
Diagnostic Utility of Auto Antibodies in Inflammatory Nerve Disorders
Abstract
A wide range of autoantibodies have been described in immune-mediated nerve disorders that target glycans borne by glycolipids and glycoproteins enriched in the peripheral nerves. Their use as diagnostic biomarkers is very widespread, despite some limitations on sensitivity and specificity, and the lack of standardized assays and access to quality assurance schemes. Although many methods have been applied to measurement, ELISA, in the form of commercial kits or in-house assays, still remains the most widely available and convenient assay methodology.
Some antibodies have a particularly robust and widely appreciated clinical significance. Thus, the anti-MAG IgM antibodies that are found in IgM paraprotein related neuropathies define a relatively uniform clinical and prognostic phenotype. IgG antibodies against gangliosides GM1 and GD1a are strongly associated with motor axonal variants of Guillain-Barré syndrome, and anti-GQ1b with Miller Fisher syndrome. In other chronic neuropathies, antibodies against disialylated gangliosides including GD1b and GD3 are detected in ataxic neuropathies, usually associated with an IgM paraprotein, and antibodies against GM1 and the complex GM1:GalC are frequently found in multifocal motor neuropathy. Unfortunately, autoantibodies strongly associated with the diagnosis of chronic inflammatory demyelinating polyneuropathies and with demyelinating forms of GBS are still lacking.
Identification of autoantibodies that map onto a specific clinical phenotype not only allows for improved classification, but also provides better understanding of the pathophysiology of inflammatory neuropathies and the potential for therapeutic interventions.
Peripheral neuropathies are one of the mostaetiologically diverse group of neurological disorders in which biomarkers and other diagnostic investigations are very widely used in both clinical classification and understanding of disease. Broadly speaking, neuropathies can be metabolic, toxic, hereditary or inflammatory, and although well recognised clinical patterns offer distinctive clues to pathological processes, diagnostic investigations also rely heavily on electrophysiological studies and biomarker screening.With respect to biomarkers, numerous categories of auto-antibodies are able to define very specific clinical phenotypes. Although they mainly target glycans borne by glycolipids and glycoproteins, some react with intracellular or membrane-associated protein antigens. The tortuous historical evolution of the neuropathy-associated autoantibody field, combined with often poor sensitivity and specificity, does lead many to conclude that their use as diagnostic biomarkers in clinical practice is overly complicated and often unhelpful to clinical care. In this article, we review the recent advance on auto-antibodies to describe their diagnostic utility in inflammatory neuropathies and attempt to summarise the useful clinical algorithms and their pitfalls.
ANTI-GANGLIOSIDE ANTIBODIES
Gangliosides are a distinct category of glycosphingolipids comprising a ceramide moiety with one or more hexose sugars that include at least one sialic acid residue as their defining feature (Fig. 1) [1]. Many other glycolipids that are not gangliosides, but nevertheless share structural similarities, are also neuropathy-associated autoantigens. The hydrophobic ceramide tail of glycolipids (including gangliosides) are inserted in the outer leaflet of the lipid bi-layer that forms the plasma membrane, with the hydrophilic oligosaccharide moiety being displayed extracellularly, where it can be recognised by specific antibodies. Since many gangliosides share common structural motifs due to common sugar sequences, a single antibody species may have the capacity to bind multiple gangliosides. Gangliosides are concentrated in cholesterol-enriched microdomains of the plasma membrane termed lipid rafts, in which they may adopt particular steric conformations that either enhance or attenuate the capacity for autoantibody recognition, depending upon the precise binding requirements for a particular antibody. Although ubiquitous in all cells types throughout the body, the major gangliosides are highly enriched in axonal membranes within the peripheral nervous system, and can be accessed by antibodies at exposed axonal regions of the neuromuscular junction and the node of Ranvier [2]. A limiting factor in antibody access is also the blood nerve/brain barrier; thus ganglioside distribution and antibody access and binding are discordant considerations. Indeed the absence of CNS pathology in anti-ganglioside autoantibody states is presumably a reflection of limited access rather than poor antibody binding capacity, as the CNS is also very highly enriched in gangliosides.
