A Premature Stop Codon in MYO18B is Associated with Severe Nemaline Myopathy with Cardiomyopathy
Abstract
Background: Nemaline myopathies (NM) are rare and severe muscle diseases characterized by the presence of nemaline bodies (rods) in muscle fibers. Although ten genes have been implicated in the etiology of NM, an important number of patients remain without a molecular diagnosis.
Objective: Here we describe the clinical and histopathological features of a sporadic case presenting with severe NM and cardiomyopathy. Using exome sequencing, we aimed to identify the causative gene.
Results: We identified a homozygous nonsense mutation in the last exon of MYO18B, leading to a truncated protein lacking the most C-terminal part. MYO18B codes for an unconventional myosin protein and it is mainly expressed in skeletal and cardiac muscles, two tissues severely affected in the patient. We showed that the mutation does not impact on mRNA stability. Immunostaining and Western blot confirmed the absence of the full-length protein.
Conclusion: We propose MYO18B as a novel gene associated with nemaline myopathy and cardiomyopathy.
INTRODUCTION
Congenital myopathies are rare monogenicdisorders characterized by distinctive morphologic abnormalities in skeletal muscle [1, 2]. Nearly half of the congenital myopathy patients still await molecular characterization and cannot benefit from adequate genetic counselling and healthcare delivery [1]. The identification of additional implicated genes is not only important for molecular diagnosis, but also suggests potential drug targets.
Nemaline myopathy (NM) is a congenital muscle disorder associating hypotonia, muscle weakness, and often skeletal deformities with the presence of numerous nemaline bodies (rods) in muscle fibers [3].The disorder has a marked clinical variability, ranging from neonatal lethality to mild and non-progressive forms with childhood and adulthood onset [4]. To date, ten genes have been implicated in NM,(ACTA1, MIM#161800; NEB, MIM#256030; TPM2, MIM#609285; TPM3, MIM#609284; TNNT1, MIM#605355; KBTBD13, MIM#609273; CFL2, MIM#610687; KLHL40 MIM#615340; KLHL41 MIM#615731, and LMOD3 MIM#616112), encoding proteins of the skeletal muscle thin filament or Kelch domain containing proteins [5–14].
Although dilated and hypertrophic cardiomyopathies have been previously reported in some nemaline myopathy patients [15–17], NM is not typically associated with cardiomyopathy. The majority of the reported cases were genetically undiagnosed, apart from three cases with de novo ACTA1 mutations, presenting congenital NM and hypertrophic cardiomyopathy (HCM), or dilated cardiomyopathy (DCM) [18–20].
With the aim to identify novel genes responsible for NM with cardiomyopathy, we performed exome sequencing in a severe sporadic case. We found a premature stop codon in the C-terminal part of the unconventional myosin MYO18B, and we confirmed the presence of a truncated protein on muscle biopsy. We propose MYO18B as a novel NM gene, which should be considered as candidate gene for severe cases, especially if a cardiomyopathy is diagnosed.
MATERIALS AND METHODS
Ethical issues, biopsy, histology, and electron microscopy
The parents of the index patient gave informed consent for the genetic analysis and muscle biopsy according to French legislation (Comité de Protection des Personnes Est IV DC-2012-1693). Genomic DNA was extracted from blood by standard methods.
The index patient underwent an open muscle biopsy from vastus lateralis at 14 days of life. For conventional histochemical techniques, 10μm thick cryostat sections were stained with modified Gomori trichrome (mGT). Haematoxylin and eosin (H&E), Periodic acid Schiff technique (PAS), Oil red O, reduced nicotinamide adenine dinucleotide dehydrogenase-tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), cytochrome c oxidase (COX), and adenosine triphosphatase (ATPase) at pH 9.4, 4.63, 4.35 staining were also performed, but are not shown. Pictures of each section were obtained with a Zeiss AxioCam HRc linked to a Zeiss Axioplan Bright Field Microscope and processed with the Axio Vision 4.4 software (Zeiss, Germany).
Resin-embedded blocks for ultrastructural analyses were not prepared at time of the muscle biopsy. We therefore fixed frozen muscle sections (40μm) in osmium tetroxide (1% ), dehydrated and embedded them in epoxy resin (EMBed-812, ElectronMicroscopy Sciences, USA). Ultra-thin sections (80 nm) were stained with uranyl acetate and lead citrate. The grids were analyzed with a Philips CM120 electron microscope (80 kV; Philips Electronics NV, Eindhoven, The Netherlands) and imaged using a Morada digital camera (Soft Imaging System, France).
