
The Angiotensin Converting Enzyme Inhibitor Lisinopril Improves Muscle Histopathology but not Contractile Function in a Mouse Model of Duchenne Muscular Dystrophy
Abstract
Background: Angiotensin converting enzyme inhibitors (ACEi) are the current standard of care treatment for cardiac dysfunction in Duchenne muscular dystrophy patients. We previously showed treatment with an ACEi plus mineralocorticoid receptor (MR) antagonist improves limb and respiratory skeletal muscles, in addition to cardiac muscles, in a dystrophic mouse model at 20 weeks-of-age.
Objective: To determine whether previously observed preclinical benefits of an ACEi plus MR antagonist on dystrophic skeletal muscles can be reproduced by increasing ACEi dosage alone. We also compared functional and histological outcome measures at 10 and 20 weeks-of-age.
Methods: Dystrophin deficient utrophin haplo-insufficient (utrn +/- ; mdx) “het” mice were treated with 10, 20, or 50 mg/kg × day of the ACEi lisinopril from 4 to 10 weeks-of-age via water bottles and compared with C57BL/10 wild-type control mice and untreated hets. Data from 10 week-old het mice were also compared to data collected from an untreated het group at 20 weeks-old. In vivo cardiac and grip strength measurements, in vitro diaphragm and extensor digitorum longus muscle force measurements, and histopathological analyses were performed. One-way ANOVA followed by Dunnett post hoc comparison was used to determine significance.
Results: ACEi treatment reduced skeletal muscle damage but had no significant effect on muscle force. Body weight, heart rate, grip strength and blood pressure were unaffected by treatment. Limb muscle histopathology was more informative at 10 than 20 weeks-of-age.
Conclusions: These results suggest increased ACEi dosage alone cannot improve all dystrophic parameters. Further optimization of MR antagonists in 20 week-old mice is warranted.
ABBREVIATIONS
ACEi | Angiotensin converting enzyme inhibitors |
MR | mineralocorticoid receptor |
utrn +/- ; mdx or “het” | dystrophin-deficient utrophin haplo-insufficient mice |
DMD | Duchenne muscular dystrophy |
ROS | reactive oxygen species |
MRI | magnetic resonance imaging |
ECG | electrocardiography |
BDM | 2,3-butanedione monoxime |
EDL | extensor digitorum longus |
H&E | Hematoxylin and Eosin |
IgG | Immunoglobulin G |
CSA | cross-sectional area |
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a progressive, degenerative striated muscle disease that results in skeletal muscle weakness and cardiomyopathy. Most patients are wheelchair bound by age 12 due to skeletal muscle degeneration [1]. Nearly all DMD patients develop dilated cardiomyopathy by age 18 and 40% succumb to heart failure [2]. Therapeutics that can target and improve both cardiac and skeletal muscle function are needed to provide the most clinical benefit to DMD patients.
Preclinical studies using the dystrophin-deficient mdx mouse model of DMD found that treatment with the angiotensin converting enzyme inhibitor (ACEi) enalapril at clinically relevant dosages, increased mouse forelimb strength and reduced reactive oxygen species (ROS) production after four weeks of treadmill running exercise [3]. Combination treatment with lisinopril (ACEi) and the mineralocorticoid receptor (MR) antagonist spironolactone in dystrophin deficient, utrophin haplo-insufficient (utrn +/- ; mdx) “het” mice significantly improved both skeletal and cardiac muscle function and reduced muscle damage when mice were treated prior to the first signs of myocardial damage [4]. Mice treated with a combination of these drugs from 4 to 20 weeks-of-age, retained 80% of normal muscle force generation in both respiratory and limb muscles compared to only 40% of normal force observed in untreated 20 week-old het mice. Treatment with both an MR antagonist and ACEi may improve function by concomitantly affecting differing points in the renin-angiotensin-aldosterone pathway [5, 6]. The ACEi lisinopril indirectly affects MR by blocking formation of angiotensin II and preventing aldosterone production, a necessary ligand for MR activation [5]. Angiotensin converting enzyme inhibitors are the current standard of care treatment for cardiomyopathy in DMD patients [7]. Our recent double-blind, placebo-controlled clinical trial has also shown the addition of the selective MR antagonist eplerenone to background ACEi therapy before cardiomyopathy complications such as heart failure and arrhythmias developed, further stabilizes cardiac function in DMD patients [8].
This study seeks to identify whether the previous preclinical skeletal muscle benefits seen with a combined treatment using an ACEi and MR antagonist can be replicated using only an ACEi at higher dosages, since ACE functions upstream of MR and ACEi are already used as standard of care for DMD cardiomyopathy. Determining whether an increased dosage of ACEi can mimic the efficacy seen from the combinatorial treatment would help clarify the importance of MR antagonism in dystrophic skeletal muscle and further characterize MR as a therapeutic target for degenerative muscle disease.
Concurrently, we assessed whether analyzing mice at 10 weeks-of-age, provides similarly informative histological and functional outcome measures as at 20 weeks-of-age, allowing more preclinical comparisons to be carried out in a similar time-frame. Fibrosis, muscle degeneration, and limb muscle force were compared between wild type and dystrophic het mice at each of the two time points to identify which one provides more informative data and should be used for further analyses.
MATERIALS AND METHODS
Mice and treatment
All protocols were approved by the Institutional Animal Care and Use Committee of The Ohio State University and are in compliance with the laws of The United States of America and conform to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health.
For this study we used a dystrophin-deficient, utrophin haplo-insufficient (utrn +/- ; mdx) “het” mouse model, which has a more severe phenotype than mdx mice [9, 10]. C57BL/10 (Harwell) wild-type control mice were also used in this study. Dystrophic and wild-type control mice were bred and dystrophic mice were genotyped, as described previously [11]. Mice were weaned at 4 weeks-of-age and males and females from each litter were housed 2 per cage and used for treatment or as untreated controls. Mice were treated for 6 weeks and then analyzed by personnel not involved in genotyping or treating the animals and blinded to the groups.
Equal numbers of female and male het mice were given water bottles containing 66, 133, or 330 mg/L of the ACEi lisinopril (SBH Medical) in reverse-osmosis water or no drug (reverse-osmosis water only). Medicated water bottles were replaced 3 times per week; mice were weighed and the volume of water consumed was recorded to ensure mice were receiving the estimated dosage (10, 20 or 50 mg/kg × day). The 2 lowest dosages were based on allometric scaling of clinical dosages to use in mice (based on the FDA: Guidance for Industry; Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers) and are commonly used for experiments in cardiomyopathic mice and rats. The lowest dosage (10 mg/kg × day) was that used in our previous combinatorial ACEi plus MR antagonist treatments shown to be efficacious in dystrophic mice [4, 12]. 50 mg/kg × day was included as a 5X normal dosage to capture any increased dose-response. In total, 5 groups of mice were included in this study: C57BL/10 wild-type control mice (C, n = 10), untreated het mice (U, n = 10), and lisinopril treated het mice at 3 different dosages (L10, L20, L50, n = 6 respectively). The combination of the three groups of het mice receiving lisinopril is referred to as total lisinopril treated mice (L) for some dataanalyses.