Anti-ganglioside antibodies can be detected by several techniques, including enzyme-linked immunosorbent assay (ELISA), immunodot-assay, flow-cytometry and cell surface binding, and glyco-array [3–6]. Wide variations in assay performance, both within a single assay and between assays, have been reported [4, 7], indicating that these techniques should ideally be standardized for consistency between different laboratories [5]. Different methods may preferentially detect different types of antibody, owing to variations in the orientation of the glycan headgroup on the immobilised surface. Thus, different techniques should not necessarily be expected to be fully concordant with each other, and there is no recognised optimal assay for detecting these antibodies, ELISA remains the most commonly used method as all laboratories are widely conversant with this standard technology. Glyco-array is useful to screen for many anti ganglioside antibodies with a small amount of serum [8].
Different anti-ganglioside antibodies are associated with different inflammatory neuropathies (Table 1). As a general but somewhat counter-intuitive rule, IgG antibody isotypes are found in acute neuropathies and IgM isotypes in chronic neuropathies. Acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN) represent about 10% of the Guillain-Barré syndrome in Western countries and are strongly associated with IgG antibodies against GM1 and GD1a, and structurally similar but quantitatively minor a-series gangliosides (e.g. GM1b and GalNAcGD1a) [9]. The diagnosis of acute neuropathy with a pharyngeal-cervical-brachial pattern of weakness (PCB) or pure orophayngeal palsy is supported by the detection of IgG antibodies against GT1a that may or may not also react with GQ1b. Miller Fisher syndrome, characterized by acute-onset areflexia, ataxia, ophthalmoplegia is associated with IgG anti-GQ1b antibodies [10]. Bickerstaff brainstem encephalitis and incomplete forms of Miller Fisher syndrome (MFS), as acute ophthalmoplegia, are also associated with IgG anti GQ1b antibodies. Antibodies found in acute sensory ataxic neuropathy are directed against either GQ1b or GD1b [11]. The strong association of anti-GQ1b and related disialylated ganglioside antibodies with the above regional forms of GBS have led to the concept of ‘the anti-GQ1b antibodies syndromes’ as an umbrella term for MFS and its myriad of forms frustes [10–12]. It is noteworthy that false positive anti-GQ1b antibody assays, occurring in other disease or control populations are extremelyuncommon.
Multifocal motor neuropathy (MMN) is a chronic neuropathy featuring pure motor weakness and motor conduction blocks on neurophysiological testing [13]. IgM antibodies against GM1 are found in around half of the MMN sera. A high titre may be associated with a more severe disease [14]. The diagnosis of CANOMAD (chronic ataxic neuropathy, ophthalmoplegia, IgM paraprotein, cold agglutinins and anti disialosyl antibodies) rests on the detection of an IgM monoclonal gammopathy reacting against disialosyl gangliosides (principally GD3, GD1b, GT1b and GQ1b) [15]. The gammopathy may be of a small amount, requiring immunofixation of the sera to be detected. Intravenous immunoglobulins may be therapeutically effective in MMN and in chronic sensory ataxic neuropathies associated with IgM anti GD1b antibodies, with or without IgM monoclonal gammopathy [16]. Antibodies involved in MFS and PCB tend to preferentially react with the terminal disialosyl structure shared by GQ1b and GT1a (Fig. 1), whereas antibodies involved in CANOMAD also react with the internal disialosyl structure [11, 15].
The relevance of antibodies against complexes of gangliosides has been recently stressed and this remains a highly active field of research. As new data emerges, this difficult field will hopefully undergo some clarification. Ganglioside complexes in this context are defined as interacting partnerships between 2 structurally distinct ganglisoides (e.g. GM1 and GD1a) that create new antibody binding sites when in heteromeric complex, that are not present in either individual ganglioside when presented alone [8]. Anti-ganglioside complex antibodies are more frequently detected in Guillain-Barré syndrome than antibodiesagainst single gangliosides [17–19]. Antibodies against complexes of GM1 and galactocerebroside (GM1:GalC) appear to be a more sensitive marker than antibodies against GM1 alone for the diagnosis of MMN [6, 20–22]. The precise mechanisms underlying antibody-complex interaction require further study; however the currently held view is that complexes of gangliosides may enhance antibody detection by one or both of two mechanisms. Either both component of the complex of gangliosides can form a heterodimer that generates a new epitope, or the cis-interaction between the gangliosides can result in a preferential presentation of an epitope present on one or other of the partners in the complex [23].
