
Effects of high fat diet and perinatal dioxin exposure on development of body size and expression of platelet-derived growth factor receptor β in the rat brain
Abstract
Environmental exposure to dioxins, consumption of a high fat diet, and platelet-derived growth factor receptor β signaling in the brain affect feeding behavior, which is an important determinant of body growth. In the present study, we investigated the effects of prenatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin and high fact diet after weaning on body growth and expression of platelet-derived growth factor receptor β in the brain in rat pups. Subjects from the control and dioxin exposure groups were assigned to 1 of 3 different diet groups: standard diet, high fat diet in the juvenile period, or high fat diet in adulthood. Body weight gain rate in the juvenile high fat diet group and the length gain rate in the adult high fat diet group were greater than the corresponding values in the standard diet group only in male offspring, although the effects of dioxin exposure on growth were not significant. Consumption of a high fat diet decreased platelet-derived growth factor receptor β levels in the amygdala and hippocampus in both sexes compared to control groups, while 2,3,7,8-tetrachlorodibenzo-p-dioxin decreased platelet-derived growth factor receptor platelet-derived growth factor receptor β levels in the amygdala and striatum only in females receiving an high fat diet. Furthermore, platelet-derived growth factor receptor β levels in the hippocampus and platelet-derived growth factor receptor β striatum were inversely correlated with increases in body length, while changes in platelet-derived growth factor receptor β in the amygdala and nucleus accumbens were significantly correlated to body weight gain or body mass index. In conclusion, these findings suggest that these 2,3,7,8-tetrachlorodibenzo-p-dioxin and high fat diet-induced changes in body growth and feeding behaviors might be partially mediated by changes in brain platelet-derived growth factor receptor β levels.
Abbreviations
HFD | food such as high fat diet |
NAc | nucleus accumbens |
SC | superior colliculus |
EDC | endocrine-disrupting chemicals |
TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
PDGF | platelet-derived growth factor |
b.w. | body weight |
GD | gestational day |
BMI | body mass index |
GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
TBS-T | tris-buffered saline with tween 20 |
mTOR1 | rapamycin complex 1 |
ER | endoplasmic reticulum |
IRS | insulin receptor substrates |
PI3K | phosphoinositide 3-kinase |
Akt | protein kinase B |
GH | growth hormone |
DA | dopamine |
1.Introduction
Environmental factors such as diet and environmental pollution influence human health and well-being [1]. The World Health Organization (WHO) estimated that 24% of the global burden of disease was due to modifiable environmental factors [2]. The consumption of palatable food such as that in a high fat diet (HFD) is rewarding, and activates neural circuits for motivated behavior in the mesolimbic system, including the nucleus accumbens (NAc) and amygdala [3]. Activation of these neural circuits promotes overconsumption of food beyond nutritional requirements [4]. Perinatal exposure to endocrine-disrupting chemicals (EDCs) including dioxins, may influence neural circuits for appetite and feeding behavior in the developing brain, leading to obesity later in life [5]. Perinatal exposure to dioxins, of which 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the most toxic dioxin congener, affects growth and neurodevelopment in infants and children [6–9], and influences eating behavior, particularly in girls aged 3 years (under submission).
In animal studies, maternal exposure to TCDD reduces both body and brain weight in fetuses and reduces litter size and birth weight in offspring [10–12]. In adult rats, TCDD exposure reduces feeding and consequently produces a decline in body weight (wasting syndrome) [13]. The adverse effects of TCDD on feeding behavior were greater when TCDD was administered intracerebroventricularly rather than peripherally, suggesting that the adverse effects of TCDD on feeding behavior may be mediated through effects on the brain [14]. Furthermore, TCDD had a minimal impact on gene induction in the hypothalamus, which has a central role in feeding behavior and energy balance, suggesting that TCDD affects brain regions other than the hypothalamus in the neural networks influencing feeding behavior [15].
Recent findings have indicated that two receptor subtypes of platelet-derived growth factor (PDGF; PDGFR-α and -β) can form mature dimeric receptor complexes that bind ligands with different affinities [16]. PDGFR-α is largely expressed in oligodendroglial progenitors, while PDGFR-β is predominantly expressed in neurons [17], and is also upregulated in the neonatal rat brain [18]. Recent studies have reported the involvement of PDGF in feeding behavior; consumption of high fat or regular powder diets increased PDGF levels in the cortex and cerebrospinal fluid in rats [19,20], and plasma PDGF levels were elevated in genetically obese mice [21]. Furthermore, intracerebroventricular infusion of PDGF decreased feeding behavior [19].
Therefore, evidence suggests that both TCDD and PDGF signaling affects feeding behavior, and consequently modulates body growth and development of obesity. In the present study, we investigated the impact of an HFD and perinatal exposure to TCDD on body growth and PDGFR-β levels of rat offspring in 7 brain regions, including the limbic system.
2.Materials and methods
2.1.Animals and exposure to chemicals
On gestation day (GD) 15, TCDD dissolved in corn oil (0.2 μg/mL; MP Biomedicals LLC, CA, USA) at a dose of 1.0 μg/kg body weight (b.w.) (1–1.2 mL) was gavaged to 5 pregnant Wistar rats (12 weeks of age) that served as the TCDD-exposed group. This protocol of TCDD exposure has been validated in a previous study to increase TCDD levels in pup brain tissues and to produce adverse developmental effects on endocrine function in the brain [22]. The same volume of pure corn oil was gavaged to 5 pregnant Wistar rats in the control group. All dams gave birth on gestational day (GD) 22. After birth, litter size was adjusted to 7–8 pups per litter, in order to ensure adequate nutrition. A total of 30 pups (16 males and 14 females) in the control group, and 26 pups (12 males and 14 females) in the TCDD group were available for assignment to diet groups after weaning, which occurred on postnatal day (PD) 21.
Rats were individually housed after weaning, with food and water ad libitum in a temperature-controlled room. Light/dark cycles were reversed to enable observation of behavior during typical daytime hours (lights were turned on and off at 20:00 and 08:00, respectively).
