A de novo Mutation in the SCN4A Gene Causing Sodium Channel Myotonia
Abstract
We describe the case of a six year old boy with findings consistent with myotonia congenita: muscular hypertrophy, stiffness when commencing movements and typical warm-up signs. The most prominent symptom was myotonia of the eyelid muscles with apparent swelling around the eyes. Even though the pronounced warm-up phenomena in our patient suggested a chloride channel-associated myotonia congenita, the myotonia of his eyelid muscles indicated an involvement of sodium channels. Screening for mutations in the underlying CLCN1 gene was negative, however, in the SCN4A gene, we identified the missense mutation c.2108T>C; p.Leu703Pro for which there is strong evidence of pathogenicity because it arose de novo in the index patient.
INTRODUCTION
The non-dystrophic myotonias belong to the group of muscle ion channelopathies. Traditionally two clinical groups have been described: myotonia congenita (MC) and paramyotonia congenita (PMC), caused by a defect in the chloride channel and the sodium channel, respectively.
The typical clinical picture of MC is muscle stiffness (myotonia) and muscle hypertrophy. The myotonia is most pronounced when movement is initiated but decreases when the movement is repeated several times, the so-called warm-up phenomenon. The autosomal dominant MC, also called Thomsen disease, characteristically has an early onset (infancy to early childhood) while the recessively inherited form may have a later onset but is more severe [1]. These disorders are caused by mutations in the CLCN1 gene, which encodes the major muscle voltage-gated chloride channel [2].
Sodium channelopathies are also associated with myotonia and were previously thought to include three disorders: Paramyotonia congenita, which is precipitated by cold and aggravated by exercise; Potassium-aggravated myotonia (PAM); Hyperkalemic periodic paralysis. All three can be caused by mutations in the SCN4A gene, which is located on chromosome 17 and codes for the alpha subunit of the voltage-dependent sodium channel. Lately it has become clear that mutations in the SCN4A gene can give rise to myotonia that is not aggravated by exercise nor influenced by potassium. These are named sodium channel myotonias (SCM) [3].
CASE REPORT
A 6-year old boy was referred for a consultation at the Oslo University Hospital due to recurrent episodes of “involuntary eyelid closing”. These episodes had increased in frequency with age and could be provoked by tiredness, stress or fright. During an episode, his eyelids remained closed for several seconds but he could use his fingers to manually open them. In addition, episodes with localized swelling around one or both eyes were observed. The swelling would build up over several seconds and then disappear spontaneously. Furthermore, he had difficulties initiating movement after periods of inactivity but he did not complain of pain related to the stiffness. His parents reported that his speech articulation and mastication could also be affected. The symptoms did not appear to be influenced by temperature.
He was the eldest child of unrelated parents. His mother had hearing impairment, but the parents were otherwise healthy and there was no history of neuromuscular disorders in the family. Attainment of motor developmental milestones was described as normal; he sat independently at 7 months and walked at 15 months. On direct questioning, however, his parents reported that his movements appeared “clumsy” from the time he began to walk and that the symptoms related to his eyes were already present when he was an infant. However, they had never registered episodes of strained respiration or respiratory arrest.
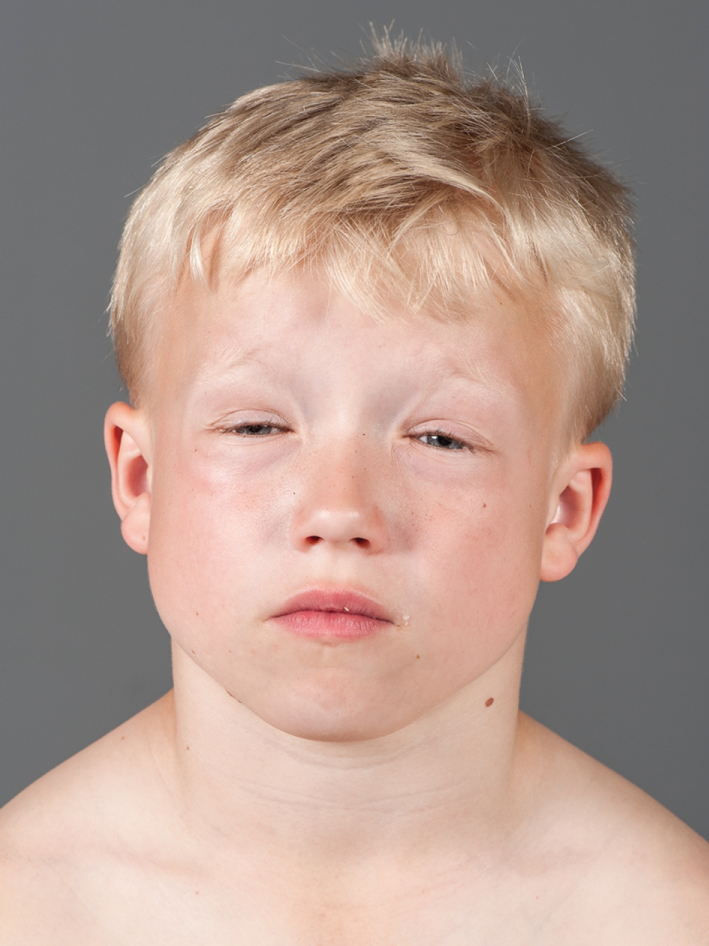
On clinical examination there was generalized muscle hypertrophy, especially around his torso and neck, with unusually prominent trapezius muscles (Fig. 1). He had increased lumbar lordosis. During the examination he had several episodes of swelling, both unilateral and bilateral around his eyes. After closing his eyes tight, it would take several seconds before he could open them again (Fig. 2). Except for eyelid myotonia, his eye motility was normal.
