
Immunohistochemical Characterization of FacioscapulohumeralMuscular Dystrophy Muscle Biopsies
Abstract
Background: Posited pathological mechanisms in Facioscapulohumeral Muscular Dystrophy (FSHD) include activation in somatic tissue of normally silenced genes, increased susceptibility to oxidative stress, and induction of apoptosis.
Objective: To determine the histopathological changes in FSHD muscle biopsies and compare to possible pathological mechanisms of disease.
Methods: We performed a cross-sectional study on quadriceps muscle biopsies from 32 genetically confirmed FSHD participants, compared to healthy volunteers and myotonic dystrophy type 1 as disease controls. Biopsies were divided into groups to evaluate apoptosis rates, capillary density, myonuclear and satellite cell counts.
Results: Apoptosis rates were increased in FSHD (n = 10, 0.74% ) compared to myotonic dystrophy type 1 (n = 10, 0.14% , P = 0.003) and healthy volunteers (n = 14, 0.13% , P = 0.002). Apoptosis was higher in FSHD patients with the smallest residual D4Z4 fragments. Capillary density was decreased in FSHD1 (n = 10, 316 capillaries/mm2) compared to healthy volunteers (n = 15, 448 capillaries/mm2, P = 0.001). No differences were seen in myonuclear or satellite cell counts.
Conclusions: Preliminary evidence for increased apoptosis rates and reduced capillary density may reflect histopathological correlates of disease activity in FSHD. The molecular-pathological correlates to these changes warrants further investigation.
ABBREVIATIONS
BSA | bovine serum albumin |
CON | healthy control |
CSS | clinical severity score |
DAPI | 4,6-diamidino-2-phenylindole |
DM1 | myotonic dystrophy type 1 |
FSHD | Facioscapulohumeral muscular dystrophy |
IQR | interquartile range |
MHC-s | myosin heavy chain slow |
PBS | phosphate buffered saline |
PFA | paraformaldehyde |
SROM | shoulder range of motion |
TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
INTRODUCTION
Facioscapulohumeral Muscular Dystrophy (FSHD) is one of the most common progressive muscular dystrophies (prevalence 1:15000) [1, 2]. Recent studieshave suggested that both Facioscapulohumeral muscular dystrophy (FSHD) types 1 and 2 operate through a common downstream genetic mechanism of de-repression of a retrogene DUX4, not normally expressed in somatic muscle tissue [3, 4]. Gene expression profiling from FSHD muscle has shown altered regulation of large number of genes associated with myogenesis, increased susceptibility to oxidative stress, angiogenensis, vascular smooth muscle or endothelial cells, muscle isoenzymes, cholesterol metabolism, and the actin cytoskeleton [5–8]. Expression of DUX4 in vitro inhibits myogenesis, increases oxidative stress in myogenic precursors, and induces apoptosis [9–11]. DUX4 is a transcription factor normally expressed in the luminal cells of the testis, which activates genes involved in the germline and early stem cell development [12]. One line of thinking posits induction of a mitotic program in a post-mitotic cell results in apoptosis. Another line of thinking proposes DUX4 may affect satellite cell renewal. The muscle immunohistochemical correlates to these posited pathological mechanisms are unknown. A better understanding of the muscle pathology in FSHD can be important in two ways: 1) providing insights into the patho-mechanism in FSHD, and 2) providing potential biomarkers for proof of concept or early phase clinical studies.
Here we perform a retrospective cross-sectional histopathological study in genetically confirmed FSHD muscle biopsies. We quantify the apoptotic rates, satellite cell counts, and nuclear and capillary density and compare to healthy volunteers (CON) and myotonic dystrophy type 1 (DM1) as a diseasecontrol.
MATERIALS AND METHODS
We performed a cross-sectional observational study of prospectively collected muscle biopsies from genetically confirmed FSHD participants seen at the University of Rochester Medical Center (URMC) between 2002 to 2013. The study was approved by institutional review board and written and informed consent was obtained from all participants.
