PABPN1 (GCN)11 as a Dominant Allele in Oculopharyngeal Muscular Dystrophy –Consequences in Clinical Diagnosis and Genetic Counselling
Abstract
Oculopharyngeal muscular dystrophy (OPMD) is mainly characterized by ptosis and dysphagia. The genetic cause is a short expansion of a (GCN)10 repeat encoding for polyalanine in the poly(A) binding protein nuclear 1 (PABPN1) gene to (GCN)12–17 repeats. The (GCN)11/Ala11 allele has so far been described to be either a polymorphism or a recessive allele with no effect on the phenotype in the heterozygous state. Here we report the clinical and histopathological phenotype of a patient carrying a single (GCN)11/Ala11 heterozygous allele and presenting an atypical form of OPMD with dysphagia and late and mild oculomotor symptoms. Intranuclear inclusions were observed in his muscle biopsy. This suggests a dominant mode of expression of the (GCN)11/Ala11 allele associated with a partial penetrance of OPMD.
INTRODUCTION
Oculopharyngeal muscular dystrophy (OPMD) (MIM #164300) is an autosomal dominant inherited muscular dystrophy. It was first described in a French-Canadian family in 1915 [1] and referenced as OPMD in 1962 [2]. The clinical onset is usually in the fifth or sixth decade of life and OPMD patients are characterized by ptosis and dysphagia with a slow progressive course [3, 4]. In later stages of the disease other skeletal muscle symptoms can occur such as proximal weakness and atrophy of limb-girdle muscles [5–7], impairment of eye movements,diplopia and nasal voice [8]. Homozygous patients show an earlier onset of clinical symptoms and cognitive decline can be observed [9–12]. OPMD has a world-wide distribution, in Europe, the estimated prevalence is 1:100 000 and the largest OPMD cluster is in the French-Canadian population, where the estimated prevalence is 1:1000 [13]. OPMD is usually transmitted as an autosomal dominant trait with complete penetrance. In 1998, the locus was mapped to the polyadenylate-binding protein nuclear 1 gene (PABPN1) on chromosome 14q11.2–q13 [14]. The normal PABPN1 gene (NG_008239) has at the 5’end of the first exon, a (GCN)10 repeat encoding a polyalanine (polyA) stretch, while in OPMD patients this repeat is expanded to (GCN)12–17. In contrast with most triplet repeat expansion diseases, OPMD represents one of the few diseases caused by a very short and meiotically stable trinucleotide-repeat expansion which does not increase in mutation size from generation to generation [14]. The mutated expanded PABPN1 proteins accumulate as insoluble tubulofilamentous intranuclear inclusions (INIs) in muscles of OPMD patients. These INIs are the hallmark of this disease [15]. Despite several studies on the distribution of PABPN1 alleles in large cohorts [4, 16–19], no clear correlation has been made between the size of the expansion and the severity of the phenotype. Patients homozygote for the (GCN)11 allele have been described and this has been shown to lead to an autosomal recessive form of OPMD [10]. This (GCN)11 allele in the PABPN1 gene has a 1% –2% prevalence in North America, Europe, and Japan [8]. It has been described to have no effect on phenotype in the heterozygous state, nevertheless, in compound heterozygous patients carrying one expanded (GCN)12–17 allele and one (GCN)11 allele, (GCN)11 acts as a modifier resulting in a more severe phenotype [14, 20]. The only two genetically proven recessive OPMD cases to date were homozygous carriers with two (GCN)11 PABPN1 alleles [21]. Here we describe a clinical, morphological and genetic analysis of a patient heterozygous carrier of the (GCN)10/(GCN)11 genotype in PABPN1.
MATERIAL AND METHODS
Muscle histology
Muscle biopsies were mounted on gum tragacanth and snap frozen in liquid N2-cooled isopentane. Staining was carried out on transverse serial cryosections of muscles (5 μm). For the assessment of tissue morphology and visualization of connective tissue, muscle sections were stained with hematoxylin and eosin for light microscopic examination. Changes in fiber architecture and structural abnormalities were assessed by Gomori trichrome staining.