In vivo cardiac and forelimb grip strength measurements
All in vivo measurements were performed by individuals blinded to the genotype and treatment of the mice. Each in vivo measurement was performed bythe same individual to limit variability. Within 4 days of the animals reaching 10 weeks-of-age, het untreated and C57BL/10 wild-type control mice were anesthetized (isoflurane) and magnetic resonance imaging (MRI) was performed on a 9.4 or 11.7 Tesla 30 mm bore system (Bruker Biospin) with electrocardiographic (ECG) leads while under body temperature control (37°C), as described previously [4]. Myocardial strain and strain rate were computed using vector-based tracking software (Vector Velocity Imaging, Siemens).
Blood pressure was measured non-invasively in conscious mice by the tail cuff method using a 6-channel CODA High Throughput Acquisition System (Kent Scientific Corporation). Measurement recording sessions included 10 acclimation cycles followed by 10 measurement cycles. The average of accepted measurement cycles for each mouse was used for systolic, diastolic, and mean arterial blood pressure [13]. Measurements were done between 10:00 AM and 12:00 PM in untrained mice at least one day before ECG, grip strength, and in vitro muscle force experiments.
On the day of sacrifice, the body weight of each mouse was recorded and resting, non-anesthetized, and non-invasive electrocardiographic recordings were taken using the ECGenie system (Mouse Specifics Inc.) as described previously [4]. Briefly, mice were placed on the ECG platform in a quiet room and allowed sufficient time (typically 5– 10 minutes) for heart rate to level after handling. ECG tracings were continuously recorded for 30 minutes using LabChart7 (AD Instruments), and later analyzed using ECG e-Mouse 9 software (Mouse Specifics Inc). Analysis was done using time intervals when the paws were in contact with the electrodes (typically 10– 15 seconds) and heart rate (HR) remained consistent. ECG signals were used to assess: QT-interval, QT-interval:heart rate, and heart rate variability.
Forelimb grip strength was then assessed in conscious, non-anesthetized mice. The grip strength meter (Columbus Instruments) consists of a wire mesh grid attached to a force transducer atop a sturdy aluminum plate. Mice were suspended so that their front paws were able to grasp the center edge of the wire mesh. Once the mouse had established a grip without body movement, it was swiftly and uniformly pulled backwards in plane with the transducer to break the clasp [3] and as described in Treat-NMD SOP DMD_M.2.2.001. This procedure was repeated three times with one-minute rests between measurements. The highest value (N) of the three was reported. All grip strength measurements were conducted by the same investigator to limit variability.
In vitro diaphragm and extensor digitorum longus (EDL) contraction force measurements
All force measurements were performed by individuals blinded to the genotype and treatment of the mice. Measurement of each muscle type was performed by the same individual to limit variability. Mice were sacrificed by cervical dislocation. The thorax was opened and the diaphragm and ribcage were removed and placed in a modified Krebs-Henseleit solution [95% O2/5% CO2 (pH 7.4), 5 mM KCl, 137 mM NaCl, 1.2 mM NaH2PO4, 1.2 mM MgSO4, 20 mM NaHCO3, 10 mM D-glucose, and 0.25 mM CaCl2 ] with 20 mM 2,3-butanedione monoxime (BDM) added to prevent muscle damage during dissection. All regents were purchased from Sigma Aldrich.
Two linear strips of muscle approximately 2-3 mm in width were carefully dissected from the center of each diaphragm so that one end of the muscle had rib attached. This allowed the diaphragm strip to be held in place, inside the muscle bath, by inserting the muscle through a stainless steel basket connected to a KG2 force transducer (World Precision Instruments). The central tendon on the interior end of the diaphragm strip was then pierced over a hook connected to a linear micro-manipulator. The muscle bath contained oxygenated Krebs-Henseleit solution as described above (without BDM and CaCl2 raised to 2.0 mM) at 37°C, since this is the temperature at which the in vivo diaphragm operates [14]. Single electrical stimulation pulses (180 Hz for 4 ms) were delivered via two parallel platinum-iridium electrodes surrounding the muscle to determine the “optimal length” for measuring maximum twitch force. The muscles were subsequently allowed to rest for 10 min, and then subjected to a series of tetanic contractions. Six tetanic contractions, one at each 20, 50, 80, 120, 150, and 180 Hz (250 ms duration each) were performed with 2 minutes of rest between stimulations. 5 minutes after the final tetanic measurement, a fatigue protocol (stimuli at a frequency of 100 Hz and 250 ms duration each second for a total of 66 seconds) was performed. After a 15 minute rest the protocol was repeated.
For comparative purposes, all force measurements are expressed per unit of cross-sectional area (CSA) (normalized isometric force or tension: mN/mm2). CSA was calculated using the following equation: muscle mass in mg/[(optimal length in mm) × (muscle density in mg/mm3)], where muscle density is assumed to be 1.056 mg/mm3. Mass was determined after all force measurements by blotting the muscle between two Kimwipes beneath a 10 g weight for 10 seconds [12].
The EDL muscle from each leg was carefully excised under a dissection microscope. Before removal, two pieces of suture were used to tie a knot on each tendon. The muscle was then removed and placed in the modified Krebs-Henseleit solution (with BDM) while 5– 8 additional knots were tied onto each tendon using the original suture ends described above. Sutures were tied around the tendons on each end of the muscle so that knots could be placed over two hooks (one attached to a force transducer, the other a linear micromanipulator) in a muscle bath through which oxygenated Krebs-Henseleit (without BDM and 2.0 mM CaCl2 added) at 30°C was continuously cycled. This multi-knot attachment method removes any attachment compliance and ensures that all length changes are directly reflecting changes in the muscle length. Muscles were stretched to optimal length using twitch contractions (evolked by a single 4 ms pulse) as previously described [12, 14]. After 10 minutes, a tetanus contraction was performed (150 Hz for 250 ms). After another 5 min rest period, 10 eccentric contractions (150 Hz for 450 ms, subjected to a 3% stretch for the final 200 ms of contraction) were done with two minutes of rest between stimulations. Following force measurements, the sutures were removed, and muscles were weighed as described above. Forces are expressed per unit of cross-sectional area (CSA).
Force recordings and analysis were done using custom-made LabView (National Instruments) programs. If more than one diaphragm strip or both EDL muscles were assessed from the same mouse, values were averaged for each muscle type and treated as n = 1 for statistical analyses [12].
Histopathology and quantitation
All histological assessments were performed by individuals blinded to genotypes and treatments of the mice. Each measurement was performed by the same individual to limit variability. From each mouse used for all of the above measurements, additional striated muscle tissues were embedded in optimal-cutting temperature (OCT) medium and frozen on liquid-nitrogen cooled isopentane for histological analyses. These samples included heart, quadriceps, rectus abdominis, and the remaining portions of the diaphragm muscle. 8μm sections were cut using Cryostat model OTF 5000 (Bright). One section from each tissue block was stained with Hematoxylin and Eosin (H&E), using standard methods to assess overall histopathology. An additional section from each tissue block was used for immunofluorescence experiments for analysis of ongoing muscle damage, with an antibody against mouse IgG, as previously described (Alexa 488 goat anti-mouse IgG, 1:200; Life Technologies) [4].
