduction of sarcoglycan expression on biopsies and genetic testing. Imaging studies have shown that in comparison to the thigh and shoulder girdle muscles, the calf muscles are fairly well preserved in patients with sarcoglycanopathies [86]. (See ‘Fig. 2’)
LGMD2G (telethoninopathy)
Telethoninopathy is described in only a small number of patients [39, 87] and onset of disease seems tomost frequently start in adolescence, with proximal anddistal limb weakness. Serum CK is moderately raised (up to 10 times normal levels). The diagnosis can be suggested by the lack of telethonin expression in bio-psies, which typically show a dystrophic pattern [88].
LGMD2I, 2K, 2M, 2N, 2O, 2P, 2T, 2U (The dystroglycanopathies)
Dystroglycanopathies are caused by defects in the dystroglycan complex, mainly through abnormalglycosylation of α-dystroglycan. Dystroglycan is present in many tissues throughout the body and is essential in function of myocytes and membrane stability [89]. The phenotypic spectrum of disease is highly variable, and presentation may be anywhere from the first to the fourth decade of life. LGMD2I is the most common of the dystroglycanopathies. Respiratory failure and cardiomyopathy are common features of this group, with myoglobinuria and myalgia also reported [90]. Serum CK is dependent on subtype, but ranges from moderately raised (5–10 times normal) to very high levels (up to 50 times normal). MRI studies have shown that the most severely affected muscles are: gastrocnemius, soleus, long head of the biceps femoris, semimembranosus and semitendinosus. The vastus lateralis, gracilis and sartorius are relatively spared [11]. On muscle biopsies most patients show an abnormal expression of α-dystroglycan.
LGMD2J (titinopathy)
The TTN gene has the largest number of exons and has the longest coding sequence of any known gene. Only recent advances in next generation sequencing have enabled diagnosis of more patients with mutations in this gene. Titinopathy is thought to be under-recognised due to the difficulties in diagnosis, and may be far more common than previously believed. Originally titinopathy was described in a family from Finland [91] with presentation of severe weakness in the first to third decades of life. Cardiomyopathy was not described in the original Finnish family; [91] however other mutations in titin have predisposed patients to cardiac involvement. Serum CK can be highly raised. Patients with mutations in the TTN gene may also present with distal weakness, severe respiratory involvement and characteristic features of a myofibrillar myopathy [92]. It can still be very challenging to confirm the diagnosis of LGMD2J and other titin-associated myopathies.
LGMD2L
LGMD2L is one of the most common forms of LGMD in Northern Europe and Canada. Onset of symptoms may be from the third decade of life onwards [12, 93]. Unlike most subtypes of LGMD, LGMD2L predominantly affects males although it is not fully understood why this appears to be the case. No cardiac involvement has been reported [93]. Serum CK levels are typically highly elevated and the diagnosis needs to be established genetically. An example of MRI findings is seen in ‘Fig. 2’.
Other LGMD subtypes
LGMD1D, 1F, and 1G are all described in very small numbers, and sometimes within single families. The age of presentation is varied, but typically onset is with proximal muscle weakness in adulthood. Serum CK in these subtypes may be normal or increased by ten times. The role of cardiac and respiratory involvement is not yet clear in these LGMD subtypes [26–28, 37, 94–96]. Patients with LGMD1D show similar features to patients with myofibrillar myopathies.
LGMD2H, 2Q, 2S, 2V, 2W are very rare LGMD subtypes and have only been described in a few cases, although LGMD2H is frequently diagnosed in the Hutterites due to a common founder mutation [40]. These subtypes typically present in the first to second decades. All of these subtypes give a rise in serum CK; in some cases as much as fifty times normal [8, 9, 53, 55, 97].
Differential diagnosis
LGMD is a group of highly heterogeneous disorders with many distinguishing features. A wide differential diagnostic spectrum should be considered, as several conditions may have similar presentations. Below are some conditions which have certain features in common with some of the LGMD subtypes.
DMD and BMD are more common than LGMD and are often suspected first. Dystrophinopathies may present with similar onset and patterns of weakness to some of the LGMD. Female carriers may also manifest mild proximal weakness and are easily misdiagnosed with LGMD [98]. It is likely that there are more female carriers of DMD and BMD than most of the LGMD subtypes. Genetic studies identifying the mutation in the DMD gene or immunohistochemical analysis are diagnostic.
In facioscapulohumeral muscular dystrophy (FSHD) the pattern of muscle weakness can be variable, and progression to facial features may not be seen for many years, leading to suspicions of LGMD. Confirmation of FSHD is through genetic testing [99].
Patients with spinal muscular atrophy (SMA) types II and III can present similarly to LGMD with progressive proximal muscle weakness. Diagnosis is confirmed with genetic testing to identify deletions of SMN1.
Patients with Emery Dreifuss muscular dystrophy (EDMD) also form an important differential diagnosis to LGMD. The most prominent sign of EDMD is cardiac involvement and arrhythmias and patients commonly present with progressive joint contractures, typically in elbow flexion, and hip girdle related weakness in the first decade of life [100].
Pompe disease is another differential diagnosis that needs to be considered in patients with limb girdle weakness. Pompe disease is highly variable in presentation and may present at any age from childhood to adulthood. It is diagnosed by a deficiency or reduction of glucosidase enzyme activity [101].
Bethlem myopathy, caused by mutations in any of the three genes for collagen VI, may present in early childhood or develop later in life and can form an important differential diagnosis to LGMD. Contractures are common and patients frequently present with skin changes that are suggestive of a collagen VI related disorder [102].
Acquired muscle diseases, such as polymyositis, dermatomyositis and inclusion body myositis can present with signs of proximal muscle weakness and elevated serum CK levels and may mimic a form of LGMD. History of skin changes, arthritis and elevation in inflammatory markers should be ascertained. Histological changes on muscle biopsy can be similar to types of LGMD, in particular LGMD2A, 2B and 2L, as all may show inflammatory infiltrates [103, 104]. Patients with refractory cases of inflammatory myositis, i.e. unresponsive to immunosuppression, should be investigated for LGMD as misdiagnosis is possible [105].
Diagnostic investigations
Thorough history and examination is vital in the first line of investigating patients with suspected neuromuscular disease. Features such as: age of onset, presence of cardiomyopathy or arrhythmia, ‘rippling’ muscles, or symptoms of rigid spine may give clues as to the subtype of LGMD. When considering any genetic condition, thorough family history is vital.
