
Diagnostic use of Massively Parallel Sequencing in Neuromuscular Diseases: Towards an Integrated Diagnosis
Abstract
Massively parallel sequencing is revolutionizing the genetic testing in diagnosis laboratories, replacing gene-by-gene investigations with a “gene panel” strategy. This new approach is particularly promising for the diagnosis of neuromuscular disorders affecting children as well as adults, which is constrained by strong clinical and genetic heterogeneity. While it leads to a strong improvement in molecular diagnosis, this new approach is dramatically changing the whole diagnosis process, establishing new decision trees and requiring integrated strategies between clinicians and laboratories. To have an overview of the implementation and benefit of these novel sequencing strategies for the diagnosis of neuromuscular disorders, we surveyed the current literature on the application of targeted genes panel sequencing, exome sequencing and genome sequencing. We highlight advantages and disadvantages of these different strategies in a diagnosis setting, discuss about unresolved cases, and point potential validation approaches and outcomes of massively parallel sequencing. It appears important to integrate such novel strategies with clinical, histopathological and imaging investigations, for a faster and more accurate diagnosis and patient care, and to foster research projects and clinical trials.
INTRODUCTION
Inherited neuromuscular disorders (NMD) form a group of genetic diseases which result in chronic long term disability posing a significant burden to the patients, their families and public health care. NMD are often severe and include more than 200 monogenic disorders with a total incidence exceeding 1 in 3000.
The molecular diagnosis of muscular disorders is often necessary to confirm a clinical diagnosis and contribute to specific healthcare. It also allows the access to genetic counselling and specific therapies. The precise diagnosis of NMD requires a conjunction of extensive clinical examination and targeted complementary tests. Previous/current routine genetic diagnosis is mainly done on a gene-by-gene basis, starting from the most pertinent one. This strategy is often difficult to implement as specific clinical or histological signs of the disease are sometimes not present, particularly at the beginning of the pathology or in affected newborns. Diagnosis bottlenecks are also caused by genetic and clinical heterogeneity, and lack of segregation data in sporadic cases. As an example, limb-girdle muscular dystrophies (LGMD) implicate more than 30 genes [1]. Also, large genes, like Titin (TTN) with 363 exons, are not entirely Sanger sequenced on routine even if previously linked to neuromuscular diseases [2, 3]. As a consequence, clinical tests are multiplied, including invasive investigations. DNA is sent to different laboratories making the molecular diagnosis laborious, time-consuming and expensive. In addition, the gene-by-gene strategy usually stops at the first probable mutated gene, while it would have been preferable to clearly discard additional genes. Often, the genetic diagnosis is completed after several months or even years, and the molecular causes of NMD are still unknown for about 40% of patient despite tremendous research and clinical efforts.
Recently, massively parallel sequencing (MPS) using high throughput sequencing technologies has emerged as a successful approach to interrogate several genes simultaneously [4]. The introduction of MPS in diagnosis laboratories is not only a revolution at the technical level, but also in terms of organisation as it necessitates a multidisciplinary integration between molecular biologists, expert in bioinformatics, clinicians and histopathology specialists.
Concerning neuromuscular disorders, Lupski and colleagues and Montenegro and colleagues respectively used Genome Sequencing and Exome Sequencing in patients with Charcot-Marie-Tooth neuropathy (CMT) on a research setting to provide the proof-of-principle that these approaches may be adapted for diagnosis [5, 6]. In both cases, they analysed a single family and focused their variants ranking on known CMT genes. In 2012, we have established the proof-of-principle that nearly all the known NMD genes can be tested at once using Targeted Sequencing [7], and since then several groups have reported results of the implementation of MPS in diagnosis setting. We review here the applications of MPS for NMD diagnosis and focus on expected advantages and possible improvements that should be anticipated, and we highlight important points to consider for the adequate implementation and success of MPS within an integrated diagnostic strategy.
PRINCIPLES OF DIFFERENT MPS STRATEGIES IN A DIAGNOSIS SETTING
Three approaches using MPS technology have been developed: Targeted Sequencing (TS) that focuses on genes panel, Exome Sequencing (ES) and Genome Sequencing (GS) (Fig. 1). Although they are widely used, we would like to argue against the use of the terms whole ES (WES) and whole GS (WES) considering that neither exome nor genome sequence can be completely recovered due to technical and data analysis issues (see Unresolved cases). The exome represents about 1% of the genome and it is estimated it contains 85% of the disease-causing mutations [8]. TS has mainly been used to target the coding regions of a panel of genes or genomic regions [9, 10],with preliminary enrichment of the regions to be sequenced by an amplicon-based method (PCR) or a hybridisation-based method (capture). GS is not biased by preliminary enrichment and retrieves sequencevariants in non-coding and intergenic regions as well.
This is not the scope of this review to assess the performance of each method, which is linked to numerous parameters in the MPS process including targets, protocols, sequencing platforms and bioinformatics parameters (Fig. 2) as well as biological and clinical interpretation. These different aspects are well documented and discussed in several Guidelines for the implementation of MPS in clinical diagnosis [11–15] as well as concerning variants interpretation and reporting [11, 16].
Whatever the MPS strategy, there are three established principles. First, it is mandatory to focus on genes with clear clinical significance. Secondly, it is necessary to control the performance efficiency of a MPS test prior its implementation in a routine setting [17, 18]. Finally, a MPS investigation should report the calculated sensitivity and specificity, the targeted regions and mutations, and the coverage and depth for each targeted gene (with possibly supplementation with second-tiers Sanger sequencing for full coverage) (Fig. 1). Furthermore, it is appropriate to verify the presence of pathogenic variants detected by MPS through Sanger sequencing (or dosage analysis) to counter-check for potential sample mixing errors in case no genotyping identification is embedded in the MPS strategy, and to validate the Sanger protocol that will be used for future genetic counselling in the family.
OUTCOMES OF MPS FOR NMD IN A DIAGNOSIS SETTING
Several studies concerning the validation of different approaches on DNAs with known mutations have been reported [19–22]. We review here several approaches reported in a diagnosis setting, focusing on their efficiency (solved cases rate) and discussing several points which have to be considered in unresolved cases.
Resolved cases
Several studies using different MPS approaches for NMD molecular diagnosis are presented in Table 1. Taken together, these studies show that both ES and gene panel TS approaches reach definite molecular diagnosis in about 27.5% of explored patients (228 out of 827). However, many patients in these pilot projects have been enrolled after excluding frequent implicated genes for the related diseases, i.e. common forms of spinocerebellar ataxia [23, 24], CMT [25], non syndromic muscle disorders [26], LGMD and distal myopathies [27, 28], polyneuropathies [29, 30], or early onset NMD [31]. This has increased the possibility that disease-causing mutations lie in previously undiscovered genes or in intergenic regions. ES has been considered either as a direct first tier [19] or second-tier diagnostic approach, i.e. after initial exclusion of gene(s) based on single gene analysis [27]. Although a second tiers approach is indicated when frequent mutations can be rapidly tested in one or a few genes (spinocerebellar ataxia genes as an example), it is likely that the overall diagnostic rate of MPS approaches will increase as this technology is used as the first test of choice for genetic testing.