Anti-ganglioside antibodies are thought to be the principle pathogenic driver of the disease in which they are found, on the basis of a substantial body of evidence accumulated over many years, which is briefly summarised here. Cases of AMAN have been reported after the administration of ganglioside [24]. Campylobacter jejuni, the most common predisposing agent in GBS and MFS, has surface lipooligosaccharides (LOS) that are structural mimics of mammalian gangliosides including GM1, GD1a and GT1a/GQ1b [25, 26]. This strongly favours the hypothesis that molecular mimicry between LOS and gangliosides underpins the autoimmune process [27]. Sensitization of experimental rabbits with GD1b or GM1 has induced an experimental inflammatory neuritis with ataxic or motor dominant components respectively, mirroring the human clinical counterparts [28, 29]. Anti ganglioside antibodies are able to bind the nerve roots, the pre-synaptic motor nerve terminal, the nodes of Ranvier and dorsal root ganglion neuronal cell bodies [30, 31]. Binding of the antibodies activates complement leading to the formation of the highly neurotoxic membrane attack complex. Voltage-gated sodium channels clusters disappear through calpain cleavage, axo-glial junctions are disrupted at the nodes of Ranvier and failure of conduction occurs (reversible conduction block). If the immunopathological process progresses, axonal degeneration may occur. There is growing opinion that many features of the anti-ganglioside antibody-mediated neuropathies can be encompassed in a new categorisation referred to as the nodo-paranodopathy [32, 33]. As our experimental knowledge grows, it is becoming increasing difficult to clinically and electrophysiologically distinguish reversible axonal conduction block from that caused by paranodal demyelination, especially since both may occur concurrently in the same nerve fibre.
ANTI-MYELIN ASSOCIATED GLYCOPROTEIN (MAG) ANTIBODIES
Anti-MAG antibodies are detected in half of IgM paraproteinaemic neuropathy cases [34]. In the remaining cases there is no uniform autoantibody specificity. Patients with anti-MAG neuropathy have highly characteristic distal, chronic, slowly progressive sensory involvement with ataxia and often tremor. Muscular weakness is mild or absent even in the presence of motor demyelination [35, 36]. Some patients may have atypical clinical features, as proximal weakness and sub-acute progression, which can mimic chronic inflammatory demyelinating polyneuropathy (CIDP) [36]. Nerve conduction studies show a predominantly distal demyelinating neuropathy with prolonged distal motor latencies and generally absent sensory nerve action potentials [37].
ELISA is considered more sensitive than Western blot to detect anti MAG antibodies [38]. However, ELISA may be less specific if the titre is between 1000 and 10000, possibly because of some cross-reactivity with GM1 and disialosyl gangliosides [39]. There is no association between the anti-MAG antibodies titres and the clinical features of the patients [35, 40, 41]. In general, anti-MAG neuropathy does not respond well to any treatment, and many patients remain untreated, often after several trials of failed or insufficiently successful therapy [42–44]. The correlation between anti-MAG antibody titre and the efficiency of Rituximab therapy (an anti-CD20) is an equivocal finding, as improvement after therapy has been associated with either high or low anti MAG titres [44, 45]. There is an expectation that once new therapies emerge that target the long lived plasma cells that are believed to be the source of anti-MAG antibodies, there may be a reasonable prospect of a successful treatment regime. This would also apply to other paraproteinaemic neuropathies.
MAG is an integral membrane glycoprotein enriched in periaxonal Schwann cell membranes, paranodal loops and Schmit-Lanterman incisures, and member of the immunoglobulin superfamily. The antigenic region of the MAG molecule for the IgM antibodies found in affected humans is the human natural killer-1 (HNK-1) carbohydrate epitope, which comprises an unusual glucoronic acid that is 3-sulphated. The HNK-1 epitope is also present in other peripheral nerve glycoproteins, including P0, PMP-22 and phosphocan and thus the in vivo target for the human antibodies may reside on multiple nerve molecules. Furthermore, 2 peripheral nerve glycolipids, sulphated glucuronyl paragloboside (SGPG), and its higher lactosaminyl homologue (SGLPG) also bear the antigenic determinant. Therefore, patients with anti MAG neuropathy may have antibodies against SGPG and other glycoconjugated structures of the myelin sheaths. Extension of antibody reactivity to various HNK-1 bearing proteins other than MAG, might be associated with treatment resistance [40]. Some ‘anti-MAG’ antibodies also react with sulfatides (3-sulphated galactocerebroside) [46]. It is also noteworthy that some cases of CIDP may harbour anti-SGPGantibodies [47].