Male and female offspring rats from each litter in the control and dioxin exposure groups were assigned to 1 of 3 different diet groups: standard diet, the HFD in juvenile period group (juvenile HFD: PD 23 to PD 36), or the HFD in adulthood group (adult HFD: PD 49 to PD 58). The standard diet included normal rat chow (LaboMR-stock chow; Sankyo Labo Service Cooperation, Inc., Tokyo, Japan), whereas the HFD consisted of a rodent diet with 45 kcal% fat content (D12451; Research Diets Inc., New Brunswick, NJ, USA).
All animals were treated in strict compliance with the policies of the National Institutes of Health on the Care of Humans and Laboratory Animals and the Guidelines for the Care and Use of Laboratory Animals at Kanazawa Medical University. All experimental procedures were approved by the Ethics Committee at Kanazawa Medical University.
2.2.Body size measurement
Body weight and length (nose-to-anus) were measured daily at the same time (09:30–10:30 h). Body mass index (BMI) was calculated at the end of the observation period (PD 58) using the following formula: BMI = Weight (g)/Length (cm)2. Growth velocity of body size measurements is considered a good marker of infant growth, thus we calculated the ratios of the increase in weight or length in the juvenile period from PD 23 to PD 36/weight or length at PD 23, and those in adulthood from PD 49 to PD 58/weight or length at PD 49.
2.3.Western blotting study: PDGFR-β measurement
At 14 weeks of age, the rats were anesthetized with pentobarbital (60 mg/kg, i.p.) and sacrificed by decapitation. Brain areas including the medial prefrontal cortex (mPFC), hippocampus, nucleus accumbens (NAc), striatum excluding the NAc, parietal cortex, amygdala, and superior colliculus (SC) were then quickly dissected on ice under a stereoscopic microscope. The samples were weighed (4–18 mg), then immediately frozen in liquid nitrogen, and stored at −80°C until analyses. For western blot analysis of PDGF receptor expression, proteins were extracted by homogenization using a multibeads shocker (Shake Master NEO, BMS-M10N21 Biomedical Science, Tokyo, Japan) for 20 s in protein lysate buffer (using cOmplete Ultra Tablet mini EDTA free, Roche, Switzerland). Lysate samples were centrifuged at 20000 rpm for 30 min at 4°C, and samples extracted from the supernatant were run on a 12% SDS-polyacrylamide gel electrophoresis, following protein concentration analyses using a BCA protein assay kit (Thermo Scientific, Rockford, IL, USA). Separated proteins were transferred to polyvinylidene fluoride membranes. The membranes were blocked using Odyssey blocking solution (Odyssey, Li-cor, Lincoln, NE, USA) for 60 min at room temperature and incubated with primary antibodies against PDGFR-β (Cell Signaling Technology, Tokyo) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore, USA) overnight at 4°C. After the membranes were washed with Tris-buffered saline with Tween 20 (TBS-T), the blots were incubated with anti-rabbit and anti-chicken secondary antibodies for 1 h. Band densities were analyzed using Image studio software (Li-cor). All data for PDGFR-β expression were normalized to the GAPDH expression level and expressed as the relative expression level.
2.4.Data analysis
SPSS (ver.21.0) software package for Windows (IBM; NY, USA) was used for statistical analyses. Body size, weight and length gains, as well as PDGFR-β levels in brain tissue were analyzed using 2-way ANOVA with “dioxin exposure” and “diet” as factors followed by Fisher’s least significant difference (LSD). Correlations between PDGF levels and body size were analyzed using Spearman’s rho.
3.Results
3.1.Effects of an HFD and TCDD on body size and BMI
At the end of the observation period, mean body weight, length, and BMI were compared among the 3 diet groups, for both dioxin-exposed and control groups, using 2-way ANOVA with “diet” and “dioxin exposure” as factors. However, there was no significant main effect of dioxin exposure, or significant interaction between diet and dioxin exposure (data not shown). Therefore, estimated means of body sizes adjusted for the factor of dioxin exposure were compared among the 3 diet groups. In male offspring, there was a significant main effect of diet on body weight [
3.2.Effects of an HFD and TCDD on body weight and length gain
Body weight and length gain rates (gain of weight or length/value at the starting point) were used as indices of growth, and effects of the “diet” and “exposure” factors were analyzed by 2-way ANOVAs. There was no significant main effect of dioxin exposure in either sex, nor a significant interaction between diet and dioxin exposure. Therefore, the means of gain rates of weight and length were compared after adjusting for the effects of dioxin exposure (Table 2). During the juvenile period in male offspring, there was a significant main effect of diet on weight gain rate [
Table 1
Comparisons of exposure-effect adjusted body weight and BMI at the end of the observation period among the 3 different diet groups; Juvenile HFD (high fat diet (HFD) in juvenile period), Adult HFD (HFD in adulthood), and standard diet groups
| Food | Body weight (g) | Body length (cm) | BMI | |||||||||||||||||
| Control | Exposed | Marginal | Control | Exposed | Marginal | Control | Exposed | Marginal | ||||||||||||
| N | Mean | SE | N | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | |
| Male | ||||||||||||||||||||
| Standard diet | 3 | 248.2 | 9.9 | 2 | 245.5 | 12.2 | 246.8 | 7.8 | 20.7 | 0.39 | 20.8 | 0.47 | 20.8 | 0.31 | 58.0 | 1.63 | 56.6 | 2.00 | 57.3 | 1.29 |
| Juvenile HFD | 7 | 283.8 | 6.5 | 5 | 275.2 | 7.7 | 279.5 | 5.0ab | 21.4 | 0.25 | 21.6 | 0.30 | 21.5 | 0.20 | 62.0 | 1.07 | 59.2 | 1.26 | 60.6 | 0.83ab |
| Adult HFD | 6 | 266.3 | 7.0 | 5 | 257.5 | 7.7 | 261.9 | 5.2 | 21.6 | 0.27 | 21.1 | 0.30 | 21.4 | 0.20 | 57.3 | 1.15 | 57.7 | 1.26 | 57.5 | 0.86 |
| (ANOVA-P < 0.01) | (ANOVA-P < 0.05) | |||||||||||||||||||
| Female | ||||||||||||||||||||
| Standard diet | 3 | 185.2 | 8.8 | 3 | 189.2 | 8.8 | 187.2 | 6.2 | 19.2 | 0.26 | 19.3 | 0.26 | 19.2 | 0.18 | 50.3 | 1.66 | 50.8 | 1.66 | 50.5 | 1.17 |
| Juvenile HFD | 6 | 201.5 | 6.2 | 6 | 196.9 | 6.2 | 199.2 | 4.4 | 19.7 | 0.18 | 19.9 | 0.18 | 19.8 | 0.13a | 51.9 | 1.17 | 49.7 | 1.17 | 50.8 | 0.83 |
| Adult HFD | 5 | 195.8 | 6.8 | 5 | 188.6 | 6.8 | 192.2 | 4.8 | 19.6 | 0.20 | 19.6 | 0.20 | 19.6 | 0.14 | 50.9 | 1.28 | 48.9 | 1.28 | 49.9 | 0.91 |
| (ANOVA-P < 0.1) | ||||||||||||||||||||
a Significant difference from the standard food group (
b Significant difference from the HFD in adulthood group (
HFD: high fat diet.