Second-tier diagnostic approaches prevented us to determine which gene is the most frequently involved in a pathology based on MPS literature alone. Within a national project of 1000 exomes coordinated byJ. Laporte, Nebulin (NEB) mutations was found to be involved in 15 out of 43 patients (35%) with nemaline myopathy for whom most of the known genes have been previously excluded. These findings are probably due to the fact that NEB is a large gene poorly tested in routine Sanger diagnosis, and highlight NEB as a major nemaline myopathy gene that deserves more scrutiny.
Unresolved cases
Negative testing does not necessarily imply that no causative variant(s) is present in the patient DNA but may be due to technical issues, difficulties to detect specific mutations, or analysis and interpretationlimitations:
Technical issues
It is possible to miss a mutation when the coverage depth (also reported as “depth”, see Fig. 1) is not sufficient for reliable variant detection. An exon is referred as being a “low coverage exon” if at least a single nucleotide in the exon has a coverage depth below a fixed threshold (usually <20X, i.e. less than 20 reads). This implies that the minimum mean coverage depth should be much higher than 20X [11]. Valencia and colleagues reported that using ES, 10–20% had low or zero coverage; exons poorly covered may involve NMD genes [21]. Some targets are particularly prone to low coverage due to high GC-rich content impacting on enrichment or sequencing (technical challenge) or exons or genes with homologous pseudo-exons or pseudo-genes (bioinformatics challenge in sequence mapping, Fig. 2).
Savarese and colleagues [26] have compared data from TS of 93 genes and from ES on a set of 20 DNA samples belonging to patients with different NMD and presenting with 84 variants in 17 genes, previously genotyped using Sanger genotyping. They reported 20–30% more variants in Targeted versus Exome Sequencings, probably due to the higher coverage of the targeted approach.
In case of suboptimal coverage, complementary Sanger sequencing is required to achieve full sensitivity. Among the studies presented in Table 1, Sanger sequencing was added for a small subset of coding regions in 26 genes involved in Congenital Muscular Dystrophy (CMD) [10], and for 68 exons (22 genes) with a coverage <20X for completion of the exonic panel for NMD, CMD and LGMD [32]. Such complementary analysis being time-consuming and costly, the MPS protocols should be efficient enough to necessitate minimal Sanger sequencing for full coverage of the targets. GS was shown to have a more homogeneous coverage compared to ES that relies on a first enrichment step.
Detection of specific mutation types
Although Sanger sequencing cannot detect large heterozygous deletion or duplication (except hemizygous deletion on the X chromosome in a male), the detection of CNV (Copy Number Variation) using MPS would be a very valuable improvement, preventing the need for specific dosage anomaly analysis such as array Comparative Genomic Hybridization (aCGH). As an example, Ankala and colleagues have studied 41 genes by MPS and aCGH and discovered a rate of 5 CNV out of 70 patients presenting with CMD and 8 CNV out of 193 patients presenting with LGMD [32]. MPS has proven to be able to detect CNV through different approaches as for example TS of 267 NMD genes [7], TS of the whole genomic Duchenne muscular dystrophy gene (DMD) [9], TS of seven genes implicated in peripheral neuropathies [33] or TS of 26 muscular dystrophy related genes [10]. The coverage being much more variable in ES approaches, detection of CNV is more challenging. GS may result in uniform distribution of coverage and thus a betterprediction of large rearrangements. In addition, severalbioinformatics tools are being validated for CNV detection from MPS data [34–36].
The detection of expansion, i.e. repeat of tri or tetra nucleotides, involved in several NMD is also challenging due to the difficulty to map to the reference genome short reads of repeated sequences, but may be improved with longer reads technologies.
Analysis and interpretation limitations
Interpretation is straightforward in two cases: when a mutation(s) is already described (providing the effect is ascertained through actual knowledge) or when a clear pathogenic effect can be predicted (for e.g. new mutation with similar impact as known mutations). Other cases could remain unresolved if encompassed within the following possibilities:
The pathogenicity of previously reported mutation might have been based only on in silico criteria, for missense and splice site variants in particular. As an example, a variant in the Senataxin gene (SETX) reported as a putative mutation was finally confirmed as a non-pathogenic rare polymorphism [30]. Also, the implication of the gene in the pathology should be recognized, i.e. though the identification of several linked families and functional validation of the mutation impact.
In case of a variant of unknown significance (VOUS), further analyses are needed to prove the causality: familial segregation analysis of the variant, impact on RNA or protein, or functional analysis (Fig. 3). This situation is highlighted in several studies presented in Table 1 in which a potential causative gene that required further testing for definitive conclusion has been found in 26% of the 442 enrolled patients [10, 24, 26, 28–31].
In case only one mutation is found in a gene linked to an autosomal recessive disease, a second (undetected) mutation should be searched. Alternatively, this may represent an incidental finding (IF) that the patient is a carrier for another disease. Savarese and colleagues reported such cases for 26 out of 177 patients [26] and Bartoli and colleagues for 3 out of 37 patients [27]. The “second” mutation may have been missed due to low coverage in the sequencing data, difficulty to detect it (see above), or because the MPS protocol does not target the mutated region (5′ and 3′ UTR, deep intronic or intergenic mutation in ES). Alternatively, the mutated gene may not be targeted by TS or ES analysis. ES and GS may allow the reanalysis of the data in a diagnosis setting, for example when a new gene is discovered, or can be the basis of research projects aiming for new gene discovery. This is obviously not the case for TS.
In some cases, potential pathogenic mutations can be found in a gene previously linked to a different phenotype than the patient’s, and could represent a novel genotype-phenotype correlation and the discovery of an allelic disease. An integrated comparison of the genetic data with the phenotypic investigations has to be performed to eventually reorient the diagnosis (Fig. 3).
DIAGNOSIS VALIDATION AND INTEGRATED DIAGNOSIS
The use of MPS for genetic testing is not straightforward and depends from many factors such as informatics pipeline [12] and biologist interpretation, as shown by the Clarity study in which 30 international groups analysed independently the sequence of three genomes and only two groups identified the consensus candidate variants in all diseases cases [37].
Sequencing strategies (Table 2)
Only genes previously linked to the patient’ disease need to be tested in a diagnosis setting. However, the initial clinical presentation may be atypical or incomplete, misleading the clinician to select the wrong gene panel. The use of large gene panels, ES or GS can alleviate this drawback and allows the inclusion of patients in a first tier diagnosis approach, without the need for preliminary extensive clinical investigations. Such strategy should allow expanding clinical spectra and reducing the time to diagnosis. Considering patient management, the clinical utility of MPS was highlighted in several studies, as for example in congenital myasthenia syndrome in which early diagnosis is important for effective treatment [31].
In principle, GS should permit reaching a better diagnosis yield by allowing the discovery of pathogenic variants in non-coding regions. Nevertheless, the interpretation of deep intronic variants is particularly difficult concerning potential impact on splicing or expression, considering that in silico predictions need to be confirmed through RT-PCR study from a disease-relevant tissue. Furthermore, while the frequency of intronic variants in the population is a valuable indicator of the pathogenicity of a variant, it is necessary to increase the availability of such data in different ethnical groups.
The yield of positive findings is increased by including several relatives, affected or not. In two cohorts enrolling 500 and 814 families with undiagnosed genetic conditions, the diagnostic rate was significantly higher among families undergoing trio ES, i.e. both parents and their affected child sequenced simultaneously (37% and 31% respectively) as compared to ES for the patient only (21% and 22% respectively) [38, 39]. However, trio analysis implies an additional cost and parental samples are not always available, in particular for adult patients.