OTHER ANTIBODIES ASSOCIATED WITH INFLAMMATORY NEUROPATHIES
Several proteins of the nodal and paranodal domains have recently been identified as possible target in inflammatory neuropathy sera [9]. Antibodies against neurofascin (NF) 186 or gliomedin have been found in 62% of 53 MMN patients. Ten percent of these MMN sera without IgM anti GM1 reactivity had anti NF186 antibodies [48]. Antibodies against NF 186, NF 155, LM1 and contactin are detected in less than 5% of CIDP sera [49–52]. Anti-moesin antibodies have been indentified in GBS subsequent to CMV infection [53, 54]. As these antibodies are infrequent, they are not necessarily good biomarkers for screening inflammatory neuropathy sera, but they are nevertheless of major interest to explain the pathophysiology of the cases in which they are found. Extensive ongoing work is characterising these phenotypes in animal models [9, 55].
IN CLINICAL PRACTICE
The diagnostic utility of anti-nerve auto-antibodies is often limited by their modest sensitivity and by the lack of standardized assays. ELISA remains the most widely available and well-standardized assay. Some antibodies have a robust clinical significance. Thus IgG isotype antibodies against GM1, GD1a and GQ1b are very helpful in the diagnosis of respectively AMAN and MFS. Unfortunately, the results of these tests are not always available at the time of a diagnosis of such an acute neuropathy. Anti-MAG antibodies are an excellent marker in cases of IgM paraproteinemic neuropathy. Chronic ataxic neuropathy is often associated with antibodies against disialosyl gangliosides. Anti-GM1 antibodies may be helpful at the beginning of the disease to differentiate MMN from amyotrophic lateral sclerosis. Unfortunately, we are still lacking an auto-antibody profile strongly associated with the diagnosis of CIDP, or the acute demyelinating forms of GBS. The ongoing identification of anti-nerve auto-antibodies continues to allow us to develop a better understanding of the pathophysiology of inflammatory neuropathies. Further studies are needed to develop additional biomarkers and to clarify their use in clinical practice.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1 | Svennerholm L(1994) Designation and schematic structure of gangliosides and allied glycosphingolipidsProg Brain Res101: XIXIV |
2 | Plomp JJ, Willison HJ(2009) Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junctionJ Physiol587: 1639793999 |
3 | Escande-Beillard N, David MJ, Portoukalian J, Pouget J, Azulay JP, Bernard D BJ(2002) A sensitive flow cytometry method for anti-GM1 antibodies detectionJ Neuroimmunol125: 163169 |
4 | Caudie C, Quittard Pinon A, Bouhour F, Vial C, Garnier L, Fabien N(2013) Comparison of commercial tests for detecting multiple anti-ganglioside autoantibodies in patients with well-characterized immune-mediated peripheral neuropathiesClin Lab59: 11-1212771287 |