Serum CK is raised in the majority of the LGMD subtypes and should always been tested in patients with limb girdle weakness. The degree to which it is raised may provide diagnostic clues.
Genetic investigation in patients with progressive limb girdle weakness is directed by history, examination, specific family history and the results of other investigations such as electrophysiology, muscle imaging and muscle biopsy analysis.
Electrophysiology is useful to rule out some of the differential diagnoses such as neuromuscular junction disease, SMA III and neuropathies. LGMD electrophysiology findings are of nonspecific myopathic changes.
MRI is a non-invasive, highly sensitive way of assessing patients with suspected primary muscle disease, although it’s application is more useful for the differential diagnosis of congenital myopathies and myofibrillar myopathies than for LGMD. MRI is on the other hand particularly useful in assessing disease progression and in targeting muscle biopsy. Studies have investigated LGMD using MRI to investigate patterns of muscle involvement which can also help to direct genetic and immunohistochemical analysis [11, 81, 86, 94, 95].
A wide panel of immunohistochemical stains are used in analysis of muscle biopsy tissue. For many forms of LGMD muscle histology is nonspecific and may share several features with other neuromuscular disorders such as the dystrophinopathies. Histology findings include rimmed vacuoles, muscle regeneration, necrosis, macrophage infiltration and inflammation but are nonspecific to any of the LGMD subtypes. Immunohistochemical and Western blot analysis using a panel of antibodies greatly increases diagnostic capabilities and can either guide or confirm genetic testing [106]. Due to the highly population specific nature of some of the LGMD, a selection of the most common subtypes of LGMD are investigated initially in this way. Next generation sequencing (NGS) has been used to aid in identification of more genetic loci and subtypes of LGMD [107].
Management
To date, no cause-specific treatment is available for any of the LGMD subtypes. Limited evidence exists for response to immunomodulating therapies such as corticosteroids which has been reported to have some effects in some of the LGMDs [108, 109]. To date, only one large scale randomised controlled trial has assessed the effects of corticosteroids on LGMD. In 2008, Walter et al found no significant clinical benefit in giving Deflazacort to patients with LGMD 2B (dysferlinopathy) [110].
Supportive management is important with therapies being tailored to the subtype of LGMD and the individual’s needs. Specialist multidisciplinary management is vital. Physiotherapy and orthotic intervention is directed at aiding the individual to maintain independence and ambulance for as long as possible. Efforts should be focussed on prevention of contractures and orthopaedic referral where necessary [111]. Non maximal aerobic exercise should beencouraged.
Respiratory failure is a feature of many types of LGMD. Regular assessment of forced vital capacity via spirometry, and overnight pulse oximetry is very important. Interventions such as non-invasive ventilation and cough assist machines should be considered on an individual basis [112]. Several of the subtypes of LGMD are associated with cardiomyopathy and cardiac arrhythmias. Cardiologist review for those at risk is essential; this should include regular echocardiogram and ECG.
FUTURE RESEARCH
In the past, research into rare diseases has been hindered by small numbers of patients available on a regional and even national level. Multinational networks such as TREAT NMD [113] have been created to encourage research into rare neuromuscular conditions and to enable recruitment on a larger scale.
Multicentre, natural history studies are important in increasing our knowledge of LGMD. Ongoing long term follow up studies are looking at the measurable progression of LGMD 2I and LGMD 2B using both functional outcomes and MRI [67, 114]. Characterisation of the natural history of LGMD subtypes is the first step in developing LGMD subtype specific treatments with the involvement of funding agencies such as the European Union, NIH, patient organisations and pharmaceutical companies.
Patient registries are also important in research into rare disease. Registries give researchers the ability to contact and review details of patients with rare diseases across large geographical areas. Registries are available for LGMD 2A, 2B, 2C, 2D, 2I, 2K, 2M, 2N, and 2O [115–120].
Future avenues for research include gene therapy. Some success has been seen in ameliorating murine models of subtypes of LGMD [121–123]. Barriers to successful treatment with adeno-associated virus (AAVs) include difficulties in delivery to affected muscle and the size of molecule [124]. Functionality of a truncated protein in some LGMDs has been confirmed including dysferlin. Approaches such as gene transfer of ‘mini-dysferlin’ and exon skipping which can create a shortened but functional protein has been used in vitro and murine models [125, 126]. Another, non-subtype specific approach is to block myostatin, one of the body’s regulators of muscle growth [127]. Proteasome inhibition [128] and stem cell therapy are also under investigation [129]. With improved diagnostic tools, the identification of new drug targets and increased collaboration in the field or rare diseases, there is hope that more effective treatments will be developed for future use in patientswith LGMD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DISCLOSURE STATEMENT
VS is or has been a principal investigator for trials sponsored by Genzyme/Sanofi, GSK, Prosensa/Biomarin, ISIS Pharmceuticals, and Sarepta. He received speaker honoraria from Genzyme/Sanofi, is a member of the international Pompe advisory board of Genzyme/ Sanofi, and has been on advisory boards for Acceleron Pharma, Audentes, Italfarmaco S.p.A., Nicox, Pfizer, Prosensa, Santhera and TrophyNOD. He also has a research collaboration with Ultragenyx and Genzyme/Sanofi.
ACKNOWLEDGMENTS
The authors would like to thank the team at the Newcastle University John Walton Muscular Dystrophy Research Centre for their support and the MYO-MRI COST Action (BM1304) for information on MRI findings. Diagnostic facilities at the John Walton Muscular Dystrophy Research Centre are supported by the Rare Diseases Advisory Group Service for Neuromuscular Diseases (NHS England). Clinical research could not take place without close involvement of LGMD patients and their families who volunteer to participate in trials and give permission for invasive procedures. The John Walton Muscular Dystrophy Research Centre is part of the MRC Centre for Neuromuscular Diseases.