Exome sequencing, variants prioritization and mutation analysis
The nemaline myopathy genes ACTA1, NEB, TPM2, TPM3, and TNNT1, as well as spinal muscular atrophy (SMA), Prader-Willi syndrome, and myotonic dystrophy-1 (DM1) were excluded by conventional techniques. Exome sequencing was performed on DNA samples from the index patient on a HiSeq 2000 (Illumina, San Diego, USA) using the Agilent 44M v2 SureSelect Exon enrichment kit at the BGI (Shenzhen, China). Variant calling was performed with the SOAP software. The mean coverage of the exome was 57x, and the percentage of the exome covered at least 20x was 77.2% . Variants with a frequency <20% of the total reads for a specific position were excluded. Variant filtering was performed by comparison with SNV databases as dbSNP, Exome Variant Server and EXAC, and polymorphisms with a minor allele frequency (MAF) of more than 0.5% were discarded. Variant prioritization was done with the VaRank program [21]. Sanger sequencing was performed as reported [22]. The position of the mutation was numbered according to NM_032608.5 and NP_115997.5.
RT-PCR
The patient’s RNA was extracted from the muscle biopsy with TRI-Reagent (Sigma, St. Louis, USA), and reverse transcribed using the SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, USA) and random hexamer primers following the manufacturer’s protocol. MYO18B-specific primers encompassing several exons and the mutation were used to assess mRNA steady state levels. NEB primers were used as internal controls.
Immunolabeling and protein analysis
Immunofluorescence analyses were performed on frozen muscle samples from the index patient and a control biopsy without histopathological lesions. Anti-myosin 18B C-ter (Sigma, St. Louis, MO, USA), anti-myosin 18B C-ter (GTX104872, GeneTex, Euromedex, Paris France) and anti-myosin 18B N-ter polyclonal (kind gift from Professor Gerolamo Lanfranchi, University of Padua, Italy) antibodies, were applied on 10-μm-thick cryosections overnight at 4°C. The next day, the sections were incubated with appropriate conjugated secondary antibodies following routine protocols. Slides without primary antibodies were used as controls.
Western blot was performed on skeletal muscle protein extracts by routine methods using the anti-myosin 18B C-ter antibody (Sigma), as well as an anti-myosin heavy chain antibody (Sigma. St. Louis, USA) as loading control.
RESULTS
Clinical findings
The index patient was a girl born to healthy consanguineous parents of Portuguese origin. The mother reported three previous miscarriages. Pregnancy was uneventful until the discovery of a cardiomyopathy characterized by reduced left ventricle and aortic valve growth during a routine ultrasound at 22 weeks of gestation. Birth took place at term by natural vaginal delivery. The weight was 2830 grams (-2 SD),the height 49 cm, and the cranial circumference 32.5 cm. Physical examination revealed severe axial and peripheral hypotonia, reduced spontaneous peripheral movements, tendon contractures, and absence of nursing necessitating nasogastric feeding (Fig. 1A, B, and C). The patient also presented dysmorphic features including transversal enlargement of the skull, small and horizontal palpebral fissure, high-arched palate, low-set ears, pectum excavatum, large and lowly implanted thumbs (Fig. 1A), and clinodactyly of both fourth and fifth fingers of the hands. Cardiac ultrasound confirmed the presence of small and mildly hypertrophic left cavities, compensatory dilatation of right cavities, interatrial communication, and pulmonary hypertension. The patient was tachycardic (170 bt/min), and a therapy with diuretics was started. The discontinuation of diuretics at one month of age precipitated hemodynamic instability leading to the sudden development of acute lung edema promptly recuperated with high doses of diuretics and digitalis treatment. Cerebral MRI showed minimal signs of subependimal hemorrhage. EEG was normal. Karyotype was normal, and spinal muscular atrophy (SMA), Prader-Willi syndrome, and neonatal myotonic dystrophy-1 (DM1) were excluded by molecular analysis. Metabolic work-up including serum and CRL lactate assessment, mitochondrial respiratory chain complexes enzymatic assay, chromatography of blood and urinary aminoacids and urinary organic acid was normal. The course was progressive with degradation of the hemodynamic and respiratory status, and failure to thrive. The patient deceased at 4.5 months following a septic shock. Autopsy was not performed.
Muscle morphology
Muscle biopsy revealed marked fiber size variability, and a contingent of atrophic muscle fibers was observed. A few of them had a single centralized nucleus. Modified Engel-Gomori trichrome staining revealed the presence of fuchsinophilic inclusions in around 40% of the muscle fibers (Fig. 1D). The inclusions corresponded to small nemalinebodies (minirods), found both in subsarcolemmal areas and dispersed in the cytoplasm. The minirods occupied most of the cytoplasm of marked atrophic fibers (Fig. 1D; asterisk). Oxidative techniques failed to show unevenly stained areas or negative COX fibers. Ultrastructural studies confirmed the presence of minirods presenting the typical lattice structure of Z-disc material (Fig. 1E). In conclusion, the histopathological and ultrastructural findings were strongly suggestive of nemaline myopathy.
