Exosomal Protein Deficiencies: How Abnormal RNA Metabolism Results in Childhood-Onset Neurological Diseases
Abstract
Defects of RNA metabolism have been increasingly identified in various forms of inherited neurological diseases. Recently, abnormal RNA degradation due to mutations in human exosome subunit genes has been shown to cause complex childhood onset neurological presentations including spinal muscular atrophy, pontocerebellar hypoplasia and myelination deficiencies. This paper summarizes our current knowledge about the exosome in human neurological disease and provides some important insights into potential disease mechanisms.
INTRODUCTION
Neurodegenerative disorders are a group of debilitating and currently incurable disorders characterised by progressive degeneration and death of neuronal cells. Despite major advances in the factors that trigger neurodegeneration, our understanding of neuronal death pathways in the majority of neurodegenerative disorders is still limited [1]. Among several potential pathways, a precise control of messenger RNA (mRNA) processing and abundance is increasingly being recognized as critical for gene expression and cell survival, particularly in neurons [1].
The human genome is transcribed to produce an extraordinary diversity of RNA, requiring complex regulation. These regulatory events are governed by a large number of trans-acting factors found in neurons, such as RNA-binding proteins, long non-coding RNAs, micro-RNAs, which bind to specific cis-acting elements or structures within mRNAs and thus control different aspects of mRNA metabolism [1].
The degradation of mRNAs is an important regulatory step, which also controls gene expression [2]. Interactions between many different proteins are required for mRNA decay and these pathways are not yet completely understood [3]. The importance of RNA processing in neurodegeneration is highlighted with a rapidly increasing number of human diseases caused by mutations in proteins involved in mRNA metabolism [4–9]. Concentration of mRNAs depends on the balance between transcription and degradation rates [10]. On both sides of the equilibrium, synthesis and degradation show differences that have conditioned the evolution of gene regulatory mechanisms. Furthermore, pre- and posttranscriptional modifications can regulate both transcription and decay rates, thereby maintaining proper mRNA homeostasis [4].
A novel mechanism of RNA-associated neurodegeneration has been suggested by the identification of mutations in genes encoding components of the exosome, a multi-protein complex required for rapid degradation of AU-rich elements (ARE) containing RNAs [9].
mRNA degradation can be classified into two aspects: removal of faulty mRNA species and degradation of correct mRNAs to regulate protein amounts in the cell [10]. Regular cytoplasmic mRNA decay consists of five processes: decapping, 5′-to-3′ exonucleolytic decay, deadenylation, 3′-to-5′ exonucleolytic decay and endonucleolytic cleavage. Nonsense mediated decay is the prime example of a RNA degradation mechanism which acts as quality control for newly synthesised mRNAs and removes aberrant mRNAs with premature stop codons. As many mutations that cause genetic disorders introduce premature termination codons, nonsense mediated decay is an essential factor for most genetic disorders. Variability of nonsense mediated decay can also influence the disease phenotype [11]. Whereas there is a clear link between degradation of aberrant mRNA and human disease, other RNA degradation processes except degradation by the exosome have not been linked to any human genetic disorders yet.
This paper focuses on the RNA degradation by the exosome complex and endeavours to summarise the information on human neurological diseases caused by deficiency of the human exosome and also provide an overview on the potential disease mechanisms.