REFERENCES
1 | Walton JN, Nattrass FJ(1954) On the classification, natural history andtreatment of the myopathies, by Brain77: 169231 |
2 | Bushby KM(1995) Diagnostic criteria of the limb-girdle muscular dystrophies: Report of the ENMC consortium on limb-girdle dystrophiesNeuromuscul Disord5: 7174 |
3 | Pegoraro E, Hoffman EPLimb-Girdle Muscular Dystrophy OverviewPagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH(1993) GeneReviews(R)Seattle WAUniversity of Washington, Seattle |
4 | van der Kooi AJ, Barth PG, Busch HFM, de Haan R, Ginjaar HB, van Essen AJ(1996) The clinical spectrum of limb girdle muscular dystrophy A survey in the NetherlandsBrain119: 514711480 |
5 | Bushby K(1992) Report on the 12th ENMC Sponsored International Workshop — The “Limb-Girdle” Muscular DystrophiesNeuromuscular Disorders2: 135 |
6 | Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V(2009) Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic populationBrain132: Pt 1131753186 |
7 | Hicks D, Sarkozy A, Muelas N, Koehler K, Huebner A, Hudson G(2011) A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophyBrain134: Pt 1171182 |
8 | Preisler N, Lukacs Z, Vinge L, Madsen KL, Husu E, Hansen RS(2013) Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophiesMolecular Genetics and Metabolism110: 3287289 |
9 | Chardon JW, Smith AC, Woulfe J, Pena E, Rakhra K, Dennie C(2015) LIMS2 mutations are associated with a novel musculardystrophy, severe cardiomyopathy andtriangular tonguesClinical Genetics10.1111/cge.12561[Epub ahead of print] |
10 | Magri F, Bo RD, D’Angelo MG, Sciacco M, Gandossini S, Govoni A(2012) Frequency and characterisation ofanoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patientsNeuromuscular Disorders22: 11934943 |
11 | Willis TA, Hollingsworth KG, Coombs A, Sveen M-L, Andersen S, Stojkovic T(2014) Quantitative Magnetic Resonance Imaging in Limb-Girdle Muscular Dystrophy 2I: A Multinational Cross-Sectional StudyPLoS ONE9: 2e90377 |
12 | Jarry J, Rioux MF, Bolduc V, Robitaille Y, Khoury V, Thiffault I(2007) A novel autosomal recessive limb-girdle muscular dystrophywith quadriceps atrophy maps to 11p13-p12Brain130: Pt 2368380 |
13 | Hauser MA, Conde CB, Kowaljow V, Zeppa G, Taratuto AL, Torian UM(2002) myotilin Mutation Found in Second Pedigree with LGMD1AAmerican Journal of Human Genetics71: 614281432 |
14 | Yamaoka LH, Westbrook CA, Speer MC, Gilchrist JM, Jabs EW, Schweins EG(1994) Development of a microsatellitegenetic map spanning 5q31-q33 and subsequent placement of the LGMD1A locus between D5S178 and IL9NeuromusculDisord4: 5-6471475 |
15 | Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD(1988) Clinical and genetic investigation in autosomal dominantlimb-girdle muscular dystrophyNeurology38: 159 |
16 | Speer MC, Yamaoka LH, Gilchrist JH, Gaskell CP, Stajich JM, Vance JM(1992) Confirmation of geneticheterogeneity inlimb-girdle muscular dystrophy: Linkage of an autosomaldominant form to chromosome 5qAm J Hum Genet50: 612111217 |
17 | Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA(2000) Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B)Human Molecular Genetics9: 914531459 |
18 | Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L(1999) Mutations in the gene encodinglamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophyNature Genetics21: 3285288 |
19 | Gazzerro E, Bonetto A, Minetti C(2011) Caveolinopathies: Translational implications of caveolin-3 in skeletal and cardiac muscle disordersHandbook of Cinical Neurology101: 135142 |
20 | Stoppani E, Rossi S, Meacci E, Penna F, Costelli P, Bellucci A(1812) Point mutated caveolin-3 form (P104L)impairs myoblast differentiation via Akt and p38 signalling reduction, leading to an immature cell signatureBiochimica et Biophysica Acta4468479 |
21 | Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M(2012) Mutations affecting thecytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdlemuscular dystrophyNature Genetics44: 4450455S1-2 |
22 | Couthouis J, Raphael AR, Siskind C, Findlay AR, Buenrostro JD, Greenleaf WJ(2014) Exome sequencing identifies aDNAJB6 mutation in a family with dominantly-inherited limb-girdle muscular dystrophyNeuromuscul Disord24: 5431435 |
23 | Greenberg SA, Salajegheh M, Judge DP, Feldman MW, Kuncl RW, Waldon Z(2012) Etiology of limb girdle muscular dystrophy 1D/1Edetermined by laser capture microdissection proteomicsAnnals ofNeurology71: 1141145 |
24 | Goldfarb LG, Olive M, Vicart P, Goebel HH(2008) Intermediate filament diseases: DesminopathyAdvances in Experimental Medicine and Biology642: 131164 |
25 | Torella A, Fanin M, Mutarelli M, Peterle E, Del Vecchio Blanco F, Rispoli R(2013) Next-generation sequencing identifies transportin 3 as the causative gene for LGMD1FPLoS One8: 5e63536 |
26 | Gamez J, Navarro C, Andreu AL, Fernandez JM, Palenzuela L, Tejeira S(2001) Autosomal dominant limb-girdlemuscular dystrophy: A large kindred with evidence for anticipationNeurology56: 4450454 |