5 | Willison HJ, Veitch J, Swan A V, Baumann N, Comi G, Gregson NA(1999) Inter-laboratory validation of an ELISA for the determination of serum anti-ganglioside antibodiesEur J Neurol6: 17177 |
6 | Delmont E, Halstead S, Galban-Horcajo F, Yao D, Desnuelle C, Willison H(2014) Improving the detection of IgM antibodies against glycolipids complexes of GM1 and Galactocerebroside in Multifocal Motor Neuropathy using glycoarray and ELISA assaysJ Neuroimmunol278: 159161 |
7 | Van Schaik IN, Bossuyt PMM, Brand A, Vermeulen M(1995) Diagnostic value of GM1 antibodies in motor neuron disorders and neuropathies: A meta-analysisNeurology45: 815701577 |
8 | Galban-Horcajo F, Halstead SK, McGonigal R, Willison HJ(2014) The application of glycosphingolipid arrays to autoantibody detection in neuroimmunological disordersCurr Opin Chem Biol18: 7886 |
9 | Lim JP, Devaux J, Yuki N(2014) Peripheral nerve proteins as potential autoantigens in acute and chronic inflammatory demyelinating polyneuropathiesAutoimmun Rev13: 1010701078 |
10 | Wakerley BR, Uncini A, Yuki N(2014) Guillain-Barré and Miller Fisher syndromes–new diagnostic classificationNat Rev Neurol10: 9537544 |
11 | Yuki N, Uncini A(2014) Acute and chronic ataxic neuropathies with disialosyl antibodies: A continuous clinical spectrum and a common pathophysiological mechanismMuscle Nerve49: 5629635 |
12 | Willison HJ, Yuki N(2002) Peripheral neuropathies and anti-glycolipid antibodiesBrain125: 25912625 |
13 | Guimarães-Costa R, Bombelli F, Léger J-M(2013) Multifocal motor neuropathyCurr Opin Neurol26: 5503509 |
14 | Cats EA, Jacobs BC, Yuki N, Tio-Gillen AP, Piepers S, Franssen H(2010) Multifocal motor neuropathy: Association of anti-GM1 IgM antibodies with clinical featuresNeurology75: 2219611967 |
15 | Willison HJ, O’Leary CP, Veitch J, Blumhardt LD, Busby M, Donaghy M(2001) The clinical and laboratory features of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodiesBrain124: 19681977 |
16 | Attarian S, Boucraut J, Hubert AM, Uzenot D, Delmont E, Verschueren A(2009) Chronic ataxic neuropathies associated with anti-GD1b IgM antibodies: Response to IVIg therapyJ Neurol Neurosurg Psychiatry81: 16164 |
17 | Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M(2004) Ganglioside complexes as new target antigens in Guillain-Barré syndromeAnn Neurol56: 4567571 |
18 | Rinaldi S, Brennan KM, Kalna G, Walgaard C, van Doorn P, Jacobs BC(2013) Antibodies to heteromeric glycolipid complexes in guillain-barré syndromePLoS One8: 12e82337 |
19 | Shahrizaila N, Kokubun N, Sawai S, Umapathi T, Chan Y-C, Kuwabara S(2014) Antibodies to single glycolipids and glycolipid complexes in Guillain-Barré syndrome subtypesNeurology83: 2118124 |