Wide-field images were taken on a Nikon Eclipse 800 microscope through a 10X objective using a SPOT RT slider digital camera and SPOT software. The percentage of damage in cardiac, quadriceps and abdominal muscle tissue was assessed on IgG stained sections using Photoshop CS6 (Adobe) similar to that described previously [12]. The photographs were quickly composited using the automation function “Photomerge.” For all composite images, the background and any holes within the sections were painted white using the paint-bucket tool. For IgG stained sections, damaged areas were painted blue using the paint-bucket tool. Normal IgG localization in the capillaries and endomysium was left unpainted. Painted sections were analyzed using the magic-wand tool and the histogram window. The magic wand tool was used to select all blue portions of the section and the number of selected pixels was shown in the histogram. The magic wand tool was then used to select all white portions of the section. The inverse function was used to switch the selection to the entire section including damage (everything that is not white), the total number of selected pixels was shown in the histogram. Damage is reported as a percentage of the entire section. Longitudinal sections were excluded from analysis.
Data and statistical analysis
All data was included for statistical calculations and analyzed using one-way ANOVA. If the overall ANOVA indicated statistical significance, the non-parametric Dunnett post-hoc test was used to test for significant differences to all other groups compared with the untreated het mice. The untreated het mice allow for the most unambiguous interpretation as they are the only group that differ in only 1 degree of freedom (genotype or dose) from all other groups. Summary values are presented as mean ± SE. Two-tailed P values < 0.05 were accepted as significant. The percentage of damage in quadriceps muscle tissue and the percentage of EDL specific force ratio data were also compared with 20 week-old male C57BL/10 wild-type control mice and untreated het mice.
RESULTS
Lisinopril treatment does not affect mouse body weight or grip strength
Body weight was not different between treatment groups (P = 0.1572; Fig. 1A). Forelimb total grip strength (P = 0.0001) and normalized forelimb grip strength (P < 0.0001) was significantly (Dunnett) higher only in C57BL/10 compared to untreated het mice (Fig. 1B and not shown). Treatment of het mice with lisinopril did not significantly improve total or normalized forelimb grip strength at any of the three dosages tested (10, 20, or 50 mg/kg × day [L10, L20, L50]) (Fig. 1B and not shown).
Diaphragm and extensor digitorum longus (EDL) muscle forces are not improved with lisinopril treatment
Only C57BL/10 mice showed a significant difference (P = 0.0009) in maximal in vitro specific contractile force in diaphragm muscle compared with the untreated het mice, suggesting lisinopril treatment has no beneficial effect on diaphragm function (Fig. 2A). Diaphragm specific forces were 196 ± 21 mN/mm2 for C57BL/10 compared with 118 ± 8 mN/mm2 for untreated hets and 122 ± 9 mN/mm2 for all of the lisinopril-treated mice combined. The highest lisinopril dosage only resulted in a diaphragm specific force of 128 ± 13 mN/mm2. EDL specific force (Fig. 2B) was statistically similar between all groups, including C57/BL10 control mice. C57BL/10 and untreated het mice had an average EDL specific force of 343 ± 23 and 301 ± 26 mN/mm2, respectively, compared to 343 ± 46 mN/mm2 for the het mice treated with the highest lisinopril dosage (L50).
Lisinopril treatment reduces muscle damage
Histopathological analysis was performed on quadriceps, diaphragm and abdominal muscles from all treatment groups. Hematoxylin and eosin (H&E) staining was used to examine the overall pathology of the muscle. Immunofluorescence staining for accumulation of serum proteins (IgG) both intracellularly and in fibrotic areas replacing muscle fibers provided a quantifiable measurement of damage. Representative H&E staining and IgG immunofluorescence for C57BL/10, untreated het, and het mice treated with the highest dosage of lisinopril (50 mg/kg × day) are shown (Figs. 3, 4A). Het mice treated with 50 mg/kg × day of lisinopril (L50) showed reduced overall pathology in quadriceps, diaphragm, and abdominal muscles, compared with untreated het mice (Fig. 3).
A similar pattern was also observed with IgG staining. The highest dosage lisinopril treatment group showed some abnormal IgG staining in all three muscle types analyzed, but was not as severe as observed in untreated het mice (Fig. 4A). To measure the effect of lisinopril on muscle damage in het mice, IgG localization within myofibers, replacing myofibers, and abnormal staining in endomysium, was quantified as a percentage of the total muscle section area for all three muscle types. As expected, C57BL/10 wild-type controls had very little or no muscle damage in any muscle type (Figs. 3, 4). ANOVA revealed a significant difference between groups in the amount of muscle damage in quadriceps (P = 0.00071) and diaphragm (P < 0.0001) muscles. The percentage of damage in quadriceps muscle was significantly lower in C57/BL10 mice (Dunnett, P < 0.0001), all lisinopril treated het mice (L10 + L20 + L50; P = 0.013) and mice treated with both 20 (L20; P = 0.031), or 50 mg/kg × day of lisinopril (L50; P = 0.0239) compared to untreated het mice (Fig. 4B). The highest lisinopril-treatment group had 9.1 ± 1.9% IgG staining in quadriceps muscles, compared to 20.3 ± 3.8% in the untreated het group and 0.7 ± 0.1% in the C57BL/10 control group. A similar reduction was seen in diaphragm muscle, for C57/BL10 control mice (P < 0.0001) and mice treated with 50 mg/kg × day of lisinopril (L50 mg) (P = 0.0494) compared to untreated mice (Fig. 4C). L50 lisinopril treated mice had 14.9 ± 0.8% IgG staining in diaphragm muscle compared to 19.3 ± 1.5% in the untreated het and 1.9 ± 0.4% in the C57BL/10 control group.
There was also a significant difference between groups for abdominal muscle damage (P = 0.00314). However, no significant reduction was observed between lisinopril treated mice (L20, P = 0.8862; L50, P = 0.1395) and untreated het mice although there was a trend towards improvement with lisinopril treatment with a reduction from 16.4 ± 3.1% in untreated to 9.3 ± 1.9% in the L50 group. Only C57BL/10 wild type and untreated het were significantly different for abdominal muscles (P = 0.0012). These results suggest lisinopril treatment leads to a limited reduction in abdominal muscle damage (Fig. 4D).
Heart rate, and blood pressure are unaffected by lisinopril treatment, but QT-interval is lower in lisinopril treated mice
Blood pressure recordings measured by tail cuff and heart rate measurements from electrocardiogram recordings, in conscious mice, showed no significant differences between any of the groups (P = 0.17 and 0.46 respectively, Table 1). However, QT-interval was significantly lower in total lisinopril treated het mice (P = 0.001), the two higher dosage lisinopril treatment groups (L20, P = 0.0146; and L50, P = 0.0025) and C57BL/10 (P = 0.027) mice compared to the untreated het mice (Table 1). The ratio between QT – interval:heart rate (HR) was also different (P = 0.019) between total lisinopril (P = 0.0049), L20 mg (P = 0.04) and L50 mg (P = 0.0235) treated mice compared to untreated het mice (Table 1). A summary of heart rate, mean arterial pressure, QT-interval and QT-interval:heart rate ratio is shown in Table 1.
