
Congenital Myasthenic Syndromes with Predominant Limb Girdle Weakness
Abstract
Congenital myasthenic syndromes are a heterogeneous group of genetically determined disorders characterized by impaired neuromuscular transmission. They usually present from birth to childhood and are characterised by exercise induced weakness and fatigability. Genotype-phenotype correlations are difficult. However, in some patients particular phenotypic aspects may point towards a specific genetic defect.
The absence of ptosis and ophthalmoparesis in patients with limb-girdle weakness makes the diagnosis of a neuromuscular transmission defect particularly challenging (LG-CMS). This is illustrated by a well-documented case published by Walton in 1956. The diagnosis of LG-CMS is secured by demonstrating a neuromuscular transmission defect with single fibre EMG or repetitive nerve stimulation, in the absence of auto-antibodies. Ultimately, a genetic test is required to identify the underlying cause and assure counselling and optimization of treatment. LG-CMS are inherited in autosomal recessive traits, and are often associated with mutations in DOK7 and GFPT1, and less frequently with mutations in COLQ, ALG2, ALG14 and DPAGT.
Genetic characterization of CMS is of the upmost importance when choosing the adequate treatment. Some of the currently used drugs can either ameliorate or aggravate the symptoms depending on the underlying genetic defect. The drug most frequently used for the treatment of CMS is pyridostigmine an acetylcholinesterase inhibitor. However, pyridostigmine is not effective or is even detrimental in DOK7- and COLQ-related LG-CMS, while beta-adrenergic agonists (ephedrine, salbutamol) show some sustained benefit. Standard clinical trials may be difficult, but standardized follow-up of patients and international collaboration may help to improve the standards of care of these conditions.
INTRODUCTION
The congenital myasthenic syndromes (CMS) include a broad range of genetically determined conditions characterized by impaired neuromuscular transmission that results from mutations in several genes that code for constitutive proteins of the neuromuscular junction (NMJ).
The NMJ has three main compartments; the presynaptic terminal (demyelinated motor neuron with synaptic vesicles containing acetylcholine as a neurotransmitter), the synaptic cleft (20-30 nm space composed of basal lamina structured so that it allows the concentration and flux of the neurotransmitter towards the muscle) and the postsynaptic region (specialized region of the muscle cell plasma membrane were the neurotransmitter receptors are located) (Fig. 1).
Traditionally due to the structure of the NMJ the CMS were classified according to the location of the mutated protein as presynaptic, synaptic or postsynaptic (Table 1). In recent years new groups of genes, coding for ubiquitous proteins, were identified such as those involved in the glycosylation pathways (GFPT1, DPAGT1, ALG2, ALG14, and GMPPB) [1–4] or in the synthesis of propyl-oligopeptidase (PREPL) [5].
The most common genetic defects associated with CMS are mutations in the CHRNE gene resulting in primary acetylcholine receptor deficiency; other frequent defects are the CMS with mutations in DOK7 and COLQ and the CMS with mutations in the RAPSN gene [6–8].
Although CMS have been defined as clinical entities in the 1970 s, Walton published the first full clinical description of a patient resembling limb-girdle CMS well before that in 1956 [9]. Walton’s introduction to the case report states that “ ... in addition to cases which fall into recognizable categories of muscle disease, a number of less common disorders occur from time to time which do not correspond to the accepted descriptions. ... seem to fall into a borderland of either myopathy or myasthenia gravis.” The case report refers to a woman (Fig. 2) who developed muscular weakness and hypotonia in the first year of life. She had delayed motor development being able to walk only at the age of 5 years old. The ability to keep her head up remained poor and she developed marked proximal limb weakness. She experienced particular difficulties with raising her arms and climbing stairs; had a waddling gait and lumbar hyper-lordosis. The proximal muscle weakness became worse after exertion and tended to fluctuate during the day. The eye movements’ were normal. The muscle biopsy showed unspecific myopathic changes and an extensive investigation excluded a metabolic cause for the disease. The patient had an unsustained response to esterase inhibitors and a good response to ephedrine. She was re-evaluated in 1974 at the age of 54 years and she remained independent for most of her daily living activities. On examination her face was described as myopathic and it was noted that she had a “paralytic kyphoscoliosis”, severe bilateral ptosis and limitation of the upward gaze with fatigue of the elevation of the right eye. There was weakness of the neck muscles with wasting of the sternomastoid muscles. Slight wasting of the girdle muscles with marked proximal weakness was noted. Fifty years later, in 2006, the same phenotype was associated with CMS due to DOK7 mutations [10].
This review highlights the particular clinical features of LG – CMS to assist with the differential diagnosis.
CLINIC AND LABORATORY INVESTIGATIONS
The hallmark of CMS is fatigable weakness with markedly fluctuating severity of symptoms. Thedisease onset is usually from birth to the first 2 years of age although patients can present much later in life. When present from the neonatal period the most frequent manifestations relate to the cranial or respiratory muscles with poor suck and cry, choking spells, respiratory failure and ptosis. The symptoms are worsened by crying or activity. Later in life the limb muscles and spinal erector muscles are most affected with subsequent delayed motor milestones. These patients seldom learn how to run, experience difficulties with stairs, and have abnormal fatigability on exertion that does not allow them to keep with their peers. Ptosis, fixed or fluctuating ocular palsies, spinal deformities and reduced muscle bulk are other common manifestations. In contrast, eye muscle symptoms are usually subtle or completely lacking in LG-CMS.