Participants
FSHD participants were between 18 and 75 years of age and had genetic confirmation per previously published protocols [13, 14]. The residual D4Z4 fragment size on chromosome 4q35 was determined by Southern blot after double digestion with EcoRI/BlnI restriction enzymes. Participants were ineligible if they had muscle wasting making a needle biopsy impracticable. DM1 disease controls were obtained from the Myotonic Dystrophy Natural History Study being performed at URMC. All patients had genetic confirmation of DM1. Healthy volunteers were drawn from URMC or from spouses of affected FSHD participants. Disease controls were not obtained for nuclear density or satellite cell counts as no differences were seen between FSHD and the healthy volunteers.
Clinical severity
We used an age-adjusted 10-grade clinical severity scale (CSS), which takes into account the extent of weakness in various body regions and descending spread of symptoms from face and shoulders to pelvic and leg muscles typical of FSHD [15]. Higher scores are assigned to patients with involvement of pelvic and proximal lower limb muscles. The scale was adjusted for the patient’s age at examination as previously described: Age-corrected CSS = ((CSS x 2) / age at examination) × 1000 [16].
Immunohistochemistry techniques
Needle muscle biopsies were obtained from the vastus lateralis of all participants. The vastus lateralis was chosen to reduce the variability due to sampling from different muscles. Details of the procedure are available at: http://www.urmc.rochester.edu/fields-center/protocols/needle-muscle-biopsy.cfm.[center/protocols/needle-muscle-biopsy.cfm.] The his-topathologic samples were graded for the severity of their pathologic changes based on 10μm sections stained with Hematoxylin & Eosin, Trichrome, NAHD and myosin ATPase. A pathologic severity was determined using a 12-point scale giving a 0–3 score (normal, mild, moderate, severe) to each of four major histologic features: variability in fiber size, centrally located nuclei, interstitial fibrosis, and muscle fiber necrosis/regeneration.
For immunohistochemistry all samples were air dried for 20 minutes, then fixed in 2% paraformaldehyde (PFA) for 10 minutes (except capillary density, and apoptosis [4% PFA]). Slides were mounted with Vectashield (Vector Laboratories) containing 4,6-diamidino-2-phenylindole (DAPI) in all experiments except capillary density measurements. Images were taken with a fluorescent microscope (Nikon Eclipse E600, Japan) equipped with a digital camera and analyzed with Metaview software (version 6.1, Molecular Devices, Sunnyvale, CA).
The following antibodies were utilized: PAX7 serum (Developmental studies Hybridoma Bank)dilution 1:2 in bovine serum albumin (BSA); rabbit polyclonal anti-Laminin alpha-2 (Gene Tex) dilution 1:200 (dilution 1:100 for nuclear density) in BSA; fluorescein-conjugated TUNEL reaction mixture prepared as per manufacturer’s instructions (product # 11684795910, Roche Applied Science, Indianapolis); mouse Anti Caveolin-3 (from BD transduction Laboratories) dilution 1:000 in phosphate buffered saline (PBS); Ulex Europaeus Agglutinin 1 Fluorescein conjugated(Vector Laboratories, Burlingame CA) dilution 1:250 in PBS; mouse Myosin heavy chain-slow or MHC-s (Novacastra) dilution 1:20 in PBS; anti mouse Alexa fluor 350 (Invitrogen) dilution 1:400 in PBS; anti mouse Alexa Fluor 488 (Invitrogen) dilution 1:500 (dilution 1:400 for nuclear density) in BSA; and anti-rabbit alexa fluor 568 (Invitrogen) dilution 1:500 (dilution 1:400 for capillary density) in BSA.
For capillary density, sections were triple-stained with Caveolin-3, MHC-s and Ulex to measure fiber cross-sectional area, fiber type proportions and capillaries, respectively. Capillary densities were determined from digitized images from microscopic sections per previously described protocols and defined as the number of capillaries divided by the cross sectional area [17–19]. Individual average fiber cross sectional area was defined as the total area/number of fibers. Nuclear density was defined as the total number of DAPI-positive myonuclei per total number of myofibers as delineated by laminin staining. Double staining with PAX7 and laminin was used to define satellite cells as PAX7/DAPI positive nuclei below the basal lamina. Satellite cell counts were calculated as the number of PAX7 positive myonuclei per total myofibers [20, 21]. For measurement of apoptosis all sections were double-stained with laminin and TUNEL antibodies. Apoptotic rates were defined as the number of TUNEL-positive nuclei per total number of myofibers [22, 23]. Sections were also triple-stained with TUNEL, laminin, and PAX7 to determine if TUNEL positive cells corresponded to satellite cells.
