
Non-Ambulant Duchenne Patients Theoretically Treatable by Exon 53 Skipping have Severe Phenotype
Abstract
Background: Exon skipping therapy is an emerging approach in Duchenne Muscular Dystrophy (DMD). Antisense oligonucleotides that induce skipping of exon 51, 44, 45, or 53 are currently being evaluated in clinical trials. These trials were designed on the basis of data available in general DMD population.
Objectives: Our objective was to compare the clinical and functional statuses of non-ambulant DMD patients theoretically treatable by exon 53 skipping and of DMD patients with other mutations.
Methods: We first compared fifteen non-ambulant DMD patients carrying deletions theoretically treatable by exon 53 skipping (DMD-53) with fifteen closely age-matched DMD patients with mutations not treatable by exon 53 skipping (DMD-all-non-53) then with fifteen DMD patients carrying deletions not treatable by exon 53 skipping (DMD-del-non-53).
Results: We found that DMD-53 patients had a lower left ventricular ejection fraction, more contractures and they tend to have weaker grips and pinch strengths than other DMD patients. DMD-53 patients lost ambulation significantly younger than other DMD patients. This result was confirmed by comparing ages at loss of ambulation in all non-ambulant DMD patients of the DMD cohort identified in a molecular diagnostic lab.
Conclusions: These prospective and retrospective data demonstrate that DMD-53 patients have clinically more severe phenotypes than other DMD patients.
INTRODUCTION
Among gene and pharmaco-gene therapies, exon skipping therapy is an emerging approach for treatment of Duchenne Muscular Dystrophy (DMD) [1–3]. The aim of exon skipping therapy is to modify processing of the dystrophin pre-mRNA to allow expression of a shorter but functional dystrophin protein [4–7]. Exon skipping naturally occurs in some fibers of patients resulting in the so-called revertant fibers. Their number varies with age and type of deletion [8] and may even result in milder Becker-like phenotype [9]. Antisense oligonucleotides that induce skipping of exon 51, 44, 45, or 53 are currently under evaluation in clinical trials; some agents have progress to Phase III evaluation.
There are substantial data that demonstrate a phenotype-genotype correlation for intellectual function in DMD patients, with more intellectual disability in patients with mutations that occur after exon 63 [10–12]. Several studies have reported that motor and respiratory function and survival of DMD patients depend on genotype [10, 13, 14]. From analysis of a large, two-site cohort of 144 patients, Davidson et al. reported that boys with deletions in the dystrophin gene were six times more likely to stop walking before age 10 than were boys with duplications or point or unknown mutations [15]. Wong et al. showed that patients with deletions of exons 3–7 presented with a milder phenotype despite their out of frame deletion predictive of DMD [16]. Epigenetic or environmental factors also impact DMD phenotype. High levels of osteopontin, a secreted multifunction protein, have recently been reported in a large Italian cohort as associated with earlier loss of ambulation and more rapid weakness progression in DMD patients [17, 18]. In a cohort of patients in the United States, LTBP4 haplotype influenced the age of loss of ambulation [19].
Within the subgroup of patients who might potentially benefit from exon skipping therapy, it is known that the number of revertant fibers varies greatly regarding the mutated exon. For instance, patients with a deletion theoretically treatable by exon 44 skipping present with more revertant fibers than do patients with a deletion theoretically treatable by exon 51 or 53 skipping [8]. There is no published data on these patients’ natural history, and therefore the statistical plans of clinical trials in these specific populations are designed on the basis of data available from the general DMD population. Since significant changes in the patient phenotype and rate of decline may alter statistical power of these studies, we compared the clinical and functional parameters for of a cohort of non-ambulant DMD patients theoretically treatable by inducing exon 53 with a precisely age-matched overall DMD population; the same devices and the same team evaluated all subjects over the same period of time. To confirm the data, we retrospectively reviewed the UMD-DMD-Cochin database for age at loss of ambulation for patients carrying deletions theoretically treatable by exon 44, 45, 50, 51, 52, 53, or 55 skipping and compared data on these patients with data for the general DMD population.
PATIENTS AND METHODS
Prospective data
Patients were selected from among non-ambulant patients (i.e., patients not able to walk more than 10 meters without human assistance) included in two observational natural history studies (Pre-U7 and ULENAP) carried out by the Institute of Myology (Paris) (Table 1). These studies were approved by the local ERB and by the French health authority (ANSM) and were registered with www.clinicaltrials.gov (NCT01385917 and NCT00993161, respectively)
General design and preliminary data of the multi-centric study ULENAP have been already reported [20–22]. In this study, we assessed upper limb strength and function in 53 non-ambulant DMD patients (regardless of their mutation) every six months for one year. All patients were evaluated using a standardized evaluation. Age at loss of ambulation, steroid intake, left ventricular ejection fraction (LVEF) and forced vital capacity (FVC) were obtained from the available medical files. LVEF and FCV were recorded only if performed within one year before inclusion in the study. All patients underwent clinical examination and upper limb assessments as previously described [20]. These assessments included pinch and handgrip strength measurements by the sensitive devices MyoPinch and MyoGrip, respectively, and an upper hand assessment using the MoviPlate. Both arms of each patient were tested. The Motor Function Measurement (MFM) was also performed. The first assessment included test-retest evaluation.
In the Pre-U7 study conducted at the Institute of Myology, we identified 27 patients aged 6 to 20 years old, theoretically treatable by exon 53 skipping. These patients lived in Belgium, France, Romania, or Switzerland. One patient with deletion 52 was already included in an exon 51 skipping trial, two could not be contacted and one did not wish to participate. Twenty-three patients (ambulant and non-ambulant) agreed to take part in the Pre-U7 study (recruitment is ongoing).Design and assessments were the same as those used in ULENAP study, and the operating procedures were similar, except that test-retest assessments were not conducted in this study. An additional upper limb assessment using MRI, skin biopsy, and an AAV serology assessment were also conducted at the first visit and then every year. Results are not reported here. Of the 23 Pre-U7 patients theoretically treatable by exon 53 skipping (DMD-53), 15 non-ambulant were compared firstly to age-matched non-ambulant DMD patients whatever their mutations not treatable by exon 53 skipping (DMD all-non-53), and secondly to age-matched non-ambulant DMD patients with deletions not treatable by exon 53 skipping (DMD del-non-53) (Fig. 1).
Retrospective data
The “laboratoire de biochimie et génétique moléculaire” (LBGM), Cochin hospital, Paris, is one of the French molecular diagnosis laboratories dedicated to routine molecular diagnosis of dystrophinopathies. It also provides data to the UMD-DMD France database [23]. This database includes all DMD gene mutations identified in male subjects as well as symptomatic female carriers (www.umd.be/DMD/W_DMD/index.html). The clinical section of this database includes 200 fields of clinical information on each patient, including age at loss of major motor milestones. For the purpose of the present work, the age at loss of ambulation of DMD patients were extracted from the clinical data when available. These patients will be thereafter referred to as UMD-DMD-Cochin database patients.
Statistical analysis
DMD all-non-53 and DMD del-non-53 patients were precisely age-matched to DMD-53 patients on a patient/patient basis (mean of the difference: 0.2 and 0.3 year respectively, maximal difference 0.9 and 1.1 year respectively). When several patients were available for matching, the patient with the closest age was selected. The difference in the distribution of ages between compared groups was tested by performing a non-parametric Friedman ANOVA test for paired samples (Fig. 2). There were slight differences between the two study protocols, especially the test-retest assessment in the first visit of ULENAP and the MRI assessment in Pre-U7 study, which that may induce training or fatigue, respectively. In order to avoid any possible learning and fatigue effect, clinical, strength and function data from ULENAP study were those of the first visit of ULENAP, from test assessment for MyoGrip and MyoPinch and from retest assessment for MoviPlate. Data from Pre-U7 study were those measured at the second visit. Clinical data, strength and functional data were compared between groups using a non-parametric Wilcoxon test for paired samples. Nominal data were compared using a Chi squared test with Yates’s correction for continuity if needed. The limit of statistical significance was set to 0.05.