The diagnosis is secured by demonstrating a neuromuscular transmission defect using single fibre EMG (SFEMG) or repetitive nerve stimulation (RNS). SFEMG is more sensitive but less specific than RNS. The usual findings are an increased jitter in SFEMG and a decrement larger than 10% on the amplitude of the compound muscle action potential (CMAP) on RNS. If a LG-CMS is suspected, then RNS should be carried out in proximal muscles otherwise the test may potentially be misleading as normal. It is also important to verify the absence of auto-antibodies against acetylcholine receptors (AChRs) and muscle-specific tyrosine kinase (MuSK). More recently other autoantibodies have been described in the so called seronegative Myasthenia Gravis, antibodies against the low-density lipoprotein receptor-related protein 4 (LRP4) or agrin. Except for some rare forms of CMS showing tubular aggregates, conventional muscle biopsy is inconspicuous. The presence of tubular aggregates in the muscle biopsy may help to distinguish between the LG-CMS that respond to acetylcholinesterase inhibitors (GFPT1, DPAGT1) from the ones that aggravate or do not respond to this medications (DOK7; COLQ).
Most CMS are autosomal recessive with the exception of the dominant slow channel syndrome and the SYT2 -related CMS. All LG-CMS so far are recessive. Support for a genetic condition comes from a positive family history of a NMJ disorder, consanguinity and the lack of response to immunosuppressive treatment.
Although most patients respond to treatment with acetylcholinesterase inhibitors, CMS due to mutations in COLQ, DOK7 and the slow channel syndromes may deteriorate with pyridostigmine.
Recently SNAP25B and LRP4 mutations have been described in anecdotal cases with complex CMSphenotypes. [11, 12]. Although the muscle weakness in these cases was described as generalised, it may have a proximal predominance.
CONGENITAL MYASTHENIC SYNDROMES WITH PREDOMINANT LIMB GIRDLE WEAKNESS (TABLE 2)
DOK 7 mutations
DOK7 is a postsynaptic cytoplasmic protein that binds and activates the muscle specific tyrosine kinase (MUSK) and induces Rapsyn-associated AChR clustering and folding of the postsynaptic membrane [13]. DOK7 mutations interfere with the AChR (acetylcholine receptors) clustering pathway [10].
CMS due to DOK7 mutations are the most common form of limb-girdle myasthenia [6–8]. It usually starts after the first year of life with limb girdle weakness however, in younger children, the presence of feeding difficulties and vocal cord paralysis with stridor may be a clue for the diagnosis even in the absence of limb muscle weakness. These children in spite of the LG weakness achieve normal motor milestones and start walking at a normal age.
Some patients may experience respiratory crisis with need for assisted ventilation and some may develop respiratory weakness with the need for regular non-invasive ventilation. Other common sign is the presence of ptosis without ophthalmoparesis. Some patients may have tongue wasting and some nonspecific myopathic features. Characteristically the disease fluctuations do not occur on a daily basis but go on over weeks. Patients with DOK7 mutations do not experience long term benefit from acetylcholinesterase inhibitors and can even exacerbate with these drugs. They have a good response to salbutamolor ephedrine.
Neurophysiology (SFEMG and RNS) is characteristic of a postsynaptic involvement. Due to the major involvement of the proximal muscles, attention should be made that these muscles are tested.
Mutations in DOK7 affect both the size and structure of the neuromuscular junction [10]. The majority of the mutations are located in exon 7. The most common mutation is a frameshift mutation c.1124_1127dupTGCC (p.Ala378SerfsTer30) in particular in patients with central/western European ancestry. The mutation S45L is more frequently found in patients of Portuguese/Spanish origin [14].
GFPT1 and DPAGT1 mutations
These 2 autosomal recessive syndromes result from defects in genes involved in glycosylation. They give rise to CMS with limb-girdle weakness and some show tubular aggregates in the muscle biopsy. These patients do not experience ptosis or ophthalmoparesis and benefit from esterase inhibitors. CK levels may be slightly raised.
GFPT1 mutations are usually associated with slowly progressive LG muscles weakness that may begin from infancy to adulthood. These patients usually don’t experience weakness of the respiratory or bulbar muscle and they also do not have ocular or facial weakness. RNS shows a decrement of the CMAP at 3 Hz stimulation and SFEMG will show abnormal jitter compatible with a post-synaptic defect of the NMJ. To increase the accuracy, these tests should be performed in proximal muscles.
GFPT1 or glutamine-fructose-6-phosphate transaminase 1 is the rate-limiting enzyme in the biosynthesis of N-acetylglucosamine, which is essential for both O- and N-glycosylation. GFPT1 has a muscle specific isoform GFPTL1 and mutations that hamper this specific isoform although rare usually manifest with a very severe phenotype resulting both in a myopathy with autophagy and in severe impairment of the NMJ [15, 16]. However, GFPT1 mutations can occur along the entire coding sequence of the gene. Most are missense mutations and a few truncatingmutations.
UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase 1 (DPAGT1), like GFPT1, is also involved in the N-glycosilation pathway. It promotes the first step of the dolichol-linked oligosaccharide pathway for the glycoprotein biosynthesis. Like with GFPT1, mutations in DPAGT1 are responsible for a distinct form of CMS with predominant involvement of the proximal muscle of the limbs and the axial muscles. The facial, ocular, bulbar or respiratory muscles are usually not involved. The onset is usually during childhood and patients show a good response to pyridostigmine. The EMG with SFEMG and RNS shows features of a post-synaptic involvement. These patients may also have tubular aggregates in the musclebiopsy.
Most of the patients reported in literature, with CMS due to DPAGT1 mutations also have some degree of cognitive impairment that goes from mild learning difficulties to autistic spectrum disorder and this can also be seen in GFPT1-CMS patients [17, 18].
Some studies [19] have shown that the possible primary defect at the NMJ level, in both DPAGT1 and GFPT1 CMS, is a loss in the number of AChR due to deficient glycosylation of AChR subunits.
COLQ mutations
Homozygous or compound heterozygous mutations in the acetylcholinesterase collagen-like tail subunit gene (COLQ) cause synaptic basal-lamina CMS with end-plate AChE deficiency [20]. The absence of AChE does not allow for the acetylcholine to be removed from the synaptic cleft so the end-plate current outlasts the muscle refractory period and a second compound muscle action potential is generated. At the same time the prolonged end-plat currents induce an end-plate myopathy with loss of AChRs.
The classical phenotype of COLQ mutations is that of a severely affected patient, starting early in infancy with respiratory insufficiency, motor development delay and severe scoliosis. EMG with double CMAP, slow pupillary reflexes, no response or deterioration with pyridostigmine are useful clues for the diagnosis [21].
These patients may have day-time fluctuation of the symptoms and also long term fluctuation lasting weeks to months. Some patients may experience increased fatigability around puberty and during the last months of pregnancy and early post-partum period [22].
A cohort of 22 patients published in 2008 [20] points out that a broader spectrum of clinical features should be considered. Although muscle weakness is usually generalized, some patients present with LG weakness suggestive of either a myopathy or LG-CMS. A subgroup of COLQ-mutated patients has prominent proximal muscle weakness, waddling gait and muscle atrophies similar to DOK7-mutated patients.
Other frequent manifestations found in this cohort are facial weakness, impairment of ocular movements and ptosis. Delayed pupillary light response although typical of the condition was present in only a quarter of the patients.
A decremental response on RNS may be elicited in the majority of the patients. The same is true for one of the hallmarks of the disease the double CMAP as response to single nerve stimulus. Double CMAPs are also found in slow channel CMS.
Treatment with esterase inhibitors is not advisable in these patients due to the lack of response or clinical deterioration, but some patients may show an initial, transitory benefit. These patients usually respond well to ephedrine and 3, 4-diaminopyridine.
ALG2, ALG14 and GMPPB mutations
ALG2, ALG14 and GMPPB defects are the cause of extremely rare forms of autosomal recessive LG-CMS, and are therefore clinically less well defined [3]. They have also been found in more severe childhood conditions such as CDG syndromes and congenital muscular dystrophy. ALG14 forms a complex with ALG13 and DPAGT1 constituting the UDP-GlcNAc transferase that catalyses the first two steps in endoplasmic reticulum N-linked glycosylation. ALG14 is localized in the endoplasmic reticulum membrane and at the endplate region of the NMJ.
ALG14-mutated patients have slowly progressive and fatigable, proximal muscle weakness present from childhood. No facial, ocular or bulbar muscle impairment was described although strabismus was reported. The unspecific clinical findings make the differential diagnosis with other LG-CMS and with limb girdle muscular dystrophies challenging. The ancillary diagnostic tests and therapeutic response to pyridostigmine are useful tools for this. Muscle biopsies do not show tubular aggregates and have unspecific changes. RNS shows a decremental response and SFEMG shows increased jitter. There is a good response to pyridostigmine that is sustained over time.
In ALG2-mutated patients [23] the course may be slowly progressive from early infancy. Patients have delayed motor milestones and hypotonia and most of them never achieve ambulation. Muscle weakness is predominantly proximal and mild facial weakness may be present. There is no involvement of the ocular or bulbar muscles. CK levels may be normal and tubular aggregates were found in some of the few reported muscle biopsies.
GDP-mannose pyrophosphorylase B (GMPPB) cat-alyses the formation of GDP-mannose required for the glycosylation of lipids and proteins. Mutations in GMPPB are responsible for a secondary dystroglycanopathy [24]. The usual clinical phenotype extends from severe congenital muscular dystrophy [25] with or without brain abnormalities to limb girdle muscular dystrophy (LGMD2T) or episodes of rhabdomyolysis. Recently mutations in the GMPPB gene have been identified in 7 patients with CMS [4]. These patients share clinical features of other CMS associated with glycosylation defects; proximal fatigable muscle weakness with sparing of facial, eye and bulbar muscles. In these 7 cases ptosis was only present in one and the age of onset as in the case of the muscular dystrophies was quite variable from childhood to adulthood. The EMG was clearly myopathic with associated decrement of CMAP with RNS of the proximal muscles. CK levels were raised and the muscle biopsy in 2 of the patients revealed dystrophic features with necrosis, centralized nuclei and type 1 fibre predominance. Most of the patients responded to pyridostigmine in isolation or in combination with salbutamol.