20 | Nobile-Orazio E, Giannotta C, Musset L, Messina P, Léger J-M(2014) Sensitivity and predictive value of anti-GM1/galactocerebroside IgM antibodies in multifocal motor neuropathyJ Neurol Neurosurg Psychiatry85: 7754758 |
21 | Pestronk A, Choksi R, Blume G, Lopate G(1997) Multifocal motor neuropathy: Serum IgM binding to a GM1 ganglioside-containing lipid mixture but not to GMl aloneNeurology48: 411041106 |
22 | Galban-Horcajo F, Fitzpatrick AM, Hutton AJ, Dunn SM, Kalna G, Brennan KM(2013) Antibodies to heteromeric glycolipid complexes in multifocal motor neuropathyEur J Neurol20: 16270 |
23 | Rinaldi S, Brennan KM, Willison HJ(2010) Heteromeric glycolipid complexes as modulators of autoantibody and lectin bindingProg Lipid Res49: 18795 |
24 | Illa I, Ortiz N, Gallard E, Juarez C, Grau JM, Dalakas MC(1995) Acute axonal Guillain-Barré syndrome with IgG antibodies against motor axons following parenteral gangliosidesAnn Neurol38: 2218224 |
25 | Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K(2004) Carbohydrate mimicry between human gangliosideGM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndromeProc Natl Acad Sci U S A101: 311140411409 |
26 | Jacobs BC, O’Hanlon GM, Breedland EG, Veitch J, van Doorn PA, Willison HJ(1997) Human IgM paraproteins demonstrate shared reactivity between Campylobacter jejuni lipopolysaccharides and human peripheral nerve disialylated gangliosidesJ Neuroimmunol80: 1-22330 |
27 | Caporale CM, Capasso M, Luciani M, Prencipe V, Creati B, Gandolfi P(2006) Experimental axonopathy induced by immunization with Campylobacter jejuni lipopolysaccharide from a patient with Guillain-Barré syndromeJ Neuroimmunol174: 1-21220 |
28 | Kusunoki S, Shimizu J, Chiba A, Ugawa Y, Hitoshi S, Kanazawa I(1996) Experimental sensory neuropathy induced by sensitization with ganglioside GD1bAnn Neurol39: 4424431 |
29 | Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y(2001) Animal model of axonal Guillain-Barré syndrome induced by sensitization with GM1 gangliosideAnn Neurol49: 6712720 |
30 | Fewou SN, Plomp JJ, Willison HJ(2014) The pre-synaptic motor nerve terminal as a site for antibody-mediated neurotoxicity in autoimmune neuropathies and synaptopathiesJ Anat224: 13644 |
31 | Santoro M, Uncini A, Corbo M, Staugaitis SM, Thomas FP, Hays AP(1992) Experimental conduction block induced by serum from a patient with anti-GM1 antibodiesAnn Neurol31: 4385390 |
32 | Uncini A, Susuki K, Yuki N(2013) Nodo-paranodopathy: Beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathiesClin Neurophysiol124: 1019281934 |
33 | Miller JAL, Spyropoulos A, Jaros E, Galban-horcajo F, Infirmary RV, Hospitals N(2014) Anti-GQ1b ganglioside positive Miller Fisher syndrome – evidence of paranodal pathology on nerve biopsyJ Neuromuscul Dis1: 191195 |
34 | Nobile-Orazio E, Manfredini E, Carpo M, Meucci N, Monaco S, Ferrari S(1994) Frequency and clinical correlates of anti-neural IgM antibodies in neuropathy associated with IgM monoclonal gammopathyAnn Neurol36: 3416424 |