For retrospective analyses of data from the UMD-DMD-Cochin database, we first performed the Kolmogorov-Smirnov test to assess the normal distribution of the age of ambulation in patients carrying deletions theoretically treatable by exon 44, 45, 50, 51, 52, 53 or 55 skipping. Since the distribution appeared to be non-normal, we used the non-parametric Kruskal-Wallis one-way analysis of variance test and a post-hoc test of Dunn-Bonferroni after verifying the variance’s homogeneity between groups by Levene’s test. The age at loss of ambulation between DMD patients carrying a deletion treatable by exon 53 skipping and those carrying all DMD mutations was compared with the non-parametric independent samples using Mann-Whitney U Test. Patients with an age at loss of ambulation older than or equal to 15 years old did not take part to the statistical analysis.
All analyses were performed using the SPSS 19 statistical software (SPSS Inc., Chicago, IL). The limit of statistical significance was set to 0.05.
RESULTS
Selection of patients
The clinical characteristics of patients are detailed in Table 2. The distribution of ages (13.9 ± 2.9 years vs. 14.0 ± 2.8 years or 14.1 ± 2.8 years in DMD-53 and DMD-all-non-53 or DMD-del-non-53, respectively) is displayed in Fig. 2. Even if DMD-del-non-53 patients appeared to be significantly older than DMD-53 patients in a Wilcoxon test for paired samples (p = 0.015), there was no significant difference between distributions (Friedman ANOVA p-value = 0.796 and 0.071 for DMD-all-non-53 or DMD-del-non-53, respectively)
Clinical features
There were no significant difference between DMD-53 patients and other DMD regarding neither steroid and cardiac protection (angiotensin-converting-enzyme inhibitor) treatment nor physiotherapy or arthrodesis surgery (Table 3). DMD-53 patients were significantly smaller than non-53 patients (144 ± 13 cm in DMD-53 vs. 153 ± 12 cm in DMD-all-non-53 or DMD-del-non-53 patients, p = 0.006 and p = 0.023 respectively) but there was no significant difference in weight between groups (45 ± 16 kg in DMD-53 vs. 41 ± 13 kg in DMD-all-non-53 and 40 ± 12 kg in DMD-del-non-53 and, p = 0.532 and p = 0.320 respectively). DMD-53 patients lost ambulation at a significant younger age than did the other DMD patients (8.7 ± 1.6 for DMD-53 vs 10.4 ± 2.4, p = 0.031 and 10.7 ± 2.1, p = 0.011 for DMD-all-non-53 and DMD-del-non-53, respectively). The left ventricular ejection fraction was significantly lower in DMD-53 patients and the contracture score on the dominant side was significantly higher in DMD-53 patients when compared with DMD patients carrying deletion not treatable by exon 53 skipping. No significant differences were observed in the other clinical features reported (Table 4).
Strength and function
DMD-53 patients presented with lower strength on both dominant and non-dominant sides as measured with MyoGrip and MyoPinch devices in comparison with both control groups, but those differences did not reach significance level except for the pinch strength in comparison with DMD-all-non-53 patients (0.789 ± 0.440 kg vs 1.236 ± 0.759 kg, p = 0.027 and 0.866 ± 0.486 kg vs 1.408 ± 0.854 kg, p = 0.020 in the non-dominant and the dominant side respectively) (Table 5). DMD-53 patients scored lower in the MFM but the difference did not reach statistical significance. No difference was observed on the MoviPlate score. Repartition of strength and function according to duration since loss of ambulation demonstrated that those parameters tend to decrease with time spend in a wheelchair (Fig. 3).
UMD-DMD-Cochin database
Data extracted from the UMD-DMD-Cochin database (Table 6) indicated that around 6.9% were carriers of a deletion theoretically treatable by exon 53 skipping. Kruskal-Wallis one-way analysis of variance was performed to compare ages at loss of ambulation in patients carrying deletions theoretically treatable by skipping of exon 44, 45, 50, 51, 52, 53, or 55. This analysis revealed a significant difference among groups (p = 0.001). DMD-53 patients lost ambulation significantly earlier than any other group: 1.2 years earlier than patients with a deletion theoretically treatable by skipping of another exon such as 44, 45, 50, 51, 52 or 55. (8.9 ± 1.4 years vs. 10.1 ± 1.8 years, p = 0.017), 1.1 years earlier than patients with a deletion outside the exon 44-55 region (8.9 ± 1.4 years vs. 10.0 ± 2.1 years p = 0.072), and 1.1 years earlier than patients with any other kind of mutation (8.9 ± 1.4 years vs. 10.0 ± 1.9 years p = 0.021). Post hoc test with correction of Bonferroni showed that DMD-53 patients lost ambulation significantly earlier than DMD patients theoretically correctible by exon 44 skipping (p = 0.024). Given the small number of patients in other groups, differences did not reach statistical significance when DMD-53 patients were compared with patients carrying deletions theoretically treatable by exon 50 or 55 skipping (p = 0.068 and p = 0.100, respectively), who lost ambulation more than two years later than those theoretically treatable by exon 53skipping (Fig. 4).
DISCUSSION
Using a prospective methodology and standardized evaluations, we demonstrated that non-ambulant DMD patients with a deletion theoretically treatable by exon 53 skipping tend to have a more severe course than age-matched controls with DMD patients who could not be treated by exon 53 skipping whatever are their mutation. This trend is confirmed even by comparing only with DMD patients carrying a deletion. The small number of patients is one of the reason why the level of significance is not reached. Using a retrospective methodology on a large population-based database, we confirmed that patients with a deletion theoretically treatable by exon 53 skipping lost ambulation more than one year earlier than other DMD patients.
Selection bias is not a concern with the UMD-DMD-Cochin database since the inclusion of patients is exhaustive within the covered territory [23]. Nevertheless, as all patients do not have an estimation of the amount of dystrophin on muscle biopsy, we choose to limit the analysis to patients with a DMD phenotype, this being defined only on clinical ground through the age of loss of ambulation. Therefore, patients with an age of loss of ambulation older or equal to fifteen were not retained for analyses due to a phenotype being more like a Becker muscular Dystrophy (BMD) which may introduce a bias in the study. However, the 92 DMD patients carrying a deletion treatable by exon 53 skipping selected from the UMD-DMD-Cochin database represented 6.9% of the DMD patients whatever is the mutation in this database, which is consistent with previously published data of 7.7% from the largest DMD database at the Leiden Duchenne Muscular Dystrophy pages which contains 3.6 time more DMD patients [24].
In the prospective study, all but four patients from France, Switzerland, and Romania with a deletion theoretically treatable by exon 53 skipping were included, which rules out a selection bias. DMD care and management may differ between countries and steroid treatment is not used widely in all European countries. Use of steroids is more common in Western Europe (except France) than Eastern Europe; however, as we consider only non-ambulant patients, only two patients were still on steroids including a Romanian. Inversely, regarding cardiac protection, use of ACE inhibitor seems less common in Romania as 3 among the 4 DMD-53 patients without ACE inhibitor were Romanian. Concerning the number of physiotherapy sessions, 2 of the 4 Romanian patients had only 1 or 0session per week, which may introduce a negative bias in upper limb strength and function evaluation. However, the raw data showed that Romanian patients were not particularly different from the mean of the DMD-53 patients. Lastly, even arthrodesis is not a standard of care in Romania, the scoliosis status of Romanian patients did not require arthrodesis surgery.