27 | Starling A, Kok F, Passos-Bueno MR, Vainzof M, Zatz M(2004) A new form of autosomal dominant limb-girdle muscular dystrophy (LGMD1G) with progressive fingers and toes flexion limitation maps to chromosome 4p21European Journal of Human Genetics: EJHG12: 1210331040 |
28 | Vieira NM, Naslavsky MS, Licinio L, Kok F, Schlesinger D, Vainzof M(2014) A defect in the RNA-processing proteinHNRPDLcauses limb-girdle muscular dystrophy 1G(LGMD1G)HumanMolecular Genetics23: 1541034110 |
29 | Chiannilkulchai N, Pasturaud P, Richard I, Auffray C, Beckmann JS(1995) A primary expression map of the chromosome15q15 region containing the recessive form of limb-girdle muscular dystrophy (LGMD2A) geneHuman MolecularGenetics4: 4717725 |
30 | Richard I, Roudaut C, Saenz A, Pogue R, Grimbergen JEMA, Anderson LVB(1999) Calpainopathy— A Survey of Mutations and PolymorphismsThe American Journal of Human Genetics64: 615241540 |
31 | Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M(1998) A gene related to Caenorhabditis elegansspermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2BNature Genetics20: 13742 |
32 | Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C(1998) Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophyNature Genetics20: 13136 |
33 | Hack AA, Groh ME, McNally EM(2000) Sarcoglycans in muscular dystrophyMicroscopy Research and Technique48: 3-4167180 |
34 | Babameto-Laku A, Tabaku M, Tashko V, Cikuli M, Mokini V(2011) The first case of primary alpha-sarcoglycanopathyidentified in Albania, in two siblings with homozygous alpha-sarcoglycan mutationGenetic counseling (Geneva,Switzerland)22: 4377383 |
35 | Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD(1994) Missense mutations in the adhalingene linked to autosomal recessive muscular dystrophyCell78: 4625633 |
36 | Semplicini C, Vissing J, Dahlqvist JR, Stojkovic T, Bello L, Witting N(2015) Clinical and genetic spectrum inlimb-girdle muscular dystrophy type 2ENeurology84: 1717721781 |
37 | Nigro V, de Sa Moreira E, Piluso G, Vainzof M, Belsito A, Politano L(1996) Autosomal recessive limb-girdle musculardystrophy, LGMD2F, is caused by a mutation in thedelta-sarcoglycan geneNature Genetics14: 2195198 |
38 | Valle G, Faulkner G, De Antoni A, Pacchioni B, Pallavicini A, Pandolfo D(1997) Telethonin, a novel sarcomericprotein of heart and skeletal muscleFEBS letters415: 2163168 |
39 | Barresi R, Morris C, Hudson J, Curtis E, Pickthall C, Bushby K(2015) Conserved expression of truncatedtelethonin in a patient with limb-girdle muscular dystrophy 2GNeuromuscular Disorders25: 4349352 |
40 | Frosk P, Greenberg CR, Tennese AA, Lamont R, Nylen E, Hirst C(2005) The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populationsHuman Mutation25: 13844 |
41 | Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ(2005) Trim32 is a Ubiquitin Ligase Mutated in Limb GirdleMuscular Dystrophy Type 2H that Binds to Skeletal Muscle Myosin and Ubiquitinates ActinJournal of MolecularBiology354: 2413424 |
42 | Bushby K, Anderson LV, Pollitt C, Naom I, Muntoni F, Bindoff L(1998) Abnormal merosin in adults. A new form of lateonset muscular dystrophy not linked to chromosome 6q2Brain121: Pt 4581588 |
43 | Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA(2001) Mutations in the fukutin-relatedprotein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency andabnormal glycosylation of alpha-dystroglycanAm J Hum Genet69: 611981209 |
44 | Udd B, Kaarianen H, Somer H(1991) Muscular dystrophy with separate clinical phenotypes in a large familyMuscle & Nerve14: 1110501058 |
45 | Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J(2002) Tibial muscular dystrophy is atitinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titinAm J HumGenet71: 3492500 |
46 | Balci B, Uyanik G, Dincer P, Gross C, Willer T, Talim B(2005) An autosomal recessive limb girdle musculardystrophy (LGMD2) with mild mental retardation is allelic to Walker–Warburg syndrome (WWS) caused by amutation in the POMT1 geneNeuromuscular Disorders15: 4271275 |
47 | Dincer P, Balci B, Yuva Y, Talim B, Brockington M, Dincel D(2003) A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of alpha-dystroglycanNeuromuscul Disord13: 10771778 |
48 | Murakami T, Hayashi YK, Noguchi S, Ogawa M, Nonaka I, Tanabe Y(2006) Fukutin gene mutations cause dilatedcardiomyopathy with minimal muscle weaknessAnnals of Neurology60: 5597602 |
49 | van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabe D, Sabatelli P, Merlini L(2005) POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndromeJournal of Medical Genetics42: 12907912 |
50 | Clement EM, Godfrey C, Tan J, Brockington M, Torelli S, Feng L(2008) Mild POMGnT1 mutations underlie a novellimb-girdle muscular dystrophy variantArchives of Neurology65: 1137141 |
51 | Akasaka-Manya K, Manya H, Kobayashi K, Toda T, Endo T(2004) Structure-function analysis of human protein O-linkedmannose beta1,2-N-acetylglucosaminyltransferase 1, POMGnT1Biochemical and Biophysical Research Communications320: 13944 |
52 | Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, Kanagawa M, Beltran-Valero de Bernabe D, Gundesli H(2011) Adystroglycan mutation associated with limb-girdle muscular dystrophyThe New England Journal of Medicine364: 10939946 |