Table 2
Comparisons of exposure-effect adjusted gains of body weight and length during the juvenile period, adulthood, and all period among the 3 diet groups; high fat diet in juvenile period (Juvenile HFD), HFD in adulthood (Adult HFD), and standard diet groups
| Gain rate during juvenile period | Gain rate during adulthood | Gain rate during all observation period | ||||||||||||||||
| Control | Exposed | Marginal | Control | Exposed | Marginal | Control | Exposed | Marginal | ||||||||||
| Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | |
| Male | ||||||||||||||||||
| Body weight (g) | ||||||||||||||||||
| Standard diet | 1.46 | 0.05 | 1.48 | 0.07 | 1.47 | 0.04 | 0.35 | 0.02 | 0.33 | 0.03 | 0.34 | 0.02 | 4.27 | 0.21 | 4.51 | 0.25 | 4.39 | 0.16 |
| Juvenile HFD | 1.77 | 0.03 | 1.70 | 0.04 | 1.73 | 0.03a | 0.37 | 0.01 | 0.34 | 0.02 | 0.35 | 0.01 | 5.02 | 0.13 | 4.54 | 0.16 | 4.79 | 0.10a |
| Adult HFD | 1.53 | 0.03 | 1.50 | 0.04 | 1.51 | 0.03 | 0.40 | 0.02 | 0.41 | 0.02 | 0.41 | 0.01ac | 4.80 | 0.15 | 4.76 | 0.16 | 4.78 | 0.11 |
| (ANOVA-P < 0.01) | (ANOVA-P < 0.01) | (ANOVA-P = 0.103) | ||||||||||||||||
| Body length (cm) | ||||||||||||||||||
| Standard diet | 0.253 | 0.02 | 0.252 | 0.03 | 0.253 | 0.02 | 0.104 | 0.01 | 0.086 | 0.01 | 0.095 | 0.01 | 0.630 | 0.06 | 0.651 | 0.08 | 0.640 | 0.05 |
| Juvenile HFD | 0.331 | 0.02 | 0.308 | 0.02 | 0.319 | 0.01aa | 0.089 | 0.01 | 0.101 | 0.01 | 0.095 | 0.01 | 0.766 | 0.04 | 0.712 | 0.05 | 0.739 | 0.03 |
| Adult HFD | 0.302 | 0.02 | 0.283 | 0.02 | 0.293 | 0.01 | 0.115 | 0.01 | 0.101 | 0.01 | 0.108 | 0.01a | 0.788 | 0.04 | 0.764 | 0.05 | 0.776 | 0.03a |
| (ANOVA-P < 0.05) | (ANOVA-P = 0.113) | (ANOVA-P = 0.104) | ||||||||||||||||
| Female | ||||||||||||||||||
| Body weight (g) | ||||||||||||||||||
| Standard diet | 1.40 | 0.05 | 1.40 | 0.05 | 1.40 | 0.03 | 0.17 | 0.02 | 0.22 | 0.02 | 0.20 | 0.01 | 3.03 | 0.22 | 3.24 | 0.22 | 3.14 | 0.16 |
| Juvenile HFD | 1.60 | 0.03 | 1.55 | 0.03 | 1.57 | 0.02a | 0.19 | 0.01 | 0.17 | 0.01 | 0.18 | 0.01 | 3.58 | 0.16 | 3.29 | 0.16 | 3.43 | 0.11 |
| Adult HFD | 1.43 | 0.03 | 1.37 | 0.03 | 1.40 | 0.02 | 0.23 | 0.02 | 0.22 | 0.02 | 0.23 | 0.01c | 3.47 | 0.17 | 3.16 | 0.17 | 3.32 | 0.12 |
| (ANOVA-P < 0.01) | (ANOVA-P < 0.05) | |||||||||||||||||
| Body length (cm) | ||||||||||||||||||
| Standard diet | 0.191 | 0.05 | 0.180 | 0.05 | 0.185 | 0.04 | 0.056 | 0.02 | 0.055 | 0.02 | 0.056 | 0.01 | 0.560 | 0.06 | 0.569 | 0.06 | 0.564 | 0.04 |
| Juvenile HFD | 0.292 | 0.04 | 0.276 | 0.04 | 0.284 | 0.03a | 0.067 | 0.01 | 0.075 | 0.01 | 0.071 | 0.01 | 0.634 | 0.04 | 0.639 | 0.04 | 0.637 | 0.03 |
| Adult HFD | 0.287 | 0.04 | 0.255 | 0.04 | 0.271 | 0.03 | 0.069 | 0.01 | 0.076 | 0.01 | 0.072 | 0.01 | 0.679 | 0.05 | 0.647 | 0.05 | 0.663 | 0.03 |
| (ANOVA-P = 0.111) | ||||||||||||||||||
Gain, gain of weight or length/value at starting point, HFD: high fat food.
a Significant difference from the standard diet group (
b Significant difference from the HFD in adulthood group (
c Significant difference from the HFD in juvenile period group (
Similarly, in female offspring (Table 2), adjusted means of weight gain rate and length gain rate during the juvenile period were significantly greater in the juvenile HFD group compared to the standard diet group (
Fig. 1.