RNA DEGRADATION BY THE EXOSOME
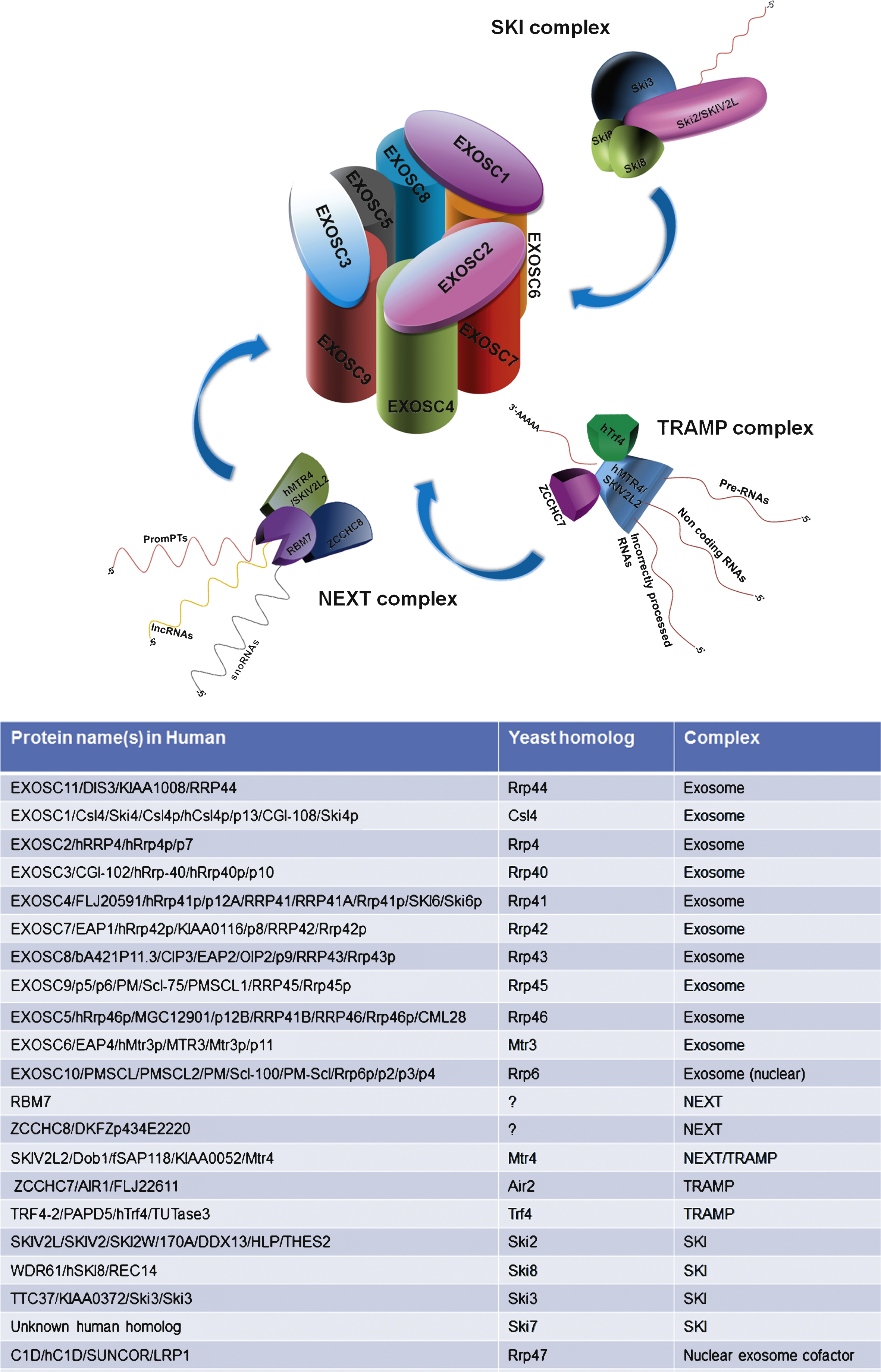
Three major degradation systems are responsible for the destruction of the defective transcripts: the exosome, the 5′-3′ exoribonucleases, and the nonsense-mediated mRNA decay machinery [2]. Some mammalian mRNAs contain ARE within their 3′-untranslated regions. Rapid degradation of ARE-containing RNAs is performed by a multi-protein complex, the exosome. The versatility and specificity of the exosome regulates the activity and maintains the fidelity of gene expression [12]. Nine proteins organize the “exosome core” in a two-layered ring (Fig. 1). The central hexamer channel is composed by six subunits (Rrp41p/EXOSC4, Rrp46p/EXOSC5, Rrp45p/PM/Scl-75/EXOSC9, Rrp42p/EXOSC7, Mtr3/EXOSC6 and Rrp43p/Oip2/EXOSC8), while the cap on the top of the ring consists of three proteins (Rrp4/EXOSC2, Rrp40/EXOSC3, Csl4/EXOSC1), involved in RNA recognition and binding [13]. The exosome is essentially a 3′–5′ exoribonuclease and degrades RNA starting at the 3′ end; in addition to functioning as an endoribonuclease in eukaryotes [14, 15]. The exosome core has no catalytic activity, but it is provided through the association with other proteins (RRP44/DIS3 and RRP6/PM/Scl-100/EXOSC10 ribonucleases).