Electron microscopy
Muscle biopsies were fixed in 2% paraformaldehyde+2% glutaraldehyde diluted in 0.1 mol/L phosphate buffer, pH 7.4. After 2% OsO4 postfixation, they were gradually dehydrated in acetone and embedded in Epon resin (EMS, Fort Washington, PA). Ultrathin sections were stained with uranyl and lead citrate and examined with a Philips CM120 electron microscope connected to an SIS Morada digital camera.
PABPN1 inclusions detection
Immunostaining were performed on 5-μm-thick cryostat muscle sections fixed in 4% paraformaldehyde. For immunodetection of PABPN1 inclusions, sections were preincubated in 1 mol/L KCl solution for 1 hour to remove soluble proteins before incubation with rabbit polyclonal anti-PABPN1 (EP2000Y, Euromedex, France) and anti-dystrophin antibody (NCL-Dys1, Novocastra). Sections were further incubated with appropriate secondary antibodies and stained with Hoechst (Sigma-Aldrich, St. Louis, MO) to visualize nuclei.
Image acquisition and analysis
Images were visualized using an Olympus BX60 microscope (Olympus Optical, Hamburg, Germany), digitized using a CCD camera (Photometrics CoolSNAP fx; Roper Scientific, Tucson, AZ), and analyzed using MetaView image analysis system (Universal Imaging, Downingtown, PA), MetaMorph imaging system (Roper Scientific) software, and ImageJ 1.44o (http://imagej.nih.gov/ij).
CASE REPORT
The patient is a man born in 1919 who suffered from dysphagia since 60 years of age. There was no known family history of OPMD, his parent’s died at 67 and 83 years of age with no apparent symptoms and he had three older sisters (status unknown). He was able to play football and tennis until 67 years of age. His first medical visit at 67 years was motivated by progressive mild dysphagia including slow swallowing, rare choking events, and regurgitation. Examination revealed no ptosis of the eyelid, no limitation in eye movements, and a discrete weakening of the voice. At 74 years of age, the dysphagia rapidly increased and a pneumopathy was reported two years later. Deterioration of swallowing was confirmed by videofibroscopy showing reduced pharyngeal propulsion and choking. First complains of some muscle weakness in the limbs was reported at the same age. The main result of electromyography was a clearly diffuse myogenic pattern. A very mild decrease of sensory potentials amplitudes evidenced in distal lower limbs, with no clinical sensory defect, was considered, in an 80 years old patient, as a possible minimal old age related neuropathy. Diabetes, dysthyroidis, dysglobulinemia, Gougerot Sjögren syndrome (normal salivary gland biopsy) were ruled out. At 80 years of age, the patient underwent a cricopharyngeal myotomia with impressive improvement of dysphagia and weight gain. At that time, there was no ptosis but a mild limitation of upper gaze with minimal diplopia. A mild weakness of psoas and flexors of the neck and the trunk was also found. At that age, a scanner of limbs was normal despite the patient complained of weakness. At 83 years of age, dysphagia reappeared and progressively worsened with daily choking but without weight loss. Axial and proximal lower limb weakness was evident: standing up from squatting was impossible, rising from a chair required help and sitting from a lying position was impossible. Except for mild biceps and finger interosseous muscle weakness, upper limbs were spared. A mild left ptosis of the eyelid and nasal voice were evidenced. CT scan of lower limbs showed a hypodense aspect of glutei, posterior thigh and leg compartment muscles. Biological tests were normal excepted for CK levels which were slightly elevated (340 UI/l). The patient died at age 84 due to severe pneumopathy consecutive to swallowing impairment. Some weeks before, he had refused a proposition of per-endoscopic gastrostomy.