Analysis at 20 weeks-of-age provides more informative outcome measures than 10 weeks-of-age for all compared parameters except ongoing quadriceps muscle damage
To determine whether 10 or 20 weeks-of-age is a better time point for analyzing treatment efficacy infuture preclinical studies, several parameters were compared between C57BL/10 and untreated het mice. Differences in EDL specific force measurements between untreated het and C57BL/10 mice at 20 weeks-of-age were more informative than at 10 weeks-of-age (Fig. 5A). In the current study, 20 week-old untreated het mice showed 59% of the specific EDL force observed in 20 week-old C57BL/10 controls, compared to 88% of wild-type force levels observed in 10 week-old untreated het mice compared to 10 week-old C57BL/10 mice. However, ongoing damage in quadriceps muscles measured by IgG staining is more informative at 10 weeks-of-age (20.3% of total muscle section area) than 20 weeks-of-age (4.4% of total muscle section area) in het untreated mice(Fig. 5B).
Importantly, analysis of cardiomyopathy is uninformative at 10 weeks-of-age in het mice. H&E and IgG staining of the entire left ventricles of hearts from 10 week-old untreated het mice did not show quantifiable damage compared to that previously observed and quantified in 20 week-old het mice (Fig. 5C, and not shown). Furthermore, cardiovascular MRI performed on untreated het mice and C57BL/10 wild-type control mice at 10 weeks-of-age showed no significant differences in peak systolic strain rate at the base of left ventricle between the 2 groups (P = 0.097, Fig. 5D), compared to previous differences observed at 20 weeks-of-age. These data suggest that cardiomyopathic changes are not yet detectable in 10 week-old het mice and preclinical studies to detect therapeutic effects on the heart will need to be carried out in 20 week or older het mice.
DISCUSSION
This study was designed with two goals. The first goal of this study was to determine whether previously observed preclinical benefits of the ACEi lisinopril plus the MR antagonist spironolactone on dystrophic skeletal muscles can be reproduced by increasing ACEi dosage alone. The second goal of this study was to determine whether comparison of utrn +/- ; mdx “het” mice with wild-type controls were informative at 10 weeks-of-age. We had previously only analyzed these mice at 20 weeks-of-age and found that physiological and histological outcome measures for both skeletal muscles and heart were informative at this time-point. However, since reducing the time-course of these experiments would have both time and cost benefits for future preclinical studies, we investigated a shorter experimental time-course. We show that both functional and quantifiable histological parameters to detect cardiomyopathy are not yet present at 10 weeks-of-age in het mice (Fig. 5), similar to previous studies in mdx mice [15]. The current study also shows that limb muscle force measurements are not as informative in 10 compared to 20 week-old mice (Fig. 5), but ongoing limb muscle damage is more informative at younger ages. The increased ongoing muscle damage is likely due to the activity of the mice and that the mice are still actively growing at this younger age. Therefore, 20 weeks-of-age is an appropriate early time-point to detect treatment effects on both skeletal and cardiac dystrophic muscle function and pathology.
Our previous studies have shown that combined lisinopril and mineralocorticoid receptor antagonist therapy shows efficacy on 20 week-old het mice in all physiological and histological parameters measured in limb, respiratory, and cardiac muscles [4]. Since both of these drugs are used routinely in heartfailure, our goal is to dissect the efficacy of these drugs on dystrophic skeletal muscles. The current study demonstrates that lisinopril, even at higher dosages, does not appear to show a similar efficacy on skeletal muscle function as assessed by grip strength or in vitro force measurements. A previous study showed that a similar ACE inhibitor, enalapril, at low dosages (1– 5 mg/kg × day) showed increases in forelimb grip strength in exercised mdx mice at similar ages to the mice used in the current study [3]. Another previously published study showed that enalapril treatment of both sedentary and exercised mdx mice improved gastrocnemius muscle force at 8 months of age [16]. However, neither of these studies assessed diaphragm function and we observe no improvement in diaphragm muscle function due to lisinopril treatment in our study, despite large differences between wild-type and untreated het forces.
One limitation of our current study was that significant differences in EDL force were not able to be detected between 10 week-old wild-type and het mice (Fig. 2). This data contrasts with some of our previous studies using young mdx mice and is likely due to improvements in our EDL measurement techniques. In previous studies we removed the EDL muscle before tying sutures to the tendons, but we have now switched to tying the sutures in situ before removing the muscles. This technical improvement leads to considerably less compliance in the preparation during the experiment, and to less muscle damage during dissection. Dystrophic muscles are probably more susceptible to dissection-induced damage, and thus may be weaker at the start of the protocol where sutures are not tied on the muscle in situ. However, this improved technique was also used in the current study to measure EDL force in 20 week-old het and wild-type mice (Fig. 5A) and was a more informative outcome measure at this later time point.
Another minor limitation is that the design of the lisinopril treatment groups as an escalation study to identify safety and benefits of increasing dosages of an ACE inhibitor resulted in small individual lisinopril groups of n = 6. For many parameters, all lisinopril dosages resulted in a similar efficacy, or lack of therapeutic effect, and we were able to analyze all lisinopril-treated mice as one n = 18 group. In other cases, the highest dosage was the most beneficial and was analyzed alone. Importantly, in all parameters, none of the dosages appeared to lead to any harm, including negative effects on blood pressure (Table 1).
Quantification of ongoing muscle damage in all three muscle types showed differences at 10 week-of-age. This contrast with some of the functional measurements likely represents less variability in histological than physiological measurements. For the first time, we included analysis of abdominal muscles in a test of a drug treatment in a DMD mouse model. Pathology of secondary respiratory muscles like diaphragm and abdominal muscles has been shown to positively correlate with pathology and dysfunction of the heart in a mouse model of limb girdle muscular dystrophy [17]. Our study demonstrates that these muscles show damage prior to measurable heart damage or dysfunction and are also an informative outcome measure in het mice. However, lisinopril treatment showed less of a therapeutic effect on respiratory muscles than limb muscles. Since respiratory muscles are crucial to therapeutically target in DMD patients, our future preclinical and mechanistic studies will further evaluate the efficacy of mineralocorticoid receptor antagonism on dystrophic skeletal muscles. Since previous studies have demonstrated that glucocorticoids demonstrate efficacy only at early stages of disease in dystrophic mice, new treatment strategies should be evaluated at 20 weeks-of-age and after one year-of-age to demonstrate continued efficacy [18].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank Anna Bratasz and Jessica Pyles of the Small Animal Imaging Core for conducting the MRI scans. This study was funded by NIH R01 HL116533 and DOD MD120063. Part of the EDL and diaphragm studies were supported by P30 core grant NINDS P30 NS045758.