ARE recognition requires certain ARE binding proteins that can interact with the exosome for recruitment, thereby promoting the rapid degradation of RNAs [16]. The human exosome also regulates gene expression via diverse RNA processing reactions [17]. Many cellular RNAs that play key roles in important cellular processes such as translation (rRNAs, tRNAs and sno(Small Nucleolar)RNAs) and mRNA splicing (sn(Small Nuclear)RNAs) are produced as precursor molecules that are trimmed from their 3′-ends by the human exosome [18]. The exosome may have different roles in the different cellular compartments (nucleus, nucleolus, cytoplasm) [19]. The exosome activity also depends on cofactors, which show specialisation in localisation and targeting. This organization provides the versatility needed for the exosome to cope with the huge variety of RNA substrates in the cell [20]. However, detailed in vivo analyses are technically challenging and many questions remain unresolved.
EXOSOMAL PROTEIN DEFICIENCY AND HUMAN DISEASE
Autosomal recessive mutations in two core exosome subunits, EXOSC3 and EXOSC8, have been identified very recently in patients with complex neurological presentations including pontocerebellar hypoplasia, spinal motor neuron dysfunction and abnormal myelination.
EXOSC3 MUTATIONS
Mutations in EXOSC3 were reported in pontocerebellar hypoplasia and spinal motor neuron abnormalities (PCH) [9]. Many reports of patients with EXOSC3 mutations have been published since the first description of EXOSC3 mutations responsible for PCH type 1 [21–26]. EXOSC3 mutations underlie about half of the PCH1 cases [25]. A broad phenotype range and many different mutations have been identified. The age of onset and the severity of the clinical presentations can be more heterogeneous than previously thought. Spasticity can be also observed and the degree of pontocerebellar or pure cerebellar hypoplasia is very variable with survival in the milder forms into adult age [26].
Different mutations, missense and frame shift/splice site mutations have been described and it has been assumed that the mutations are either null or hypomorphic alleles [9]. Genotype-phenotype correlations indicate that one missense mutation p.Asp132Ala is associated with milder phenotype if present in homozygous state [25]. The patients have a longer life expectancy and preservation of the pons compared to PCH1 patients with other EXOSC3 mutations. Notably, cerebellar cysts were present in three out of four patients compound heterozygous for p.Asp132Ala and another mutation; these patients also suffered from a more severe disease phenotype than the patients homozygous for p.Asp132Ala [25]. Another EXOSC3 mutation, p.Val80Phe, is also associated with a milder phenotype of early onset spasticity, mild intellectual disability, distal amyotrophy, and cerebellar atrophy in a patient of Bangladeshi origin [23]. However, a founder mutation p.Gly31Ala in the Czech Roma population cause a severe form and fatal before 6 months of age [24]. Recently, a novel homozygous EXOSC3 mutation, p.Gly191Cys, was reported to cause autosomal recessive hereditary spastic paraplegia in a consanguineous family of Arab origin [26]. Patients with this mutation survived until adulthood. No patients with complete loss of EXOSC3 (i.e. two null mutations) have been reported yet; a complete loss of EXOSC3 is likely to be lethal. The expression of EXOSC3 protein was not assessed in patient material (cells or tissue) by western blot. It is unclear whether the mutations indeed lead to a reduction of protein amount and if there is any difference among the individual missense mutations.
Knockdown of Exosc3 in zebrafish embryos by antisense morpholino oligonucleotides resulted in embryos of reduced body length, curved spine and a small brain [9]. The embryos displayed poor motility and died around 3 days post fertilization. A shrunken or collapsed hindbrain was detected in exosc3 morphants; whole-mount in situ hybridization further showed a lack of expression of pvalb7, a specific marker for differentiated cerebellar Purkinje neurons. The zebrafish data indicate that normal function of the EXOSC3 component is essential for the survival of cerebellar and spinal motor neurons. Although a large number of patients with EXOSC3 mutations have been described by several groups, very little functional analysis has been performed yet to understand the consequences of EXOSC3 mutations.