ANIMAL MODELS AND PATHOPHYSIOLOGY
Zebrafish are useful to examine the NMJ disruption in vivo. As vertebrates they share genetic similarity and functional homology with mammals. They are easier to care and to bread compared to rodents. Genetic changes are easily introduced and the effects can be monitored as the embryo is transparent and has external embryonic development. Although the muscle structure of the zebrafish is different from mammals the NMJ development occurs in a similar way. First a band of AChR forms in the middle region of the muscle; subsequently the axonal growth cone contacts the muscle and forms the synapse. Knockdown of neuromuscular junction proteins in zebrafish usually affects motility and swimming behaviour of the injectedembryos.
Two good examples of defect neuromuscular transmission in LG-CMS are morpholino-knockdown experiments for DOK7 and GFPT1 in zebrafish.
DOK7 is crucial in the very early stages of the NMJ formation (clustering of the AChR in the middle of the muscle fibre prior to the axonal contact). The contact of the axonal growth cone with the muscle, in morphant zebrafish with down-regulated expression of DOK7 is established; however these contacts are smaller than in the wild-type zebrafish. This aspect mimics the endplate pathology seen in patients with DOK7 mutations. There are also changes in slow muscle fibre arrangement. Due to this finding promotion of slow muscle fibres was suggested as an additional role for Dok7 [27].
GFPT1 is conserved in zebrafish. It was possible to knock-down GFPT1 and confirm that it is required for the normal NMJ formation. The knock-down of the zebrafish GFPT1 induces a reduction of GFPT1 levels at 48 hrs. post-fertilization and the embryos display altered tail morphology (Fig. 3), and impairment of swimming and touch-evoked escape response. Histologically there is abnormal muscle morphology and delayed NMJ development. Motor neuron axons lose their ability to project branches into laterally located muscle fibers and form synapses [28].
Mice are valid models to study the role of proteins in synapse formation and maintenance, to study CMS pathophysiology due to specific mutations and to understand the effects of certain drugs in the NMJ. However, the complete inactivation of NMJ genes in knockout mice is often lethal during embryonal development or shortly after birth. Interestingly, mice do not upregulate the foetal subunit of the AChR encoded by the CHRNG gene in the same way as humans do, when the adult subunit (CHRNE) is mutated [29]. Therefore it is necessary to overexpress human CHNRG in transgenic mice with a CHNRE background to model human receptor deficiency.
Several mouse models have been developed to study different subtypes of CMS including LG-CMS. A mouse model of AGRIN-associated CMS was developed by chemical mutagenesis. The affected mice (nmf380) manifested symptoms of NMJ dysfunction and whole-mount NMJ studies showed simplified post-synaptic regions and axonal sprouting beyond the post-synaptic region [30]. Other mousse models were developed to study CMS due to MuSK mutations [31], slow channel syndromes [32, 33] or DOK7 associated CMS [13]. Mice lacking Dok-7 do not form AChR clusters or NMJs, and die shortly after birth due to breathing difficulties.
CONCLUSION
The clinical heterogeneity of CMS and the difficulty in establishing correlations between genotypes and phenotypes often delay the clinical diagnosis. LG-CMS presentations are particularly challenging, as some of the hallmarks of CMS are frequently absent. Like in the case described in 1956 by Walton, the combination of LG weakness with myasthenic findings is the best clue for the diagnosis of LG-CMS. In undiagnosed patients with congenital myopathies or limb-girdle muscular dystrophies it is important to consider LG-CMS in the differential diagnosis, as, effective pharmacological therapies are already available for LG-CMS, but not for the dystrophies. However, we would like to stress that single cases of congenital myopathies may be treatable with pyridostigmine, e.g. some myotubular or centronuclear myopathies and RYR1-related myopathies with demonstrated aberrant NMJ transmission [34, 35]
ACKNOWLEDGMENTS
Work by the authors has been supported by the TREAT-NMD Alliance, the EU funded projects Neuromics (No. 305121) and RD-Connect (No. 305444) and the Medical Research Council (MRC). The contributions by patients, their families and referring clinicians are acknowledged. We also thank Yasmin Issop and Juliane Müller for help with theillustrations.