53 | Gundesli H, Talim B, Korkusuz P, Balci-Hayta B, Cirak S, Akarsu NA(2010) Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophyAm J Hum Genet87: 6834841 |
54 | Cetin N, Balci-Hayta B, Gundesli H, Korkusuz P, Purali N, Talim B(2013) A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: Distinct histopathological outcomes compared with desminopathiesJournal of Medical Genetics50: 7437443 |
55 | Bogershausen N, Shahrzad N, Chong JX, von Kleist-Retzow JC, Stanga D, Li Y(2013) Recessive TRAPPC11 mutationscause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectualdisabilityAm J Hum Genet93: 1181190 |
56 | Ning B, Elbein AD(2000) Cloning, expression and characterization of the pig liver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glc pyrophosphorylases are different proteinsEuropean Journal of Biochemistry / FEBS267: 2368666874 |
57 | Carss KJ, Stevens E, Foley AR, Cirak S, Riemersma M, Torelli S(2013) Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycanAm J Hum Genet93: 12941 |
58 | Willer T, Lee H, Lommel M, Yoshida-Moriguchi T, de Bernabe DB, Venzke D(2012) ISPD loss-of-function mutations disruptdystroglycan O-mannosylation and cause Walker-Warburg syndromeNature Genetics44: 5575580 |
59 | Legate KR, Montanez E, Kudlacek O, Fussler R(2006) ILK, PINCH and parvin: The tIPP of integrin signallingNat Rev Mol Cell Biol7: 12031 |
60 | Herrmann R, Straub V, Blank M, Kutzick C, Franke N, Jacob EN(2000) Dissociation of the dystroglycan complex in caveolin-3-deficient limb girdle muscular dystrophyHuman Molecular Genetics9: 1523352340 |
61 | Defour A, Van der Meulen JH, Bhat R, Bigot A, Bashir R, Nagaraju K(2014) Dysferlin regulates cell membranerepair by facilitating injury-triggered acid sphingomyelinase secretionCell Death & Disease5: e1306 |
62 | Goonasekera SA, Davis J, Kwong JQ, Accornero F, Wei-LaPierre L, Sargent MA(2014) Enhanced Ca(2)(+) influx fromSTIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophyHuman Molecular Genetics23: 1437063715 |
63 | Alderton JM, Steinhardt RA(2000) How calcium influx through calciumleak channels is responsible for the elevated levels ofcalcium-dependent proteolysis in dystrophic myotubesTrends inCardiovascular Medicine10: 6268272 |
64 | Tidball JG(2005) Inflammatory processes in muscle injury and repairAmerican Journal of Physiology Regulatory, Integrative and Comparative Physiology288: 2R345R353 |
65 | Meng J, Bencze M, Asfahani R, Muntoni F, Morgan JE(2015) The effect of the muscle environment on the regenerative capacity of human skeletal muscle stem cellsSkeletal Muscle5: 11 |
66 | Wallace GQ, McNally EM(2009) Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophiesAnnual Review of Physiology71: 3757 |
67 | Willis TA, Hollingsworth KG, Coombs A, Sveen ML, Andersen S, Stojkovic T(2013) Quantitative muscle MRI as an assessment tool for monitoring disease progression in LGMD2I: A multicentre longitudinal studyPLoS One8: 8e70993 |
68 | Aboumousa A, Hoogendijk J, Charlton R, Barresi R, Herrmann R, Voit T(2008) Caveolinopathy–new mutations and additional symptomsNeuromuscul Disord18: 7572578 |
69 | Hauser MA, Horrigan SK, Salmikangas P, Torian UM, Viles KD, Dancel R(2000) Myotilin is mutated in limb girdle muscular dystrophy 1AHuman Molecular Genetics9: 1421412147 |
70 | Reilich P, Krause S, Schramm N, Klutzny U, Bulst S, Zehetmayer B(2011) A novel mutation in the myotilin gene (MYOT) causes a severe form of limb girdle muscular dystrophy 1A (LGMD1A)Journal of Neurology258: 814371444 |
71 | Maggi L, D’Amico A, Pini A, Sivo S, Pane M, Ricci G(2014) LMNA-associated myopathies: The Italian experience in a largecohort of patientsNeurology83: 1816341644 |
72 | Carboni N, Mura M, Marrosu G, Cocco E, Marini S, Solla E(2010) Muscle imaging analogies in a cohort of patientswith different clinical phenotypes caused by LMNA gene mutationsMuscle & Nerve41: 4458463 |
73 | Fanin M, Nascimbeni AC, Aurino S, Tasca E, Pegoraro E, Nigro V(2009) Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypesNeurology72: 1614321435 |
74 | Guglieri M, Magri F, D’Angelo MG, Prelle A, Morandi L, Rodolico C(2008) Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patientsHuman Mutation29: 2258266 |
75 | Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M(1998) Mutations in the caveolin-3 gene causeautosomal dominant limb-girdle muscular dystrophyNature Genetics18: 4365368 |
76 | Hayashi T, Arimura T, Ueda K, Shibata H, Hohda S, Takahashi M(2004) Identification and functional analysis of acaveolin-3 mutation associated with familial hypertrophic cardiomyopathyBiochemical and Biophysical ResearchCommunications313: 1178184 |
77 | Sandell S, Huovinen S, Sarparanta J, Luque H, Raheem O, Haapasalo H(2010) The enigma of 7q36 linked autosomal dominant limb girdle muscular dystrophyJournal of Neurology, Neurosurgery, and Psychiatry81: 8834839 |
78 | Speer MC, Vance JM, Grubber JM, Lennon Graham F, Stajich JM, Viles KD(1999) Identification of a new autosomal dominantlimb-girdle muscular dystrophy locus on chromosome 7Am J HumGenet64: 2556562 |