Mutations validation
The ability to interpret sequence variants in a gene is linked to several considerations and validation steps [40, 41]:
One important point for assessing the impact of a variant is to access to sequence databases of patients and controls. Variants classified as pathogenic, of uncertain significance or as polymorphisms areavailable online in numerous databases such as theHuman Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), Universal Mutation databases (UMDs, http://www.umd.be/), Leiden Open Variation Databases (LOVDs, http://www.lovd.nl/3.0/home), the Single Nucleotide Polymorphism database (dbSNP, http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genome Browser (http://www.1000genomes.org/1000-genomes-browsers), the Exome Variant Server (http://evs.gs.washington.edu/EVS/), the Ex-ome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/). There is now a trend to make publicly accessible variants list through the development of computing tools, such as the EGL’s online tool or EmVClass [42] used by Ankala and colleagues [32]. Nevertheless, not all variants lists produced through MPS projects are available online and areoften maintained as in-house variants databasesuntil the data are published. International efforts, as PhenomeCentral (https://phenomecentral.org/), aim to provide a platform for secure data sharing.
Another important step is the study of the variant segregation among relatives (if it has not been performed using MPS), and eventually complementary analysis such as RNA or protein studies. Such complementary analyses were required to reach a diagnostic conclusion for 115 out of 442 patients (26%) enrolled in 7 studies presented in Table 1. More detailed functional analyses may be required for potential mutations that were not yet reported as pathogenic and can benefit from high throughput wet bench pipelines, for example to test the impact of variants on protein stability, localisation and interaction [43], or to correlate with phenotypes in animal models as zebrafish [44].
Single gene testing needs to be available for all genes included in a panel, either to investigate additional relatives for assessing the pathogenicity of a variant through segregation analysis, or for further genetic counselling in the family when the causal effect of the mutation is established. It is advantageous to rely on laboratories expert in a subset of genes to validate the impact of mutations found by MPS. These laboratories are usually linked to research programs through patient’s clinical and molecular databases (UMD, LOVD, Treat-NMD http://www.treat-nmd.eu/) for the development of phenotype-genotype correlation studies as well as patients recruitment into specific clinical trials.
Integrated diagnosis
The clinicians provide phenotypic and familial information to assist the laboratory in analysing and interpreting the MPS results. In any case, an integrated diagnosis is necessary to direct the analysis of MPS data to reach a fast and reliable diagnosis, and requires a strong integration of the clinical, histopathological, biochemical, imaging and genetic data (Fig. 3). As such, this integrated strategy allows appropriate data interpretation taking into account potential incomplete penetrance or variable disease expression [45–47], and may lead to reverse phenotyping, i.e. complementary medical investigations directed by the result of the genetic analysis [48, 49]. To our own experience the probability to identify potential pathogenic variants in more than one gene, when testing large panel of genes, is very low when performing such an integrated diagnosis.
MPS FOR NMD DIAGNOSIS: OTHER CONSIDERATIONS
Cost
MPS is relatively cheaper compared to Sanger sequencing when considering that the cost for targeted sequencing of about 150 genes is usually equivalent to the cost for analysing 3-4 genes of medium size using Sanger sequencing. TS is at this time the less expensive MPS approach in term of investment for equipment and purchase of consumables, albeit there is a continuing decrease in sequencing cost bringing ES and GS more accessible (Table 2).
Incidental findings
Using multi-loci analysis, a recurrent concern is indeed the possibility of finding mutations unrelated to the indication for ordering the molecular diagnosis, namely incidental findings (IF). The classification into IF can be done when the pathology presented by the patient is without any link with the mutation found (in case a new allelic disease can be formallyexcluded).
On the ethical point of view, the problematic of the disclosure of IF has been encountered and discussed previously in genetic diagnosis using aCGH, with as an example the detection of carrier females of dystrophinopathy in a cohort of females presenting with intellectual disability [50]. This issue can concern carrier diagnosis for a recessive disease, or predictive diagnosis of a genetic disease linked to late onset with partial or complete penetrance. The possibility to find an IF could be dramatically reduced by targeted all or a group of NMD genes (i.e. TS), and ethical issues raised by ES and GS may be equivalent to the ones of TS in case only genes of interest are scrutinized. Nevertheless, concerning clinical Exome and Genome Sequencings, the American College of Medical Genetics (ACMG) had issued recommendations for reporting IF of medical benefit for the patient and the family, irrespectively of the age of the patient (also named actionable IF). ACMG recommends that diagnostic laboratories actively seek and report mutations in a list of 56 genes (implicated in cancer, cardiomyopathy …) in the course of their routine clinical sequencing [51]. Several issues concerning the patient, the clinician and the laboratories are discussed in this report and remain an area of debate [52–54].
CONCLUSIONS AND PERSPECTIVES
In the context of heterogeneous diseases such as NMD, MPS provides a reliable cost and time effective alternative to Sanger sequencing, providing the genes of interest are adequately sequenced. MPS may be used on a first intention as a genetic screening, to better orient clinical tests that could be invasive, costly and necessitate patient travel, but should not substitute for clinical and histopathological investigations. We can anticipate that MPS-based identification of disease mutations will have a growing impact on the diagnosis and classification of NMD as it frequently expands the clinical spectrum or reveals allelic diseases. Furthermore, better understanding of the pathological mechanisms of NMD and standards of care as well as therapeutic research should benefit from a higher diagnostic yield.
Increase in technologies performance foresees GS as the main future strategy for genetic diagnosis. To increase the benefit of MPS in a diagnosis setting, improvement in bioinformatics, storage of data and interpretation of variants are areas that require further resource development, as well as expanded and multidisciplinary databases and analysis pipelines and tools, and increased training for biologists and clinicians. High throughput assay to assess RNA integrity [55] and protein function have been described [43] and should be further developed for functional validation of genetic variants.