Identification of a MYO18B-homozygous premature stop codon in the patient
There are currently 10 known genes implicated in nemaline myopathy. However, a large number of nemaline myopathy patients do not harbor mutations in any of those genes, indicating that additional genes remain to be discovered. Owing to the fact that the major nemaline myopathy genes ACTA1, NEB, TPM2, TPM3, and TNNT1 have previously been excluded byconventional techniques, we performed exome sequencing on DNA samples from the patient. Data analysis did not reveal any potentially pathogenic sequence aberration in any of the 10 well covered nemaline myopathy genes. We detected a homozygous nonsense mutation in MYO18B (c.6496G>T; p.Glu2166*), residing within a large homozygous region encompassing the entire MYO18B gene on chromosome 22. The variant was confirmed by Sanger sequencing of a second DNA sample from the patient. The mutation was not listed in the human variation databases as EVS, dbSNP or ExAC, or in our in-house database encompassing 676 exomes. Segregation analysis revealed that both healthy parents carry the mutation in the heterozygous state (Fig. 2A and 2B). The index patient did not have siblings, and DNA samples from the miscarriages or other relatives were not available. The mutation creates a premature stop codon in the last of the 42 coding exons of MYO18B, and is predicted to remove the last 401 amino acids out of 2567 at the protein level. In the human variation databases, MYO18B nonsense or frameshift variants leading to a potential protein truncation are rare and never found to be homozygous. This goes along with the idea that heterozygous carriers are healthy and that homozygous mutations may be associated with disease. Other rare homozygous variants detected in the patient’s exome are listed in Supplemental Table 1.
The MYO18B premature stop codon does not involve major nonsense mRNA decay
Premature stop codons most often trigger nonsense-mediated mRNA decay (NMD), except if they occur in the last or second but last exon [23]. In order to assess whether the MYO18B p.Glu2166* variant affects the mRNA steady state level, we extracted mRNA from the patient’s muscle biopsy, reverse transcribed it into cDNA, and amplified a fragment encompassing the MYO18B exons 40 to 42 and the mutation. No difference was detected in the signal intensity between the patient and an age-matched control (Fig. 2C), suggesting that the overall cDNA level was comparable and that the MYO18B mRNA is not subject to major degradation through NMD.