The control group in the prospective study was first constituted on the basis of an age-matching strategy. Assuming that patients carrying a deletion presented a more severe phenotype than patient with duplication or point mutation, we constituted a second control group based on deletion mutation and age-matching strategy. Both control group presented an age at loss of ambulation higher but comparable than the general DMD population of the UMD-DMD Cochin database (10.4 ± 2.4 and 10.7 ± 2.1 years vs. 10.0 ± 1.9, respectively). The DMD-53 patients tended to be clinically more severe than other DMD patients. They lost ambulation at a younger age than other DMD patients but, partly due to the small number of patients, this difference did not reach statistical significance for several indicators, such as the MFM, nor the forced vital capacity. Interestingly, DMD-53 patients had a lower LVEF although mostly under cardiac protection and they present more contracture on the dominant side than DMD patients carrying deletions not treatable by exon 53 skipping. This difference did not reach a significant level when comparing to DMD patients whatever is the mutation. Brooke score did not suggest any difference between the groups, which is not surprising given the poor discrimination power of this 8-point scale. Unexpectedly, the specifically designed MoviPlate did not also suggest any difference between groups. However, the highly sensitive pinch strength measure devices, demonstrated distal weakness in DMD-53 patients in the dominant and non-dominant upper limbs relative the control group whatever is the mutation. The trend was maintained comparing with control group carryingonly deletion but the statistical significance was not reached. This result means that the nature of the mutation could explain a part of the severity in DMD-53 patients. Similar results were obtained with the grip strength measure device without ever reaching a statistical significance level. Distal strength through grip and pinch and motor function of moving fingers and wrist are cardinal for non-ambulant patients’ autonomy (for commanding the wheelchair, writing, using the computer or phone).
It is generally known that DMD patients are smaller than the overall population [25]. Interestingly, with an average of nine centimeters smaller, our results confirmed that the DMD-53 patients seem to be even smaller than the DMD-non-53 controls. Difference in steroids use when patients were still ambulant may play a role. This issue should be formally investigated by getting information on the steroid use during the ambulant period of patients. The relationship between height and strength is complex, since strength depends on size [26]. In addition, steroid treatment is expected to shorten the stature and increase the strength. At the time of evaluations, use of steroids in DMD-53 and control-groups patients was stopped for most of them, so we could expect that steroid treatment previously received influence strength by reducing the height of DMD-53 patients. To our knowledge, no data regarding variations of clinical course nor muscle strength or function has been published in the specific populations of patients treatable by exon skipping. However, our findings are in line with a current UK based study on North Star data [27], and of a Dutch cohort [28], which demonstrated a less severe phenotype in patients theoretically skippable by exon 44 skipping. A recent Italian prospective study in ambulant patients failed to demonstrate any significant difference in 6 minutes walking test drop across the different groups of patients eligible for exon skipping, except for patients treatable by exon skipping 44 who present a slower drop [29].
The more than one year difference in age at loss of ambulation between DMD-53 and DMD-non-53 controls observed in the prospective and retrospective data implies a faster rate of decline during the ambulation period in DMD-53 patients which must be taken into account in the design of the clinical trials in these populations. This difference would affect not only the statistical powering of the studies but also the selection of control groups for non-placebo-controlled studies. The number of revertant fibers is known to be different between the various sub-groups of patients with genotypes treatable by exon skipping. For instance, patients eligible for exon skipping 44 present more revertant fibers than patients eligible for exon 51 skipping [8]. This is a possible explanation for differences in the age at loss of ambulation in these patients. Indeed, the classification between groups that can be established from the UMD-DMD-database regarding DMD-53 and DMD-51 patients for which the age at loss of ambulation is lower than DMD-44 patients matches the data available on revertant fiber numbers. This classification of subgroups is also consistent with the North Star data [27]. It is known that specific subgroups of DMD patients, such as those with deletions of exons 2–7, may present a milder phenotype, but it is unlikely that the inclusion of these subgroups in the general DMD population can account for the difference between DMD-53 and DMD-non-53 patients.
Mutations that are theoretically treatable by exon 53 skipping include different genotypes, such as del 52, del 50–52, and del 49–52. Probably any of the existing series include enough patients to determinate whether they present a similar phenotype. In the present dataset, no obvious differences appear between sub-groups, but although we had a very large cohort of patients theoretically treatable by inducing exon 53 skipping, this study was not statistically powered to assess this question. Pooling the data from the different nation-wide databases is required to address this question. Larger datasets would also be required to assess whether the more severe phenotype of DMD-53 patients could be related to a weaker respond to steroids, which is suggested by the higher percentage of DMD-53 than DMD-non-53 patients in our study who were treated with steroids at the time of the study, despite a more severe phenotype.
Studying non-ambulant patients allowed a retrospective overview of the natural history of the disease. It is commonly assumed that in DMD patients the age of occurrence of clinically meaningful endpoints is correlated with lifespan [30]. Replication of these findings in other large databases or in well conducted prospective studies and further mechanistic studies are required to better understand the specificity of the phenotype of 53-DMD patients. We believe that the present data are important and clinically meaningful in the context of the ongoing exon skipping trials. In conclusion, our investigation on the characterization of the phenotype of non-ambulant DMD patients theoretically treatable by inducing exon 53 skipping showed that this highly specific subgroup of patients is significantly weaker than the general DMD population. This aspect should be taken into account in the design and statistical plan of future therapeutic clinicaltrials.
CONFLICTS OF INTEREST STATEMENT
JYH is inventor of the MyoGrip and author of a patent on the MyoGrip. JYH and AM are co-inventors of the MyoPinch and authors of a patent on the MyoPinch. JYH, LS, and TV are authors of a patent on the MoviPlate.
ACKNOWLEDGMENTS
ULENAP and Pre-U7 studies were funded by ADNA (Advanced Diagnostics for New TherapeuticApproaches), a program dedicated to personalized medicine, coordinated by Institut Merieux and supported by research and innovation aid from the French public agency, OSEO. The UMD-DMD Cochin database curators wish to thank clinical contributors to the UMD-DMD Cochin database regular clinical data updating and the “Association Française contre les Myopathies” (AFM-Telethon) for its constant support. The contribution of M. Benali for patient evaluation is acknowledged.