Comparisons of PDGFR-β levels in 7 brain regions among 3 diet groups in male offspring: juvenile HFD, adult HFD, and standard diet groups. Brain regions: prefrontal cortex (mPFC) (A), hippocampus (B), nucleus accumbens (NAc) (C), striatum (D), parietal cortex (E), amygdala (F), and superior colliculus (SC) (G).
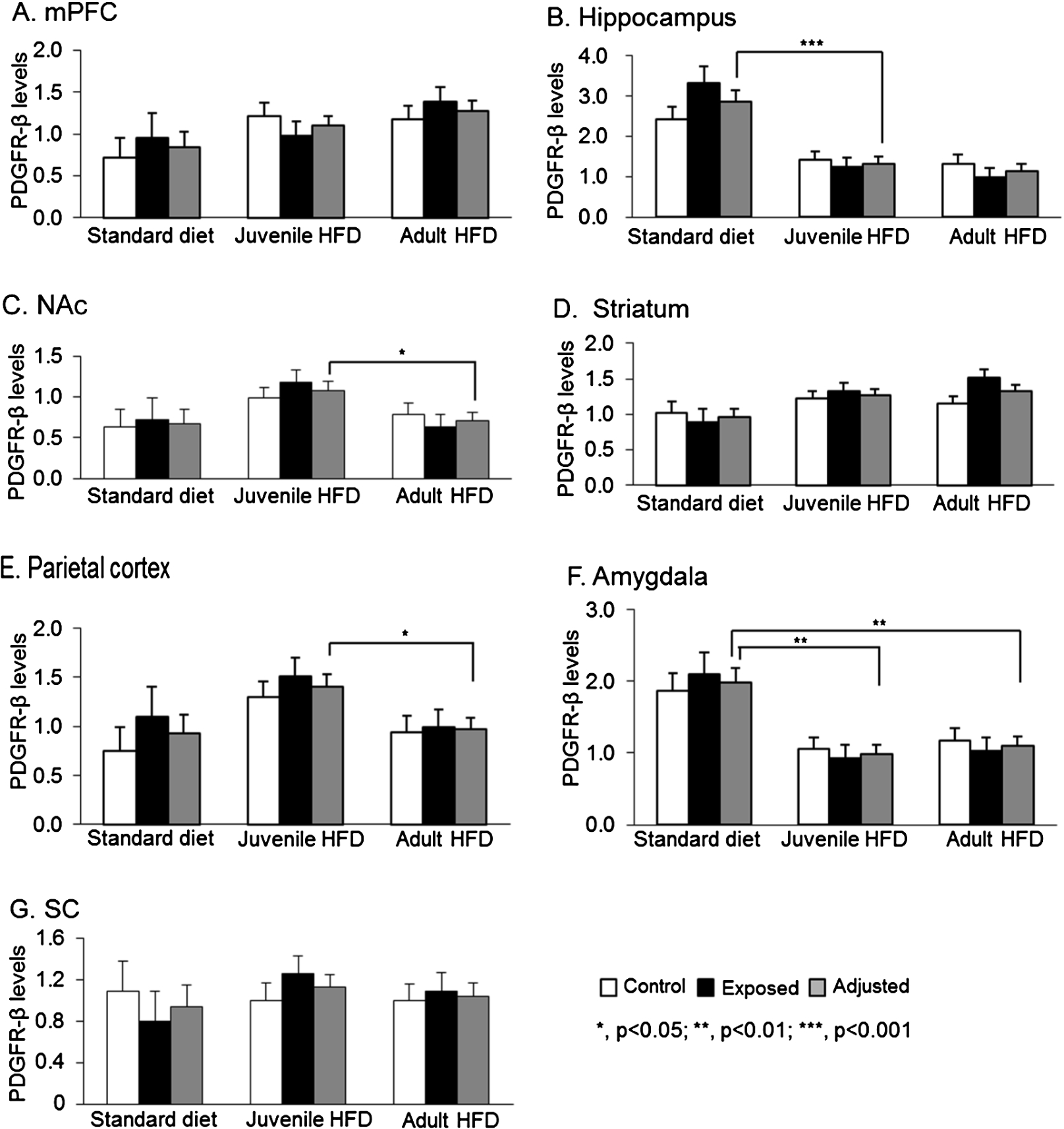
3.3.Effects of fatty food and TCDD on brain PDGFR-β levels in male offspring
Figure 1 shows PDGFR-β levels in each brain region among male offspring in the 3 diet groups. For each diet group, means of PDGFR-β levels in pups with and without TCDD exposure, and adjusted mean of PDGFR-β levels for exposure factor, as there was no significant main effect of TCDD exposure, nor significant interaction between diet and TCDD exposure in any brain regions. In the hippocampus (Fig. 1(B)), there was a significant main effect of diet [
Fig. 2.
Comparisons of PDGFR-β levels in 7 brain regions among 3 diet groups in female offspring: juvenile HFD, adult HFD, and standard diet groups. Brain regions: prefrontal cortex (mPFC) (A), hippocampus (B), nucleus accumbens (NAc) (C), striatum (D), parietal cortex (E), amygdala (F), and superior colliculus (SC) (G).