The MYO18B premature stop codon leads to a truncated protein
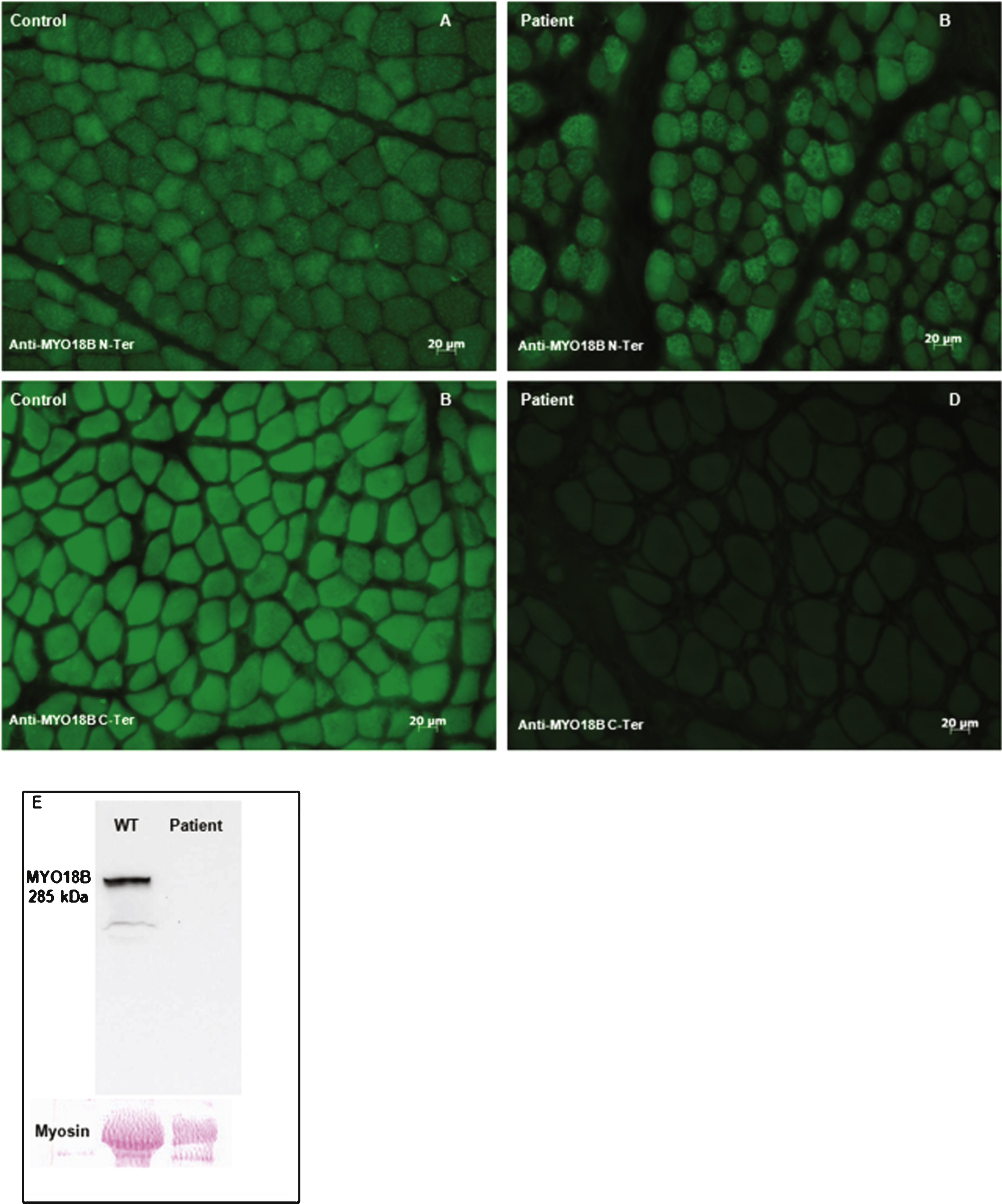
In order to verify the presence of the predicted truncated myosin 18B protein in the patient’s muscle, we performed immunofluorescence on muscle sections using N-terminal and C-terminal antibodies. Immunolabeling with an N-terminal antibody did not reveal any difference in protein localization and signal intensity between the patient and age-matched controls (Fig. 3A, and B). Myosin 18B appeared to be nuclear and cytoplasmic. In a subset of fibers, probably type 2 fibers, we detected a more intense punctate cytoplasmic signal. Immunolabeling using the C-terminal myosin 18B antibody revealed absence of staining in the patient as compared to the control (Fig. 3C, and D). Western blot analysis using theC-terminal myosin 18B antibody revealed a 285 kDa band corresponding to myosin 18B in protein extracts from an age-matched muscle biopsy, while no signal was detectable in the biopsy from the index patient. Taken together, our results suggest the presence of a truncated myosin 18B protein deprived of its most C-terminal part.
DISCUSSION
Here we propose MYO18B as novel gene associated with congenital nemaline myopathy. The index patient presented with severe hypotonia, muscle weakness and cardiomyopathy. Muscle histopathology was indicative of nemaline myopathy, and exome sequencing detected a homozygous nonsense mutation in MYO18B leading to a truncated protein. The index patient was part of a cohort analyzed by exome sequencing and encompassing 34 patients with mild nemaline myopathy, and 17 unrelated patients with severe nemaline myopathy, including two patients with additional cardiomyopathy. The index patient was the only one harboring a MYO18B mutation.
Following lines of evidence suggest the implication of the MYO18B mutation in the development of the disease: we detected a homozygous nonsense mutation in MYO18B by exome sequencing, immunolabeling confirmed the presence of a truncated protein, and Western blot using a C-terminal antibody showed the absence of the full-length protein. Both healthy parents were heterozygous for the mutation, confirming autosomal recessive disease inheritance. In accordance, homozygous MYO18B truncations were not listed in the public exome datasets. MYO18B encodes an unconventional myosin specifically expressed in skeletal and cardiac muscle, the two main affected tissues in our patient [24]. Moreover, Myo18b knockout mice were embryonic lethal, and presented a cardiac dysfunction similar to the patient [25].