REFERENCES
1 | Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J, Malik V, Shontz K, Walker CM, Flanigan KM, Corridore M, Kean JR, Allen HD, Shilling C, Melia KR, Sazani P, Saoud JB, Kaye EM, Eteplirsen Study G(2013) Eteplirsen forthe treatment of Duchenne muscular dystrophyAnn Neurol74: 5637637 |
2 | Cirak S, Feng L, Anthony K, Arechavala-Gomeza V, Torelli S, Sewry C, Morgan JE, Muntoni F(2012) Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophyMolecular therapy: The Journal of the American Society of GeneTherapy20: 2462467 |
3 | Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC(2011) Systemic administration of PRO051 in Duchenne’s muscular dystrophyThe New England Journal of Medicine364: 1615131522 |
4 | Goyenvalle A, Seto JT, Davies KE, Chamberlain J(2011) Therapeutic approaches to muscular dystrophyHum Mol Genet20: R1R69R78 |
5 | Cossu G, Sampaolesi M(2007) New therapies for Duchenne muscular dystrophy: Challenges, prospects and clinical trialsTrends Mol Med13: 12520526 |
6 | Trollet C, Athanasopoulos T, Popplewell L, Malerba A, Dickson G(2009) Gene therapy for muscular dystrophy: Currentprogress and future prospectsExpert Opin Biol Ther9: 7849866 |
7 | Warrington KHJr, Herzog RW(2006) Treatment of human disease by adeno-associated viral gene transferHum Genet119: 6571603 |
8 | Lourbakos A, Sipkens J, Beekman C, Kreuger D, Brasz L, Janson A, Campion G, van Deutekom J, de Kipme S(2011) The incidence of revertantand trace dystrophin expression in muscle biopsies of DuchenneMuscular Dystrophy patients with different exon deletionsNeuromuscular Disord21: 9-10643 |
9 | Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Howard MT, Sampson JB, Swoboda KJ, Bromberg MB, Mendell JR, Taylor LE, Anderson CB, Pestronk A, Florence JM, Connolly AM, Mathews KD, Wong B, Finkel RS, Bonnemann CG, Day JW, McDonald C, United Dystrophinopathy Project CWeiss RB(2011) Nonsense mutation-associatedBecker muscular dystrophy: Interplay between exon definition andsplicing regulatory elements within the DMD geneHuman Mutation32: 3299308 |
10 | Desguerre I, Christov C, Mayer M, Zeller R, Becane HM, Bastuji-Garin S, Leturcq F, Chiron C, Chelly J, Gherardi RK(2009) Clinical heterogeneity of duchennemuscular dystrophy (DMD): Definition ofsub-phenotypes and predictive criteria by long-term follow-upPloS One4: 2e4347 |
11 | Muntoni F, Torelli S, Ferlini A(2003) Dystrophin and mutations: One gene, several proteins, multiple phenotypesLancetNeurology2: 12731740 |
12 | Daoud F, Angeard N, Demerre B, Martie I, Benyaou R, Leturcq F, Cossee M, Deburgrave N, Saillour Y, Tuffery S, Urtizberea A, Toutain A, Echenne B, Frischman M, Mayer M, Desguerre I, Estournet B, Reveillere C, Penisson B, Cuisset JM, Kaplan JC, Heron D, Rivier F, Chelly J(2009) Analysis of Dp71 contribution in theseverity of mental retardation through comparison of Duchenne andBecker patients differing by mutation consequences on Dp71expressionHumanMolecular Genetics18: 2037793794 |
13 | Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, Turconi AC, Sciacco M, Ciscato P, Bordoni A, Tedeschi S, Fortunato F, Lucchini V, Bonato S, Lamperti C, Coviello D, Torrente Y, Corti S, Moggio M, Bresolin N, Comi GP(2011) Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-uJournal of Neurology258: 916101623 |
14 | Humbertclaude V, Hamroun D, Bezzou K, Berard C, Boespflug-Tanguy O, Bommelaer C, Campana-Salort E, Cances C, Chabrol B, Commare MC, Cuisset JM, de Lattre C, Desnuelle C, Echenne B, Halbert C, Jonquet O, Labarre-Vila A, N’Guyen-Morel MA, Pages M, Pepin JL, Petitjean T, Pouget J, Ollagnon-Roman E, Richelme C, Rivier F, Sacconi S, Tiffreau V, Vuillerot C, Picot MC, Claustres M, Beroud C, Tuffery-Giraud S(2012) Motor and respiratory heterogeneity inDuchenne patients: Implication for clinical trialsEuropean journal of paediatric neurology: EJPN: Official Journal of the European Paediatric Neurology Society16: 2149160 |
15 | Davidson ZE, Kornberg AJ, Ryan MM, Sinclair K, Cairns A, Walker KZ, Truby H(2012) Deletions in the dystrophin gene predict loss of ambulation before 10 years of age in boys with Duchenne muscular dystrophyNeuromuscular Disord22: 9-10835 |
16 | Wong BL, Hu SY, Morehart P, Cripe LH, Walker ME(2011) Phenotypic profile of dystrophinopathy patients with deletion of exons 3-7 of the dystrophin geneNeuromuscular Disord21: 9-10642 |
17 | Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, Bello L, Soraru G, Pacchioni B, Bonifati MD, Lanfranchi G, Angelini C, Kesari A, Lee I, Gordish-Dressman H, Devaney JM, McDonald CMCooperative International Neuromuscular Research G(2011) SPP1 genotype is a determinant of disease severity in Duchennemuscular dystrophyNeurology76: 3219226 |
18 | Piva L, Gavassini BF, Bello L, Fanin M, Soraru G, Barp A, Ermani M, Angelini C, Hoffman EP, Pegoraro E(2012) TGFBR2but not SPP1 genotype modulates osteopontin expression in Duchenne muscular dystrophy muscleThe Journal ofpathology228: 2251259 |
19 | Flanigan KM, Ceco E, Lamar KM, Kaminoh Y, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Gappmaier E, Howard MT, Day JW, McDonald C, McNally EM, Weiss RB, Project UD(2013) LTBP4 genotype predicts age of ambulatory loss in duchenne muscular dystrophyAnn Neurol73: 4481488 |
20 | Servais L, Deconinck N, Moraux A, Benali M, Canal A, Van Parys F, Vereecke W, Wittevrongel S, Mayer M, Desguerre I, Maincent K, Themar-Noel C, Quijano-Roy S, Serari N, Voit T, Hogrel JY(2013) Innovative methods to assess upper limb strength and function in non-ambulant Duchenne patientsNeuromuscular Disorders: NMD23: 2139148 |
21 | Seferian AM, Moraux A, Annoussamy M, Canal A, Decostre V, Diebate O, Le Moing AG, Gidaro T, Deconinck N, Van Parys F, Vereecke W, Wittevrongel S, Mayer M, Maincent K, Desguerre I, Themar-Noel C, Cuisset JM, Tiffreau V, Denis S, Jousten V, Quijano-Roy S, Voit T, Hogrel JY, Servais L(2015) Upper limb strength and function changes during a one-year follow-up in non-ambulant patients with duchenne muscular dystrophy: An observational multicenter trialPloS One10: 2e0113999 |
22 | Seferian AM, Moraux A, Canal A, Decostre V, Diebate O, Le Moing AG, Gidaro T, Deconinck N, Van Parys F, Vereecke W, Wittevrongel S, Annoussamy M, Mayer M, Maincent K, Cuisset JM, Tiffreau V, Denis S, Jousten V, Quijano-Roy S, Voit T, Hogrel JY, Servais L(2015) Upper limb evaluation and one-year follow up of non-ambulant patients with spinal muscular atrophy: An observational multicenter trialPloS One10: 4e0121799 |
23 | Tuffery-Giraud S, Beroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, Moizard MP, Bernard R, Cossee M, Boisseau P, Blayau M, Creveaux I, Guiochon-Mantel A, de Martinville B, Philippe C, Monnier N, Bieth E, Khau Van Kien P, Desmet FO, Humbertclaude V, Kaplan JC, Chelly J, Claustres M(2009) Genotype-phenotype analysis in 2,405 patients with adystrophinopathy using the UMD-DMD database: A model of nationwideknowledgebaseHuman Mutation30: 6934945 |
24 | Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, den Dunnen JT(2009) Theoretic applicability ofantisense-mediated exon skipping for Duchenne muscular dystrophymutationsHum Mutat30: 3293299 |
25 | Nagel BH, Mortier W, Elmlinger M, Wollmann HA, Schmitt K, Ranke MB(1999) Short stature in Duchenne muscular dystrophy: A study of 34 patientsActa Paediatrica88: 16265 |
26 | Hogrel JY, Decostre V, Alberti C, Canal A, Ollivier G, Josserand E, Taouil I, Simon D(2012) Stature is an essential predictor of muscle strength in childrenBMC Musculoskeletal Disorders13: 176 |
27 | Ricotti V, Ridout DA, Mercuri E, Quinlivan R, Robb SA, Manzur AY, Muntoni F(2013) P.2.1 The northStar ambulatory assessment in duchenne muscular dystrophy: Considerations for the design of clinical trialsNeuromuscular Disord23: 9-10748 |
28 | van den Bergen JCG, Niks HB, Aartsma-Rus EH, Verschuuren AJJGM(2014) Prolonged ambulation in Duchenne patients with a mutationamenable to exon 44 skippingJournal of Neuromuscular Dideases1: 19194 |
29 | Pane M, Mazzone ES, Sormani MP, Messina S, Vita GL, Fanelli L, Berardinelli A, Torrente Y, D’Amico A, Lanzillotta V, Viggiano E, D’Ambrosio P, Cavallaro F, Frosini S, Bello L, Bonfiglio S, Scalise R, De Sanctis R, Rolle E, Bianco F, Van der Haawue M, Magri F, Palermo C, Rossi F, Donati MA, Alfonsi C, Sacchini M, Arnoldi MT, BaranelloG , Mongini T, Pini A, Battini R, Pegoraro E, Previtali SC, Napolitano S, Bruno C, Politano L, Comi GP, Bertini E, Morandi L, Gualandi F, Ferlini A, Goemans N, Mercuri E(2014) 6 Minute walk test in Duchenne MD patients with differentmutations: 12 month changesPloS one9: 1e83400 |
30 | Henricson EK, Abresch RT, Cnaan A, Hu F, Duong T, Arrieta A, Han J, Escolar DM, Florence JM, Clemens PR, Hoffman EP, McDonald CM, Investigators C(2013) The cooperative internationalneuromuscular research group Duchenne natural history study:Glucocorticoid treatment preserves clinically meaningfulfunctional milestones and reduces rate of disease progression asmeasured by manual muscle testing and other commonly used clinicaltrial outcome measuresMuscle & Nerve48: 15567 |
Figures and Tables
Fig.1
Flowchart of analyzed patients.