REFERENCES
1 | Senderek J, Müller JS, Dusl M, Strom TM, Guergueltcheva V, Diepolder I, Laval SH, Maxwell S, Cossins J, Krause S, Muelas N, Vilchez JJ, Colomer J, Mallebrera CJ, Nascimento A, Nafissi S, Kariminejad A, Nilipour Y, Bozorgmehr B, Najmabadi H, Rodolico C, Sieb JP, Steinlein OK, Schlotter B, Schoser B, Kirschner J, Herrmann R, Voit T, Oldfors A, Lindbergh C, Urtizberea A, von der Hagen M, Hübner A, Palace J, Bushby K, Straub V, BeesonD , Abicht A, Lochmüller H(2011) Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defectAm J Hum Genet88: 162172 |
2 | Belaya K, Finlayson S, Slater CR, Cossins J, Liu WW, Maxwell S, McGowan SJ, Maslau S, Twigg SR, Walls TJ, PascualPascual SI, Palace J, Beeson D(2012) Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome withtubular aggregatesAm J Hum Genet91: 1193201 |
3 | Cossins J, Belaya K, Hicks D, Salih MA, Finlayson S, Carboni N, Liu WW, Maxwell S, Zoltowska K, Farsani GT, Laval S, Seidhamed MZ(2013) Congenital myasthenic syndromes due tomutations in ALG2 and ALG14Brain136: Pt 3944956 |
4 | Belaya K, Rodríguez Cruz PM, Liu WW, Maxwell S, McGowan S, Farrugia M, Petty R, Walls TJ, Sedghi M, Basiri K, Yue W, Sarkozy A, Bertoli M, Pitt M, Kennett R, Schaefer A, Bushby K, Parton M, Lochmüller H, Palace J, Muntoni F, Beeson D(2015) Mutations in GMPPB cause congenital myasthenic syndrome and bridgemyasthenic disorders with dystroglycanopathiesBrainJun 30. pii: awv185. [Epub ahead of print] PMID: 26133662 |
5 | Régal L, Shen XM, Selcen D, Verhille C, Meulemans S, Creemers JW, Engel AG(2014) PREPL deficiency with or without cystinuria causes a novel myasthenic syndromeNeurology82: 1412541260 |
6 | Abicht A, Dusl M, Gallenmüller C, Guergueltcheva V, Schara U, Della Marina A, Wibbeler E, Almaras S, Mihaylova V, von der Hagen M, Huebner A, Chaouch A, Müller JS, Lochmüller H(2012) Congenital myasthenicsyndromes: Achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: Astudy of 680 patientsHum Mutat33: 1014741484 |
7 | Finlayson S, Beeson D, Palace (2013) Congenital myasthenic syndromes: An updatePract Neurol13: 28091 |
8 | Engel AG, Shen XM, Selcen D, Sine SM(2015) Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatmentLancet Neurol14: 4420434 |
9 | Geschwind N, Simpson JA, Walton JN(1956) Benign congenital myopathy with myasthenic featuresJ Neurol NeurosurgPsychiatry19: 3224231 |
10 | Beeson D, Higuchi O, Palace J, Cossins J, Spearman H, Maxwell S, Newsom-Davis J, Burke G, Fawcett P, Motomura M, Muller JS, Lochmuller H, Slater C, Vincent A, Yamanashi Y(2006) Dok-7 mutations underlie a neuromuscular junction synaptopathyScience313: 19751978 |
11 | Shen XM, Selcen D, Brengman J, Engel AG(2014) Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia,and intellectual disabilityNeurology83: 2422472255 |
12 | Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, Ito M, Masuda A, Ito Y, Engel AG, Ohno K(2014) LRP4third β-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSKsignaling in a position-specific mannerHum Mol Genet23: 718561868 |
13 | Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y(2006) The Muscle Protein Dok-7 Is Essential for Neuromuscular SynaptogenesisScience312: 18021805 |
14 | Müller JS, Herczegfalvi A, Vilchez JJ, Colomer J, Bachinski LL, Mihaylova V, Santos M, Schara U, Deschauer M, Shevell M, Poulin C, Dias A, Soudo A, Hietala M, Aärimaa T, Krahe R, Karcagi V, Huebner A, Beeson D, Abicht A, Lochmüller H(2007) Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromesBrain130: Pt 614971506 |
15 | Zoltowska K, Webster R, Finlayson S, Maxwell S, Cossins J, Müller J, Lochmüller H, Beeson D(2013) Mutations in GFPT1 that underlie limb-girdle congenital myasthenic syndrome result in reduced cell-surface expression of muscle AChRHum Mol Genet22: 1429052913 |
16 | Selcen D, Shen XM, Milone M, Brengman J, Ohno K, Deymeer F, Finkel R, Rowin J, Engel AG(2013) GFPT1-myasthenia:Clinical, structural, and electrophysiologic heterogeneityNeurology81: 4370378 |
17 | Klein A, Robb S, Rushing E, Liu WW, Belaya K, Beeson D(2015) Congenital myasthenic syndrome caused by mutations inDPAGTNeuromuscul Disord25: 3253256 |
18 | Selcen D, Shen XM, Brengman J, Li Y, Stans AA, Wieben E, Engel AG(2014) DPAGT1 myasthenia and myopathy: Genetic, phenotypic, and expression studiesNeurology82: 2018221830 |