Interestingly, a recent paper described two patients with Klippel-Feil anomaly and severe neonatal muscle weakness harboring a homozygous MYO18B nonsense mutation [26]. Muscle examinations detected the presence of nemaline bodies, confirming MYO18B as novel gene for nemaline myopathy. Additional clinical features, in particular cervical spine fusion, were diagnosed during childhood for the Klippel-Feil patients, while our patient deceased at 4.5 months of age. A cardiomyopathy as in our patient was however not diagnosed in the Klippel-Feil patients, and this difference might be mutation-specific. Indeed, we detected a truncated myosin 18B protein in muscle extracts of our patient, while the Klippel-Feil mutation was shown to involve nonsense-mediated mRNA decay (NMD), resulting in total myosin 18B protein loss. These findings suggest a toxic effect of truncated myosin 18B in heart, but the exact pathomechanisms remain to be uncovered.
An association of rods with cardiomyopathy has been reported in less than 20 patients, all presenting general skeletal muscle involvement as primary manifestations [20]. Ten patients developed cardiac failure in adulthood, and six manifested cardiac involvement in infancy or childhood. A molecular diagnosis was available only for three pediatric patients [18–20], all carrying dominant de novo mutations in ACTA1. Two of them manifested hypertrophic cardiomyopathy (HCM) [18, 19], and one childhood-onset dilated cardiomyopathy [20]. Intriguingly, HCM was the revealing symptom in the patient reported by D’Amico et al. [18].
The cardiac disease encountered in our case was rather peculiar and consisted of antenatal reduced growth of the left ventricle and aortic valve. Soon after birth, the presence of small and mildly hypertrophic left cavities, compensatory dilatation of right cavities, and interatrial communication were noted by ultrasound. If the muscle symptoms, as frequently observed in congenital nemaline myopathy, remained stable, the cardiac phenotype progressed rapidly with the development of congestive heart failure. The presence of a complex cardiac disorder combining multiple malformations and HCM suggested a cardiac myogenesis/development dysfunction. Our patient deceased more than twenty years ago and, histologic studies on autoptic cardiac muscle were not performed.
In addition to the cardiac involvement, dysmorphic features and clinodactyly may represent specific features related to the MYO18B truncation, and these signs might be helpful in orientating molecular investigations towards MYO18B in patients with nemaline myopathy. MYO18B codes for an unconventional muscle myosin heavy chain identified for the first time in 2003 [24]. The protein is mainly expressed in human cardiac and skeletal muscle, and at lower level in testis. In mature human skeletal and cardiac muscle,it is localized in both nucleus and cytoplasm [24]. Although myosin 18B contains two overlapping C-terminal nuclear localization signals, we did not observe any difference in nuclear versus cytoplasmic localization between the patient’s and the control biopsy. Conventional myosins are known to be localized at the A-bands and function as a molecular motor for muscle contraction. In contrast, myosin 18B was found at the Z-lines of myofibrils in murine striated muscles [25]. Noteworthy, most proteins previously found mutated in nemaline myopathy are located at the Z-lines and are implicated in the F-actin pathway. The sarcomeric localization of myosin 18B suggests a possible role in functioning and/or maintenance of the contractile apparatus, and might explain the presence of nemaline bodies encountered in the patient.
Several of the most recent genes implicated in nemaline myopathy encode proteins presumably implicated in the protein degradation pathway, as KLHL40 and KLHL41. Both belong to the BTB-BACK-kelch (BBK) family of proteins, some of which have been shown to promote degradation of their substrates [12, 27]. It should be noted that myosin 18B interacts with the proteasomal subunit Sug1 and is degraded by the ubiquitin-proteasome pathway [28], supporting the hypothesis that myosin 18B may link the contractile apparatus and protein degradation, representing the two main pathways implicated in nemaline myopathy to date.
In conclusion, our clinical, histological, ultrastructural, genetic, immunolabeling, and protein studies point to MYO18B as a novel nemaline myopathy gene. The identification and clinical comparison of additional patients with MYO18B mutations will be needed to clearly define the clinical spectrum of this novel entity.
Databases
Following public databases were used to assess the prevalence of the identified MYO18B mutation:ExAC (http://exac.broadinstitute.org/), EVS (http://evs.gs.washington.edu/EVS/), and dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/).
FUNDING
This work was supported in part by the Assistance Publique-Hôpitaux de Paris (AP-HP), the Institut National de la Santé et de la Recherche Médicale (INSERM), the Association Française contre les Myopathies (AFM), the Association Institut de Myologie (AIM), the Agence Nationale de la Recherche (ANR-11-BSV1-026).
None of the authors reports conflict of interest.
ACKNOWLEDGMENTS
We thank L. Manéré, (Institut de Myologie, Unité de Morphologie Neuromusculaire), as well as C. Kretz and R. Schneider (IGBMC) for their excellent technical help. We thank the BGI for the exome sequencing, G. Lanfranchi for the anti-myosin 18B N-ter antibody, and M. Ney (IGBMC) for the control cDNA. We furthermore thank N. Laing, C. Wallgren-Pettersson,J. Lunardi, and N. Monnier for the molecular screening of known nemaline myopathy genes.