79 | Schramm N, Born C, Weckbach S, Reilich P, Walter MC, Reiser MF(2008) Involvement patterns in myotilinopathy and desminopathy detected by a novel neuromuscular whole-body MRI protocolEuropean Radiology18: 1229222936 |
80 | Vajsar J, Becker LE, Freedom RM, Murphy EG(1993) Familial desminopathy: Myopathy with accumulation of desmin-type intermediate filamentsJournal of Neurology, Neurosurgery, and Psychiatry56: 6644648 |
81 | Mercuri E, Bushby K, Ricci E, Birchall D, Pane M, Kinali M(2005) Muscle MRI findings in patients with limbgirdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contracturesNeuromuscul Disord15: 2164171 |
82 | Passos-Bueno MR, Vainzof M, Moreira ES, Zatz M(1999) Seven autosomalrecessive limb-girdle muscular dystrophies in the Brazilianpopulation: From LGMD2A to LGMD2GAmerican Journal of MedicalGenetics82: 5392398 |
83 | Stramare R, Beltrame V, Dal Borgo R, Gallimberti L, Frigo AC, Pegoraro E(2010) MRI in the assessment of muscular pathology: Acomparison between limb-girdle muscular dystrophies, hyaline bodymyopathies and myotonic dystrophiesLa Radiologia Medica115: 4585599 |
84 | Fanin M, Duggan DJ, Mostacciuolo ML, Martinello F, Freda MP, Soraru G(1997) Genetic epidemiology of muscular dystrophies resulting from sarcoglycan gene mutationsJournal of Medical Genetics34: 12973977 |
85 | Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C(1997) Mutations in the sarcoglycan genes in patients with myopathyThe New England Journal of Medicine336: 9618624 |
86 | Lodi R, Muntoni F, Taylor J, Kumar S, Sewry CA, Blamire A(1997) Correlative MR imaging and 31P-MR spectroscopy study in sarcoglycan deficient limb girdle muscular dystrophyNeuromuscul Disord7: 8505511 |
87 | Moreira ES, Vainzof M, Marie SK, Sertie AL, Zatz M, Passos-Bueno MR(1997) The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11-12Am J Hum Genet61: 1151159 |
88 | Olive M, Shatunov A, Gonzalez L, Carmona O, Moreno D, Quereda LG(2008) Transcription-terminating mutation in telethonin causing autosomal recessive muscular dystrophy type 2G in a European patientNeuromuscul Disord18: 12929933 |
89 | Michele DE, Campbell KP(2003) Dystrophin-glycoprotein complex: Post-translational processing and dystroglycan functionThe Journal of Biological Chemistry278: 181545715460 |
90 | Mathews KD, Stephan CM, Laubenthal K, Winder TL, Michele DE, Moore SA(2011) Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2INeurology76: 2194195 |
91 | Udd B, Rapola J, Nokelainen P, Arikawa E, Somer H(1992) Nonvacuolar myopathy in a large family with both late adult onset distal myopathy and severe proximal muscular dystrophyJournal of the Neurological Sciences113: 2214221 |
92 | Pfeffer G, Sambuughin N, Olive M, Tyndel F, Toro C, Goldfarb LG(2014) A new disease allele for the C1Rmutation in titin causing hereditary myopathy with earlyrespiratory failureNeuromuscul Disord24: 3241244 |
93 | Penttila S, Palmio J, Suominen T, Raheem O, Evila A, Muelas Gomez N(2012) Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5Neurology78: 12897903 |
94 | Sandell SM, Mahjneh I, Palmio J, Tasca G, Ricci E, Udd BA(2013) ‘Pathognomonic’ muscle imaging findings in DNAJB6 mutated LGMD1DEuropean journal of neurology: The official Journal of the European Federation of Neurological Societies20: 1215531559 |
95 | Peterle E, Fanin M, Semplicini C, Padilla JJ, Nigro V, Angelini C(2013) Clinical phenotype, muscle MRI and muscle pathology of LGMD1FJournal of Neurology260: 820332041 |
96 | Bisceglia L, Zoccolella S, Torraco A, Piemontese MR, Dell’Aglio R, Amati A(2010) A new locus on 3p23-p25 for an autosomal-dominant limb-girdle muscular dystrophy, LGMD1HEuropean journal of human genetics: EJHG18: 6636641 |
97 | Weiler T, Greenberg CR, Zelinski T, Nylen E, Coghlan G, Crumley MJ(1998) A gene for autosomal recessive limb-girdle muscular dystrophy in Manitoba Hutterites maps to chromosome region 9q31-q Evidence for another limb-girdle muscular dystrophy locusAm J Hum Genet63: 1140147 |
98 | Hoffman EP, Pegoraro E, Scacheri P, Burns RG, Taber JW, Weiss L(1996) Genetic counseling of isolated carriersof Duchenne muscular dystrophyAmerican Journal of Medical Genetics63: 4573580 |
99 | Sacconi S, Camano P, de Greef JC, Lemmers RJ, Salviati L, Boileau P(2012) Patients with a phenotype consistent with facioscapulohumeral muscular dystrophy display genetic and epigenetic heterogeneityJournal of Medical Genetics49: 14146 |
100 | Koch AJ, Holaska JM(2014) Emerin in health and diseaseSeminars in Cell & Developmental Biology29: 95106 |
101 | Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J(2014) Methods of diagnosis of patients with Pompe disease: Data from the Pompe RegistryMolecular Genetics and Metabolism113: 1-28491 |
102 | Deconinck N, Richard P, Allamand V, Behin A, Laforet P, Ferreiro A(2014) Bethlem myopathy: Long-term follow-up identifies COL6mutations predicting severe clinical evolutionJournal of Neurology, Neurosurgery, and Psychiatry10.1136/JNNP-2013-307245[Epub ahead of print] |