19 | Belaya K, Finlayson S, Cossins J, Liu WW, Maxwell S, Palace J, Beeson D(1275) Identification of DPAGT1 as a new genein which mutations cause a congenital myasthenic syndromeAnn N Y Acad Sci2935 |
20 | Mihaylova V, Müller JS, Vilchez JJ, Salih MA, Kabiraj MM, D’Amico A, Bertini E, Wölfle J, Schreiner F, Kurlemann G, Rasic VM, Siskova D, Colomer J, Herczegfalvi A, Fabriciova K, Weschke B, Scola R, Hoellen F, Schara U, Abicht A, Lochmüller H(2008) Clinical and molecular genetic findings in COLQ-mutant congenital myasthenicsyndromesBrain131: Pt 3747759 |
21 | Engel AG, Ohno K, Sine SM(2003) Sleuthing molecular targets for neurological diseases at the neuromuscular junctionNat Rev Neurosci4: 339352 |
22 | Wargon I, Richard P, Kuntzer T, Sternberg D, Nafissi S, Gaudon K, Lebail A, Bauche S, Hantaï D, Fournier E, Eymard B, Stojkovic T(2012) Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQmutationsNeuromuscul Disord22: 4318324 |
23 | Monies DM, Al-Hindi HN, Al-Muhaizea MA, Jaroudi DJ, Al-Younes B, Naim EA, Wakil SM, Meyer BF, Bohlega S(2014) Clinical and pathological heterogeneity of a congenital disorder of glycosylationmanifesting as a myasthenic/myopathic syndromeNeuromusc. Disord.24: 353359 |
24 | Maeda Y, Kinoshita T(1780) Dolichol-phosphate mannose synthase: Structure, function and regulationBiochim Biophys Acta861868 |
25 | Carss KJ, Stevens E, Foley AR, Cirak S, Riemersma M, Torelli S(2013) Mutations in GDP-mannose pyrophosphorylaseB cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycanAm J Hum Genet93: 2941 |
26 | Cabrera-Serrano M, Ghaoui R, Ravenscroft G, Johnsen RD, Davis MR, Corbett A, Reddel S, Sue CM, Liang C, Waddell LB, Kaur S, Lek M, North KN, MacArthur DG, Lamont PJ, Clarke NF, Laing NG(2015) Expanding the phenotype of GMPPB mutationsBRAIN29 |
27 | Müller JS, Jepson CD, Laval SH, Bushby K, Straub V, Lochmüller (2010) Dok-7 promotes slow muscle integrity as well as neuromuscular junction formation in a zebrafish model of congenital myasthenic syndromesH Hum Mol Genet19: 917261740 |
28 | Senderek J, Müller JS, Dusl M, Strom TM, Guergueltcheva V, Diepolder I, Laval SH, Maxwell S, Cossins J, Krause S, Muelas N, Vilchez JJ, Colomer J, Jimenez Mallebrera C, Nascimento A, Nafissi S, Kariminejad A, NilipourY , Bozorgmehr B, Najmabadi H, Rodolico C, Sieb JP, Steinlein OK, Schlotter B, Schoser B, Kirschner J, Herrmann R, Voit T, Oldfors A, Lindbergh C, Urtizberea A, von der Hagen M, Hübner A, Palace J, Bushby K, Straub V, Beeson D, Abicht A, Lochmüller H(2011) Hexosamine Biosynthetic Pathway Mutations Cause Neuromuscular TransmissionDefectAm J Hum Genet88: 2162172 |
29 | Cossins J, Webster R, Maxwell S, Burke G, Vincent A, Beeson D(2004) A mouse model of AChR deficiency syndrome with a phenotype reflecting the human conditionHum Mol Genet13: 2329472957 |
30 | Bogdanik LP, Burgess RW(2011) A valid mouse model of Agrin-associated congenital myasthenic syndromeHum Mol Genet20: 2346174633 |
31 | Chevessier F, Girard E, Molgó J, Bartling S, Koenig J, Hantaï D, Witzemann V(2008) Hum Mol Genet17: 2235773595 |
32 | Webster RG, Cossins J, Lashley D, Maxwell S, Liu WW, Wickens JR, Martinez-Martinez P, de Baets M, Beeson D(2013) A mouse model of the slow channel myasthenic syndrome: Neuromuscular physiology and effects of ephedrine treatmentExp Neurol248: 286298 |
33 | Chevessier F, Peter C, Mersdorf U, Girard E, Krejci E, McArdle JJ, Witzemann V(2012) A new mouse model for theslow-channel congenital myasthenic syndrome induced by the AChR ɛL221F mutationNeurobiol Dis45: 3851861 |
34 | Illingworth MA, Main M, Pitt M, Feng L, Sewry CA, Gunny R, Vorstman E, Beeson D, Manzur A, Muntoni F, Robb SA(2014) RYR1-related congenital myopathy with fatigable weakness, responding to pyridostigimineNeuromuscul Disord24: 8707712 |
35 | Robb SA, Sewry CA, Dowling JJ, Feng L, Cullup T, Lillis S, Abbs S, Lees MM, Laporte J, Manzur AY, Knight RK, Mills KR, Pike MG, Kress W, Beeson D, Jungbluth H, Pitt MC, Muntoni F(2011) Impaired neuromuscular transmission andresponse to acetylcholinesterase inhibitors in centronuclear myopathiesNeuromuscul Disord21: 6379386 |
Figures and Tables
Fig.1
NMJs of the mouse extensor digitorum longus muscle. Motor axons and pre-synaptic region labelled with anti-neurofilament antibody; post-synaptic region labelled with anti-AChR antibody. Synaptic vesicle membrane proteins labelled with anti-synaptophysin antibody.