Appendices
The supplementary table is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-150085.
REFERENCES
1 | Romero NB, Clarke NF(2013) Congenital myopathiesHandb Clin Neurol113: 132136 |
2 | Nance JR, et al(2012) Congenital myopathies: An updateCurr Neurol Neurosci Re12: 216574 |
3 | Malfatti E, et al(2014) Muscle histopathology in nebulin-related nemaline myopathy: Ultrastrastructural findings correlated to disease severity and genotypeActa Neuropathol Commun2: 44 |
4 | Romero NB, Sandaradura SA, Clarke NF(2013) Recent advances in nemaline myopathyCurr Opin Neurol26: 551926 |
5 | Nowak KJ, et al(1999) Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathyNat Genet23: 220812 |
6 | Pelin K, et al(1999) Mutations in the nebulin gene associated with autosomal recessive nemaline myopathyProc Natl Acad Sci USA96: 5230510 |
7 | Donner K, et al(2002) Mutations in the beta-tropomyosin (TPM2) gene–a rare cause of nemaline myopathyNeuromusculDisord12: 21518 |
8 | Laing NG, et al(1995) A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathyNat Genet9: 1759 |
9 | Johnston JJ, et al(2000) A novel nemaline myopathy in the Amish caused by a mutation in troponin T1Am J Hum Genet67: 481421 |
10 | Agrawal PB, et al(2007) Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2Am J Hum Genet80: 11627 |
11 | Sambuughin N, et al(2010) Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with coresAm J Hum Genet87: 68427 |
12 | Ravenscroft G, et al(2013) Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathyAm J Hum Genet93: 1618 |
13 | Gupta VA, et al(2013) Identification of KLHL41 Mutations Implicates BTB-Kelch-Mediated Ubiquitination as an Alternate Pathway to Myofibrillar Disruption in Nemaline MyopathyAm J Hum Genet93: 6110817 |
14 | Yuen M, et al(2014) Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathyJ Clin Invest124: 114693708 |
15 | Van Antwerpen CL, Gospe SM Jr, Dentinger MP(1988) Nemaline myopathy associated with hypertrophic cardiomyopathyPediatr Neurol4: 53068 |
16 | Skyllouriotis ML, et al(1999) Nemaline myopathy and cardiomyopathyPediatr Neurol20: 431921 |
17 | Nakajima M, et al(2008) An infant with congenital nemaline myopathy and hypertrophic cardiomyopathyJ Nippon Med Sch75: 63503 |
18 | D’Amico A, et al(2006) Fatal hypertrophic cardiomyopathy and nemaline myopathy associated with ACTA1 K336E mutationNeuromuscul Disord16: 9-1054852 |
19 | Kim SY, et al(2011) Nemaline myopathy and non-fatal hypertrophic cardiomyopathy caused by a novel ACTA1 E239K mutationJ Neurol Sci307: 1-21713 |
20 | Gatayama R, et al(2013) Nemaline myopathy with dilated cardiomyopathy in childhoodPediatrics.e131: 6198690 |
21 | Geoffroy V, et al(2015) VaRank: A simple and powerful tool for ranking genetic variantsPeerJ.e3: 796 |
22 | Bohm J, et al(2014) Adult-onset autosomal dominant centronuclearmyopathy due to BIN1 mutationsBrain137: Pt 12316070 |
23 | Maquat LE, Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics(2004) Nat Rev Mol Cell Biol5: 28999 |
24 | Salamon M, et al(2003) Human MYO18B, a novel unconventional myosinheavy chain expressed in striated muscles moves into the myonucleiupon differentiationJ Mol Biol326: 113749 |
25 | Ajima R, et al(2008) Deficiency of Myo18B in mice results in embryonic lethality with cardiac myofibrillar aberrationsGenes Cells13: 1098799 |
26 | Alazami AM, et al(2015) A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18BJ Med Genet52: 64004 |
27 | Garg A, et al(2014) KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathyJ Clin Invest124: 8352939 |
28 | Inoue T, et al(2006) MYO18B interacts with the proteasomal subunit Sug1 and is degraded by the ubiquitin-proteasome pathwayBiochem Biophys Res Commun342: 382934 |
Figures and Tables
Fig.1
Clinical and histological features of the nemaline myopathy patient. A) Clinical examination of the patient revealed a floppy infant with pectus excavatum, and severely reduced active movements. B) The patient showed severe axial muscle weakness, and C) absence of antigravity reflexes, impossible neck extension or brief head control. D) Modified Engel-Gomori trichrome staining revealed the presence of marked fiber size variation, and the presence of nemaline bodies in 40-50% of fibers. The asterisk indicates a severely atrophic fiber filled by minirods.E) Electron microscopy confirmed the presence of small nemaline bodies (minirods).