REFERENCES
1 | Emery AE(2002) The muscular dystrophiesLancet.359: 687695 |
2 | Verhaert D, Richards K, Rafael-Fortney JA, Raman SV(2011) Cardiac involvement in patients with muscular dystrophies: Magnetic resonance imaging phenotype and genotypic considerationsCirc Cardiovasc Imaging4: 6776 |
3 | Cozzoli A, Nico B, Sblendorio VT, Capogrosso RF, Dinardo MM, Longo V, Gagliardi S, Montagnani M, De Luca A(2011) Enalapril treatmentdiscloses an early role of angiotensin II in inflammation- andoxidative stress-related muscle damage in dystrophic mdx micePharmacol Res64: 482492 |
4 | Rafael-Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfin DA, Janssen PM, Raman SV(2011) Early treatment withlisinopril and spironolactone preserves cardiac and skeletalmuscle in duchenne muscular dystrophy miceCirculation124: 582588 |
5 | Delyani JA, Rocha R, Cook CS, Tobert DS, Levin S, Roniker B, Workman DL, Sing YL, Whelihan B(2001) Eplerenone: A selective aldosterone receptor antagonist (SARA)Cardiovascular Drug Reviews19: 185200 |
6 | Tamargo J, Solini A, Ruilope LM(2014) Comparison of agents that affect aldosterone actionSeminars in Nephrology34: 285306 |
7 | Goemans N, Buyse G(2014) Current treatment and management of dystrophinopathiesCurrent Treatment Options in Neurology16: 287 |
8 | Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael-Fortney JA, Lowe J, Roble SL, Cripe LH(2015) Eplerenone for early cardiomyopathy inDuchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trialLancet Neurol14: 153161 |
9 | van Putten M, Kumar D, Hulsker M, Hoogaars WM, Plomp JJ, vanOpstal A, van Iterson M, Admiraal P, van Ommen GJ, t Hoen PA, Aartsma-Rus A(2012) Comparison of skeletal muscle pathology and motorfunction of dystrophin and utrophin deficient mouse strainsNeuromuscul Disord22: 406417 |
10 | Zhou L, Rafael-Fortney JA, Huang P, Zhao XS, Cheng G, Zhou X, Kaminski HJ, Liu L, Ransohoff RM(2008) Haploinsufficiency of utrophin gene worsens skeletal muscleinflammation and fibrosis in mdx miceJ Neurol Sci264: 106111 |
11 | Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE(1997) Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophyCell90: 717727 |
12 | Janssen PM, Murray JD, Schill KE, Rastogi N, Schultz EJ, Tran T, Raman SV, Rafael-Fortney JA(2014) Prednisolone attenuates improvementof cardiac and skeletal contractile function and histopathology bylisinopril and spironolactone in the mdx mouse model of Duchennemuscular dystrophyPLoS One9: e88360 |
13 | Elnakish MT, Schultz EJ, Gearinger RL, Saad NS, Rastogi N, Ahmed AA, Mohler PJ, Janssen PM(2015) Differential involvement of varioussources of reactive oxygen species in thyroxin-induced hemodynamicchanges and contractile dysfunction of the heart and diaphragmmusclesFree Radic Biol Med83: 252261 |
14 | Murray JD, Canan BD, Martin CD, Stangland JE, Rastogi N, Rafael-Fortney JA, Janssen PM(2012) The force-temperature relationship in healthy and dystrophic mouse diaphragm; implications for translational study designFront Physiol3: 422 |
15 | Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS(2004) Evolution of the mdx mouse cardiomyopathy: Physiological and morphological findingsNeuromuscul Disord14: 491496 |
16 | Morales MG, Cabrera D, Cespedes C, Vio CP, Vazquez Y, Brandan E, Cabello-Verrugio C(2013) Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2)Cell and Tissue Research353: 173187 |
17 | Gardner BB, Swaggart KA, Kim G, Watson S, McNally EMCardiac function in muscular dystrophy associates with abdominal muscle pathologyJournal of Neuromuscular Diseases2: 3949 |
18 | Sali A, Guerron AD, Gordish-Dressman H, Spurney CF, Iantorno M, Hoffman EP, Nagaraju K(2012) Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mousePLoS One7: e34204 |
Figures and Tables
Fig.1
Body weight and grip strength analysis. A) Body weight was consistent among all treatment groups. B) Forelimb normalized grip strength was significantly higher in C57BL/10 mice compared to untreated het mice. C: C57BL/10 wild-type control mice (n = 10); U: untreated het mice (n = 10); L: all 3 groups of lisinopril-treated het mice (n = 18); and L10, L20, L50 = groups of het mice treated with lisinopril at 3 different (10, 20, and 50 mg/kg × day) dosages (n = 6 per group). Data are shown as means ± SEM. *ANOVA followed by a Dunnett post-hoc test indicated a significantly higher value compared to the untreated het group, P≤0.0001.
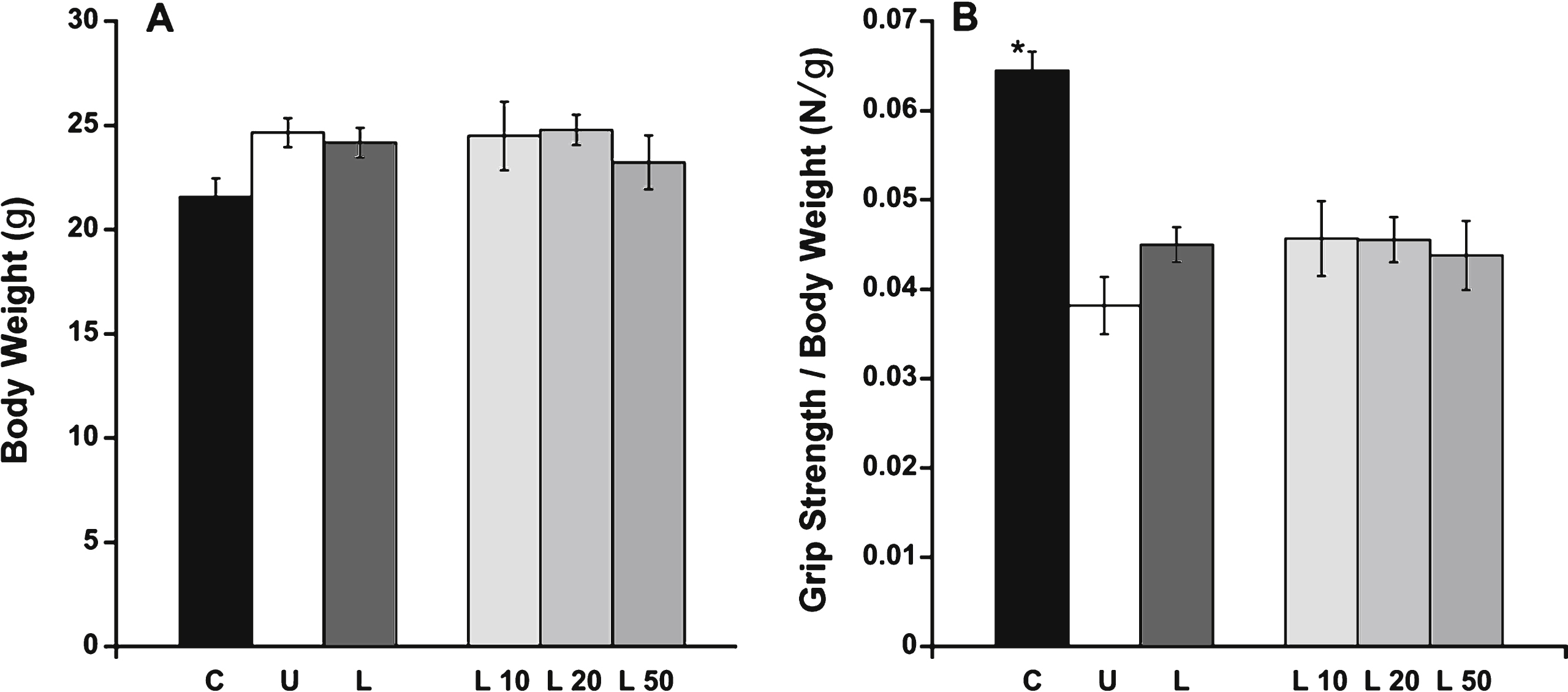
Fig.2
EDL and diaphragm force measurements. A) Normalized maximal contractile force in diaphragm muscle was only significantly higher in C57BL/10 compared with untreated het mice. B) EDL specific force showed no significant differences between the treatment groups. C: C57BL/10 wild-type control mice (n = 10); U: untreated het mice (n = 10); L: all 3 groups of lisinopril-treated het mice (n = 18); and L10, L20, L50 = groups of het mice treated with lisinopril at 3 different (10, 20, and 50 mg/kg × day) dosages (n = 6 per group). Data are shown as means ± SEM. *indicates a significantly higher value compared to the untreated het group, via ANOVA followed by a Dunnett post-hoc test, P = 0.0009.