EXOSC8 MUTATIONS
Our group identified mutations in another exosome component, EXOSC8, in children with hypomyelination, spinal muscular atrophy and cerebellar hypoplasia [27]. Two homozygous missense mutations, p.Ser272Thr and p.Ala2Val, were identified in patients from three independent families, two of Hungarian Roma origin and one consanguineous Arab–Palestinian family, respectively. These are to date the only two EXOSC8 mutations that have been reported. EXOSC8 mutations seem less frequent than EXOSC3 mutations. One of the mutations, p.Ser272Thr, could be a founder mutation in the Hungarian Roma population. The other mutation was identified in a Palestinian family. No null mutations have been described so far. The patients have slightly different clinical phenotypes but genotype-phenotype correlation is difficult given the low number of patients and mutations.
We have shown by Western Blot analysis of patient myoblasts and fibroblasts that both EXOSC8 mutations lead to a reduction of EXOSC8 protein amount in patient cells, indicating that the missense mutations act as hypomorphic mutations [27].
There is significant overlap of the clinical phenotype between EXOSC3 and EXOSC8 deficient patients but clinical phenotypes are not identical although the two proteins are thought to function together in the same complex. It might be that the missense mutations alter the RNA binding abilities of the exosome and different mutations change binding affinities for a different subset of RNAs. We showed that EXOSC8 is an essential protein of the exosome core, as its depletion caused a severe growth defect in yeast. Experimental down-regulation of EXOSC8 in human oligodendroglia cells and in zebrafish induced a specific increase in the ARE mRNAs encoding myelin proteins. This imbalanced supply of the different myelin proteins caused a disruption of myelin formation and supports the clinical presentation. Downregulation of the EXOSC8 paralog in zebrafish by antisense morpholino oligonucleotide injection resulted in morphants with abnormal head and tail appearance and impaired movement. Examination of the brain and nervous system in exosc8 morphants revealed loss of myelination of axons and a disrupted structure of cranial motor neurons in the morphants with the most severe phenotype. Like previously shown for EXOSC3, the absence of EXOSC8 in zebrafish embryos recapitulated many of the disease characteristics observed in patients.
EXOSOME – ASSOCIATED COMPLEXES
Exosome activity in the nucleus and in the cytosol is assisted by different cofactors and associated proteins; these cofactors determine the specificity of the exosome and are important for distinguishing between RNA molecules that need to be degraded and others that need to be processed. RNA substrates are guided to the exosome with the help of the two associated complexes, the nuclear TRAMP complex (Trf4–Air2–Mtr4 polyadenylation complex) or the cytoplasmic Ski (superkiller) complex (Fig. 1) [28, 29]. The helicase components of these two complexes, Mtr4 and Ski2, are important exosome regulators. Ski2 associates with Ski3 and Ski8 to form the so-called Ski complex; the role of the Ski complex is exosome-mediated cytoplasmic mRNA degradation and mRNA surveillance [29]. The best characterized cofactor of the nuclear exosome in yeast is Trf4p/5p-Air1p/2p- Mtr4p polyadenylation (TRAMP) complex [30]. The TRAMP complex targets nuclear RNA (incorrectly processed RNAs, rRNAs tRNAs, as well as non-coding RNAs) to the exosome for degradation or processing.
In mammals, two distinct nuclear exosome cofactors have so far been characterized, which are the nucleolar- localized hTRAMP complex [31] and the nucleoplasm-specific Nuclear Exosome Targeting (NEXT) complex [32]. hMTR4 is shared between the two complexes. The proteins RBM7, hMTR4 and ZCCHC8, form the NEXT complex [33], which is specifically involved in the exosomal degradation of PROMoter uPstream Transcripts (PROMPTs) [34]. These sequences are produced only upstream of the promoters of active protein coding genes and are rapidly turned over by the exosome complex [34]. Although the general function of PROMPTs remains unknown, it seems they might be involved in fine tuning of gene expression [34, 35].