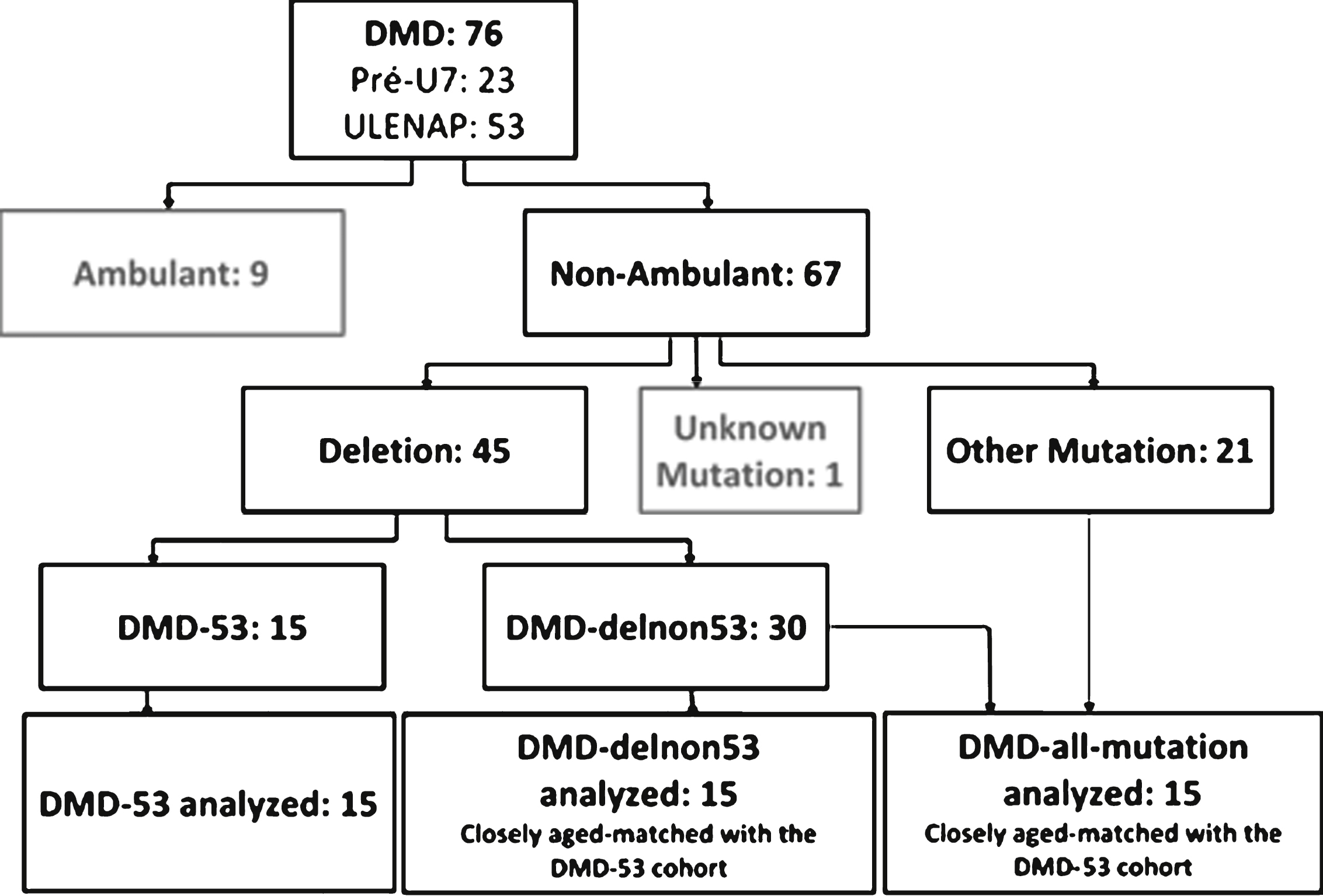
Fig.2
Distribution of ages of patients included in the analyses.