Other potential applications than diagnosis of monogenic diseases are under discussion or proposal, in particular with the idea of early diagnosis for optimal clinical management or disease prevention: preconception screening [56], non-invasive prenatal diagnosis using maternal circulating nucleic acids [57], new born screening [58] and predictive testing.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
J.L. was supported by INSERM.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1 | Nigro V., Savarese M.(2014) Genetic basis of limb-girdle muscular dystrophies: The updateActa Myol33: 112 |
2 | Chauveau C., Rowell J., Ferreiro A.(2014) A rising titan: TTN review and mutation updateHum Mutat35: 10461059 |
3 | Bang M.L., Centner T., Fornoff F., Geach A.J., Gotthardt M., McNabb M., Witt C.C., Labeit D., Gregorio C.C., Granzier H., Labeit S.(2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking systemCirc Res89: 10651072 |
4 | Ng S.B., Nickerson D.A., Bamshad M.J., Shendure J.(2010) Massively parallel sequencing and rare diseaseHum Mol Genet19: R119R124 |
5 | Lupski J.R., Reid J.G., Gonzaga-Jauregui C., Rio Deiros D., Chen D.C., Nazareth L., Bainbridge M., Dinh H., Jing C., Wheeler D.A., McGuire A.L., Zhang F., Stankiewicz P., Halperin J.J., Yang C., Gehman C., Guo D., Irikat R.K., Tom W., Fantin N.J., Muzny D.M., Gibbs R.A.(2010) Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathyN Engl J Med362: 11811191 |
6 | Montenegro G., Powell E., Huang J., Speziani F., Edwards Y.J., Beecham G., Hulme W., Siskind C., Vance J., Shy M., Zuchner S.(2011) Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth familyAnn Neurol69: 464470 |
7 | Vasli N., Bohm J., Le G.ras S., Muller J., Pizot C., Jost B., Echaniz-Laguna A., Laugel V., Tranchant C., Bernard R., Plewniak F., Vicaire S., Levy N., Chelly J., Mandel J.L., Biancalana V., Laporte J.(2012) Next generation sequencing for molecular diagnosis of neuromuscular diseasesActa Neuropathol124: 273283 |
8 | Rabbani B., Tekin M., Mahdieh N.(2014) The promise of whole-exome sequencing in medical geneticsJ Hum Genet59: 515 |
9 | Lim B.C., Lee S., Shin J.Y., Kim J.I., Hwang H., Kim K.J., Hwang Y.S., Seo J.S., Chae J.H.(2011) Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: Comprehensive mutational search in a single platformJ Med Genet48: 731736 |
10 | Lim B.C., Lee S., Shin J.Y., Hwang H., Kim K.J., Hwang Y.S., Seo J.S., Kim J.I., Chae J.H.(2013) Molecular diagnosis of congenitalmuscular dystrophies with defectiveglycosylation of alpha-dystroglycan using next-generationsequencing technologyNeuromuscul Disord23: 337344 |
11 | Rehm H.L., Bale S.J., Bayrak-Toydemir P., Berg J.S., Brown K.K., Deignan J.L., Friez M.J., Funke B.H., Hegde M.R., Lyon E.(2013) ACMG clinical laboratory standards for next-generation sequencingGenet Med15: 733747 |
12 | O’Rawe J., Jiang T., Sun G., Wu Y., Wang W., Hu J., Bodily P., Tian L., Hakonarson H., Johnson W.E., Wei Z., Wang K., Lyon G.J.(2013) Low concordance of multiple variant-calling pipelines: Practical implications for exome and genome sequencingGenome Med5: 28 |
13 | van E.l CG., Cornel M.C., Borry P., Hastings R.J., Fellmann F., Hodgson S.V., Howard H.C., Cambon-Thomsen A., Knoppers B.M., Meijers-Heijboer H., Scheffer H., Tranebjaerg L., Dondorp W., de Wert G.M.(2013) Whole-genome sequencing in health care. Recommendations of the European society of human geneticsEur J Hum Genet. 21 SupplS1: 15 |
14 | Lek M., MacArthur D.(2014) The Challenge of Next Generation Sequencing in the Context of Neuromuscular DiseasesJ Neuromuscular Diseases1: 135149 |
15 | Xue Y., Ankala A., Wilcox W.R., Hegde M.R.(2014) Solving themolecular diagnostic testing conundrum for Mendelian disorders inthe era of next-generation sequencing: Single-gene, gene panel, orexome/genome sequencingGenet MedSep 18. doi: 10.1038/gim.2014.122 |
16 | Richards C.S., Bale S., Bellissimo D.B., Das S., Grody W.W., Hegde M.R., Lyon E., Ward B.E.(2008) ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007Genet Med10: 294300 |
17 | Gargis A.S., Kalman L., Berry M.W., Bick D.P., Dimmock D.P., Hambuch T., Lu F., Lyon E., Voelkerding K.V., Zehnbauer B.A., Agarwala R., Bennett S.F., Chen B., Chin E.L., Compton J.G., Das S., Farkas D.H., Ferber M.J., Funke B.H., Furtado M.R., Ganova-Raeva L.M., Geigenmuller U., Gunselman S.J., Hegde M.R., Johnson P.L., Kasarskis A., Kulkarni S., Lenk T., Liu C.S., Manion M., Manolio T.A., Mardis E.R., Merker J.D., Rajeevan M.S., Reese M.G., Rehm H.L., Simen B.B., Yeakley J.M., Zook J.M., Lubin I.M.(2012) Assuring the quality of next-generation sequencing in clinicallaboratory practiceNat Biotechnol30: 10331036 |
18 | Aziz N., Zhao Q., Bry L., Driscoll D.K., Funke B., Gibson J.S., Grody W.W., Hegde M.R., Hoeltge G.A., Leonard D.G., Merker J.D., Nagarajan R., Palicki L.A., Robetorye R.S., Schrijver I., Weck K.E., Voelkerding K.V.(2015) College of Americanpathologists’ laboratory standards for next-generation sequencingclinical testsArch Pathol Lab Med139: 4481493 |
19 | Dias C., Sincan M., Cherukuri P.F., Rupps R., Huang Y., Briemberg H., Selby K., Mullikin J.C., Markello T.C., Adams D.R., Gahl W.A., Boerkoel C.F.(2012) An analysis of exome sequencingfor diagnostic testing of the genes associated with muscle diseaseand spastic paraplegiaHum Mutat33: 614626 |
20 | Valencia C.A., Rhodenizer D., Bhide S., Chin E., Littlejohn M.R., Keong L.M., Rutkowski A., Bonnemann C., Hegde M.(2012) Assessment of target enrichment platforms using massively parallel sequencing for the mutation detection for congenital muscular dystrophyJ Mol Diagn14: 233246 |
21 | Valencia C.A., Ankala A., Rhodenizer D., Bhide S., Littlejohn M.R., Keong L.M., Rutkowski A., Sparks S., Bonnemann C., Hegde M.(2013) Comprehensive mutation analysis for congenital muscular dystrophy: A clinical PCR-based enrichmentand next-generation sequencing panelPLoS One8: e53083 |
22 | Sawyer S.L., Schwartzentruber J., Beaulieu C.L., Dyment D., Smith A., Warman Chardon J., Yoon G., Rouleau G.A., Suchowersky O., Siu V., Murphy L., Hegele R.A., Marshall C.R., Bulman D.E., Majewski J., Tarnopolsky M., Boycott K.M.(2014) Exome sequencing as a diagnostic tool for pediatric-onset ataxiaHum Mutat35: 4549 |