Statistical considerations
Standard statistical methods were used to describe all groups (FSHD1, healthy volunteers, and DM1), including the calculation of the median and the first and third quartiles (i.e., interquartile range, IQR). The test for differences in distribution in apoptotic rates, capillary density, nuclear and satellite counts between FSHD1 and CON or DM1 employed the Kruskal-Wallis test for factors that were either continuous data, or ordered data with more than 7 levels (e.g. pathology grade 0–12). As age and gender were not completely matched between groups, effects of age and gender on histopathological outcomes were evaluated using a mixed effects model; however no group affects due to differences in age or gender were detected. We used a linear mixed effects model to evaluate the associations of apoptotic rates or capillary density (dependent variables) to pathology grade, residual D4Z4 residual fragment size, age, and gender (independent variables). Adjusted values are from the least means squares estimates with associated 95% confidence limits. Correlations between histopathological findings and measures of disease severity used Pearson correlation. All P-values presented are two-tailed. Descriptive analysis and statistical tests were conducted using SAS version 9.3 (SAS Institute Inc., Cary, NC), and STATA version 11.2 (StataCorp,College Station, TX).
RESULTS
A total of 32 FSHD participants were recruited, 43.8 % male, with a median age of 47.0 years (IQR 40.8–55.3, Table 1). As not enough muscle tissue was collected to perform all procedures on each participant, they were divided into groups for analysis (Table 2). Attempts were made to match all controls by age and gender. Overall FSHD participants were older for capillary density and satellite cell counts, and younger for apoptotic rates. As mentioned in the methods interactions between age and gender and apoptotic rates, capillary density, nuclear density and satellite cell counts were investigated using a mixed effects model. As no significant interaction was found all values presented here are unadjusted.
Apoptotic rates were determined by double staining with laminin alpha-2 and TUNEL to identify nuclear fragmentation (TUNEL positive nuclei, Fig. 1) in nuclei below the basal lamina. This is to distinguish apoptosis in myonuclei from other cell types, like inflammatory cells. The number of TUNEL positive nuclei was divided into the total number of myofibers evaluated per individual (median number of myofibers per individual: FSHD 835 [IQR 556–1101]; DM1 1164 [IQR 456–1625]; and CON 877.5 [IQR 387–1453]). Participants with FSHD had increased apoptotic rates compared to CON and participants with DM1 (FSHD1 0.74% , IQR 0.38–1.27 compared to CON 0.13% , IQR 0.069–0.34, P = 0.002; or compared to DM1 0.14% , IQR 0.058–0.44, P = 0.003, Table 3). We looked to see the frequency of apoptotic nuclei which co-stained with PAX7, a satellite cell marker, and found nodifference between FSHD and CON (5.26% versus 10.53% , P = 0.45). This would suggest most of the apoptotic nuclei within the sarcolemma were mature myonuclei, not satellite cells. Although it is possible a decaying satellite cell nuclei may stop producing PAX7.
As previously reported for apoptotic rates in FSHD patients we see no association between apoptosis and pathology grade in the biopsied muscle (r = 0.30, P = 0.40), even after adjusting for age and gender [22]. We saw a trend towards an association between apoptosis and D4Z4 residual fragment size (r =−0.57, P = 0.08). Adjusting residual D4Z4 fragment size for age and gender revealed significantly higher apoptotic rates in participants with the smallest residual D4Z4 fragments (≤18 kb n = 3:1.53% , 95% CI 0.84, 2.22; compared to >18 kb n = 7:0.61% , 95% CI 0.23, 0.99, P = 0.03, Fig. 2A). This corresponded with a trend towards an association with an age-adjusted clinical severity score (r = 0.39, P = 0.26, Fig. 2B).