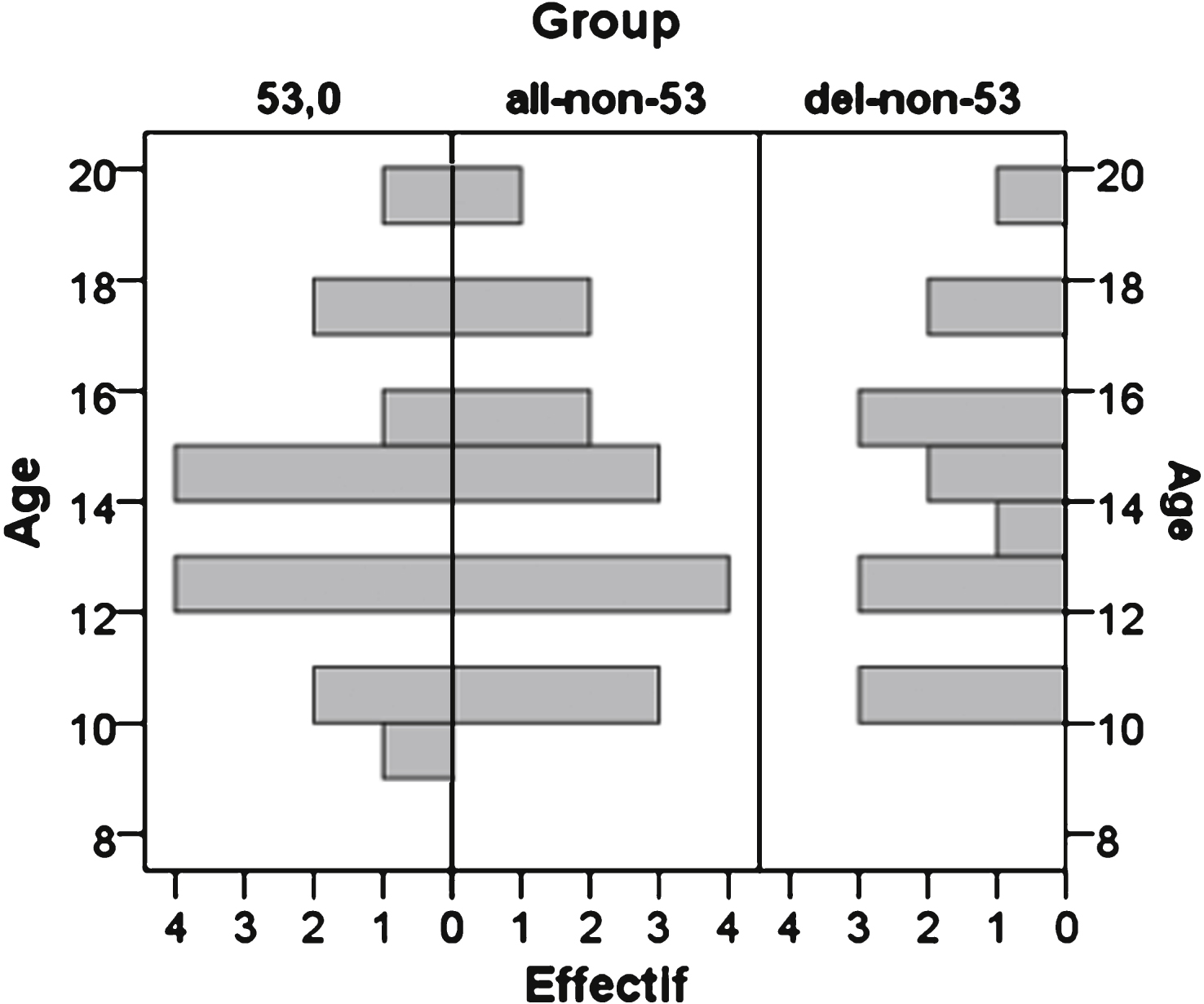
Fig.3
Dispersion of data on dominant side for strength (A and B) and motor function (C and D). The full squares represented the DMD-53 patients; the empty squares symbolized DMD-del-non-53 patients; the grey triangle represented DMD patients carrying other mutation (LoA: Loss of Ambulation).
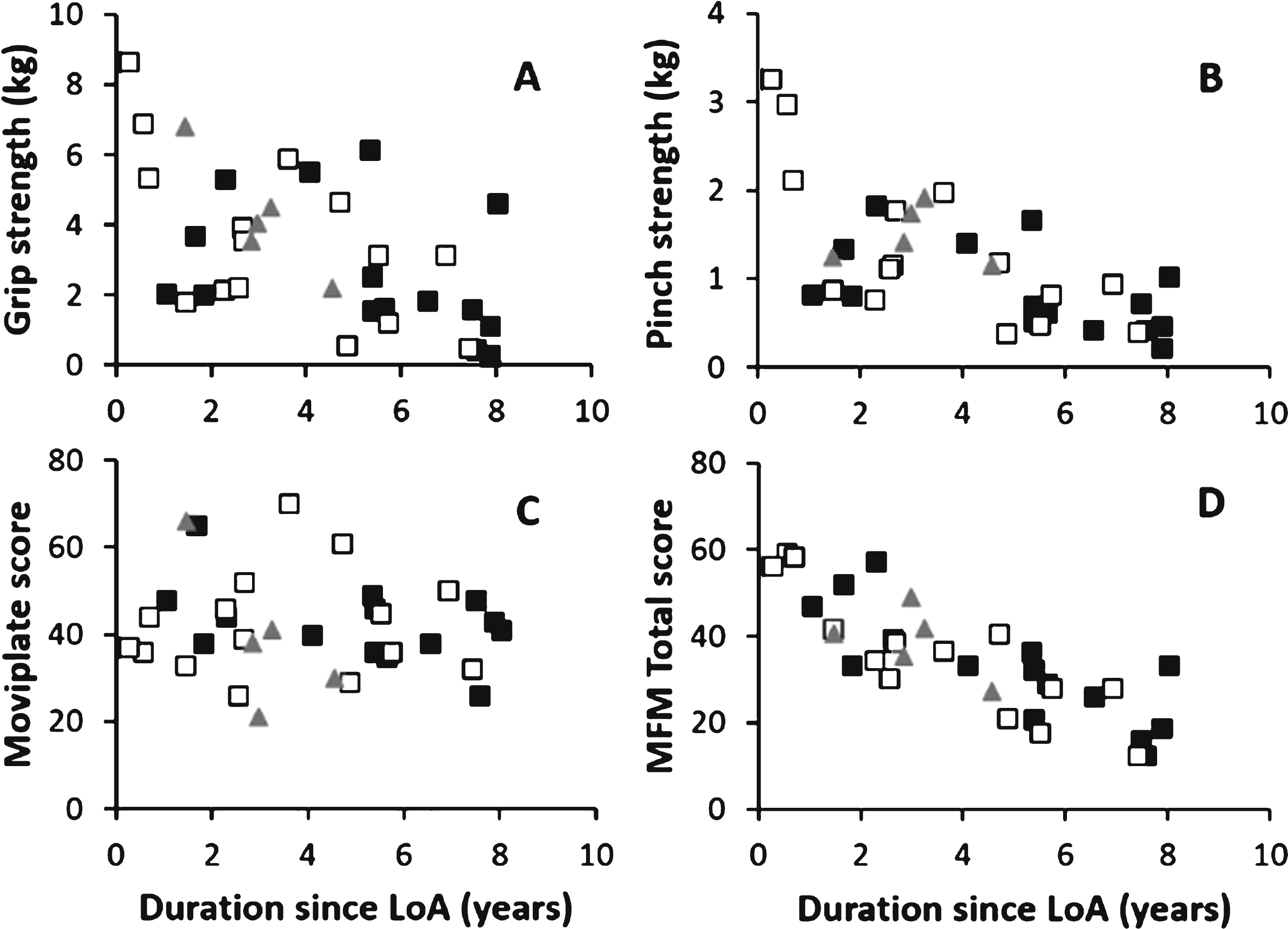
Fig.4
Box-plot of the age at loss of ambulation of DMD patients from -UMD-DMD-Cochin database (Exon 44, 45, 50, 51, 52, 53, 55: patients carrying deletions theoretically treatable by skipping of exon 44, 45, 50, 51, 52, 53 or 55; DelOut45–55: patients carrying a deletion outside the region limited by exons 45 and 55; AllDMD: all DMD mutations; rectangles represent the 25th–75th percentile, the line is the median, ∘: outliers, ★: faroutliers).