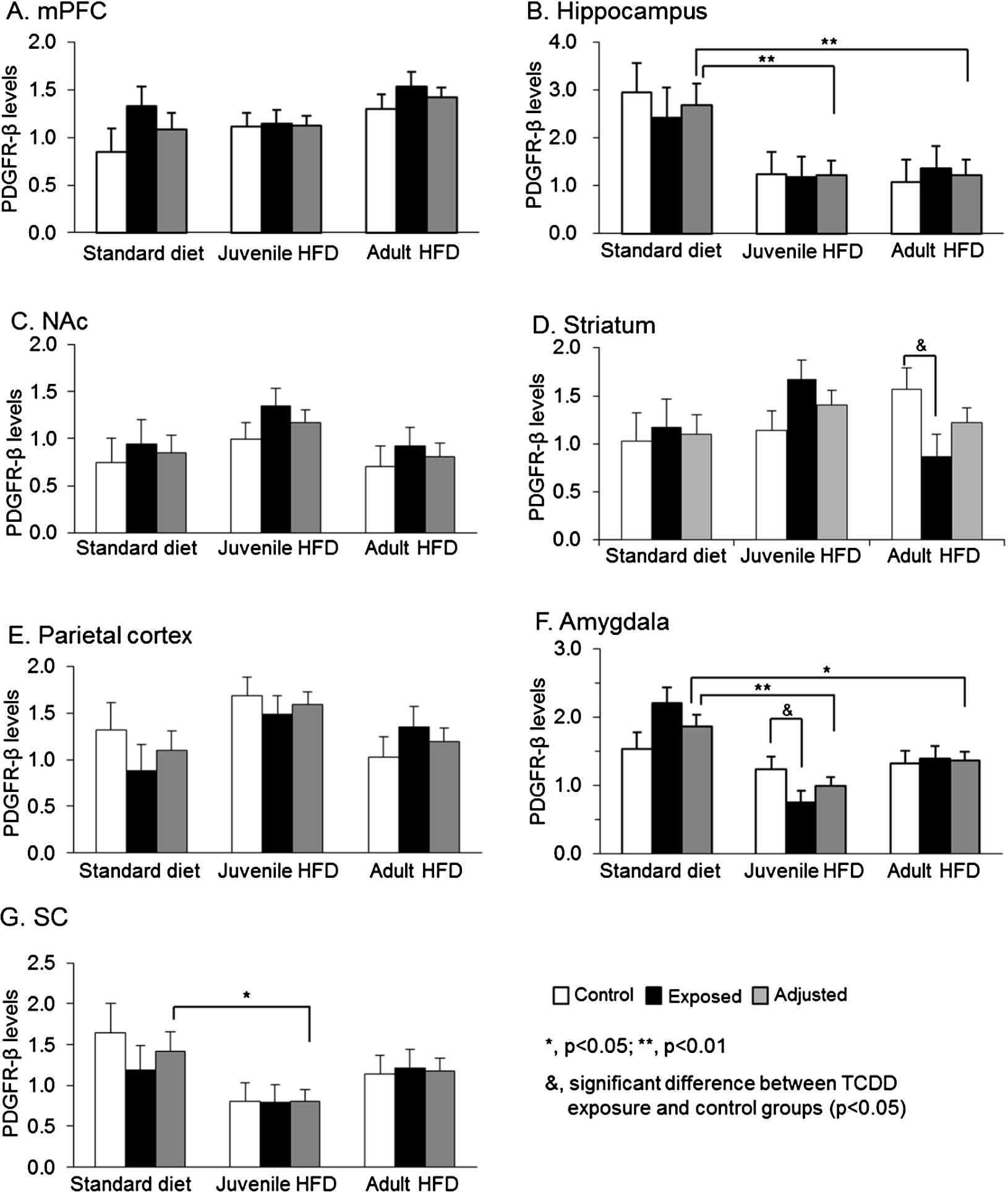
3.4.Effects of an HFD and TCDD on brain PDGFR-β levels in female offspring
Figure 2 shows PDGFR-β levels in each brain region in the 3 diet groups in female offspring. In female offspring, there were no significant main effects of TCDD exposure, nor significant interaction between diet and exposure factors on PDGFR-β levels in 5 brain regions except the striatum and amygdala. In the hippocampus (Fig. 2(B)), there was a significant main effect of diet [
In the striatum (Fig. 2(D)), there was no significant main effect of diet, but there was a significant interaction between diet and TCDD exposure [
3.5.Relationships between body size gain rates and BMI, and brain PDGFR-β levels
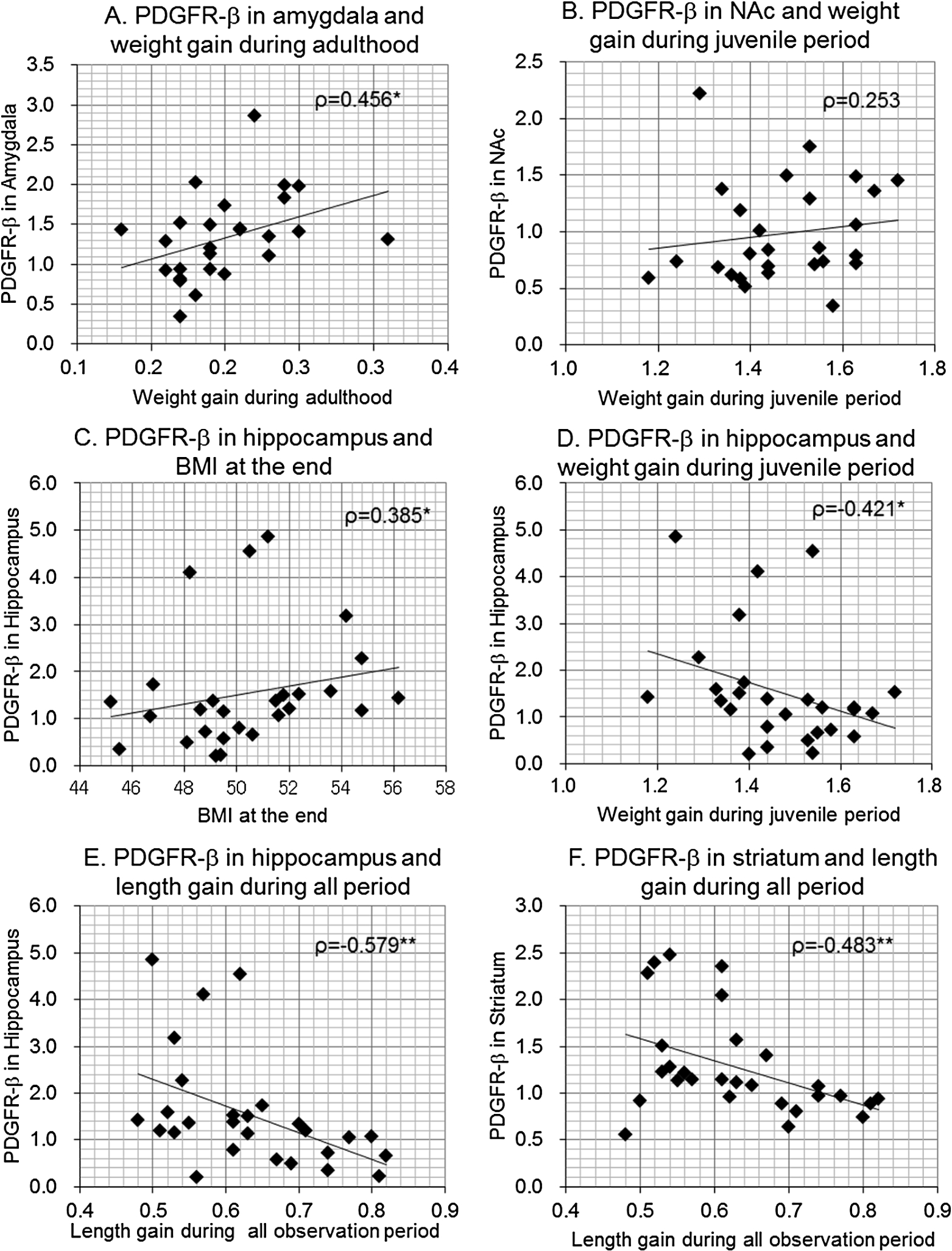
In order to investigate the relationship between body growth and PDGFR-β levels in each brain region, we analyzed correlations of weight and length gain during the juvenile period, adulthood and overall observation period, as well as BMI at the end of observation, with PDGFR-β levels in the mPFC, hippocampus, NAc, striatum, parietal cortex, amygdala, and SC. Correlation coefficients in males and females are shown in Table 3. Additionally, several scatter plots between PDGFR-β levels and body size parameters with significant correlations are illustrated in Fig. 3 for males and in Fig. 4 for females.
Table 3
Correlations of body size gain rates and BMI with PDGFR-β levels in 7 brain regions
| mPFC | Hippo | NAc | Striatum | P-cortex | Amygdala | SC | |
| Male | |||||||
| Weight gain rate during juvenile period | −0.222 | −0.240 | 0.553** | −0.273 | 0.030 | −0.041 | −0.074 |