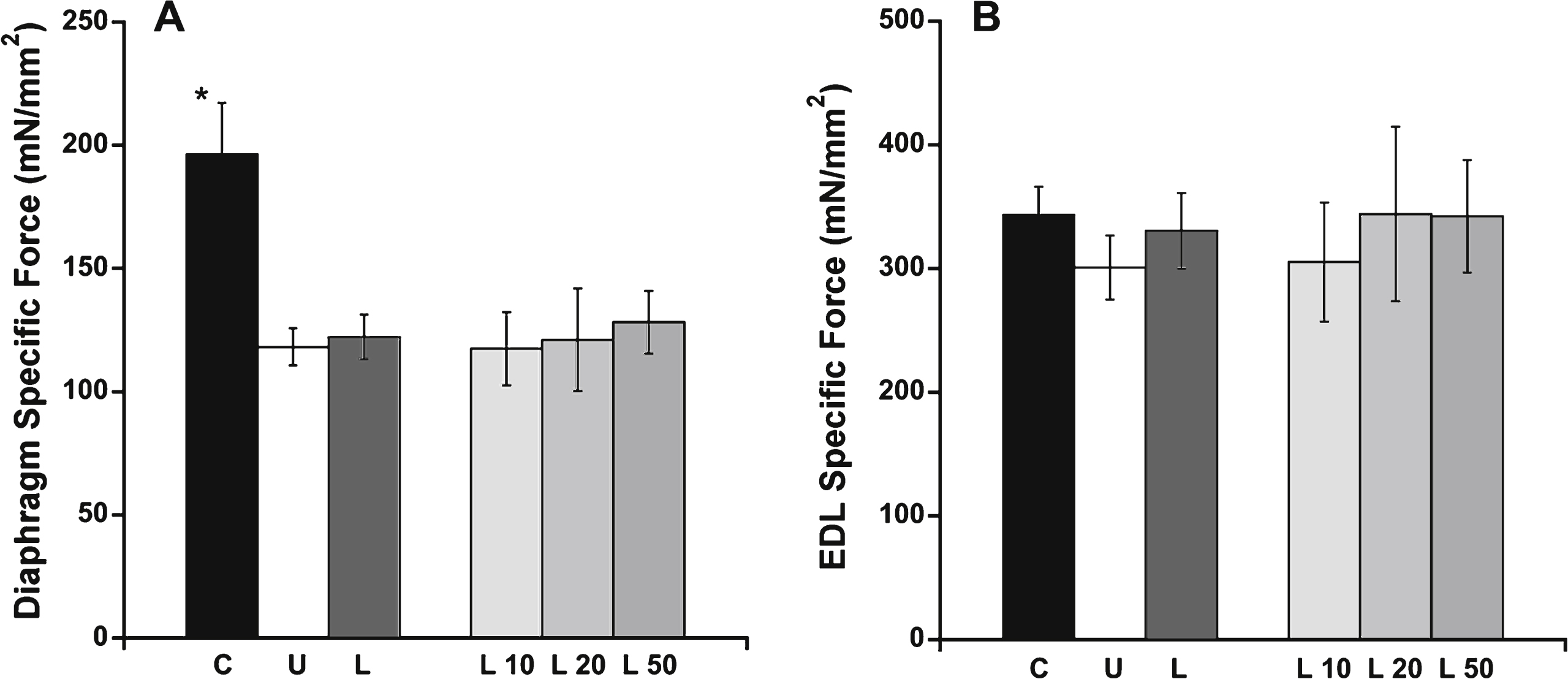
Fig.3
Hematoxylin and eosin stained representative quadriceps, diaphragm and abdominal sections. Het mice treated with 50 mg/kg × day of lisinopril had less observable overall muscle damage in quadriceps (Quad), diaphragm (Dia) and abdominal (Abd) sections compared to untreated het mice. As expected, C57BL/10 wild-type control mice had no observable muscle damage in any of the muscle types. C57: C57BL/10 wild-type control mice (n = 10); Het: untreated het mice (n = 10); L50: het mice treated with 50 mg/kg × day of lisinopril (n = 6). Bar = 100μm.
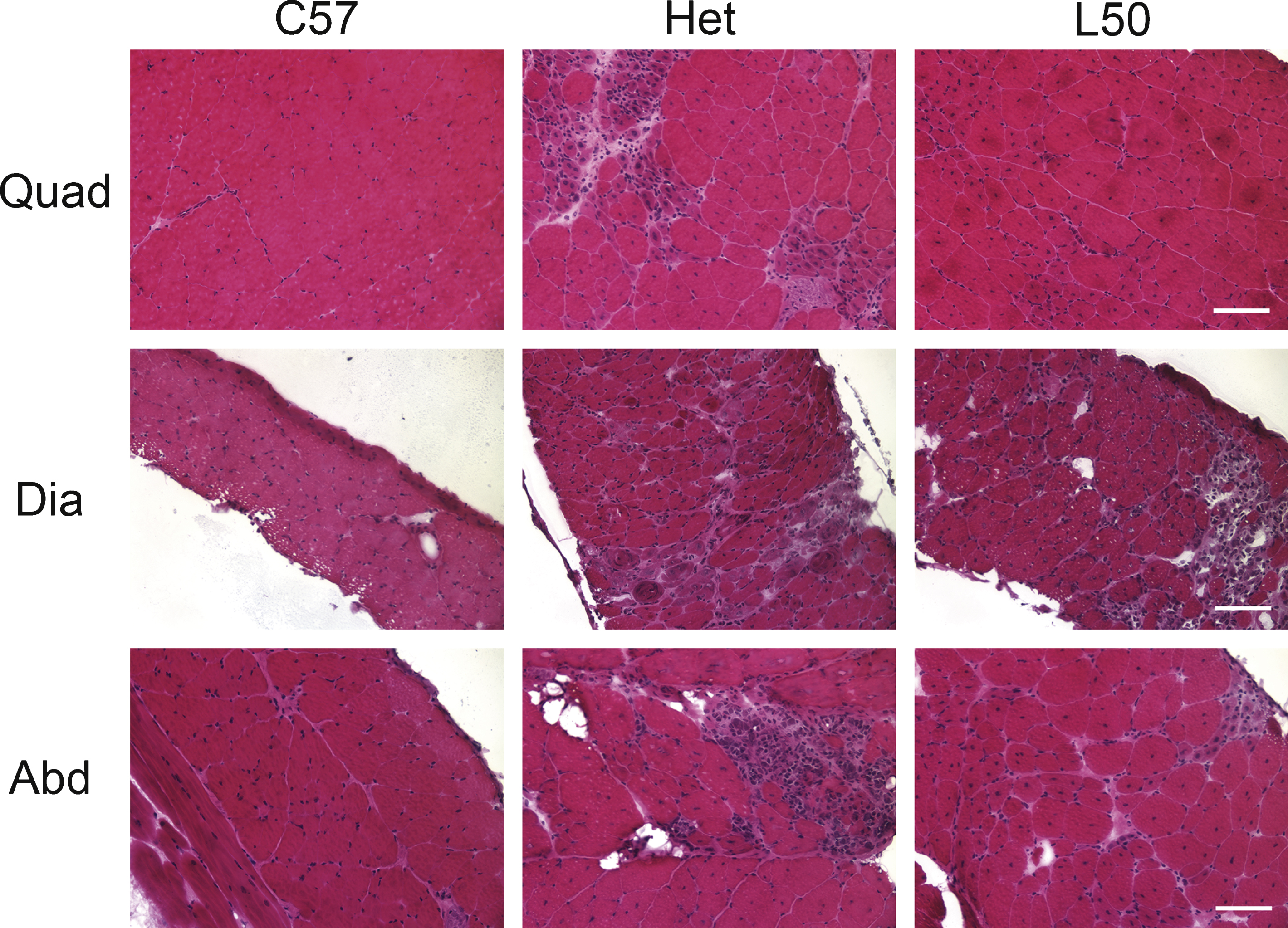
Fig.4
Quantification of muscle damage in quadriceps, diaphragm and abdominal sections of treated and untreated het mice. A) Immunofluorescence staining for serum IgG on representative quadriceps, diaphragm and abdominal sections. Mice treated with 50 mg/kg × day of lisinopril had substantially less ongoing muscle damage than untreated het mice. C57BL/10 wild-type control had no observable muscle damage. C57: C57BL/10 wild-type control mice (n = 10); Het: untreated het mice (n = 10); L50: het mice treated with 50 mg/kg × day of lisinopril (n = 6). Bar = 100μm. B) Damage in the quadriceps muscle, quantified by the percentage of IgG staining out of the entire muscle section area, was significantly lower in lisinopril treated mice overall (L), in the individual L20 and L50 groups, and C57BL/10 controls, compared to untreated het mice. C) Damage in diaphragm muscle quantified by IgG staining was reduced by the highest lisinopril dosage (L50) and in C57BL/10 controls, compared to untreated het mice. D) An overall reduction in abdominal damage was detected using ANOVA but a Dunnett post hoc test, comparing lisinopril treated to untreated mice, found no significant reduction. Only C57BL/10 compared to untreated hets were significantly reduced. However, the lower percentage of IgG staining in the highest dosage of lisinopril-treated compared to untreated hets, suggests a trend towards