23 | Nemeth A.H., Kwasniewska A.C., Lise S., Parolin Schnekenberg R., Becker E.B., Bera K.D., Shanks M.E., Gregory L., Buck D., Zameel Cader M., Talbot K., de Silva R., Fletcher N., Hastings R., Jayawant S., Morrison P.J., Worth P., Taylor M., Tolmie J., O’Regan M., Valentine R., Packham E., Evans J., Seller A., Ragoussis J.(2013) Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a modelBrain136: 31063118 |
24 | Fogel B.L., Lee H., Deignan J.L., Strom S.P., Kantarci S., Wang X., Quintero-Rivera F., Vilain E., Grody W.W., Perlman S., Geschwind D.H., Nelson S.F.(2014) Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxiaJAMA Neurol71: 12371246 |
25 | Choi B.O., Koo S.K., Park M.H., Rhee H., Yang S.J., Choi K.G., Jung S.C., Kim H.S., Hyun Y.S., Nakhro K., Lee H.J., Woo H.M., Chung K.W.(2012) Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth diseaseHum Mutat33: 16101615 |
26 | Savarese M., Di Fruscio G., Mutarelli M., Torella A., Magri F., Santorelli F., Comi G., Bruno C., Nigro V.(2014) MotorPlexprovides accurate variant detection across large muscle genes bothin single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun2: 100 |
27 | Bartoli M., Desvignes J.P., Levy N., Krahn M.(2014) Exome sequencingas a second-tier diagnostic approach for clinically suspecteddysferlinopathy patientsMuscle Nerve50: 610071010 |
28 | Sevy A., Cerino M., Gorokhova S., Dionnet E., MathieuY , Verschueren A., Franques J., Maues de Paula A., Figarella-Branger D., Lagarde A., Desvignes J.P., Beroud C., Attarian S., Levy N., Bartoli M., Krahn M., Campana-Salort E., Pouget J.(2016) Improving molecular diagnosis of distal myopathiesby targeted next-generation sequencingJ Neurol Neurosurg PsychiatryMar 17. pii: jnnp-2014-309663. doi: 10.1136/jnnp-2014-309663. |
29 | Klein C.J., Middha S., Duan X., Wu Y., Litchy W.J., Gu W., Dyck P.J., Gavrilova R.H., Smith D.I., Kocher J.P., Dyck P.J.(2014) Application of whole exome sequencing in undiagnosed inherited polyneuropathiesJ Neurol Neurosurg Psychiatry85: 12651272 |
30 | Drew A.P., Zhu D., Kidambi A., Ly C., Tey S., Brewer M.H., Ahmad-Annuar A., Nicholson G.A., Kennerson M.L.(2015) Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencingMol Genet Genomic Med3: 143154 |
31 | Chae J.H., Vasta V., Cho A., Lim B.C., Zhang Q., Eun S.H., Hahn S.H.(2015) Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disordersJ Med Genet52: 208216 |
32 | Ankala A., da Silva C., Gualandi F., Ferlini A., Bean L.J., Collins C., Tanner A.K., Hegde M.R.(2015) A comprehensive genomicapproach for neuromuscular diseases gives a high diagnostic yieldAnn Neurol77: 206214 |
33 | Goossens D., Moens L.N., Nelis E., Lenaerts A.S., Glassee W., Kalbe A., Frey B., Kopal G., De Jonghe P., De Rijk P., Del-Favero J.(2009) Simultaneous mutation and copy number variation (CNV) detection by multiplex PCR-based GS-FLX sequencingHum Mutat30: 472476 |
34 | Hayes J.L., Tzika A., Thygesen H., Berri S., Wood H.M., Hewitt S., Pendlebury M., Coates A., Willoughby L., Watson C.M., Rabbitts P., Roberts P., Taylor G.R.(2013) Diagnosis of copy number variation byIllumina next generation sequencing is comparable in performanceto oligonucleotide array comparative genomic hybridisationGenomics102: 174181 |
35 | Retterer K., Scuffins J., Schmidt D., Lewis R., Pineda-Alvarez D., Stafford A., Schmidt L., Warren S., Gibellini F., Kondakova A., Blair A., Bale S., Matyakhina L., Meck J., Aradhya S., Haverfield E.(2014) Assessing copy number fromexome sequencing and exome array CGH based on CNV spectrum in alarge clinical cohortGenet MedNov 6. doi: 10.1038/gim.2014.160. |
36 | Zhao M., Wang Q., Wang Q., Jia P., Zhao Z.(2013) Computational tools for copynumber variation (CNV) detection using next-generation sequencing data: Features and perspectivesBMC Bioinformatics14 Suppl 11: S1 |
37 | Brownstein C.A., Beggs A.H., Homer N., Merriman B., Yu T.W., Flannery K.C., DeChene E.T., Towne M.C., Savage S.K., Price E.N., Holm I.A., Luquette L.J., Lyon E., MajzoubJ , Neupert P., McCallie D.Jr, Szolovits P., Willard H.F., Mendelsohn N.J., Temme R., Finkel R.S., Yum S.W., Medne L., Sunyaev S.R., Adzhubey I., Cassa C.A., de Bakker P.I., Duzkale H., Dworzynski P., Fairbrother W., Francioli L., Funke B.H., Giovanni M.A., Handsaker R.E., Lage K., Lebo M.S., Lek M., Leshchiner I., MacArthur D.G., McLaughlin H.M., Murray M.F., Pers T.H., Polak P.P., Raychaudhuri S., Rehm H.L., Soemedi R., Stitziel N.O., Vestecka S., Supper J., Gugenmus C., Klocke B., Hahn A., Schubach M., Menzel M., Biskup S., Freisinger P., Deng M., Braun M., Perner S., Smith R.J., Andorf J.L., Huang J., Ryckman K., Sheffield V.C., Stone E.M., Bair T., Black-Ziegelbein E.A., Braun T.A., Darbro B., DeLuca A.P., Kolbe D.L., Scheetz T.E., Shearer A.E., Sompallae R., Wang K., Bassuk A.G., Edens E., Mathews K., Moore S.A., Shchelochkov O.A., Trapane P., Bossler A., Campbell C.A., Heusel J.W., Kwitek A., Maga T., Panzer K., Wassink T., Van Daele D., Azaiez H., Booth K., Meyer N., Segal M.M., Williams M.S., Tromp G., White P., Corsmeier D., Fitzgerald-Butt S., Herman G., Lamb-Thrush D., McBride K.L., Newsom D., Pierson C.R., Rakowsky A.T., Maver A., Lovrecic L., Palandacic A., Peterlin B., Torkamani A., Wedell A., HussM , Alexeyenko A., Lindvall J.M., Magnusson M., Nilsson D., Stranneheim H., Taylan F., Gilissen C., Hoischen A., vanBon B., Yntema H., Nelen M., Zhang W., Sager J., Zhang L., Blair K., Kural D., Cariaso M., Lennon G.G., Javed A., Agrawal S., Ng P.C., Sandhu K.S., Krishna S., Veeramachaneni V., Isakov O., Halperin E., Friedman E., Shomron N., GlusmanG , Roach J.C., Caballero J., Cox H.C., Mauldin D., Ament S.A., Rowen L., Richards D.R., San Lucas F.A., Gonzalez-Garay M.L., Caskey C.T., Bai Y., Huang Y., Fang F., Zhang Y., Wang Z., Barrera J., Garcia-Lobo J.M., Gonzalez-Lamuno D., Llorca J., Rodriguez M.C., Varela I., Reese M.G., De LaVega F.M., Kiruluta E., Cargill M., Hart R.K., Sorenson J.M., Lyon G.J., Stevenson D.A., Bray B.E., Moore B.M., Eilbeck K., Yandell M., Zhao H., Hou L., Chen X., Yan X., Chen M., Li C., Yang C., Gunel M., Li P., Kong Y., Alexander A.C., Albertyn Z.I., Boycott K.M., Bulman D.E., Gordon P.M., Innes A.M., Knoppers B.M., Majewski J., Marshall C.R., Parboosingh J.S., Sawyer S.L., Samuels M.E., Schwartzentruber J., Kohane I.S., Margulies D.M.(2014) Aninternational effort towards developing standards for bestpractices in analysis, interpretation and reporting of clinicalgenome sequencing results in the CLARITY ChallengeGenome Biol15: R53 |