The Ski complex is an obligatory co-factor of the RNA exosome in the cytoplasm in yeast; the components of the human SKI complex have not yet been well characterised. Mutations in the SKI complex components SKIV2L or TTC37 (thought to be the human paralogs of Ski2 and Ski3, respectively) cause a rare genetic condition named trichohepatoenteric syndrome; SKIV2L and TTC37 mutations have recently been reviewed in Fabre et al. [29]. No mutations in TRAMP or NEXT complex components have been identified yet in human disease.
CONCLUSIONS
Early-onset and progressive neurological presentations affecting motor neurons, cerebellar Purkinje cells and oligodendroglia are typical for mutations in exosomal proteins and highlight the importance of exosome function in developing neurons. Despite the complex and central role of the exosome and its associated complexes in RNA metabolism in all cell types it is so far not known why and how only neuronal cells are affected by exosomal protein deficiency. The exosome may be also important for degradation of various populations of non-coding RNA [36, 37]. Accumulation of certain non-coding RNAs in addition to mRNAs might contribute to disease mechanism. Non-coding RNA expression is even more cell type specific than mRNA expression, which could explain why some neuronal cell types are more affected by exosome mutations. Identifying the exact biochemical pathways of RNA metabolism alterations due to different types of exosome dysfunction may explain why mutations in separate components of the exosome cause different disease presentations, which is surprising given the fact that exosomal components function together in a single protein complex. Other genetic forms of pontocerebellar hypoplasias are also caused by mutations in RNA processing molecules, such as tRNA splicing endonuclease subunit genes (PCH2, PCH4, PCH5) or mitochondrial tRNA synthetases (PCH6) [8]. Splicing of the pre-mRNAs by the spliceosome depends on small nuclear ribonucleoproteins (snRNPs), which require Spinal Motor Neuron 1 (SMN1) protein for their assembly and defect of SMN1 results in spinal muscular atrophy (SMA), a leading cause of infantile mortality [6]. The fact that defects in RNA processing and degradation can cause severe neurological disorders emphasises the role of the RNA metabolism for the development and maintenance of cells in the nervous system. It remains to be determined why neuronal cells are more vulnerable to changes in RNA levels than other cell types.
ACKNOWLEDGMENTS
JM is supported by the MRC Confidence in Concept funding. MG is supported by the Mitochondrial European Educational Training (MEET), ITN MARIE CURIE PEOPLE, (317433). RH is supported by the Medical Research Council (UK) (G1000848) and the European Research Council (309548).
REFERENCES
1 | Smith R, Rathod RJ, Rajkumar S, Kennedy D(2014) Nervous translation, do you get the message? A review of mRNPs, mRNA-protein interactions and translational control within cells of the nervous systemCell Mol Life Sci71: 2039173937 |
2 | Lehner B, Sanderson CM(2004) A protein interaction framework for humanmRNA degradationGenome Res14: 713151323 |
3 | Estevez AM, Lehner B, Sanderson CM, Ruppert T, Clayton C(2003) The roles of intersubunit interactions in exosome stabilityJ Biol Chem278: 373494334951 |
4 | Pérez-Ortín JE, Alepuz P, Chávez S, Choder M(2013) Eukaryotic mRNA decay: Methodologies, pathways, and links to other stages of gene expressionJ Mol Biol425: 2037503775 |
5 | Ascano MJr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, Williams Z, Ohler U, Tuschl T(2012) FMRP targetsdistinct mRNA sequence elements to regulate protein expressionNature492: 7429382386 |