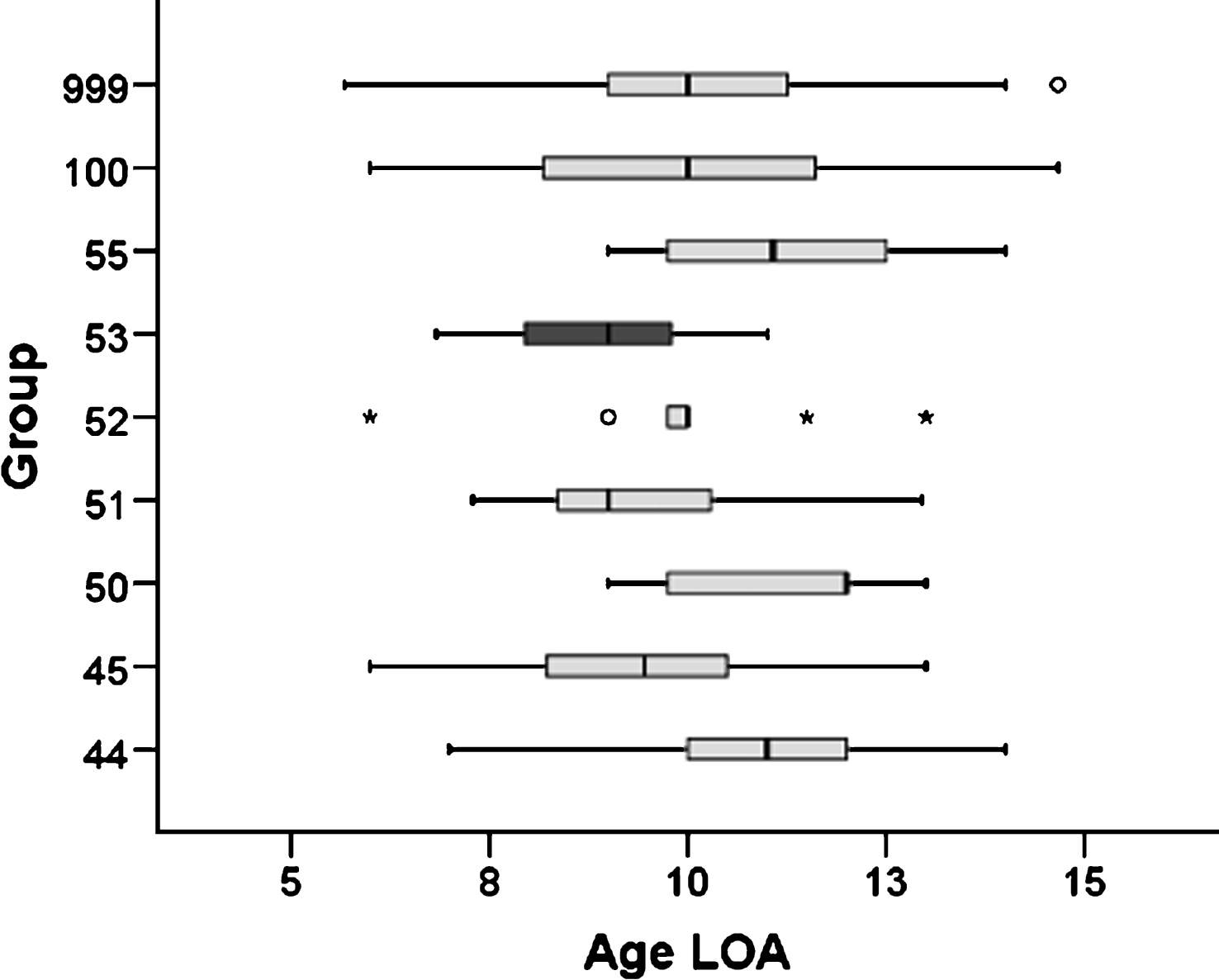
Table 1
Main selection criteria of Pre-U7 and ULENAP studies from which patients were selected
| Pre-U7 | ULENAP |
| - DMD theoretically treatable by exon 53 skipping | - Neuromuscular disease confirmed by molecular analysis |
| - Age between 6 and 20 | - Age between 5 and 30 |
| - Able to understand rules of assessments | - Able to understand rules of assessments |
| - Ambulant and non-ambulant | - Non-ambulant |
| - Signed informed consent | - Signed informed consent |
Table 2
Demographic data of patients
| Group | Age (years) | Mutation | Height (cm) | Weight (Kg) | Age at loss of ambulation (years) | Steroid 0: no 1: yes (mg per day) | ACE inhibitor 0: no 1: yes | PT (nb/week) | Scoliosis 0: no 1: yes 2: arthrosesis | Contractures 0: no 1: yes | VEF | FVC (% of predicted value) | Brooke score (#) |
| 53 ** | 10,3 | del48-52 | 125 | 30 | 8 | 0 | 0 | 0 | 0 | 0 | NA | NA | 5 |
| 53 ** | 10,6 | del52 | 135 | 60 | 6 | 0 | 0 | 1 | 0 | 1 | 36 | 180 | 4 |
| 53 | 9,1 | del45-52 | 124 | 34 | 8 | 0 | 1 | 4 | 0 | 0 | 64 | 60 | 3 |
| 53 | 12,3 | del52 | 137 | 38 | 10 | 1 (20) | 0 | 1 | 0 | 0 | 45 | 73 | 2 |
| 53 ** | 12,3 | del45-52 | 153 | 69 | 7 | 0 | 0 | 3 | 1 | 1 | NA | 74 | 5 |
| 53 | 12,5 | del49-52 | 143 | 53 | 6 | 0 | 1 | 4 | 1 | 1 | NA | 33 | 5 |
| 53 ** | 12,7 | del45-52 | 130 | 27 | 11 | 1 (10) | 1 | 5 | 0 | 0 | 45 | NA | 1 |
| 53 | 14,4 | del45-52 | 154 | 62 | 9 | 0 | 1 | 3 | 2 | 1 | 47 | 30 | 5 |
| 53 | 14,4 | del49-52 | 147 | 26 | 9 | 0 | 1 | 4 | 1 | 1 | NA | 51 | 5 |
| 53 | 14,6 | del45-52 | 166 | 40 | 9 | 0 | 1 | 3 | 2 | 1 | 55 | 49 | 5 |
| 53 | 14,9 | del52 | 145 | 27 | 7 | 0 | 1 | 3 | 2 | 1 | 50 | NA | 6 |
| 53 | 15,5 | del45-52 | 153 | 57 | 8 | 0 | 1 | 4 | 1 | 1 | NA | 27 | 5 |
| 53 | 17,6 | del50-52 | 161 | 28 | 10 | 0 | 1 | 2 | 2 | 1 | NA | NA | 6 |
| 53 | 17,9 | del45-52 | 151 | 58 | 10 | 0 | 1 | 7 | 2 | 1 | 60 | 25 | 6 |
| 53 | 19,0 | del48-52 | NA | 60 | 11 | 0 | 1 | 3 | 1 | 0 | 52 | NA | 3 |
| non53 *a | 10,0 | c.7392delC | 140 | 30 | 7 | 0 | 1 | 5 | 0 | 0 | 71 | 104 | 4 |
| p.Leu2465 * | |||||||||||||
| non53 *a | 12,2 | dup8-9 | 158 | 55 | 9 | 1 (30) | NA | NA | NA | NA | 61 | 65 | 4 |
| non53 a | 12,8 | c.9459_9462del | 156 | 48 | 10 | 0 | 1 | 2 | 1 | NA | 68 | 60 | 4 |
| p.Cys3153 * | |||||||||||||
| non53 *a | 14,4 | c.7657C>T | 155 | 29 | 10 | 0 | 1 | 3 | 2 | 1 | 64 | 41 | 5 |
| p.Arg2553 * | |||||||||||||
| non53 *a | 15,5 | c.998C>A | 166 | 40 | 14 | 0 | 1 | 3 | 2 | 1 | 52 | 40 | 5 |
| p.Ser333 * | |||||||||||||
| non53 *b | 10,9 | del53 | 134 | 33 | 10 | 1 (NA) | 1 | 2 | 0 | 1 | 64 | 90 | NA |
| non53 *b | 12,1 | del3-44 | 161 | 39 | 10 | 0 | 0 | 1 | 1 | 1 | NA | 43 | 5 |
| non53 b | 13,1 | del48-50 | 162 | 50 | 11 | 0 | 0 | 5 | 1 | NA | 66 | 68 | 5 |
| non53 *b | 15,7 | del42-54 | 155 | 29 | 13 | 0 | 1 | 3 | 2 | 1 | NA | 33 | 4 |
| non53 *b | 15,7 | del10-11 | 150 | 30 | 10 | 0 | 1 | 3 | 2 | 1 | 65 | 30 | 5 |
| non53 a,b | 10,2 | del21 | 135 | 21 | 9 | 0 | 1 | 3 | 0 | 1 | 65 | 56 | 4 |
| non53 a,b | 10,6 | del8-13 | 135 | 43 | 10 | 1 (25) | 0 | 3 | 1 | 1 | 62 | 77 | 2 |
| non53 a,b | 12,3 | del48-54 | NA | 34 | 12 | 1 (15) | 0 | 2 | 0 | 0 | NA | NA | 2 |
| non53 *a,b | 12,9 | del46-49 | 140 | 34 | 10 | 0 | 1 | 3 | 2 | 1 | 55 | 48 | 5 |
| non53 a,b | 14,2 | del5-7 | 162 | 47 | 10 | 0 | 1 | 3 | 0 | 1 | 71 | 61 | 5 |
| non53 a,b | 14,4 | del24-43 | 165 | 39 | 7 | 0 | 1 | 3 | 2 | 1 | 55 | 47 | NA |
| non53 *a,b | 15,2 | del8-9 | 171 | 52 | 8 | 0 | 0 | 2 | 0 | 1 | 60 | 64 | 5 |
| non53 a,b | 17,6 | del46-47 | 165 | 49 | 14 | 0 | 1 | 5 | 2 | 1 | 72 | 65 | 4 |
| non53 a,b | 17,9 | del5-7 | 160 | 70 | 13 | 0 | 1 | 4 | 2 | NA | 74 | 61 | 5 |
| non53 a,b | 19,5 | del32 | 160 | 32 | 14 | 0 | 0 | 5 | 2 | 1 | ND | 26 | 5 |
NA: Not available; ACE: Angiotensin conversion enzyme; PT: Physiotherapy session; VEF: Ventricular ejection fraction; FVC: Forced vital capacity; *: patients previously described in [20]; **: Romanian patients; a,b: patients used in statistical comparison between 53 and non53-all mutation (a) or non53-deletion (b).