| Weight gain rate during adulthood | 0.057 | −0.468* | 0.065 | −0.119 | −0.333 | 0.164 | 0.028 |
| Weight gain rate during all period | −0.238 | −0.308 | 0.440* | −0.373 | −0.234 | 0.169 | −0.180 |
| Length gain rate during juvenile period | −0.084 | −0.514** | 0.318 | −0.533** | −0.229 | 0.264 | −0.232 |
| Length gain rate during adulthood | −0.219 | −0.446* | 0.302 | −0.386* | −0.254 | 0.113 | 0.096 |
| Length gain rate during all period | −0.088 | −0.535** | 0.204 | −0.532** | −0.286 | 0.407* | −0.242 |
| BMI at the end | 0.134 | 0.196 | 0.237 | 0.361 | 0.416* | −0.584** | 0.211 |
| Female | |||||||
| Weight gain rate during juvenile period | −0.324 | −0.421* | 0.253 | 0.050 | 0.153 | −0.095 | −0.206 |
| Weight gain rate during adulthood | 0.131 | −0.037 | −0.140 | −0.275 | −0.295 | 0.456* | −0.031 |
| Weight gain rate during all period | −0.205 | −0.470* | 0.132 | −0.177 | −0.074 | 0.058 | −0.264 |
| Length gain rate during juvenile period | 0.084 | −0.620** | −0.082 | −0.531** | −0.199 | 0.016 | −0.393* |
| Length gain rate during adulthood | −0.035 | −0.524** | 0.172 | −0.338 | −0.109 | −0.138 | −0.136 |
| Length gain rate during all period | 0.126 | −0.579** | 0.035 | −0.483** | −0.296 | 0.077 | −0.277 |
| BMI at the end | −0.244 | 0.385* | −0.073 | 0.275 | 0.144 | 0.043 | −0.160 |
*
**
mPFC, medical prefrontal cortex; Hippo, hippocampus; NAc, nucleus of accumbens; P-cortex, parietal cortex; SC, superior colliculus.
Fig. 3.
Scatterplots between body size gains or BMI and brain PDGFR-β levels in male offspring. Correlation coefficients (Spearman’s ρ) were shown with p-values.