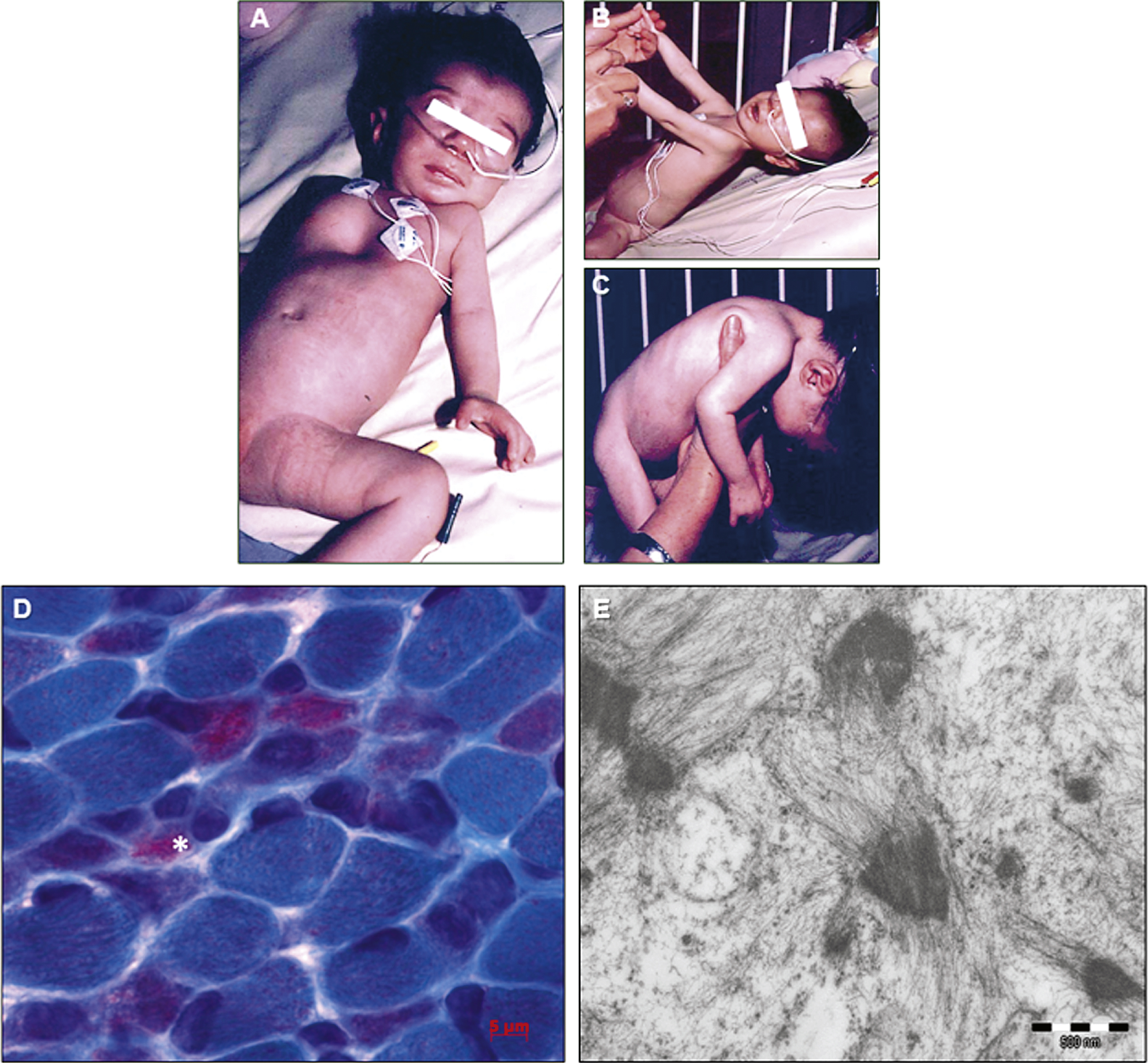
Fig.2
Identification of a homozygous nonsense mutation in MYO18B. A) Pedigree of the consanguineous family. B) Chromatopherograms showing the segregation of the MYO18B c.6496G>T mutation. Both healthy parents (1 and 2) are heterozygous carriers of the MYO18B mutation; the patient (3) is homozygous. C) RT-PCR analysis of skeletal muscle cDNA. The MYO18B amplicon encompassing the MYO18B mutation was detected at comparable levels in the patient and an age-matched control. A NEB amplicon of similar size was used as internal control.