Morphological investigations of the skeletal muscle biopsy were performed as classically described [15]. Electron microscopy analysis on the quadriceps muscle biopsy at 68 years of age revealed that several muscle fibres contained myonuclei with well limited clear areas without chromatin. Inside these areas we observed many inclusions consisting of tubular filaments. Often these tubular filaments with 7-8 nm outer diameter were converging to form structures resembling the characteristic palisades initially described [15] (Fig. 1A). Frozen sections of a sternocleidomastoid muscle biopsy at 80 years of age showed a high variability in fibre size with many internalised myonuclei and endomysial connective tissue, and a few fibers with rimmed vacuoles (Fig. 1B-C). The presence of KCl-insoluble PABPN1 intranuclear inclusions was observed by immunofluorescent staining [22] in 6% of myonuclei (Fig. 1D-D’).
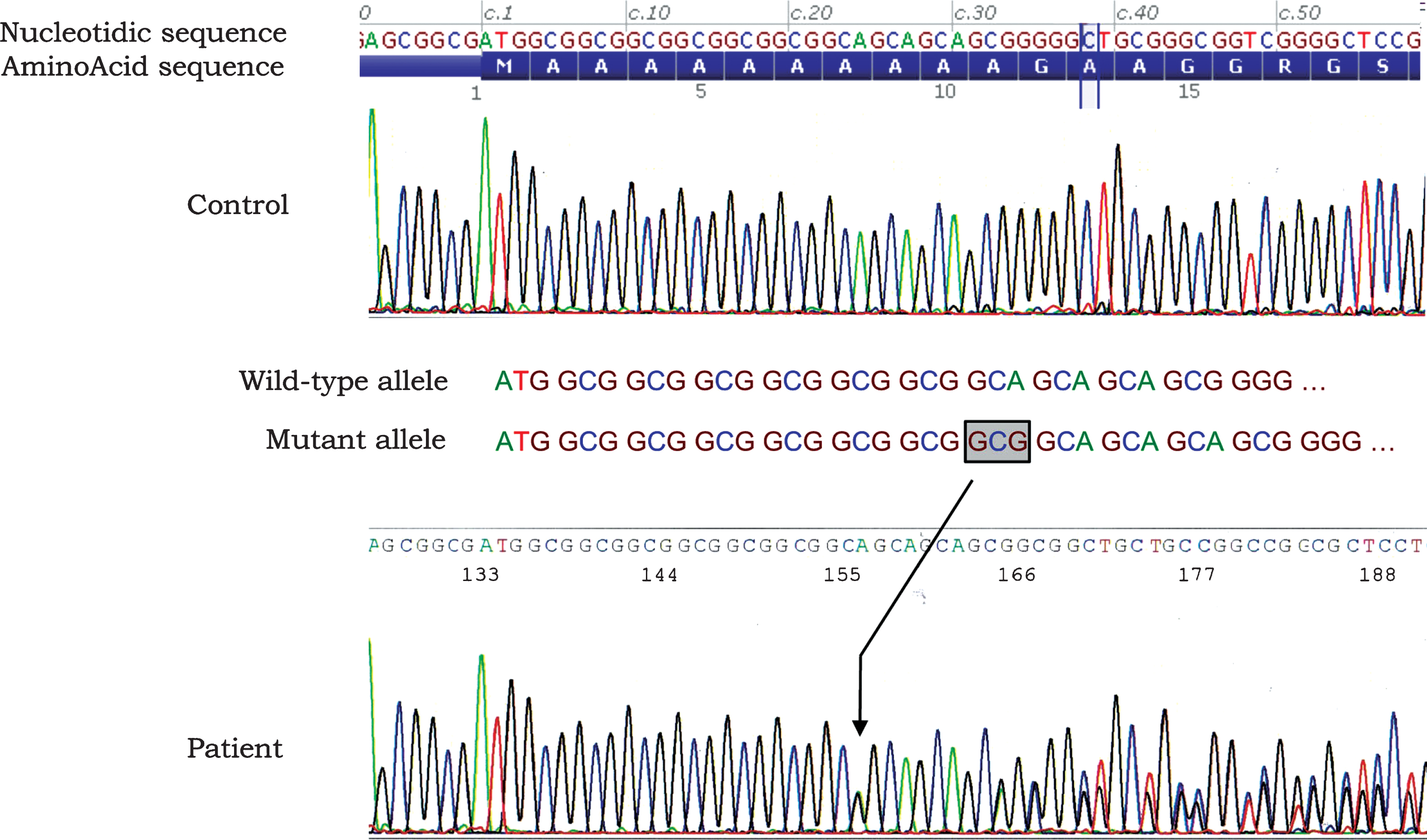
Genotyping was performed on DNA extracted from a blood sample after the informed written consent had been signed according to the French legislation. The sequence of exon 1 containing the GCN repeat sequence in the PABPN1 gene was amplified by PCR as described by Brais et al. [14] and allele sizing was done using Genescan software. The patient was found to be heterozygous for an allele of 246 base pairs corresponding to 10 (GCN) repeats and an allele of 249 base pairs corresponding to 11 (GCN) repeats (Fig. 2). In addition, the complete sequencing of the PABPN1 sequence was performed and confirmed that no additional mutation were identified.
DISCUSSION
The expanded (GCN)11/Ala11 allele in PABPN1 gene was first described as a polymorphism [4], then a recessive allele acting only in the homozygote state [20, 21, 23] and also a disease modifying allele causing a more severe phenotype when associated with another expanded allele [14]. In the French-Canadian population, the (GCN)11 allele was reported to have a background frequency of 2% . In 2007, Hebbar et al. [21] described two siblings affected with a late onset form of OPMD and homozygous for the (GCN)11 allele as a recessive case of OPMD. However, the strong family history of ptosis in the mother, aunt and cousins, might be rather in favour of a dominant familial form.
In this report, we describe a case found to carry a (GCN)11 PABPN1 recessive mutation with a potential dominant phenotype. The Ala11 expansion was known to be recessive. We cannot argue if it is a de novo mutation. The patient’s parents are deceased with no clinical information available. The patient presents an atypical and confusing clinical form of OPMD, including progressive dysphagia, which persists until 80 years of age, minimal