A warm-up sign/phenomenom was demonstrated when initiating walking, and when testing his ability to open his eyes after repetitive, intense closure. Cooling down the orbicularis oculi with ice had no evident effect on his clinical myotonia.
Ophtalmological and neurological examination was otherwise normal. He had normal creatine kinase levels. Neurophysiological examination was limited due to the patient’s lack of cooperation. Electromyographic (EMG) investigation of his right tibial muscle demonstrated profound myotonia. There were no clear signs of myopathic motor units. Nerve conduction study was only possible in one peroneal nerve, and this was normal. Neither of his parents had clinical myotonia and EMG studies were normal in both.
Direct sequencing of the SCN4A gene (Ref.Seq: NT_010783; NM_000334.4) revealed a heterozygous T > C transition at nucleotide 2108 resulting in substitution of a leucine by a proline at codon 703 (c.2108T>C; p.Leu703Pro). This sequence variant was not found in the following population databases: dbSNP, 1000 Genomes Project, and ESP. Thus, the variant is unlikely to be a normal variant. A segregation analysis in the family revealed that the variant was not present in either unaffected parent supporting a de novo occurrence in the index patient. The CLCN1 gene was sequenced and no mutation known to cause MC was found.
DISCUSSION
The mutation c.2108T>C; p.Leu703Pro in SCN4A identified in our patient has not been described as a pathogenic mutation or as a polymorphism in the literature or in international databases. At the protein level the resulting amino acid exchange is located in the fifth membrane spanning segment of domain II of the protein. Both bioinformatic prediction and segregation analysis with a de novo occurrence of the mutation in the index patient, strongly support a pathogenicity of the mutation as disease-causing for a sodium channel-associated condition.
A diagnosis of MC could be suspected in this patient as clinical examination revealed generalized muscle hypertrophy, myotonia and warm-up phenomena. Furthermore, his symptoms were not influenced by cold temperatures, a finding that has been reported in up to 79% of myotonias associated with SCN4A mutations [4]. As neither parent had abnormal findings on clinical or neurophysiological examination, a recessive variant of myotonia congenita could be suspected. Warm-up phenomena is, however, also reported in sodium channel myotonia [5, 6], and moreover, the most striking symptom in our patient was myotonia of his eyelid muscles causing delayed orbicular relaxation after closing his eyes tightly. More frequent and severe myotonia in the eyelid muscles has been described in sodium channel myotonias as compared to that in myotonia congenita [3, 5, 7] and may be the presenting or indeed only symptom in these patients. Stunnenberg and colleagues published two families with several members in each, displaying isolated myotonia of eyelid closure. In both families a L250P mutation in the SCN4A gene was found to be pathogenic [8]. Also, a father and son with extraocular muscle hypertrophy and general myotonia in addition to eyelid-myotonia was first published as a dominant myotonia congenita [9], but was later confirmed to have a mutation in the SCN4A gene [10].
Pain has been reported to be more prominent in patients with sodium channelopathies and in a larger study including 30 patients with SCN4A mutations, 57% reported pain [11]. Our patient did not report pain; however this could be related to his young age.