Table 3
Treatment analyses
| Steroid | N | P (% ) | CI |
| DMD-53 | 15 | 13.33 | [1.66–40.46] |
| DMD-all-non-53 | 15 | 20.00 | [4.33–48.09] |
| DMD-del-non-53 | 15 | 20.00 | [4.33–48.09] |
| ACE Inhibitor | N | P (% ) | CI |
| DMD-53 | 15 | 66.67 | [38.38–88.18] |
| DMD-all-non-53 | 15 | 60.00 | [32.29–83.66] |
| DMD-del-non-53 | 14 | 71.43 | [41.90–91.61] |
| PT | N | mean (nb/week) | SD |
| DMD-53 | 15 | 3.13 | 1.76 |
| DMD-all-non-53 | 14 | 3.29 | 1.07 |
| DMD-del-non-53 | 15 | 3.13 | 1.19 |
| Arthrodesis | N | P (% ) | CI |
| DMD-53 | 15 | 33.33 | [11.82–61.62] |
| DMD-all-non-53 | 14 | 50.00 | [23.04–76.96] |
| DMD-del-non-53 | 15 | 46.67 | [21.27–73.41] |
N: number of subjects; P: proportion; CI: confidence interval; SD: standard deviation; ACE: Angiotensin conversion enzyme; PT: Physiotherapy session.
Table 4
Clinical features analyses
| Group effect | Group effect | ||||||||
| 53 | all-non-53 | del-non-53 | 53 vs all-non-53 | 53 vs del-non-53 | |||||
| N | Mean | SD | Mean | SD | Mean | SD | p-value | p-value | |
| Duration since loss of ambulation (years) | 15 | 5.2 | 2.5 | 3.5 | 2.1 | 3.5 | 2.3 | 0.047 * | 0.023 * |
| Contractures-Score-ND | 11 | 5.4 | 4.5 | 4.7 | 3.6 | 4.9 | 3.0 | 0.624 | 0.624 |
| Contractures-Score-D | 11 | 7.0 | 4.2 | 4.7 | 3.5 | 5.2 | 3.1 | 0.068 | 0.046 * |
| Brooke | 14 | 4.4 | 1.6 | 4.2 | 1.1 | 4.3 | 1.1 | 0.718 | 0.905 |
| Left ventricular ejection fraction (% ) | 8 | 50.3 | 9.1 | 63.6 | 7.4 | 66.7 | 5.0 | 0.018 * | 0.028 * |
| Forced Vital capacity in sitting position | 8 | 56.9 | 51.4 | 61.4 | 21.2 | 57.1 | 17.2 | 0.327 | 0.483 |
| (% of predictive value) | |||||||||
D: dominant arm; ND: non-dominant arm; N: number of subjects; SD: standard deviation; *: significant differences.
Table 5
Force and function data analyses
| Group effect | Group effect | ||||||||
| 53 | all-non-53 | del-non-53 | 53 vs all-non-53 | 53 vs del-non-53 | |||||
| N | Mean | SD | Mean | SD | Mean | SD | p-value | p-value | |
| MyoGrip-ND | 15 | 2.45 | 1.93 | 3.67 | 2.20 | 3.45 | 2.23 | 0.053 | 0.281 |
| MyoGrip-D | 15 | 2.70 | 1.89 | 4.01 | 2.33 | 3.58 | 2.36 | 0.061 | 0.281 |
| MyoPinch-ND | 15 | 0.789 | 0.440 | 1.236 | 0.759 | 1.167 | 0.808 | 0.027 * | 0.112 |
| MyoPinch-D | 15 | 0.866 | 0.486 | 1.408 | 0.854 | 1.351 | 0.893 | 0.020 * | 0.078 |
| MoviPlate-ND | 14 | 38 | 9 | 38 | 16 | 38 | 14 | 0.433 | 0.594 |
| MoviPlate-D | 14 | 43 | 9 | 41 | 15 | 42 | 12 | 0.484 | 0.826 |
| MFM-D1 (% ) | 14 | 1 | 3 | 2 | 3 | 3 | 3 | 0.293 | 0.307 |
| MFM-D2 (% ) | 14 | 43 | 25 | 50 | 27 | 48 | 29 | 0.300 | 0.551 |
| MFM-D3 (% ) | 14 | 71 | 15 | 79 | 16 | 78 | 16 | 0.245 | 0.278 |
| MFM-Total (% ) | 14 | 32 | 13 | 37 | 14 | 36 | 15 | 0.221 | 0.414 |
D: dominant arm; ND: non-dominant arm; N: number of subjects; SD: standard deviation; *significant differences.
Table 6
Mean age at loss of ambulation between DMD patients carrying different type of mutation from UMD/DMD-Cochin database
| Exon53 | Exon51 | Exon45 | Exon52 | DelOut 45–55 | AllDMD | Exon44 | Exon50 | Exon55 | |
| N patients | 92 | 103 | 110 | 49 | 294 | 1340 | 82 | 54 | 33 |
| N non-ambulant patients | 26 | 43 | 37 | 11 | 100 | 442 | 31 | 12 | 8 |
| Mean age at loss of | 8.9 ± 1.4 | 9.3 ± 1.6 | 9.4 ± 1.8 | 9.9 ± 1.8 | 10.0 ± 2.1 | 10.0 ± 1.9 | 10.9 ± 1.7 | 11.2 ± 1.6 | 11.2 ± 1.8 |
| ambulation ± SD (years) | [13] * | [26] * | [24] * | [10] * | [68] * | [292] * | [21] * | [9] * | [8] * |
Exon44, 45, 50, 51, 52, 53, 55: patients carrying deletions theoretically treatable by skipping of exon 44, 45, 50, 51, 52, 53 or 55; DelOut45–55: patients carrying a deletion outside the region limited by exons 45 and 55; AllDMD: all DMD mutations; SD: standard deviation; *: number of subjects for which age at loss of ambulation is available.