38 | Farwell K.D., Shahmirzadi L., El-Khechen D., Powis Z., Chao E.C., Davis B.T., Baxter R.M., Zeng W., Mroske C., Parra M.C., Gandomi S.K., Lu I., Li X., Lu H., Lu H.M., Salvador D., Ruble D., Lao M., Fischbach S., Wen J., Lee S., Elliott A., Dunlop C.L., Tang S.(2014) Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: Results from 500 unselected families with undiagnosed genetic conditionsGenet Med |
39 | Lee H., Deignan J.L., Dorrani N., Strom S.P., Kantarci S., Quintero-Rivera F., Das K., Toy T., Harry B., Yourshaw M., Fox M., Fogel B.L., Martinez-Agosto J.A., Wong D.A., Chang V.Y., Shieh P.B., Palmer C.G., Dipple K.M., Grody W.W., Vilain E., Nelson S.F.(2014) Clinical exome sequencing for genetic identification of rare Mendelian disordersJama312: 18801887 |
40 | Duzkale H., Shen J., McLaughlin H., Alfares A., Kelly M.A., Pugh T.J., Funke B.H., Rehm H.L., Lebo M.S.(2013) A systematic approach to assessing the clinical significance of genetic variantsClin Genet84: 453463 |
41 | MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A., Barrett J.C., Biesecker L.G., Conrad D.F., Cooper G.M., Cox N.J., Daly M.J., Gerstein M.B., Goldstein D.B., Hirschhorn J.N., Leal S.M., Pennacchio L.A., Stamatoyannopoulos J.A., Sunyaev S.R., Valle D., Voight B.F., Winckler W., Gunter C.(2014) Guidelines for investigating causality of sequence variants in human diseaseNature508: 469476 |
42 | Bean L.J., Tinker S.W., da Silva C., Hegde M.R.(2013) Free the data: One laboratory’s approach to knowledge-based genomic variant classification and preparation for EMR integration of genomic dataHum Mutat34: 11831188 |
43 | Wei X., Das J., Fragoza R., Liang J., Bastos de Oliveira F.M., Lee H.R., Wang X., Mort M., Stenson P.D., Cooper D.N., Lipkin S.M., Smolka M.B., Yu H.(2014) A massively parallel pipeline to clone DNA variants and examine molecular phenotypes of human disease mutationsPLoS Genet10: e1004819 |
44 | Davis E.E., Frangakis S., Katsanis N.(1842) Interpreting human genetic variation with in vivo zebrafish assaysBiochim Biophys Acta19601970 |
45 | Hennekam R.C., Biesecker L.G.(2012) Next-generation sequencing demands next-generation phenotypingHum Mutat33: 884886 |
46 | Bohm J., Vasli N., Malfatti E., Le Gras S., Feger C., Jost B., Monnier N., Brocard J., Karasoy H., Gerard M., Walter M.C., Reilich P., Biancalana V., Kretz C., Messaddeq N., Marty I., Lunardi J., Romero N.B., Laporte J.(2013) An integrateddiagnosis strategy for congenital myopathiesPLoS One8: 67527 |
47 | Biesecker L.G., Green R.C.(2014) Diagnostic clinical genome and exome sequencingN Engl J Med370: 24182425 |
48 | Schulze T.G., McMahon F.J.(2004) Defining the phenotype in human genetic studies: Forward genetics and reverse phenotypingHum Hered58: 131138 |
49 | Arif B., Kumar K.R., Seibler P., Vulinovic F., Fatima A., Winkler S., Nurnberg G., Thiele H., Nurnberg P., Jamil A.Z., Bruggemann A., Abbas G., Klein C., Naz S., Lohmann K.(2013) A novel OPA3 mutation revealed by exome sequencing: An example of reversephenotypingJAMA Neurol70: 783787 |
50 | Nguyen K., Putoux A., Busa T., Cordier M.P., Sigaudy S., Till M., Chabrol B., Michel-Calemard L., Bernard R., Julia S., Malzac P., Labalme A., Missirian C., Edery P., Popovici C., Philip N., Sanlaville D.Incidental findings on array comparative genomic hybridization: Detection of carrier females of dystrophinopathy without any family historyClin Genet2014 |
51 | Green R.C., Berg J.S., Grody W.W., Kalia S.S., Korf B.R., Martin C.L., McGuire A.L., Nussbaum R.L., O’Daniel J.M., Ormond K.E., Rehm H.L., Watson M.S., Williams M.S., Biesecker L.G.(2013) ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencingGenet Med15: 565574 |
52 | Evans B.J.(2013) Minimizing liability risks under the ACMG recommendations for reporting incidental findings in clinical exome and genome sequencingGenet Med15: 915920 |
53 | Smith L.A., Douglas J., Braxton A.A., Kramer K.(2014) Reportingincidental findings in clinical whole exome sequencing:incorporation of the ACMG recommendations into currentpractices of genetic counselingJ Genet CounsNov 18. [Epub ahead of print] DOI 10.1007/s10897-014-9794-4 |
54 | Scheuner M.T., Peredo J., Benkendorf J., Bowdish B., Feldman G., FleisherL , Mulvihill J.J., Watson M., Herman G.E., Evans J.(2015) Reporting genomic secondary findings: ACMG members weigh inGenet Med17: 2735 |
55 | Kergourlay V., Rai G., Blandin G., Salgado D., Beroud C., Levy N., Krahn M., Bartoli M.(2014) Identification of splicing defects caused by mutations in the dysferlin geneHum Mutat35: 15321541 |
56 | Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D., Sheth V., Woodward J.E., Peckham H.E., Schroth G.P., Kim R.W., Kingsmore S.F.(2011) Carrier testing for severe childhood recessive diseases by next-generation sequencingSci Transl Med3: 65ra4 |
57 | Xu L., Shi R.(2014) Noninvasive prenatal diagnosis using next-generation sequencingGynecol Obstet Invest77: 7377 |
58 | Beckmann J.S.(2015) Can we afford to sequence every newborn baby’s genome?Hum Mutat36: 283286 |
Figures and Tables
Fig.1
Coverage and depth in MPS. a) Targeted Sequencing (TS) and ExomeSequencing (ES) target exons of a panel of genes or of most genesrespectively. Genome Sequencing (GS) targets the genome and inparticular exons and introns of all genes. TS may be used for thestudy of genomic regions. b) Each region of interest istheoretically sequenced several times by overlapping sequencefragments called reads. When considering a sequence of 10 bases(in blue), in case 7 out of the 10 bases are covered by 5different reads, the coverage of this region is 70% at 5X depth.A potential single nucleotide variant (SNV; C to G transition) isshown at the heterozygous state.