6 | Sleeman J(2013) Small nuclear RNAs and mRNAs: Linking RNA processing and transport to spinal muscular atrophyBiochem Soc Trans41: 4871875 |
7 | Ihara R, Matsukawa K, Nagata Y, Kunugi H, Tsuji S, Chihara T, Kuranaga E, Miura M, Wakabayashi T, Hashimoto T, Iwatsubo T(2013) RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathyHum Mol Genet22: 2244748444 |
8 | Namavar Y, Barth PG, Poll-The BT, Baas F(2011) Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasiaOrphanet J Rare Dis6: 50 |
9 | Wan J, Yourshaw M, Mamsa H, Rudnik-Schöneborn S, Menezes MP, Hong JE, Leong DW, Senderek J, Salman MS, Chitayat D, Seeman P, von Moers A, Graul-Neumann L, Kornberg AJ, Castro-Gago M, Sobrido MJ, Sanefuji M, Shieh PB, Salamon N, Kim RC, Vinters HV, Chen Z, Zerres K, Ryan MM, Nelson SF, Jen JC(2012) Mutations in the RNA exosomecomponent gene EXOSC3 cause pontocerebellar hypoplasia and spinalmotor neuron degenerationNat Genet44: 6704708 |
10 | Schoenberg DR1, Maquat LE(2012) Regulation of cytoplasmic mRNA decayNat Rev Genet13: 4246259 |
11 | Miller JN, Pearce DA(2014) Nonsense-mediated decay in genetic disease:Friend or foe?Mutat Res Rev Mutat Res762: 5264 |
12 | Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M(2001) AU binding proteinsrecruit the exosome to degrade ARE-containing mRNAsCell107: 4451464 |
13 | Makino DL, Halbach F, Conti E(2013) The RNA exosome and proteasome: Common principles of degradation controlNat Rev Mol Cell Biol14: 10654660 |
14 | Evguenieva-Hackenberg E, Roppelt V, Finsterseifer P, Klug G(2008) Rrp4and Csl4 are needed for efficient degradation but not forpolyadenylation of synthetic and natural RNA by the archaealexosomeBiochemistry47: 501315813168 |
15 | Hartung S, Niederberger T, Hartung M, Tresch A, Hopfner KP(2010) Quantitative analysis of processive RNA degradation by thearchaeal RNA exosomeNucleic Acids Res38: 1551665176 |
16 | Niederberger T, Hartung S, Hopfner KP, Tresch A(2011) Processive RNAdecay by the exosome: Merits of a quantitative Bayesian samplingapproachRNA Biol8: 15560 |
17 | Coy S, Volanakis A, Shah S, Vasiljeva L(2013) The Sm complex isrequired for the processing of non-coding RNAs by the exosomePLoS One8: 6e65606 |
18 | Milligan L, Torchet C, Allmang C, Shipman T, Tollervey D(2005) A nuclear surveillance pathway for mRNAs with defective polyadenylationMol Cell Biol25: 22999610004 |
19 | Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, Jacquier A, Libri D(2012) Extensive degradation of RNA precursors bythe exosome in wild-type cellsMol Cell48: 3409421 |
20 | Schneider C, Tollervey D(2013) Threading the barrel of the RNA exosomeTrends Biochem Sci38: 10485493 |
21 | Rudnik-Schöneborn S, Senderek J, Jen JC, Houge G, Seeman P, Puchmajerová A, Graul-Neumann L, Seidel U, Korinthenberg R, Kirschner J, Seeger J, Ryan MM, Muntoni F, Steinlin M, Sztriha L, Colomer J, Hübner C, Brockmann K, Van Maldergem L, Schiff M, Holzinger A, Barth P, Reardon W, Yourshaw M, Nelson SF, Eggermann T, Zerres K(2013) Pontocerebellar hypoplasia type Clinical spectrum and relevance of EXOSC3 mutationsNeurology80: 5438446 |
22 | Biancheri R, Cassandrini D, Pinto F, Trovato R, Di Rocco M, Mirabelli-Badenier M, Pedemonte M, Panicucci C, Trucks H, Sander T, Zara F, Rossi A, Striano P, Minetti C, Santorelli FM(2013) EXOSC3 mutations in isolated cerebellar hypoplasia and spinal anterior horn involvementJ Neurol260: 718661870 |
23 | Zanni G, Scotton C, Passarelli C, Fang M, Barresi S, Dallapiccola B, Wu B, Gualandi F, Ferlini A, Bertini E, Wei W(2013) Exome sequencing in a family with intellectual disability, early onset spasticity, and cerebellar atrophy detects a novel mutation in EXOSC3Neurogenetics14: 3-4247250 |