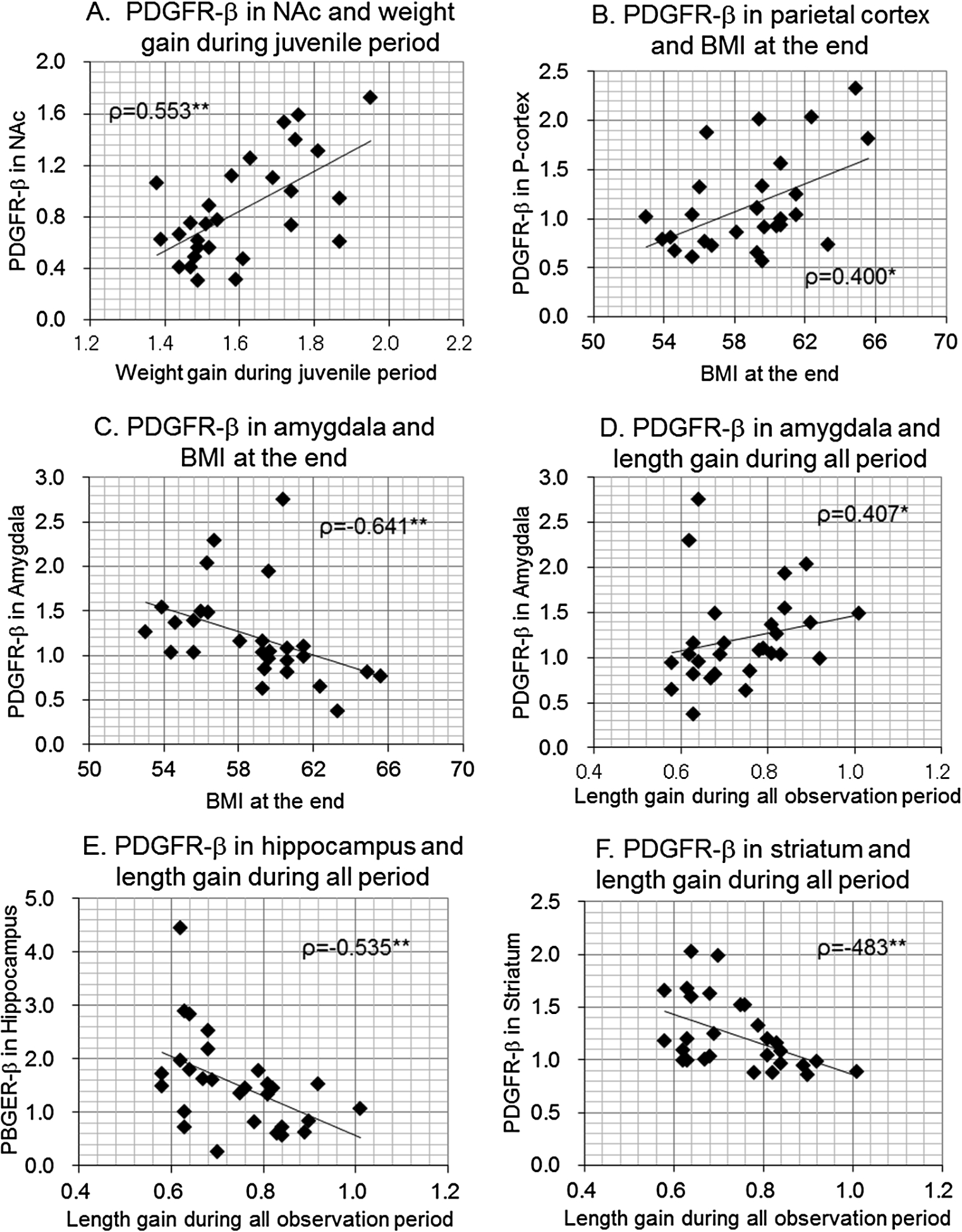
Fig. 4.
Scatterplots between body size gains or BMI and brain PDGFR-β levels in female offspring. Correlation coefficients (Spearman’s ρ) were shown with p-values.
In male offspring, PDGFR-β levels in the NAc were significantly correlated with weight gain rates during the juvenile period (Fig. 3(A)) and during overall observation period but PDGFR-β levels in the parietal cortex were significantly correlated only with BMI at the end of observation (Fig. 3(B)). PDGFR-β levels in the amygdala were significantly and inversely correlated with BMI (Fig. 3(C)), while it was positively correlated with length gain rate during overall observation period (Fig. 3(D)). PDGFR-β levels in the hippocampus (Fig. 3(E)) and striatum (Fig. 3(F)) were significantly and inversely correlated with weight gain during adulthood and length gain during the juvenile period, adulthood, and overall observation period.
In female offspring, PDGFR-β levels in the amygdala were significantly correlated with weight gain rate during adulthood (Fig. 4(A)), but the positive correlation between PDGFR-β levels in the NAc and weight gain rate during the juvenile period was not significant in females, in contrast to male subjects (Fig. 4(B)). PDGFR-β levels in the hippocampus were correlated with BMI at the end of the observation (Fig. 4(C)), but were significantly and inversely correlated with weight gain rate during the juvenile period (Fig. 4(D)) and adulthood, as well as length gain during the juvenile period, adulthood, and overall observation period (Fig. 4(E)). PDGFR-β levels in the striatum were also inversely correlated with length gain during the juvenile and overall observation period (Fig. 4(F)). Finally, PDGFR-β levels in the SC were inversely correlated with length gains during the juvenile period, which was observed in only females.
These results indicate that in male offspring, PDGFR-β levels in the hippocampus and striatum were most strongly associated with increased body length, while PDGFR-β levels in the NAc and parietal cortex, or amygdala were primarily associated with body weight gain or BMI. Similar but weaker trends were observed in the hippocampus and striatum in female offspring, while PDGFR-β levels in the amygdala were associated with body weight gain only in females.
4.Discussion
4.1.Effects of an HFD and TCDD on body size
In the present study, body weight gain rates of pups exposed to TCDD after adjusting HFD intake were smaller than controls during the juvenile period in both sexes, but the differences were not statistically significant. In a previous study, TCDD exposure reduced feeding, particularly fatty food intake such as chocolate and cheese, and consequently produced a decline in body weight of adult rats (wasting syndrome) [23,24]. The lack of a significant effect on body size in the present study may be ascribed to the dose of TCDD (1.0 μg/kg), which was much smaller than the dose previously used to induce wasting syndrome (>500 μg/kg) [23,24]. The currently employed dose (1.0 μg/kg) also had significant effects on cognitive and brain functions in the previous studies; the administration of TCDD induced deficits in social behaviors [25], with a concurrent reduction of parvalbumin-positive neurons in the limbic system in adult rats [26], as well as deficits in avoidance learning in adult rats [12]. These previous results suggest that prenatal exposure to TCDD at a low dose induces deficits in cognitive brain functions in adulthood, without significant reduction in feeding and body growth.
In the present study, in male rats, HFD intake during the juvenile period increased body weight and BMI during adulthood, even after stopping the HFD, compared to consumption of an HFD in adulthood or a standard diet across all time points. A previous study in mice reported that lean offspring suckled by dams that were fed an HFD had increased body weight and adipose tissue in adulthood [27]. The present findings provide additional knowledge on the adverse effects of HFD from the lactation period to the post-weaning period, and suggest that the consumption of an HFD during post-weaning is a risk for developing obesity later in life, particularly in males, in accordance with a previous study demonstrating that even pups with low birth weights showed catch-up growth [28].