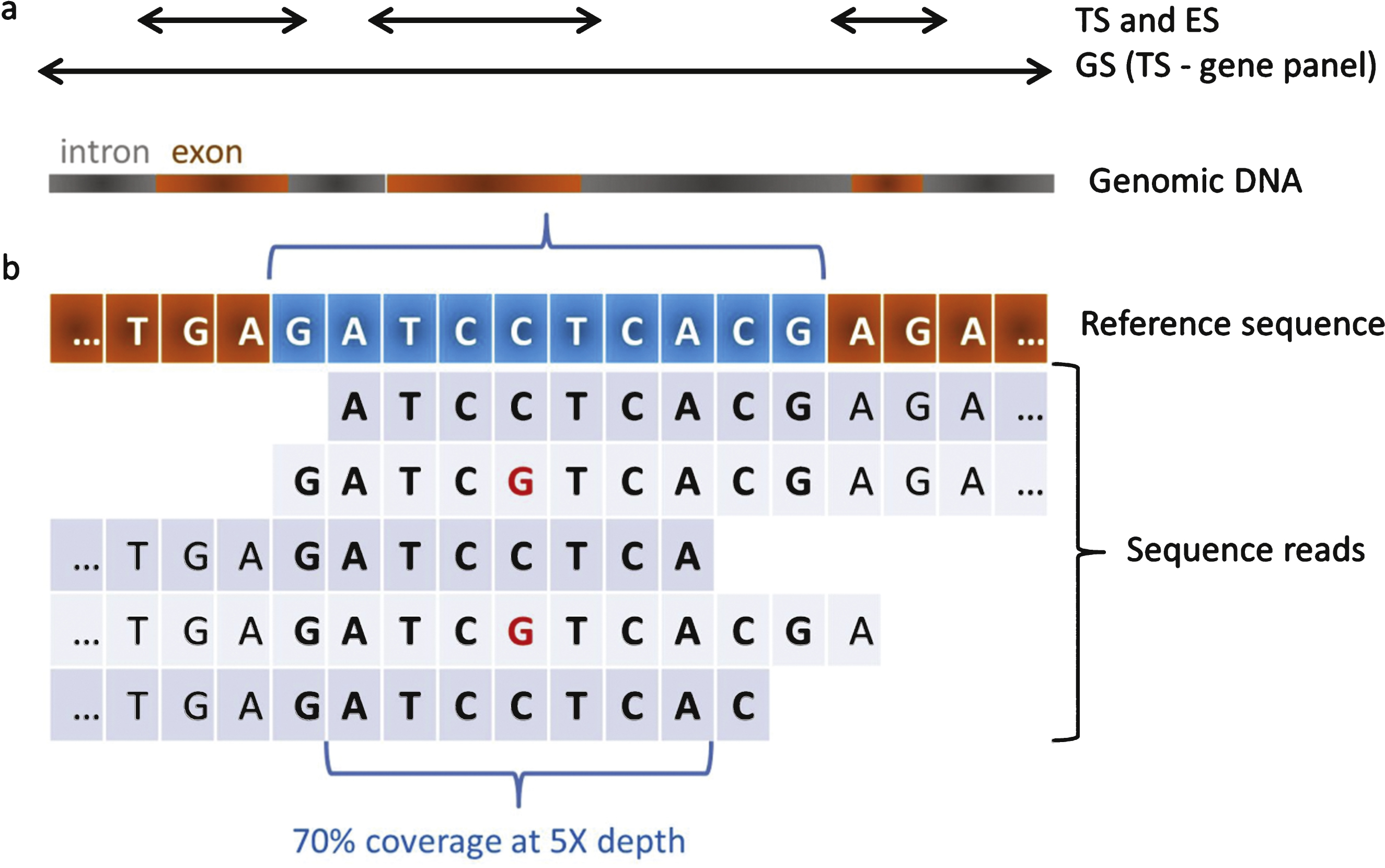
Fig.2
Workflow of informatic analysis following MPS. Each step is monitored with quality scores. Sequence reads are used for mapping to a reference genome, variant calling, filtering and ranking. This unique pipeline is nevertheless prone to end in different conclusions depending on the sequencing platform, algorithms and softwares used, as well as the training and expertise of the users.
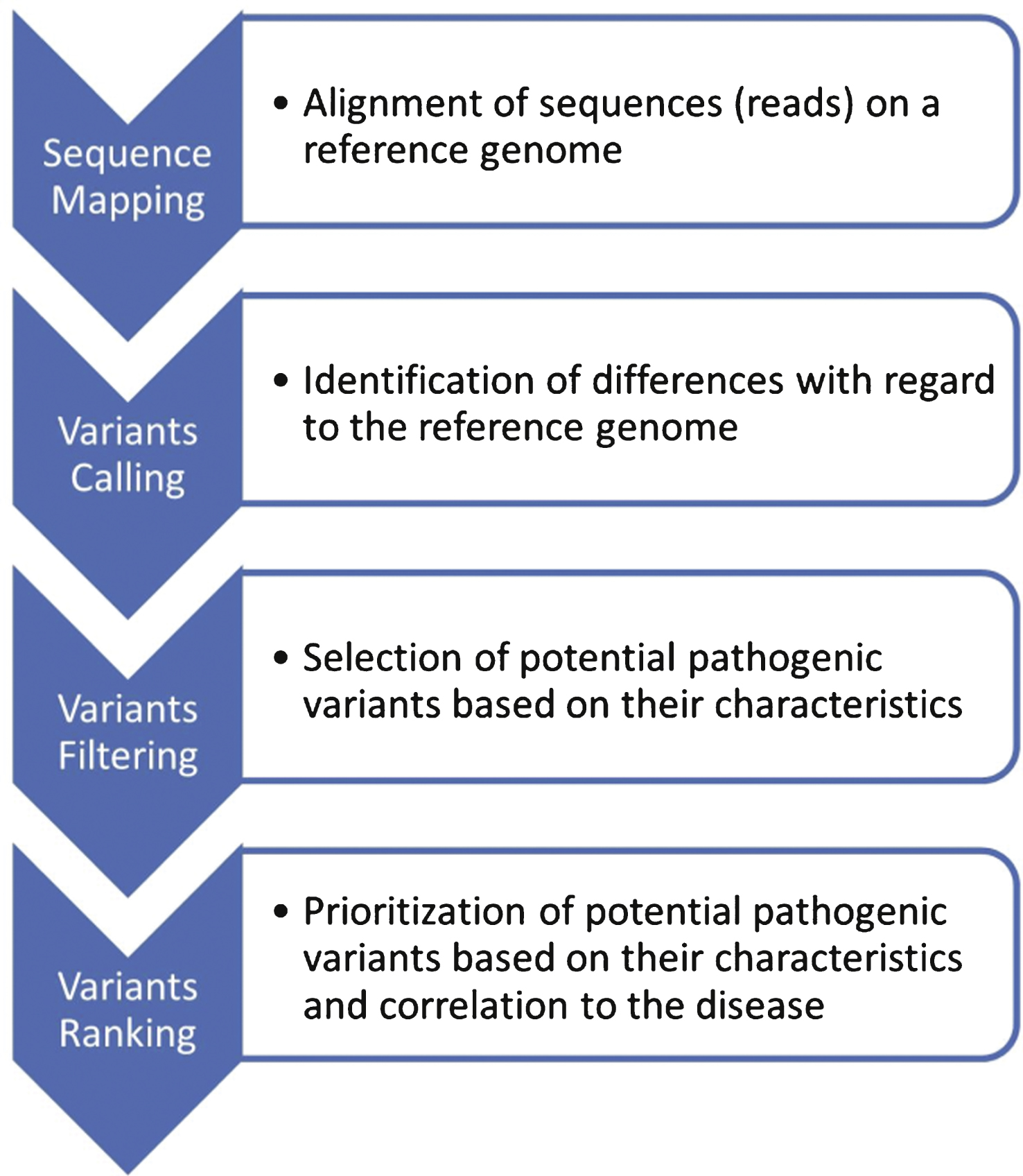
Fig.3
Close integration of clinical, biochemical, medical imaging, histopathological and genetic data for an integrated diagnosis. The whole process implies a great expertise in each disease/gene and in particular may require an expert molecular laboratory for final interpretation of sequence variants. The diagnosis may result in specific patient management including potential existing therapies, and should allow in any case genetic counselling within the family (carrier, predictive, prenatal or pre-implantation testing). Inclusion of data into molecular and clinical databases is a pre-requisite for patients recruitment in clinical trials and to allow development of research projects on the disease.