We calculated capillary density as the total number of capillaries per total myofiber cross sectional area (Median myofibers analyzed per individual per group: FSHD 430 [IQR 379–606]; DM1 424 [IQR 361–610]; and CON 311 [IQR 248–380]. Median fiber cross sectional area per individual per group: FSHD 3977μm2 [IQR 3037, 5435]; DM1 3748μm2 [IQR 3047, 5138]; and CON 3615μm2 [IQR 2959, 3968]). We found capillary density was reduced in FSHD compared to CON (FSHD1 316 capillaries/mm2, IQR 233–400, compared to CON 448 capillaries/mm2, IQR 391–469, P = 0.001, Table 3), but not DM1. Capillary density was directly related to the type 1 fiber proportion for FSHD (rho = 0.77, P = 0.006, Fig. 2C). There was a trend towards lower capillary density and higher muscle pathology grade (r =−0.62, P = 0.14). No relationship was seen between the capillary density and residual D4Z4 fragment size or age adjusted clinical severity scores.
We found no differences in nuclear density or satellite cell counts between FSHD and CON (Table 2).
DISCUSSION
This study presents preliminary evidence that FSHD participants had increased apoptosis in muscle biopsies compared to CON and DM1, and decreased capillary density compared to CON.
DUX4 encodes a double homeobox protein, a transcription factor normally expressed abundantly in human testis [12, 24]. Additionally, DUX4 is a double-homeobox protein that shares homology with other PRD class single homeobox transcription factors, including PAX3 and PAX7, and Otx1/2, which are important in the development of skeletal muscle and brain [9, 10, 25, 26]. DUX4 is toxic to mouse skeletal muscle and muscle fibers in cell culture, a process be-lieved to occur through a p53 mediated pathway. Forced expression of DUX4 in transfected cells leads to a redistribution of emerin, increased caspase 3/7 activity, fragmented chromatin, and cell death [10]. Our study demonstrated an increase in apoptosis by TUNEL staining in FSHD muscle tissue compared to both CON and DM1. Prior studies have shown increased apoptosis in Duchenne muscular dystrophy which corresponded to overall muscle pathology [22]. In the same study, increased apoptosis in FSHD was seen only in selected individuals, but they found a high degree of variability between participants not explained by differences in muscle pathology. Here we also do not see a relationship between apoptosis and muscle pathology grade, but we see a trend toward an association with the residual D4Z4 fragment size, and significantly higher apoptotic rates in participants with residual fragments ≤18 kb (1–4 repeats), suggesting the size of the D4Z4 contraction may explain the variability in FSHD apoptotic rates seen between participants. The apoptotic nuclei were most likely mature myonuclei versus satellite cells, possibly reflecting the stochastic burst-like expression of DUX4 described previously [24]. Further studies will be required to determine the exact relationship of TUNEL positive nuclei to DUX expression, or activation of downstream targets.
The association between DUX4 mediated toxicity and anomalies of microvascular development remains unclear. Patients with FSHD are clearly at risk for an exudative retinopathy due to bilateral retinal telangiectasias [28]. Gene expression profiles in FSHD patients identified a subset of genes involved in angiogenesis and endothelial growth upregulated in FSHD [6]. In our study, we found a decrease in capillary density in FSHD muscle. Inflammation has been observed in the eyes of patients with exudative retinopathy, although it is uncertain if it is a cause for the pathology or a consequence of extensive lipid deposits [29]. Inflammation has been reported in up to 1/3 of skeletal muscle biopsies from patients with FSHD –and unlike other dystrophies inflammation in FSHD tends to be perivascular [30, 31]. Whether the decreased capillary density seen here represents a fundamental defect in FSHD muscle, or a downstream effect related to an inflammatory milieu remains to bedetermined.
Skeletal muscle myogenesis occurs post-natally in precursor cells known as satellite cells [32]. DUX4 regulated genes are expressed in FSHD muscle and downstream targets of DUX4 include genes involved in myogenesis [12]. It has been proposed the mechanism for this may involve DUX4 interactingcompetitively with Pax7 to affect satellite cell renewal [25]. However, our current study does not support this hypothesis: we see no difference in satellite cell number seen between FSHD patient biopsies and CON.