In our patient, the myotonia was present at an early age; the parents described that the orbicular myotonia was already present in infancy. That SCN4A mutations can be causative for infantile phenotypes has been emphasized by recent reports of neonates with severe neonatal episodic laryngospasm that developed generalized muscle hypertrophy with clinical or electrical myotonia only later in life [12, 13]. In several of these cases the patients were succesfully treated with carbamazepine [13]. Mexiletine has been shown to be effective treatment for stiffness in a randomized controlled trial on a larger group of adult patients with non-dystrophic myotonias [14]. So far the symptoms in our patient have been mild and pharmacological treatment has not been initiated.
The finding of a de novo mutation of the SCN4A gene as the probable cause of this patient’s myotonia, confirms that patients with general myotonia and warm-up phenomena might very well have a sodium-channel myotonia. The clinical picture of this subgroup of non-dystrophic myotonias is therefore quite diverse. When myotonic lid lag is present, it is advisable to look for mutations in the SCN4A gene [15].
The classical sodium channelopathy associated with myotonia is an autosomal dominant disorder with positive family history in most cases [7]. However, awareness of infantile and early phenotypes of SCN4A caused by de novo mutations in sporadic patients will help to identify this potentially treatable condition.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1 | Colding-Jorgensen E(2005) Phenotypic variability in myotonia congenitaMuscle Nerve32: 1934 |
2 | Duno, M., Colding-Jorgensen, E. Myotonia Congenita. In: Pagon, R. A., Adam, M. P., Ardinger,H. H., Bird,T. D., Dolan, C. R., Fong, C. T., Smith, R. J. H., Stephens, K., Editors. GeneReviews |
3 | Suetterlin K, Männikkö R, Hanna MG(2014) Muscle channelopathies: Recent advances in genetics, pathophysiology and therapyCurr Opin Neurol27: 583590 |
4 | Dupré N, Chrestian N, Bouchard J, Rossignol E, Brunet D, Sternberg D, Bernard B, Mathieu J, Puymirat J(2009) Clinical, electrophyisologic, and genetic sudy of non-dystrophic myotonia in French-CanadiansNeuromuscular Disorders19: 330334 |
5 | Trip J, Drost G, Ginjaar HB, Nieman FH, van der Kooi AJ, de Visser M, van Engelen BG, Faber CG(2009) Redefining the clinical phenotypes of non-dystrophic myotonic syndromesJ Neurol Neurosurg Psychiatry80: 647652 |
6 | Lee SC, Kim HS, Park YE, Choi YC, Park KH, Kim DS(2009) Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium ChannelopathyJ Clin Neurol5: 186191 |
7 | Matthews E, Fialho D, Tan SV, Venance SL, Cannon S C, Sternberg D, Fontaine B, Amato AA, Barohn RJ, Griggs RC, Hanna MGCINCH investors(2010) The non-dystrophic myotonias: Molecular pathogenesis, diagnosis and treatmentBrain133: 922 |
8 | Stunnenberg BC, Ginjaar HB, Trip J, Faber CG, van Engelen BG, Drost G(2010) Isolated eyelid closure myotonia in two families with sodium channel myotoniaNeurogenetics11: 257260 |
9 | Wakeman B, Babu D, Tarleton J, Macdonald IM(2008) Extraocular muscle hypertrophy in myotonia congenitaJ AAPOS12: 294296 |
10 | Wakeman B, MacDonald IM, Ginjaar I, Tarleton J, Babu D(2009) Extraocular muscle hypertrophy in myotonia congenita: Mutation identified in the SCN4A gene (V445M)J AAPOS13: 526527 |
11 | Trip J, de Vries J, Drost G, Ginjaar HB, van Engelen BG, Faber CG(2009) Health status in non-dystrophic myotonias: Close relation with pain and fatigueJ Neurol256: 939947 |
12 | Lion-Francois L, Mignot C, Vicart S, Manel V, Styernberg D, Landrieu P, Lesca G, Broussolle E, Billette de Villemeur T, Napuri S, des Portes V, Fontaine B(2010) Severe neonatal episodic laryngospasm due to de novo SCN4A mutations: A new treatable disorderNeurology75: 641645 |
13 | Singh RR, Tan SV, Hanna MG, Robb SA, Clarke A, Jungbluth H(2014) Mutations in SCN4A: A Rare but Treatable Cause of Recurrent Life-Threatening LaryngospasmPediatrics134: e1447e1450 |
14 | Statland JM, Bundy BN, Wang Y, Rayan JR, Salajegheh MK, Venance SL, Ciafaloni E, Matthews E, Meola G, Herbelin L, Griggs RC, Barohn RJ, Hanna MG(2012) Mexiletine for symptoms and signs of myotonia in nondystrophic myotoniaJAMA308: 13571365 |
15 | Trip J, Drost G, Verbove DJ, van der Kooi AJ, Kuks JB, Notermans NC, Verschuuren JJ, de Visser M, van Engelen BG, Faber CG, Ginjaar IB(2008) In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotoniaEur J Hum Genet16: 921929 |
Figures and Tables
Fig.1
A and B. The patient had prominent muscles and this was specifically apparent around his neck.