103 | Rowin J, Meriggioli MN, Cochran EJ, Sanders DB(1999) Prominent inflammatory changes on muscle biopsy in patients with Miyoshi myopathyNeuromuscul Disord9: 6-7417420 |
104 | Schroder T, Fuchss J, Schneider I, Stoltenburg-Didinger G, Hanisch F(2013) Eosinophils in hereditary and inflammatory myopathiesActamyologica: Myopathies and cardiomyopathies: Official Journal ofthe Mediterranean Society of Myology / edited by the Gaetano ConteAcademy for the Study of Striated Muscle Diseases32: 3148153 |
105 | Vinit J, Samson MJr, Gaultier JB, Laquerriere A, Ollagnon E, Petiot P(2010) Dysferlin deficiency treated like refractorypolymyositisClinical Rheumatology29: 1103106 |
106 | Barresi R(2011) From proteins to genes: Immunoanalysis in the diagnosis of muscular dystrophiesSkeletal Muscle1: 124 |
107 | Nigro V, Savarese M(2014) Genetic basis of limb-girdle muscular dystrophies: The updateActa myologica:Myopathies and cardiomyopathies: Official Journal of the Mediterranean Society of Myology / edited by theGaetano Conte Academy for the Study of Striated Muscle Diseases33: 1112 |
108 | Darin N, Kroksmark AK, Ahlander AC, Moslemi AR, Oldfors A, Tulinius M(2007) Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2IEuropean journal of paediatric neurology: EJPN: Official Journal of the European Paediatric Neurology Society11: 6353357 |
109 | Godfrey C, Escolar D, Brockington M, Clement EM, Mein R, Jimenez-Mallebrera C(2006) Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophyAnnals of Neurology60: 5603610 |
110 | Walter MC, Reilich P, Thiele S, Schessl J, Schreiber H, Reiners K(2013) Treatment of dysferlinopathy with deflazacort: A double-blind, placebo-controlled clinical trialOrphanet Journal of Rare Diseases8: 26 |
111 | Skalsky AJ, McDonald CM(2012) Prevention and management of limb contractures in neuromuscular diseasesPhysical medicine and rehabilitation clinics of North America23: 3675687 |
112 | Simonds AK(2006) Recent advances in respiratory care for neuromuscular diseaseChest130: 618791886 |
113 | group T-NE. TREAT-NMD: Advancing diagnosis, care and treatment for people with neuromuscular disease around the world 2015 [cited 2015 15th of May] |
114 | JAIN. International Clinical Outcome Study for Dysferlinopathy (LGMD 2B/Miyoshi) JAIN foundation 2015 [cited 2015 15th of May]. Available from: http://www.jain-foundation.org/dysferlinoutcomestudy |
115 | CtCC. LGMD 2A Registry 2015. Available from: http://www.lgmd2a.org/ |
116 | The International Dysferlinopathy Registry Hearing before the Jain Foundation(2015). |
117 | Foundation KP. Kurt & Peter Foundation | Patient Registry 2015 [cited 2015 21st of May 2015]. Available from: www.lgmd2cregistry.org |
118 | Foundation LD. Patient Resources | LGMD 2D Foundation 2015 [cited 2015 21st of May 2015]. Available from: www.lgmd2d.org/patient-resources |
119 | Registry GF. Home - Global FKRP Registry 2015 [cited 2015 21st of May 2015]. Available from: www.fkrpregistry.org |
120 | CMD C. PatientCrossroads CMD 2015 [cited 2015 21st of May 2015]. Available from: www.cmdir.org |
121 | Sondergaard PC, Griffin DA, Pozsgai ER, Johnson RW, Grose WE, Heller KN(2015) AAV.Dysferlin Overlap VectorsRestore Function in Dysferlinopathy Animal ModelsAnnals of Clinical and Translational Neurology2: 3256270 |
122 | Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G(2009) Limb-girdle musculardystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteinsAnnals of Neurology66: 3290297 |
123 | Xu L, Lu PJ, Wang CH, Keramaris E, Qiao C, Xiao B(2013) Adeno-associated virus 9 mediated FKRP gene therapyrestores functional glycosylation of alpha-dystroglycan and improves muscle functionsMolecular therapy: TheJournal of the American Society of Gene Therapy21: 1018321840 |
124 | Lostal W, Bartoli M, Bourg N, Roudaut C, Bentaib A, Miyake K(2010) Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transferHuman Molecular Genetics19: 1018971907 |
125 | Azakir BA, Di Fulvio S, Salomon S, Brockhoff M, Therrien C, Sinnreich M(2012) Modular dispensability of dysferlin C2 domains reveals rational design for mini-dysferlin moleculesThe Journal of Biological Chemistry287: 332762927636 |
126 | Barthelemy F, Wein N, Krahn M, Levy N, Bartoli M(2011) Translational research and therapeutic perspectives in dysferlinopathiesMolecular Medicine (Cambridge, Mass)17: 9-10875882 |
127 | Murphy KT, Ryall JG, Snell SM, Nair L, Koopman R, Krasney PA(2010) Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx miceThe American Journal of Pathology176: 524252434 |
128 | Azakir BA, Di Fulvio S, Kinter J, Sinnreich M(2012) Proteasomal inhibition restores biological function of mis-sense mutated dysferlin in patient-derived muscle cellsThe Journal of Biological Chemistry287: 131034410354 |
129 | Meregalli M, Navarro C, Sitzia C, Farini A, Montani E, Wein N(2013) Full-length dysferlin expression driven by engineered humandystrophic blood derived CD133+ stem cellsThe FEBS Journal280: 2360456060 |
130 | Cotta A, Carvalho E, da-Cunha-Júnior AL, Paim JF, Navarro M, Valicek J(2014) Common recessive limb girdlemusculardystrophies differential diagnosis: Why and how? ArqNeuro-Psiquiatr72: 9721734 |