Limitations to the current study include the small sample size. The histopathological studies are currently work-intensive which will limit their use to smaller studies, or early phase clinical trials. The conclusions from any such analysis are limited by the degree to which a small sample from a single muscle can be said to represent the overall progression of disease in a given participant.
In summary, we found increased apoptosis in FSHD muscle without influencing overall satellite cell counts or myonuclear density. FSHD muscle also shows reduced capillary fiber density reminiscent of the simplification of the retinal microvasculature. These histopathological changes may prove to be an important mechanistic marker of disease in FSHD patient muscle biopsies. Whether there is a connection between apoptosis and reduced capillary density, and the exact relationship to underlying DUX4 expression will be an important question for future studies.
DISCLOSURE
Dr. Tawil is a consultant for Cytokinetics andNovartis.
AUTHOR CONTRIBUTIONS
Jeffrey Statland and Rabi Tawil: drafting/revising the manuscript for content; study concept or design; analysis or interpretation of data; in addition acquisition of data and statistical analysis.
Karen Odrzywolski, Bharati Shah, Don Henderson, and Alex Fricke: drafting/revising the manuscript for content; analysis or interpretation of data; in addition acquisition of data.
Stephen J Tapscott and Silvère M van der Maarel:drafting/revising the manuscript for content; analysis or interpretation of data.
ACKNOWLEDGMENTS
We would like to thank all the FSHD participants and their family members without whose support this study would not have been possible. The Cellular and Molecular Pathophysiology Study in FSHD has been funded in whole or in part by the National Institutes of Health (grant# 1PO1NS069539-01) and the Fields Center for FSHD and Neuromuscular Research. The project described in this publication was supported by the University of Rochester CTSA award number UL1 RR024160 from the National Center for Research Resources and the National Center for Advancing Translational Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Dr. Statland’s work on this project was supported by a CTSA grant from NCATS awarded to the University of Kansas Medical Center for Frontiers: The Heartland Institute for Clinical and Translational Research # KL2TR000119.
REFERENCES
1 | Flanigan K.M., Coffeen C.M., Sexton L., Stauffer D., Brunner S., Leppert M.F.(2001) Genetic characterization of a large,historically significant Utah kindred with facioscapulohumeraldystrophyNeuromuscul Disord.11: 525529 |
2 | Padberg G.W., Frants R.R., Brouwer O.F., Wijmenga C., Bakker E., Sandkuijl L.A.(1995) Facioscapulohumeral muscular dystrophy in the Dutch populationMuscle Nerve2: S81S84 |
3 | Lemmers R.J., Tawil R., Petek L.M.(2012) Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2Nat Genet44: 13701374 |
4 | Lemmers R.J., van der Vliet P.J., Klooster R.(2010) A unifying genetic model for facioscapulohumeral muscular dystrophyScience329: 16501653 |
5 | Celegato B., Capitanio D., Pescatori M.(2006) Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genesProteomics6: 53035321 |
6 | Osborne R.J., Welle S., Venance S.L., Thornton C.A., Tawil R.(2007) Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophyNeurology68: 569577 |
7 | Rahimov F., King O.D., Leung D.G.(2012) Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkersProc Natl Acad Sci U S A109: 1623416239 |
8 | Winokur S.T., Chen Y.W., Masny P.S.(2003) Expression profiling of FSHD muscle supports a defect inspecific stages of myogenic differentiationHum Mol Genet.12: 28952907 |
9 | Bosnakovski D., Lamb S., Simsek T.(2008) DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiationExp Neurol214: 8796 |