a reduction in ongoing damage in abdominal muscles. C: C57BL/10 wild-type control mice (n = 10); U: untreated het mice (n = 10); L: all 3 groups of lisinopril-treated het mice (n = 18); and L10, L20, L50 = groups of het mice treated with lisinopril at 3 different (10, 20, and 50 mg/kg × day) dosages (n = 6 per group). Data are shown as means ± SEM for B, C, and D. *indicates a significantly lower value compared to the untreated het group, as assessed by ANOVA followed by a Dunnett post-hoc test, P < 0.05.
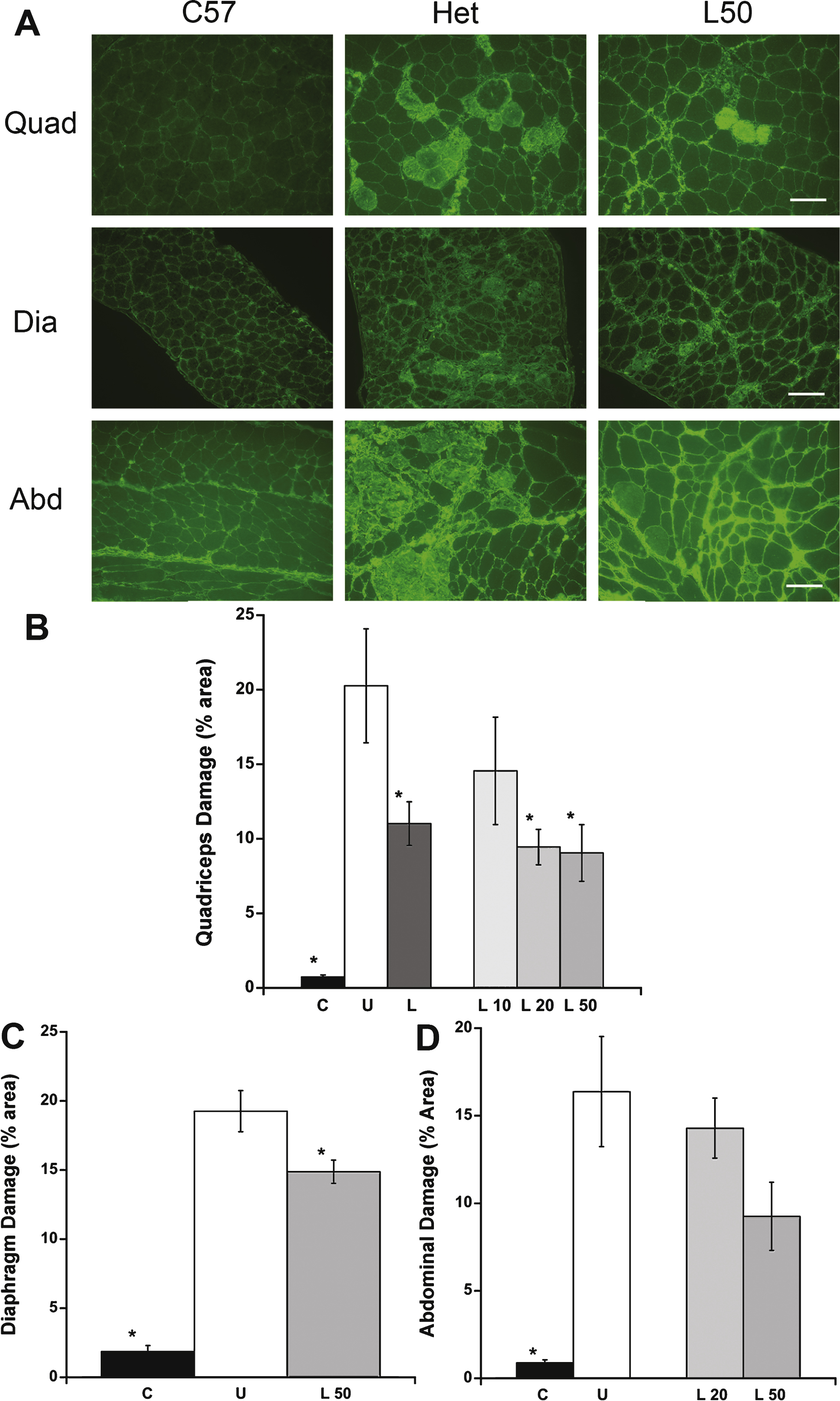
Fig.5
Outcome measure comparison between 10 and 20 week-old untreated het versus C57/BL10 mice. A) Differences in EDL specific force ratio between untreated het mice and C57 BL/10 wild-type control mice at 20 weeks-of-age were more informative than at 10 weeks-of-age. Wild-type levels were set as 100% ± SEM. B) Percentage of ongoing myofiber damage in untreated het quadriceps muscle was more informative at 10 weeks compared to 20 weeks-of-age. C) Hematoxylin and Eosin staining of heart left ventricular tissue showed muscle degeneration only at 20 weeks-of-age. Only very minimal damage can occasionally be observed in hearts from 10 week-old het mice and is not quantifiable since it represents a very small percentage of the total section area. The left panels show a transverse section through the entire left ventricle. The center and right panels (1,2,3,4) show enlarged views of the areas boxed in the left panels as labeled. Bars = 100μm. D) Myocardial peak systolic strain rate at the base of the left ventricle showed no significant differences between C57 BL/10 and het mice at 10 weeks-of-age. C-10 w: C57BL/10 wild-type control mice at 10 weeks-of-age (n = 10); U– 10 w: untreated het mice (n = 10) at 10 weeks-of-age; C-20 w: C57BL/10 mice at 20 weeks-of-age (n = 18); U– 20 w: untreated het mice 20 weeks-of-age (n = 17). Myocardial peak systolic strain rate was assessed on C-10 w (n = 7) and U– 10 w (n = 6). Data are shown as means ± SEM or percentage ± SEM.