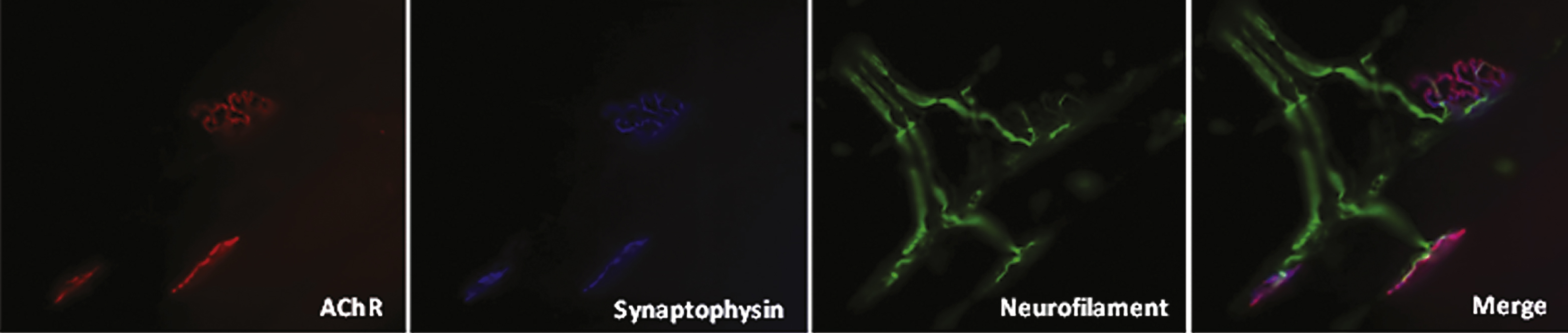
Fig.2
Bilateral ptosis and moderate atrophy of the shoulder girdle and arm muscles (figure reprinted with permission).
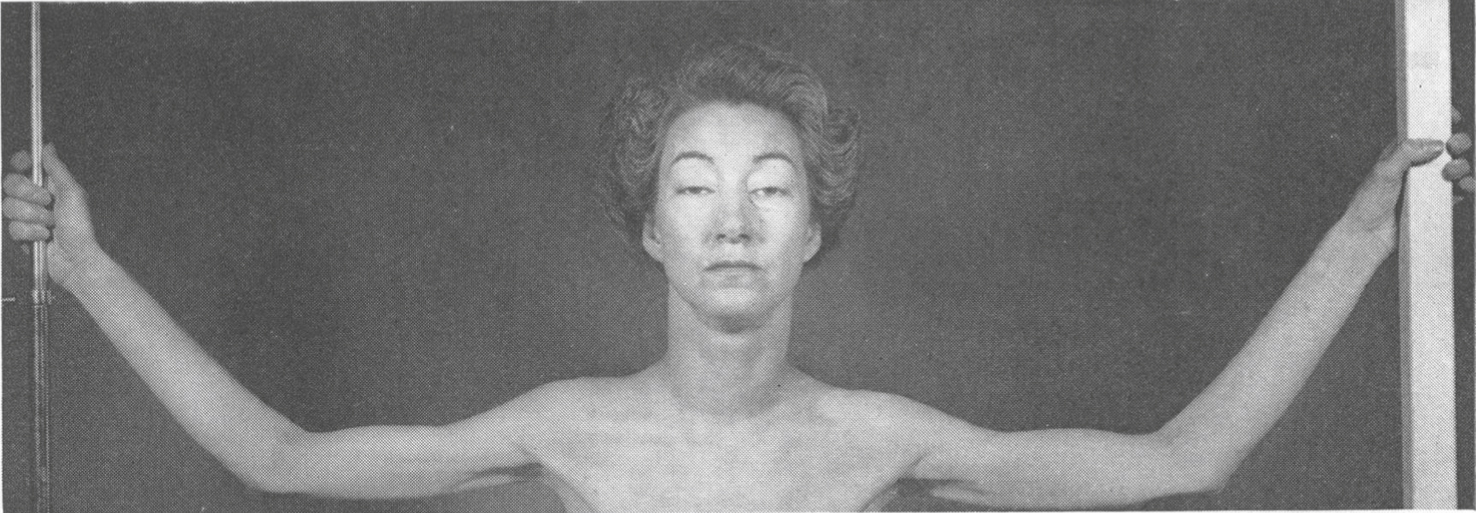
Fig.3
Zebrafish a) Control; b) GFPT1 morpholino, showing altered tail morphology.