10 | Kowaljow V., Marcowycz A., Ansseau E.(2007) The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic proteinNeuromuscul Disord17: 611623 |
11 | Snider L., Asawachaicharn A., Tyler A.E.(2009) RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: New candidates for the pathophysiology of facioscapulohumeral dystrophyHum Mol Genet18: 24142430 |
12 | Geng L.N., Yao Z., Snider L.(2012) DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophyDev Cell22: 3851 |
13 | de Greef J.C., Lemmers R.J., Camano P.(2010) Clinical features of facioscapulohumeral muscular dystrophy 2Neurology75: 15481554 |
14 | Wijmenga C., Hewitt J.E., Sandkuijl L.A.(1992) Chromosome 4q DNA rearrangements associated withfacioscapulohumeral muscular dystrophyNat Genet2: 2630 |
15 | Ricci E., Galluzzi G., Deidda G.(1999) Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotypeAnn Neurol45: 751757 |
16 | van Overveld P.G., Enthoven L., Ricci E.(2005) Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophyAnn Neurol58: 569576 |
17 | Kern P.A., Simsolo R.B., Fournier M.(1999) Effect of weight loss on muscle fiber type, fiber size, capillarity, and succinate dehydrogenase activity in humansJ Clin Endocrinol Metab84: 41854190 |
18 | Lewis M.I., Fournier M., Wang H.(2012) Metabolic and morphometric profile of muscle fibers in chronic hemodialysis patientsJ Appl Physiol112: 7278 |
19 | Lillioja S., Young A.A., Culter C.L.(1987) Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in manJ Clin Invest80: 415424 |
20 | Kottlors M., Kirschner J.(2010) Elevated satellite cell number in Duchenne muscular dystrophyCell Tissue Res340: 541548 |
21 | Mackey A.L., Kjaer M., Charifi N.(2009) Assessment of satellite cell number and activity status in human skeletal muscle biopsiesMuscle Nerve40: 455465 |
22 | Sandri M., El Meslemani A.H., Sandri C.(2001) Caspase 3 expression correlates with skeletal muscle apoptosis in Duchenne and facioscapulo human muscular dystrophy. A potential target for pharmacological treatment?J Neuropathol Exp Neurol60: 302312 |
23 | Tews D.S., Goebel H.H.(1997) DNA-fragmentation and expression of apoptosis-related proteins in muscular dystrophiesNeuropathol Appl Neurobiol23: 331338 |
24 | Snider L., Geng L.N., Lemmers R.J.(2010) Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed genePLoS Genet6: e1001181 |
25 | Bosnakovski D., Xu Z., Gang E.J.(2008) An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologiesEMBO J27: 27662779 |
26 | Holland P.W., Booth H.A., Bruford E.A.(2007) Classification and nomenclature of all human homeobox genesBMC Biol5: 47 |
27 | Wallace L.M., Garwick S.E., Mei W.(2011) DUX4, a candidategene for facioscapulohumeral musculardystrophy, causes p53-dependent myopathy in vivo Ann Neurol69: 540552 |
28 | Shields C.L., Zahler J., Falk N.(2007) Neovascular glaucoma from advanced Coats disease as the initial manifestation of facioscapulohumeral dystrophy in a 2-year-old childArch Ophthalmol125: 840842 |
29 | Bindoff L.A., Mjellem N., Sommerfelt K.(2006) Severe fascioscapulohumeral muscular dystrophy presenting with Coats’ disease and mental retardationNeuromuscul Disord16: 559563 |
30 | Arahata K., Ishihara T., Fukunaga H.(1995) Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): Immunocytochemical and genetic analysesMuscle Nerve2: S56S66 |
31 | Carpenter SK S.K.G.(2001) Pathology of Skeletal MuscleSecond Edition edNew YorkOxford University Press |
32 | Wernig A., Bone M., Irintchev A., Schafer R., Cullen M.(2004) M-cadherin is a reliable marker of quiescent satellitecells in mouse skeletal muscleBasic Appl Myol14: 161168 |
Figures and Tables
Fig.1
Apoptosis. A) DAPI stains nuclei blue. B) TUNEL stained degenerating nuclei green. C) Overlay stained with DAPI and TUNEL shows degenerating myonuclei. Laminin alpha-2 shows outline of myofiber in red. DAPI = 4,6-diamidino-2-phenylindole; TUNEL = terminal deoxynucleotidyl transferase dUTP nick end labeling.