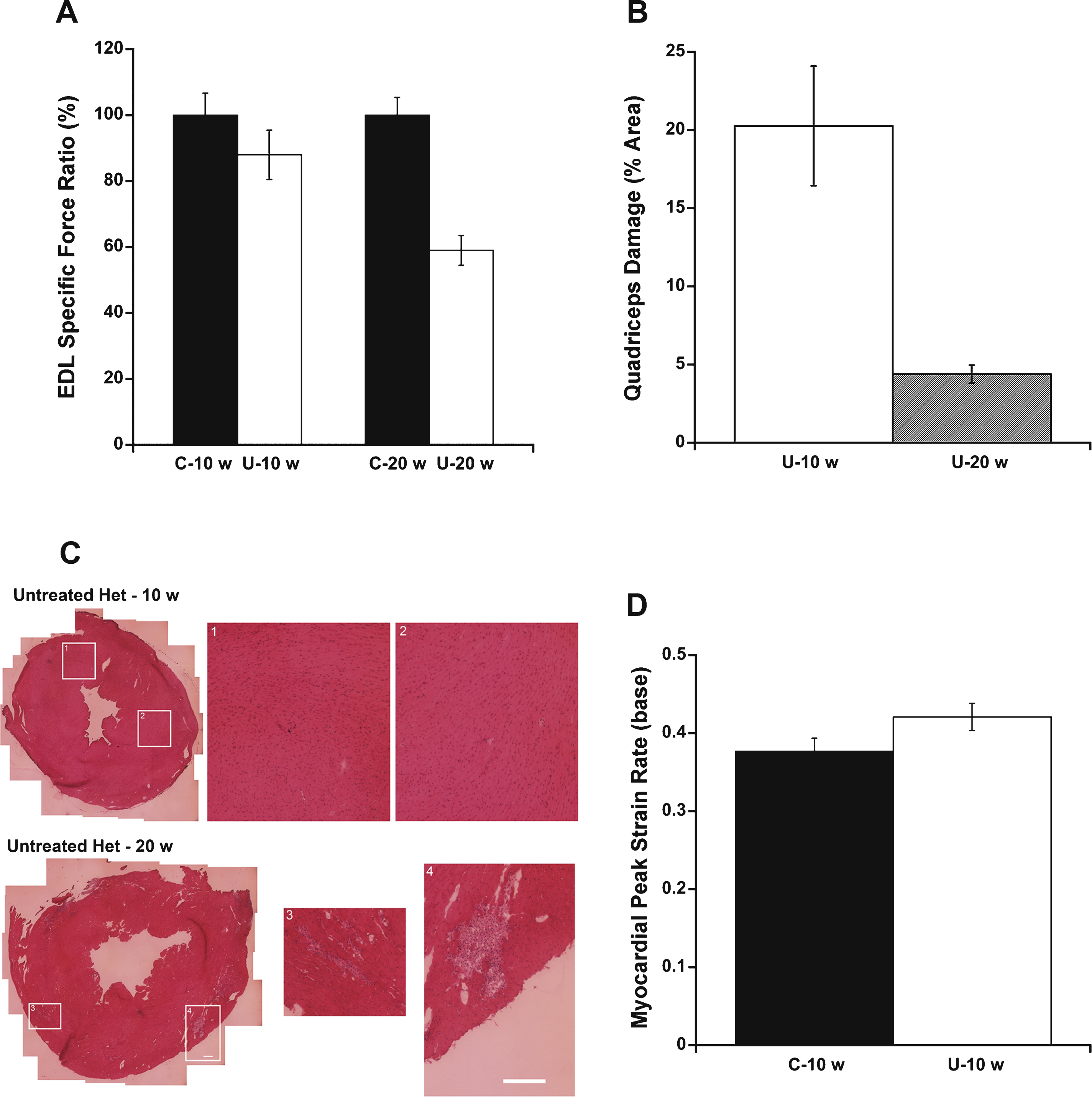
Table 1
ECG parameters. Heart rate (HR), mean arterial pressure (MAP), QT-interval and the ratio between QT-interval:heart rate were measured during conscious ECG recordings in all treatment groups. Only QT-interval and the ratio between QT-interval:heart rate was significantly different between the treatment groups. C57BL/10: wild-type control mice (n = 10); Untreated: untreated het mice (n = 10); Lisinopril: all 3 groups of lisinopril-treated het mice (n = 18); and L10, L20, L50 = lisinopril treated het mice at 3 different dosages (n = 6 per group)
| Treatment Group Abbreviation | n | HR (bpm) | MAP (mmHg) | QT (ms) | QT/HR (ms/bpm) |
| C57BL/10 | 10 | 686 ± 16 | 109 ± 4 | 43.6 ± 0.7 * | 0.06 ± 0.002 |
| Untreated | 10 | 678 ± 34 | 112 ± 6 | 47.5 ± 2.1 | 0.08 ± 0.009 |
| Lisinopril | 18 | 703 ± 11 | 107 ± 4 | 42.3 ± 0.8 * | 0.06 ± 0.002 * |
| L 10mg | 6 | 708 ± 22 | 109 ± 8 | 43.8 ± 1.3 | 0.06 ± 0.004 |
| L 20mg | 6 | 706 ± 17 | 120 ± 6 | 42.2 ± 1.4 * | 0.06 ± 0.004 * |
| L 50mg | 6 | 696 ± 21 | 96 ± 3 | 40.8 ± 1.6 * | 0.06 ± 0.002 * |
| ANOVA | P = 0.46 | P = 0.17 | P = 0.003 | P = 0.019 |
*indicates a significant difference compared to the untreated het group, via ANOVA followed by a Dunnett post-hoc test, P < 0.05. Data are shown as means ± SEM.