35 | Launay M, Delmont E, Benaím C, Sacconi S, Butori C, Desnuelle C(2009) Les polyneuropathies avec IgM monoclonale anti-MAG: étude descriptive clinique, biologique, électrophysiologique et anatomopathologique d’une cohorte de 13 patientsRev Neurol (Paris)165: 1210711079 |
36 | Joint Task Force of the EFNS and the PNS(2010) European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve SocietJ Peripher Nerv Syst185195 |
37 | Attarian S, Azulay JP, Boucraut J, Escande N, Pouget J(2001) Terminal latency index and modified F ratio in distinction of chronic demyelinating neuropathiesClin Neurophysiol112: 3457463 |
38 | Kuijf ML, Eurelings M, Tio-Gillen a P, van Doorn P a, van den Berg LH, Hooijkaas H(2009) Detection of anti-MAG antibodies in polyneuropathy associated with IgM monoclonal gammopathyNeurology73: 9688695 |
39 | Caudie C, Kaygisiz F, Jaquet P, Petiot P, Gonnaud P-M, Antoine JC(2006) [Diagnostic value of autoantibodies to MAG by ELISA Bühlmann in 117 immune-mediated peripheral neuropathies associated with monoclonal IgM to SGPG/SGLPG]Ann Biol Clin (Paris)64: 4353359 |
40 | Hamada Y, Hirano M, Kuwahara M, Samukawa M, Takada K, Morise J(2014) Binding specificity of anti-HNK-1 IgMM-protein in anti-MAG neuropathy: Possible clinical relevanceNeurosci Res16 |
41 | Planche V, Arnaud L, Viala K, Hospital Marie Anne, Miraya M, Leger Jean-Marc, Neil J, Leblond V, Musset L(2014) Heavy/Light Chain Ratio as a Biomarker for Monitoring Patients with IgM Monoclonal Gammopathy and Anti-MAG NeuropathyJ Hematol Thrombo Dis2: 3142 |
42 | Lunn MPT, Nobile-Orazio E(2012) Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathiesCochrane database Syst Rev5: CD002827 |
43 | Léger J-M, Viala K, Nicolas G, Créange A, Vallat J-M, Pouget J(2013) Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathyNeurology80: 22172225 |
44 | Dalakas MC, Rakocevic G, Salajegheh M, Dambrosia JM, Hahn AF, Raju R(2009) Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathyAnn Neurol65: 3286293 |
45 | Benedetti L, Briani C, Grandis M, Vigo T, Gobbi M, Ghiglione E(2007) Predictors of response to rituximab in patients with neuropathy and anti-myelin associated glycoprotein immunoglobulin MJ Peripher Nerv Syst12: 2102107 |
46 | Giannotta C, Di Pietro D, Gallia F, Nobile-Orazio E(2015) Anti-sulfatide IgM antibodies in peripheral neuropathy: To test or not to test?Eur J Neurol10.1111/ene.12658[Epub ahead of print] |
47 | Yuki N, Tagawa Y, Handa S(1996) Autoantibodies to peripheral nerve glycosphingolipids SPG, SLPG, and SGPG in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathyJ Neuroimmunol70: 116 |
48 | Notturno F, Di Febo T, Yuki N, Fernandez Rodriguez BM, Corti D, Nobile-Orazio E(2014) Autoantibodies to neurofascin-186 and gliomedin in multifocal motor neuropathyJ Neuroimmunol276: 1-2207212 |