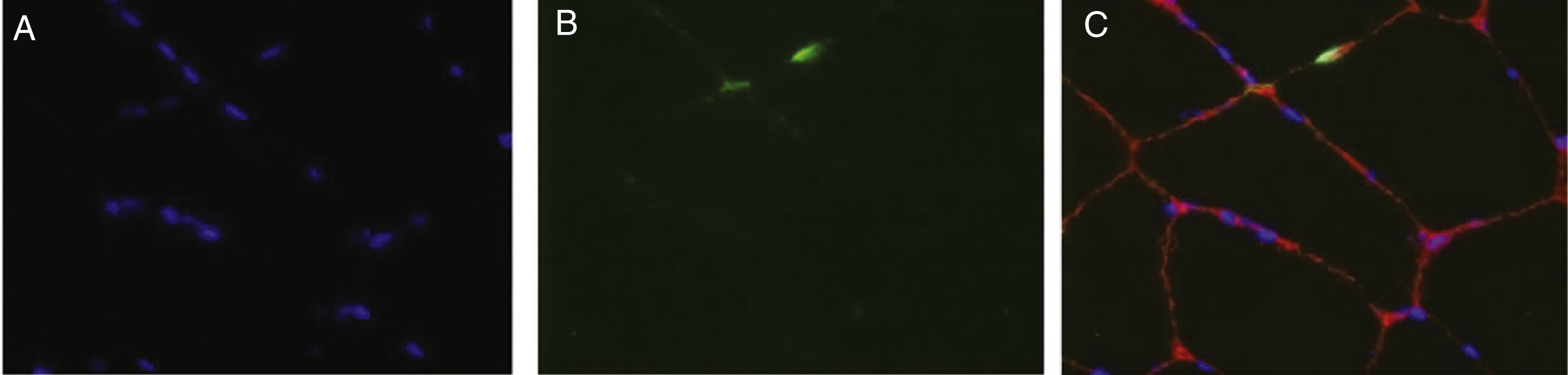
Fig.2
Relationships of histopathology findings. A) Relationship of apoptosis rate to genetic mutation (≤18kb [n = 3]; 19–27 kb [n = 4];and >27 kb [n = 3]); B) Relationship of apoptosis rate to age adjusted clinical severity score; and C) Relationship of capillary density measurement to the fiber type ratio.
![Relationships of histopathology findings. A) Relationship of apoptosis rate to genetic mutation (≤18kb [n = 3]; 19–27 kb [n = 4];and >27 kb [n = 3]); B) Relationship of apoptosis rate to age adjusted clinical severity score; and C) Relationship of capillary density measurement to the fiber type ratio.](https://content.iospress.com:443/media/jnd/2015/2-3/jnd-2-3-jnd150077/jnd-2-3-jnd150077-g002.jpg)
Table 1
Individual FSHD participant demographics
| Participant | Age (Years) | Gender | D4Z4 (kb) | CSS | Pathology score | Procedures |
| 1 | 59 | F | 31 | 3 | 2 | C, N |
| 2 | 47 | M | 25 | 3 | 2 | C |
| 3 | 53 | F | 31 | 6 | 3 | C, N |
| 4 | 47 | M | 26 | 0 | 2 | C |
| 5 | 43 | M | 27 | 8 | 4 | A |
| 6 | 51 | F | 27 | 8 | 5 | A |
| 7 | 58 | F | 28 | 6 | 2 | A, N, S |
| 8 | 50 | M | 13 | 6 | 4 | A |
| 9 | 31 | F | 25 | 5 | 6 | A |
| 10 | 28 | M | 13 | 8 | 5 | A |
| 11 | 24 | M | 16 | 5 | 4 | A |
| 12 | 43 | F | 34 | 6 | 4 | C |
| 13 | 67 | F | 34 | 2 | 1 | C |
| 14 | 53 | F | 26 | 5 | 6 | C |
| 15 | 45 | M | 18 | 6 | 4 | C |
| 16 | 56 | F | 24 | 6 | 5 | C |
| 17 | 41 | M | 31 | 6 | 3 | C |
| 18 | 56 | M | 26 | 4 | 3 | C |
| 19 | 50 | M | 30 | 5 | 3 | A |
| 20 | 46 | F | 15 | 1 | 2 | N |
| 21 | 47 | F | 35 | 1 | 7 | S |
| 22 | 30 | F | 23 | 3 | 3 | A |
| 23 | 26 | F | 15 | 7 | 5 | S |
| 24 | 55 | F | 15 | 6 | 4 | S |
| 25 | 29 | F | 12 | 9 | 3 | N |
| 26 | 59 | F | 10 | 7 | 3 | N |
| 27 | 47 | F | 19 | 1 | 2 | S |
| 28 | 59 | M | 26 | 5 | 1 | S |
| 29 | 48 | M | 17 | 3 | 4 | N, S |
| 30 | 60 | F | 26 | 3 | 2 | N, S |
| 31 | 40 | M | 29 | 2 | 2 | N, S |
| 32 | 29 | M | 35 | 2 | 2 | A |
CSS = clinical severity score; A = apoptosis, C = capillary density, N = nuclear density, S = satellite cell count.