131 | Laval SH, Bushby KMD(2004) Limb-girdle muscular dystro-phies from genetics to molecular pathologyNeuropathology and Applied Neurobiology30: 291105 |
Figures and Tables
Fig.1
A depiction of the organisation of the sarcolemma and LGMD subtypes associated with a defect in each protein. Of particular importance to the stability of the myocyte is – the dystrophin-glycoprotein complex, a highly glycosylated oligomeric structure that links the extracellular matrix to the sub-sarcolemmal cytoskeleton. (Amalgamated from Cotta [130], Laval [131]).
![A depiction of the organisation of the sarcolemma and LGMD subtypes associated with a defect in each protein. Of particular importance to the stability of the myocyte is – the dystrophin-glycoprotein complex, a highly glycosylated oligomeric structure that links the extracellular matrix to the sub-sarcolemmal cytoskeleton. (Amalgamated from Cotta [130], Laval [131]).](https://content.iospress.com:443/media/jnd/2015/2-s2/jnd-2-s2-jnd150105/jnd-2-s2-jnd150105-g001.jpg)
Fig.2
The images above show T1 weighted axial magnetic resonance images of the pelvic girdle and legs muscles of patients with LGMD2A, 2D and 2L. In all patients a selective pattern of muscle pathology can be seen, with advanced changes in the LGMD2A patient, well preserved calf muscles in the LGMD2D patients and well preserved gluteal muscles in the LGMD2L patient.
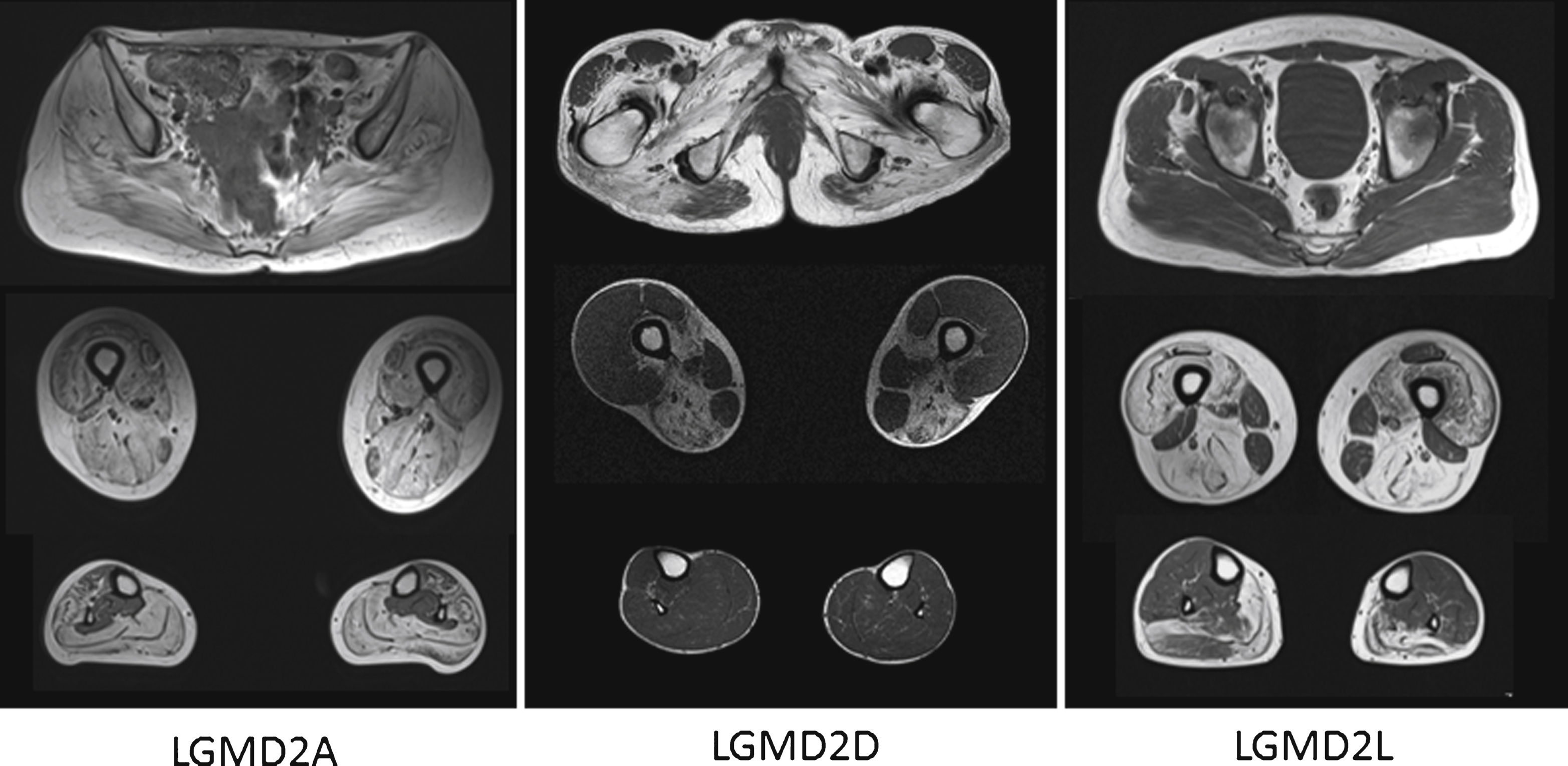
Table 1
A table to show current understanding of LGMD subtypes, genes and protein functions. Only LGMD subtypes with known genes are listed
| LGMD | Gene/Locus | Protein | Suggested function of protein | References |
| 1A | 5q 22 – 34 (TTID) | Myotilin | Sarcomeric stabilisation of actin bundles | [13–16] |
| 1B | 1q 11–21 (LMNA) | Lamin A/C | Nuclear membrane stabilisation and transcriptional regulation | [17, 18] |
| 1C | 3p25 (CAV3) | Caveolin 3 | Stabilisation of the sarcolemmal membrane, regulates cellular signal traffic | [19, 20] |
| 1D | 7q (DNAJB6) | HSP40 | Molecular chaperone involved in proteomic and autophagic turnover | [21, 22] |
| 1E | 6q23 (DES) | Desmin | intermediate filament regulating sarcomere and cytoskeletal architecture | [23, 24] |
| 1F | 7q32 (TNPO3) | Transportin 3 | Nuclear importing receptor | [25, 26] |
| 1G | 4q21 (HNRNPDL) | Heterogeneous Nuclear Ribonucleoprotein D-like protein | pre-mRNA processing | [27, 28] |
| 2A | 15q15–21 (CAPN3) | Calpain 3 | Implicated in cytoskeletal repair mechanisms, binds titin | [29, 30] |
| 2B | 2p13 (DYSF) | Dysferlin | Regulation of vesicle fusion, receptor trafficking and repair of damaged membranes | [31, 32] |
| 2C | 13q12 (SGCG) | γ-sarcoglycan | Connects the sarcolemma to the extracellular matrix, stabilisation of the dystroglycan complex | [33] |
| 2D | 17q 12–21 (SGCA) | α-sarcoglycan | Connects the sarcolemma to the extracellular matrix, stabilisation of the dystroglycan complex | [33–35] |
| 2E | 4q12 (SGCB) | β-sarcoglycan | Connects the sarcolemma to the extracellular matrix, stabilisation of the dystroglycan complex | [33, 36] |
| 2F | 5q33–34 (SGCD) | δ-sarcoglycan | Connects the sarcolemma to the extracellular matrix, stabilisation of the dystroglycan complex | [33, 37] |
| 2G | 17q11-12 (TCAP) | Telethonin | Binds to titin, T tubule organisation | [38, 39] |
| 2H | 9q31–34 (TRIM 32) | Tripartite Motif containing 32 | Binds to myosin, may ubiquitinate actin | [40, 41] |
| 2I | 19q13 (FKRP) | Fukutin related protein | Glycosylation of α-dystroglycan | [42, 43] |
| 2J | 2q (TTN) | Titin | Multiple binding sites for other proteins, connects the Z line to the M line in the sarcomere | [44, 45] |
| 2K | 9q34 (POMT1) | Protein-O-mannosyl transferase1 | Involved in glycosylation of α-dystroglycan | [46, 47] |
| 2L | 11p12-13 (ANO5) | Anoctamin 5 | Not completely understood – may act as a chloride channel | [12] |
| 2M | 9q31 (FKTN) | Fukutin | Involved in glycosylation of α-dystroglycan | [48] |
| 2N | 14q24 (POMT2) | Protein-O-mannosyl transferase 2 | Involved in glycosylation of α-dystroglycan | [49] |
| 2O | 1p34 (POMGnT1) | Protein-O-linked mannose beta 1,2 Nacetylglucosaminyl transferase | Involved in glycosylation of α-dystroglycan | [50, 51] |
| 2P | 3p21 (DAG1) | Dystroglycan | Key basement membrane receptor and component of the dystrophin-glycoprotein complex, | [47, 52] |
| 2Q | 8q24 (PLEC1) | Plectin | Structural linkage between sarcomere and sarcolemma | [53] |
| 2R | 2q35 (DES) | Desmin | Intermediate filament regulating sarcomere and cytoskeletal architecture | [54] |
| 2S | 4q35 (TRAPPC11) | Transport protein particle complex 11 | Membrane trafficking | [55] |
| 2T | 3p21 (GMPPB) | GDP-mannose pyrophosphorylase B | Involved in glycosylation of α-dystroglycan | [56, 57] |
| 2U | 7p21 (ISPD) | Isoprenoid synthase domain | Aids in o-mannosylation of α-dystroglycan | [58] |
| 2V | 17q25 (GAA) | Alpha-1,4 glucosidase | Lysosomal enzyme hydrolysing glycogen | [8] |
| 2W | 2q14 (LIMS2) | Lim and senescent cell antigen-like domains 2 | Part of the integrin-actin cytoskeleton, signalling | [9, 59] |