49 | Querol L, Nogales-Gadea G, Rojas-Garcia R, Martinez-Hernandez E, Diaz-Manera J, Suárez-Calvet X(2013) Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathyAnn Neurol73: 3370380 |
50 | Ng JKM, Malotka J, Kawakami N, Derfuss T, Khademi M, Olsson T(2012) Neurofascin as a target for autoantibodies in peripheral neuropathiesNeurology79: 2322412248 |
51 | Kuwahara M, Suzuki H, Samukawa M, Hamada Y, Takada K, Kusunoki S(2013) Clinical features of CIDP with LM1-associated antibodiesJ Neurol Neurosurg Psychiatry84: 5573575 |
52 | Querol L, Nogales-Gadea G, Rojas-Garcia R, Diaz-Manera J, Pardo J, Ortega-Moreno A, Sedano MJ, Gallardo E, Berciano J, Blesa R, Dalmau J, Illa I(2014) Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIgNeurology82: 10879886 |
53 | Sawai S, Satoh M, Mori M, Misawa S, Sogawa K, Kazami T(2014) Moesin is a possible target molecule for cytomegalovirus-related Guillain-Barré syndromeNeurology83: 2113117 |
54 | Willison H, Scherer SS(2014) Ranvier revisited: Novel nodal antigens stimulate interest in GBS pathogenesisNeurology83: 2106108 |
55 | Desmazieres A, Zonta B, Zhang A, Wu L-MN, Sherman DL, Brophy PJ(2014) Differential stability of PNS and CNS nodal complexes when neuronal neurofascin is lostJ Neurosci34: 1550835088 |
Figures and Tables
Fig.1
Structure of typical gangliosides involved in inflammatory neuropathies. Antibodies involved in MFS and PCB tend to preferentially react with the terminal disialosyl structure shared by GQ1b and GT1a (dashed box), whereas antibodies involved in CANOMAD react with the internal disialosyl structure (solid box).
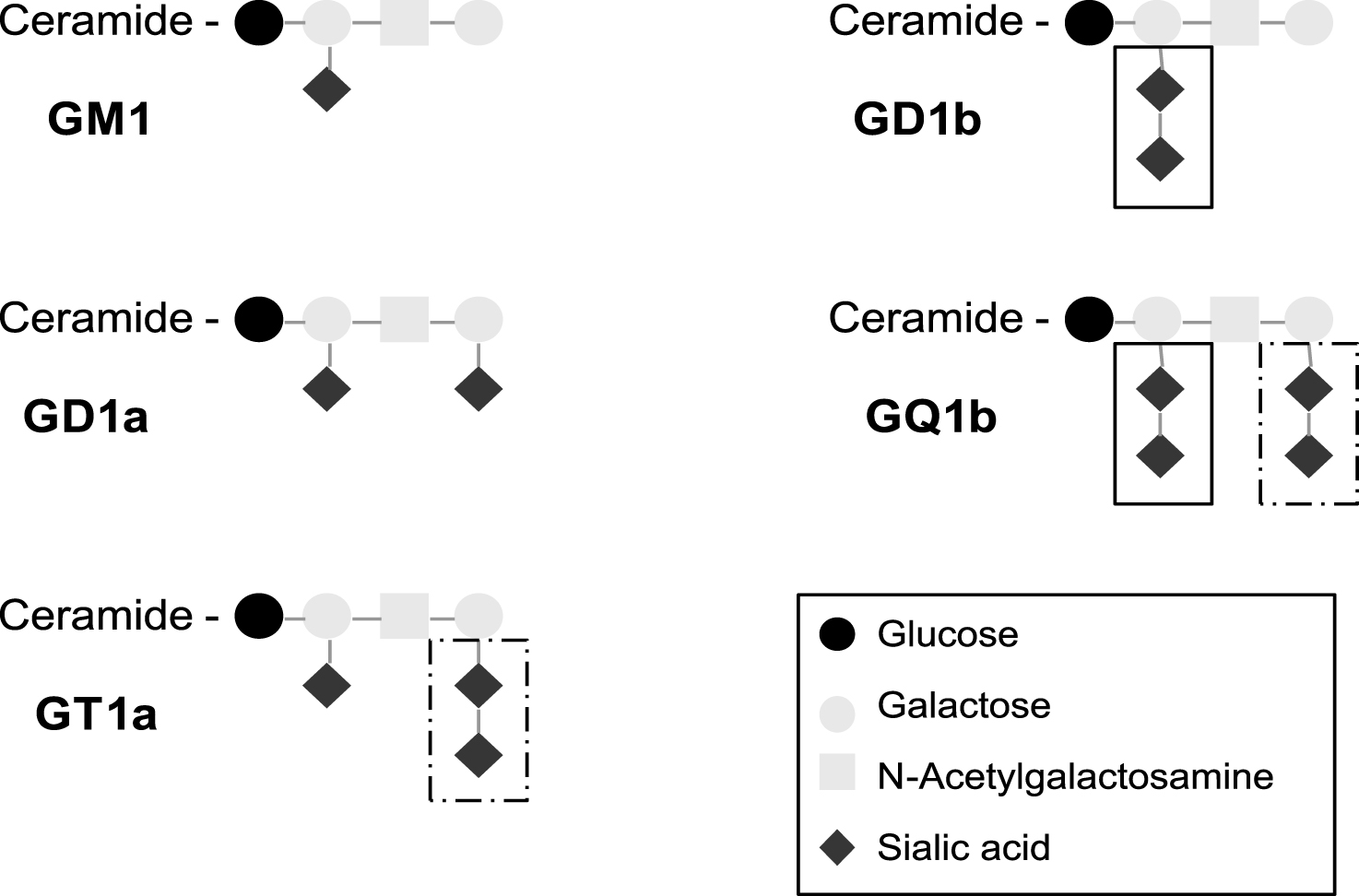
Table 1
Auto antibodies associated with inflammatory neuropathies
| Neuropathy | Main clinical features | Associated antibodies | |
| AMAN | Acute | Motor | IgG anti GM1, GD1a |
| Miller Fisher syndrome | Ataxia and ophthalmoplegia | IgG anti GQ1b, GT1a | |
| Acute sensory ataxic neuropathy | Sensory, ataxia | IgG anti GD1b or GQ1b | |
| PCB | Motor | IgG anti GT1a>GQ1b | |
| MMN | Chronic | Motor | IgM anti GM1, complex GM1:GalC |
| CANOMAD and CANDA | Sensory, ataxia | IgM anti GD3, GD1b, GT1b, GQ1b | |
| CIDP | Sensory motor | Anti NF155, NF186, contactin | |
| Paraneoplastic neuropathy | Sensory ataxia or sensory motor | Anti Hu, anti CV2 | |
| Anti MAG neuropathy | Sensory, ataxia | Monoclonal IgM anti MAG |
AMAN, acute motor axonal neuropathy; PCB, pharyngeal-cervical-brachial weakness; MMN multifocal motor neuropathy with conduction blocks; CANOMAD, chronic ataxic neuropathy ophthalmoplegia IgM paraprotein anti disialosyl antibodies; CANDA, chronic ataxic neuropathy with disialosyl antibodies; CIDP, chronic inflammatory demyelinating polyneuropathy; GM:GalC complex of GM1 and galactocerebroside (GalC); NF, neurofascin; MAG, myelin associated glycoprotein.