and very late oculomotor involvement and no known family history of OPMD. The muscle biopsy showed PABPN1 intranuclear inclusions. This strongly suggests that, the allele with eleven repeats act as a dominant allele because it is associated with the classical clinical and pathological expression of OPMD. This is to our knowledge the only case to date to be described of (GCN)11 carrier with associated symptoms. The minimal and late onset ptosis observed, which is one of the commonest first symptoms, could explain that heterozygous patients carrying a (GCN)11 allele associated with the normal (GCN)10 allele are probably under or misdiagnosed, in a context where in some countries OPMD is still nowadays under-diagnosed [24–26] with a high variability in symptoms worldwide [5–7]. The under-representation of (GCN)11 at the heterozygous status is also supported by the fact that cohort studies described several (GCN)11/(GCN)11 homozygous patients but no heterozygous (GCN)10/(GCN)11 genotypes [4, 14, 15, 17, 18]. This study highlights the importance of an OPMD diagnosis when one is faced by an atypical clinical presentation and confirms that the presence of PABPN1 inclusions are still a major histopathological marker for OPMD. Such a clinical pattern requires an adapted strategy consisting in first, a genotyping test for PABPN1 expansion [27] and second, a muscle biopsy including electron microscopy for detecting specific tubulofilamentary inclusions and/or immunostaining for PAPBN1 inclusions [22]. There are still important unanswered questions in OPMD that remain to be elucidated [27]. We hope that this report will help to increase our understanding of the correlations between the phenotype and the genotype in OPMD, and will allow a better genetic counselling in families with a more accurate detection of late onset and atypical forms of the disease.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
The authors would like to thank the patient and his family for his helpful collaboration. We are very grateful to Valérie Jobic for PABPN1 sequencing and Dr Gillian Butler-Browne for critical reading of the manuscript. This work was supported by Assistance Publique-Hôpitaux de Paris, INSERM, CNRS and Université Pierre et Marie Curie and by grants from AFM (Association Française contre les Myopathies, OPMD Network Research Program 15123 and 17110) and Fondation de l’avenir (project ET1-622). This study was conducted with the approval of the GH Pitie-Salpêtrière, Paris, France.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1 | Taylor E(1915) Progressive vagus-glossopharyngeal paralysis with ptosis: A contribution to the group family diseasesJ Nerv Ment Dis129139 |