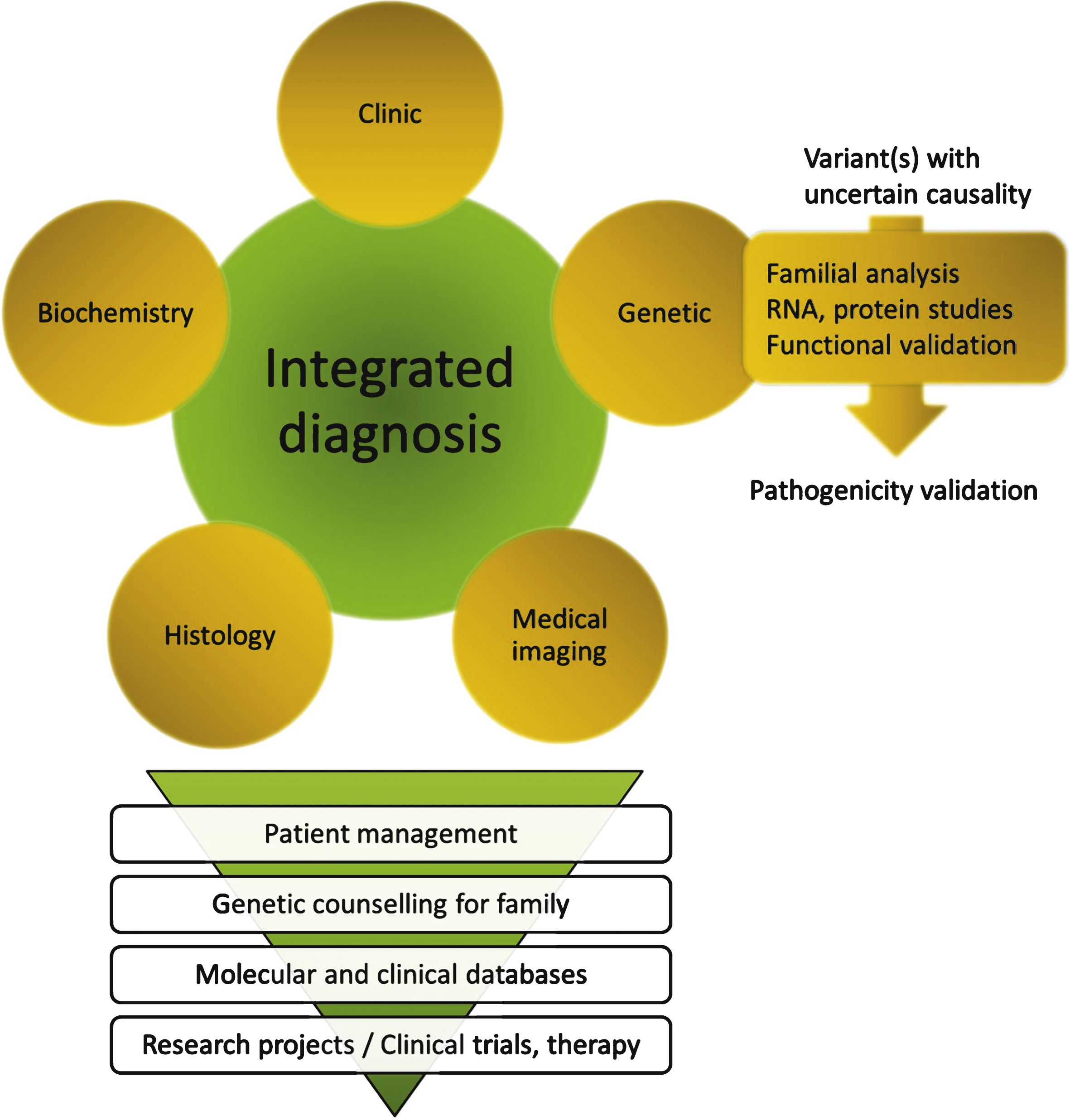
Table 1
Summary of some studies performed in a diagnosis setting. NMD: Neuromuscular Disorder; CMT: Charcot-Marie-Tooth disease; CMD: Congenital Muscular Dystrophy; LGMD: limb-girdle muscular dystrophy
| Author | Approach | Disease | Number of genes | Number unrelated patient | Number definitive diagnosis (%) | Number potential diagnosis, further test required (%) |
| Vasli et al. [7] | Exonic Custom Panel ”NMD-seq” | NMD | 267 | 8 | 4 (50%) | |
| Choi et al. [25] | Exome associated with data filtering of selected genes | CMT | 50 | 25 * | 8 (32%) | |
| Lim et al. [10] | Genomic Custom Panel | CMD with defective glycosylation of alphadystroglycan | 26 | 4 | 2 (50%) | 1 (25%) |
| Nemeth et al. [23] | Exonic Custom Panel | Ataxia | 42 ** | 50 * | 9 (18%) | |
| Savarese et al. [26] | Exonic Custom Panel “MotorPlex” | Non syndromic muscle disorders | 93 | 177 * | 52 (29%) | 56 (32%) |
| Bartoli et al. [27] | Exome associated with data filtering of selected genes | LGMD and distal myopathies | 39 | 37 * | 6 (16%) | |
| Fogel et al. [24] | Clinical Exome | Ataxia | 76 * | 16 (21%) | 30 (40%) | |
| Klein et al. [29] | Exome associated with data filtering of selected genes | Polyneuropathies | 74 | 15 * | 5 (33%) | 3 (20%) |
| Ankala et al. [32] | Exonic Custom Panel | NMD | 41 | 81 | 37 (46%) | |
| CMD | 12 | 88 | 32 (36%) | |||
| LGMD | 11 | 96 | 25 (26%) | |||
| Chae et al. [31] | Exonic Custom Panel | Early onset NMD | 579 ** | 43 * | 21 (49%) | 7 (16%) |
| Drew et al. [30] | Exome associated with data filtering of selected genes | Peripheral neuropathies | 75 | 110 * | 9 (8%) | 12 (11%) |
| Sevy et al. [28] | Exonic Custom Panel | NMD | 298 | 17 * | 2 (12%) | 6 (35%) |
*: patients for whom the best candidate geneshave been previously excluded and in particular: 17p12duplication/deletion and 3 major CMT genes: GJB1, MPZ, MFN2for the 25 patients enrolled by Choi et al.; SCA1,2,3,6,7and FXN for the 50 and 76 patients enrolled respectively byNemeth et al. and Fogel et al.; DYSF gene for the 37patients enrolled by Bartoli et al.; SMN1, DMD, MTM1 andDM1 genes for the 43 patients enrolled by Chae et al.;PMP22, MPZ, GJB1, MFN2, SPTLC1 and SPTLC2 for the110 patients enrolled by Drew et al. **: Nemeth andcolleagues included 76 supplementary candidate genes and Chae andcolleagues included candidate genes not reported as pathogenic intheir respective panels.
Table 2
Advantages and disadvantages of different MPS strategies
| Strategies | Targeted sequencing | Exome sequencing | Genome sequencing |
| Coverage depth | ++ | + | + |
| Range of mutation: | ++ | + | +++ |
| Large rearrangement, … | |||
| Interpretation | +++ | +++ | +++(exon)/+(intron) |
| Ethical issue | + | +++ | +++ |
| or + (targeted analysis) | or + (targeted analysis) | ||
| Data re-analysis for novel disease genes | NO | YES | YES |
| Cost (2015) | + | ++ | +++ |