24 | Schwabova J, Brozkova DS, Petrak B, Mojzisova M, Pavlickova K, Haberlova J, Mrazkova L, Hedvicakova P, Hornofova L, Kaluzova M, Fencl F, Krutova M, Zamecnik J, Seeman P(2013) Homozygous EXOSC3 mutation c92G⟶C, p.G31A is a founder mutation causing severe pontocerebellar hypoplasia type 1 among the Czech RomaJ Neurogenet27: 4163169 |
25 | Eggens VR, Barth PG, Niermeijer JM, Berg JN, Darin N, Dixit A, Fluss J, Foulds N, Fowler D, Hortobágyi T, Jacques T, King MD, Makrythanasis P, Máté A, Nicoll JA, O’Rourke D, Price S, Williams AN, Wilson L, Suri M, Sztriha L, Dijns-de Wissel MB, van Meegen MT, van Ruissen F, Aronica E, Troost D, Majoie CB, Marquering HA, Poll-Thé BT, Baas F(2014) EXOSC3 mutations in pontocerebellar hypoplasia type Novel mutations and genotype-phenotype correlationsOrphanet J Rare Dis9: 23 |
26 | Halevy A, Lerer I, Cohen R, Kornreich L, Shuper A, Gamliel M, Zimerman BE, Korabi I, Meiner V, Straussberg R, Lossos A(2014) Novel EXOSC3 mutation causes complicated hereditary spastic paraplegiaJ Neurol261: 1121652169 |
27 | Boczonadi V, Müller JS, Pyle A, Munkley J, Dor T, Quartararo J, Ferrero I, Karcagi V, Giunta M, Polvikoski T, Birchall D, Princzinger A, Cinnamon Y, Lützkendorf S, Piko H, Reza M, Florez L, Santibanez-Koref M, Griffin H, Schuelke M, Elpeleg O, Kalaydjieva L, Lochmüller H, Elliott DJ, Chinnery PF, Edvardson S, Horvath R(2014) EXOSC8 mutations alter mRNA metabolism andcause hypomyelination with spinal muscular atrophy and cerebellarhypoplasiaNat Commun5: 4287 |
28 | Losh JS, King AK, Bakelar J, Taylor L, Loomis J, Rosenzweig JA, Johnson SJ, van Hoof A(2015) Interaction between the RNA-dependentATPase and poly(A) polymerase subunits of the TRAMP complex ismediated by short peptides and important for snoRNA processingNucleic Acids Res43: 318481858 |
29 | Fabre A, Badens C(2014) Human Mendelian diseases related to abnormalities of the RNA exosome or its cofactorsIntractable Rare Dis Res3: 1811 |
30 | Falk S, Weir JR, Hentschel J, Reichelt P, Bonneau F, Conti E(2014) The molecular architecture of the TRAMP complex reveals the organization and interplay of its two catalytic activitiesMol Cell55: 6856867 |
31 | Fasken MB, Leung SW, Banerjee A, Kodani MO, Chavez R, Bowman EA, Purohit MK, Rubinson ME, Rubinson EH, Corbett AH(2011) Air1 zinc knuckles 4 and 5 and a conserved IWRXY motif are critical for the function and integrity of the Trf4/5-Air1/2-Mtr4 polyadenylation (TRAMP) RNA quality control complexJ Biol Chem286: 433742937445 |
32 | Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH(2011) Interaction profiling identifies the humannuclear exosometargeting complexMol Cell43: 4624637 |
33 | Preker P, Almvig K, Christensen MS, Valen E, Mapendano CK, Sandelin A, Jensen TH(2011) PROMoter uPstream Transcripts sharecharacteristics with mRNAs and are produced upstream of all threemajor types of mammalian promotersNucleic Acids Res39: 1671797193 |
34 | Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH(2008) RNA exosome depletionreveals transcription upstream of active human promotersScience322: 590918511854 |
35 | Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN, Basu U(2014) Noncoding RNA transcription targets AID to divergently transcribed loci in B cellsNature514: 7522389393 |
36 | Lubas M, Andersen PR, Schein A, Dziembowski A, Kudla G, Jensen TH(2015) The Human Nuclear Exosome Targeting Complex Is Loaded onto NewlySynthesized RNA to Direct Early RibonucleolysisCell Re10: 2178192 |
37 | Wang Y, Colonna M(2014) RNA exosomes keep endogenous RNA under the radarNat Immunol15: 9830831 |
Figures and Tables
Fig.1
Schematic representation of the human exosome and its interacting complexes and the list of genes encoding the proteins involved in these complexes.