2 | Victor M, Hayes R, Adams RD(1962) Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the evelidsN Engl J Med267: 12671272 |
3 | Bouchard JP, Brais B, Brunet D, Gould PV, Rouleau GA(1997) Recent studies on oculopharyngeal muscular dystrophy in QuebecNeuromuscul Disord1: Suppl 1S22S29 |
4 | Hill ME, Creed GA, McMullan TF, Tyers AG, Hilton-Jones D, Robinson DO(2001) Oculopharyngeal muscular dystrophy: Phenotypic and genotypic studies in a UK populationBrain124: Pt 3522526 |
5 | Witting N, Mensah A, Kober L, Bundgaard H, Petri H, Duno M(2014) Ocular, bulbar, limb, and cardiopulmonary involvement in oculopharyngeal muscular dystrophyActa Neurol Scand130: 2125130 |
6 | Van Der Sluijs BM, Hoefsloot LH, Padberg GW, Van Der Maarel SM, Van Engelen BG(2003) Oculopharyngeal muscular dystrophy with limb girdle weakness as major complaintJ Neurol250: 1113071312 |
7 | Trollet C, Gidaro T, Klein P, Perie S, Butler-Browne G, Lacau St Guily J(2010) Oculopharyngeal Muscular DystrophyPagon RA, Ardinger HHSeattle (WA)1993 |
8 | Brais B(2003) Oculopharyngeal muscular dystrophy: A late-onset polyalanine diseaseCytogenet Genome Res100: 1-4252260 |
9 | Blumen SC, Bouchard JP, Brais B, Carasso RL, Paleacu D, Drory VE(2009) Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotesNeurology73: 8596601 |
10 | Blumen SC, Brais B, Korczyn AD, Medinsky S, Chapman J, Asherov A(1999) Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the diseaseAnn Neurol46: 1115118 |
11 | Dubbioso R, Moretta P, Manganelli F, Fiorillo C, Iodice R, Trojano L(2012) Executive functions are impaired in heterozygote patients with oculopharyngeal muscular dystrophyJ Neurol259: 5833837 |
12 | Mizoi Y, Yamamoto T, Minami N, Ohkuma A, Nonaka I, Nishino I(2011) Oculopharyngeal muscular dystrophy associated with dementiaIntern Med50: 2024092412 |
13 | Tome F, Fardeau M(1994) Oculopharyngeal Muscular DystrophyMcGraw-Hil 12331245MyologyNew York |
14 | Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM(1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophyNat Genet18: 2164167 |
15 | Tome FM, Fardeau M(1980) Nuclear inclusions in oculopharyngeal dystrophyActa Neuropathol49: 18587 |
16 | Mirabella M, Silvestri G, de Rosa G, Di Giovanni S, Di Muzio A, Uncini A(2000) GCG genetic expansions in Italian patients with oculopharyngeal muscular dystrophyNeurology54: 3608614 |
17 | Muller T, Deschauer M, Kolbe-Fehr F, Zierz S(2006) Genetic heterogeneity in 30 German patients with oculopharyngeal muscular dystrophyJ Neurol253: 7892895 |
18 | Robinson DO, Hammans SR, Read SP, Sillibourne J(2005) Oculopharyngeal muscular dystrophy (OPMD): Analysis of the PABPN1 gene expansion sequence in 86 patients reveals 13 different expansion types and further evidence for unequal recombination as the mutational mechanismHum Genet116: 4267271 |