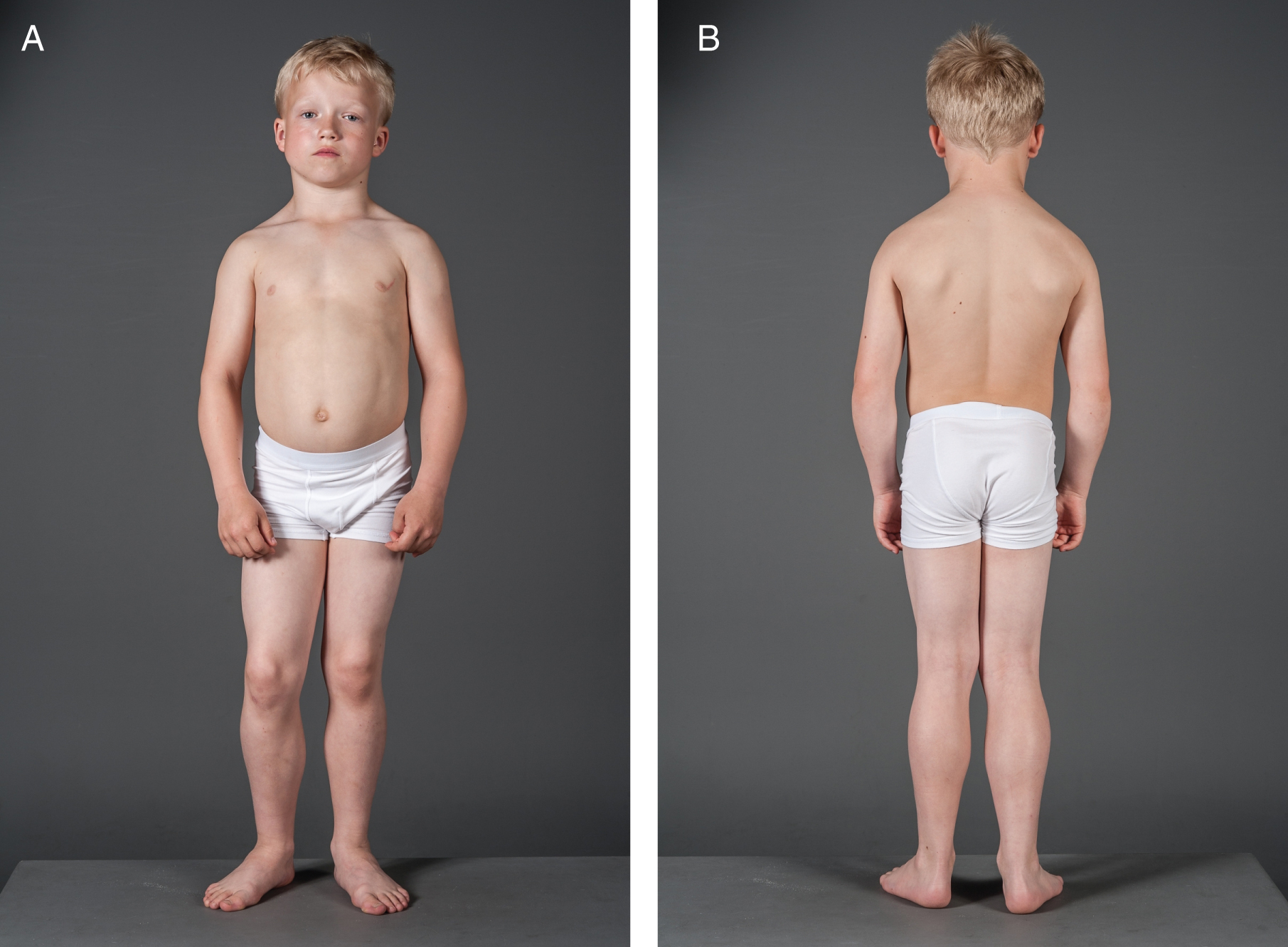
Fig.2
The patient is struggling to open his eyes after a tight closure. Note also the swelling around his eyes.