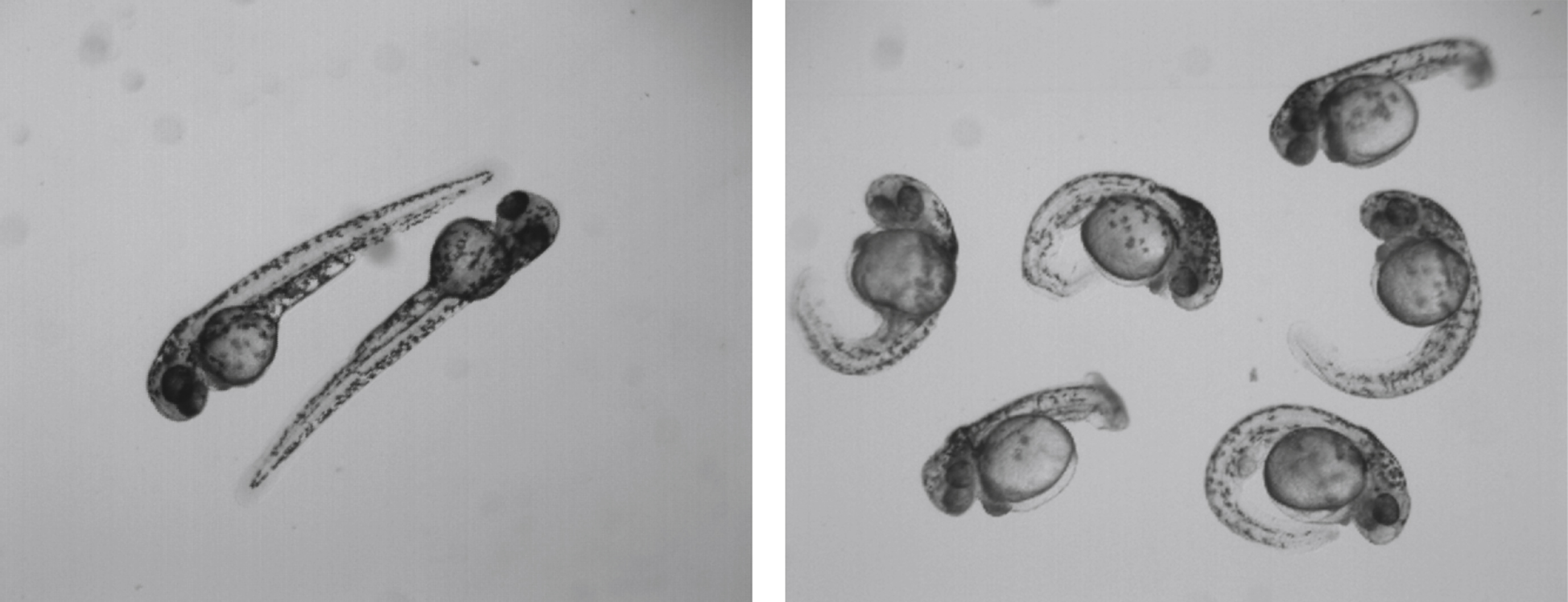
Table 1
Classification of CMS
| Gene | Pathology | ||
| Presynaptic | |||
| Choline acetyltransferase deficiency | CHAT | Reduced synthesis of ACh | |
| Mitochondrial citrate carrier | SLC25A1 | Abnormal carrier function | |
| Pre-synaptic nerve terminal abnormalities | |||
| Synaptotagmin 2 | SYT2 | Disruption of synaptic vesicle exocytosis of neurotransmitter | |
| Synaptosomal-associated protein 25 | SNAP25B | Inhibits synaptic vesicle exocytosis | |
| Synaptic | |||
| Endplate-AChE deficiency | COLQ | Reduced AChE | |
| Increased permanence of ACh in the synaptic cleft | |||
| End-plate myopathy | |||
| Laminin beta 2 | LAMB2 | Reduced β2-laminin | |
| Agrin | AGRN | Impaired NMJ formation and AChR clustering | |
| Postsynaptic | |||
| Kinetic changes of AChR / endplate-AChR deficiency | CHRNA1 | Kinetic abnormalities: slow or fast channel syndromes | |
| CHRNB1 | End-plate AChR deficiency: Reduced expression of AChR | ||
| CHRND | |||
| CHRNE | |||
| Rapsyn (impaired cluster of AChR) | RAPSN | Impaired clustering of AChR | |
| Muscle-specific kinase | MUSK | Impaired NMJ formation and AChR clustering | |
| Docking Protein 7 | DOK7 | Small and simplified NMJ | |
| Kinetic defect in voltage-gated Sodium channel 1.4 | SCN4A | Altered sodium channel function | |
| Low density lipoprotein receptor-related protein 4 | LRP4 | Expressed on the surface of the postsynaptic membrane | |
| Is a receptor for neural agrin | |||
| Reduction in size of the nerve terminal and in the postsynaptic region | |||
| Congenital defects of Glycosylation | GFPT1 | Abnormal glycosylation of NMJ components | |
| DPAGT1 | |||
| ALG2 | |||
| ALG14 | |||
| GMPPB | |||
| Other | |||
| PREPL deletion syndrome | PREPL | ||
| Plectin deficiency | PLEC1 | Abnormal muscle fibre morphology |
Table 2
LG–CMS distinctive features, (?) no data or insufficient data
| Gene (AR) | 1st symptoms | CK level | Double CMAP | Tubular aggregates | Stridor/vocal cord paralysis, neo-natal period | Ptosis | Ophthalmoparesis | Respiratory weakness | Bulbar weakness | Tongue atrophy | Motor development delay | Pyridostigmine |
| DOK7 | >1st year | Raised (occasionally) | No | No | Yes | Yes | No | Yes | yes | Yes | No | Deterioration |
| COLQ | Early infancy | Normal | Yes | No | No | Yes | Yes (+ slow pupillary response to light) | Yes | Yes | No | Yes | Deterioration |
| DPAGT1 | Childhood | Raised (occasionally) | No | Yes | No | No | No | No | No | No | In the most severe cases | Benefit |
| GFPT1 | Infancy to adulthood | Raised (occasionally) | No | Yes | No | No | No | No | No | No | In the most severe cases | Benefit |
| ALG2 | Childhood | Normal | No | No | No | No | No | No | No | No | No | Benefit |
| ALG14 | Infancy | Normal | No | Yes | No | No | No | No | No | No | Yes | ? |
| GMPPB | Infancy to adulthood | Raised | No | No | No | No | No | No | No | No | Yes | Benefit |