Table 2
Demographics by group
| Demographics | FSHD * | DM1 * | Healthy volunteers * |
| Apoptotic Rates | |||
| n | 10 | 10 | 10 |
| Male (% ) | 60.0 | 20.0 | 40.0 |
| Median Age (Q1, Q3) | 37.5 (29.3, 50.0) | 38.5 (29.8, 47.3) | 54.0 (46.0, 56.0) |
| Capillary Density | |||
| n | 11 | 17 | 15 |
| Male (% ) | 45.5 | 52.9 | 60.0 |
| Median Age (Q1, Q3) | 53.0 (46.0, 56.0) | 36.0 (31.5, 52.0) | 28.0 (22.0, 37.0) |
| Satellite Cell Counts | |||
| n | 10 | – | 10 |
| Male (% ) | 30.0 | – | 40.0 |
| Median Age (Q1, Q3) | 50.5 (47.0, 58.8) | – | 39.0 (35.0, 49.0) |
| Nuclear Density | |||
| n | 10 | – | 10 |
| Male (% ) | 30.0 | – | 50.0 |
| Median Age (Q1, Q3) | 50.5 (42.3, 58.8) | – | 45.5 (33.3, 53.3) |
*FSHD total n = 32; DM1 total n = 17; Healthy Volunteers total n = 30. FSHD = facioscapulohumeral muscular dystrophy; DM1 = myotonic dystrophy type 1; n = number; Q1 = first quartile; Q3 = third quartile.
Table 3
Histopathological findings in FSHD quadriceps biopsies
| P-Value | ||||||
| Item | Healthy volunteers | DM1 | FSHD | FSHD v Healthy Volunteers | FSHD v DM1 | DM1 v Healthy Volunteers |
| n | 14 # | 10 | 10 | |||
| Median % Tunel + nuclei/myofiber (Q1, Q3) | 0.13 (0.069, 0.34) | 0.14 (0.058, 0.44) | 0.74 (0.38, 1.27) | 0.002 | 0.003 | 0.95 |
| n | 15 | 17 | 11 | |||
| Median Capillary/area (mm2) (Q1, Q3) | 448 (391, 469) | 362 (314, 435) | 316 (233, 400) | 0.001 | 0.36 | 0.02 |
| n | 10 | – | 10 | |||
| Median Myonuclei/myofiber (Q1, Q3) | 1.21 (1.01, 1.40) | – | 1.17 (0.82, 1.35) | 0.62 | – | – |
| N | 10 # | – | 10 | |||
| Median Satellite cells/myofiber (Q1, Q3) | 0.19 (0.17, 0.20) | – | 0.20 (0.17, 0.23) | 0.36 | – | – |
*Significance level taken from kruskal-wallistest. Bold significant after Bonferroni correction for multipletesting. Differences between groups due to age and gender wereevaluated using a linear mixed effects model, but did not affecthistopathological outcomes. # Familial controls.DM1 = myotonic dystrophy type 1; FSHD = Facioscapulohumeralmuscular dystrophy; Q1 = first quartile; Q3 = thirdquartile;v = versus.