19 | Tondo M, Gamez J, Gutierrez-Rivas E, Medel-Jimenez R, Martorell L(2012) Genotype and phenotype study of 34 Spanish patients diagnosed with oculopharyngeal muscular dystrophyJ Neurol259: 815461552 |
20 | Semmler A, Kress W, Vielhaber S, Schroder R, Kornblum C(2007) Variability of the recessive oculopharyngeal muscular dystrophy phenotypeMuscle Nerve35: 5681684 |
21 | Hebbar S, Webberley MJ, Lunt P, Robinson DO(2007) Siblings with recessive oculopharyngeal muscular dystrophyNeuromuscul Disord17: 3254257 |
22 | Gidaro T, Negroni E, Perie S, Mirabella M, Laine J, Lacau St Guily J(2013) Atrophy, Fibrosis, and Increased PAX7-Positive Cells in Pharyngeal Muscles of Oculopharyngeal Muscular Dystrophy PatientsJ Neuropathol Exp Neurol72: 3234243 |
23 | Marsh EA, Robinson DO(2008) A case of rare recessive oculopharyngeal muscular dystrophy (OPMD) coexisting with hereditary neuropathy with liability to pressure palsies (HNPP)Clin Neurol Neurosurg110: 5525528 |
24 | Agarwal PK, Mansfield DC, Mechan D, Al-Shahi Salman R, Davenport RJ, Connor M(2012) Delayed diagnosis of oculopharyngeal muscular dystrophy in ScotlandBr J Ophthalmol96: 2281283 |
25 | Chien YY(2012) Oculopharyngeal muscular dystrophy –an under-diagnosed disease in China? Report a China-born Chinese with PABPN1 mutation and epidemiology review of the literatureJ Formos Med Assoc111: 7397402 |
26 | Ruegg S, Lehky Hagen M, Hohl U, Kappos L, Fuhr P, Plasilov M(2005) Oculopharyngeal muscular dystrophy - an under-diagnosed disorder? Swiss Med Wkly135: 39-40574586 |
27 | Raz V, Butler-Browne G, van Engelen B, Brais B(2013) 191st ENMC International Workshop: Recent advances in oculopharyngeal muscular dystrophy research: From bench to bedside 8-10 June Naarden, The NetherlandsNeuromuscul Disord23: 6516523 |
Figures and Tables
Fig.1
A. Electronic microscopy of the quadriceps muscle biopsy showing a central lighter area (see also inset box) with typical tubulo-filamentous inclusions in a nucleus. Magnification x50 000. Scale bars 0.1μm. B. Gomori Trichrome staining revealing the presence of rimmed vacuoles (arrow). Bar scale = 50 μm. C. Hematoxylin and eosin staining showing endomysial connective tissue (asterisk) and internally localised nuclei (around 10% , arrows) and no sign of abnormalities. Bar scale = 50 μm. D and D’. PABPN1 immunostaining following KCl treatment on the sternocleidomastoid muscle biopsy revealing PABPN1 intranuclear inclusions (arrow, see also inset box). Blue, nuclei counterstained with Hoechst; green, PABPN1; red, Dystrophin). The percentage of intranuclear inclusions was 6.9% counted on 463 nuclei of the muscle biopsy. Bar scale = 50 μm.