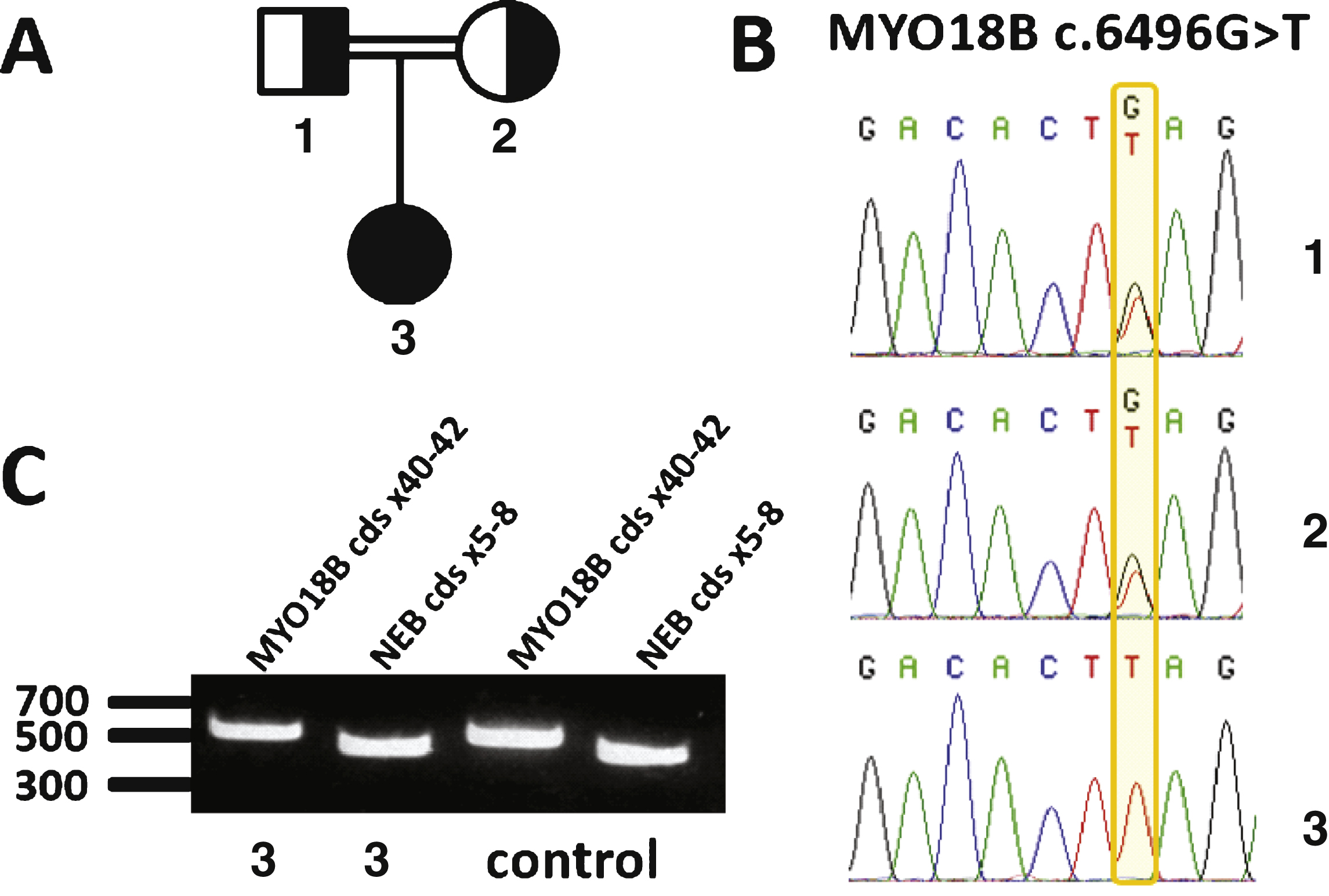
Fig.3
Impact of the MYO18B premature stop codon. Immunolabeling of the muscle biopsy of the index patient and an age-matched control using N-terminal and C-terminal myosin 18B antibodies. A) and B) Immunostaining with N-terminal myosin 18B antibodies showed a similar pattern of cytoplasmic staining in frozen muscle sections from the patient and the control. C) and D) Immunostaining using a C-ter myosin 18B antibody demonstrated the absence of staining in the patient’s section compared to the control, validating the presence of a truncated myosin 18B in the patient. E) Western blot using the C-terminal anti-myosin 18B antibody shows absence of signal in muscle extracts from the patient.