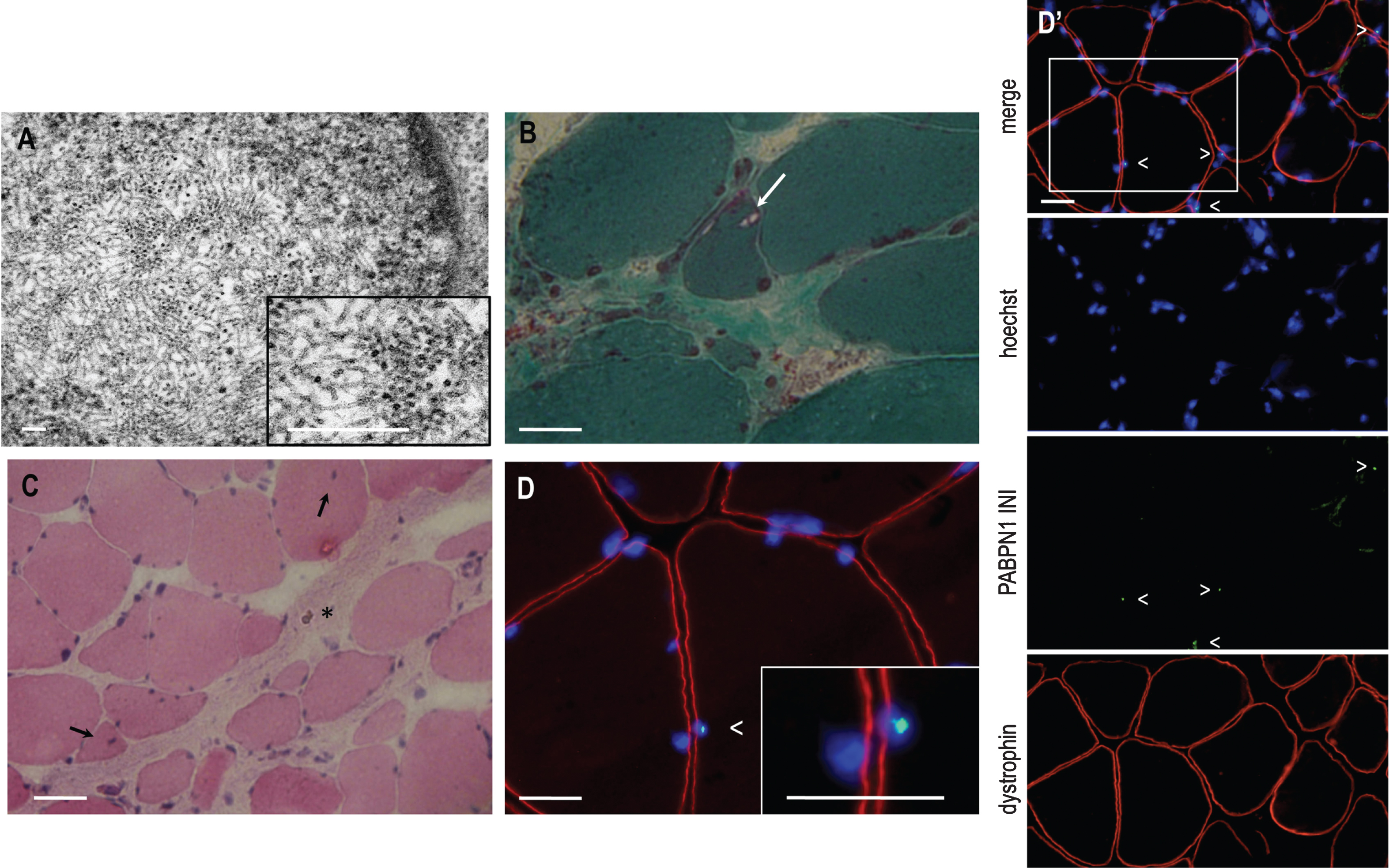
Fig.2
Electrophoregram of a partial sequence of exon 1 showing the expansion of a triplet (GCN). A simple schematic indicates the single GCG expansion in the mutated allele in the patient (surrounded text and black arrow). The absence of any other mutation that could explain the phenotype has been confirmed by full sequencing of the whole PABPN1 gene (data not shown).