However, the effects of an HFD on body size in female offspring were time-limited and mild compared to the effects in male offspring. Pettersson et al. [29] reported that female mice are resistant to HFD-induced metabolic change and to developing inflammation of intra-abdominal adipose tissue, in accordance with our observed sex differences in developing obesity with the consumption of an HFD.
4.2.Effects of an HFD and TCDD on brain PDGFR-β levels
In the present study, PDGFR-β levels were decreased in the hippocampus and amygdala in the juvenile and adult HFD groups in both males and females. Levels were also decreased in the SC of the juvenile HFD group in females, whereas PDGFR-β levels were increased in the NAc and parietal cortex in male offspring in the juvenile HFD group. This is the first report describing the effects of an HFD on PDGFR-β levels in the brain. Previous research has found that the consumption of an HFD induces leptin and insulin resistance in the brain, which deactivates rapamycin complex 1 (mTOR1) [30]. Given that mTOR1 reduces expression of PDGFR-β in vitro [31], increased PDGFR-β levels in the NAc and parietal cortex may result from deactivation of mTOR1 by an HFD in the juvenile period. These results suggest that brain changes induced by an HFD earlier in life are maintained in later life, as evidenced by the observation of high BMIs in adulthood in male offspring. However, our observed decrease in PDGFR-β levels in the hippocampus, amygdala, and SC were unexpected. Recent studies have reported that an HFD induces endoplasmic reticulum (ER) stress, which suppresses global mRNA translation [32]. Therefore, ER stress due to an HFD may decrease PDGFR-β levels, and further studies are required in order to investigate the mechanisms of reduced PDGFR-β expression.
In the present study, perinatal exposure to TCDD decreased PDGFR-β levels in the striatum and amygdala in female offspring that consumed an HFD, but not in rats fed a standard diet. Since both HFD and TCDD exposure induce ER stress [33], HFD consumption may enhance TCDD toxicity only in female offspring. Previous animal studies have reported that exposure to TCDD altered taste preference only in female rats [7,34], and the amygdala and striatum are considered to be important in encoding hedonic values of taste quality and food rewards [35,36]. Therefore, the present results might contribute to the understanding of the neural mechanisms of sexually dimorphic changes in taste preference due to dioxin exposure.
4.3.Relationships between brain PDGFR-β levels and body size parameters
In the present study, PDGFR-β levels in the amygdala were negatively correlated with BMI in male offspring. Since the amygdala has an essential contribution to peripheral metabolism and feeding behavior (see below), these findings suggest that PDGF signaling in the amygdala may decrease degree of obesity. In the hypothalamus and amygdala, insulin acts to reduce food intake by activating the insulin receptor substrates (IRS)-1 and IRS-2/phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway, which leads to a decrease in feeding behavior [37,38]. However, HFD induces ER stress and inflammatory reactions in the brain, leading to inactivation of IRS-1/2, which inhibits the IRS/PI3K/Akt pathway in the brain, including in the amygdala [39,40]. On the other hand, PDGFR-β signaling exerts protective effects against ER stress [41,42], and could activate PI3K independently from IRS-1/2 [43]. These findings suggest that decreases in PDGFR-β signaling may decrease Akt activity in the amygdala, as well as the hypothalamus, thereby increasing BMI via these two mechanisms.
In the hippocampus and striatum, PDGFR-β levels were decreased, and inversely correlated with various parameters of body size gain, particularly body length gains. Growth hormone (GH) receptors have been identified in the hippocampus, striatum, and hypothalamus, and peripheral GH may cross the blood brain barrier to exert effects on neurons in the brain [44]. Furthermore, electrical stimulation of the limbic system, including the hippocampus, modulates plasma GH levels [45]. These findings suggest that the extra-hypothalamic areas, including the hippocampus and striatum, are involved in GH release from the pituitary gland. Our finding of an inverse correlation between PDGFR-β levels in these areas and body length gain suggests that PDGFR-β signaling in these areas may exert an inhibitory influence on GH release in both female and male offspring.
In the NAc, PDGFR-β levels were increased in male offspring in the juvenile HFD group, and levels were positively correlated with body weight gain during the juvenile and overall observation period. The mesolimbic dopamine (DA) system is involved in motivation and reward-related behavior, including feeding behavior, in which DA is released in response to food reward [46]. In obese animals, dopaminergic functions including DA release are impaired in the NAc due to insulin and leptin resistance, and these animals are hyperphasic [46,47]. Since DA signaling is partially mediated by PDGFR-β signaling [48,49], increases in PDGFR-β levels may compensate for disturbed DA signaling in the NAc, thereby promoting feeding behavior.
4.4.Conclusions
Previous studies have reported that both dioxin and HFD affect feeding behaviors. The present results indicate that both TCDD and HFD modulate the expression of PDGFR-β in the amygdala, hippocampus, striatum, and NAc of rats, which was associated with body growth and BMI. Furthermore, PDGFR-β signaling affects synaptic transmission mediated by type A gamma-aminobutyric (GABAA), N-methyl-D-aspartate (NMDA), and alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors [50–52] as well as synaptic plasticity [53–55]. Together with findings from previous studies, the results suggest that the changes in body growth and feeding behaviors due to TCDD and HFD may be partially mediated by changes in brain PDGFR-β levels.
Ethics approval
All experimental procedures using animals were approved by the Committee on Animal Experiments at Kanazawa Medical University.
Consent for publication
Not applicable
Availability of data and materials
Please contact author for the data request.
Conflicts of interest
All authors have no conflicts of interest for this paper.
Funding
This work was supported partly by the Ministry of Education, Science, Sports and Culture, Grant-in-Aid for Scientific Research (B) (25305024) and for Challenging Exploratory Research (15K15244). The founders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript and they have no competing interest.
Author contribution
His.N., and M.N. conceived and designed research; A.B. and N.N.T. performed research; Q.V.L., Y.T., J.M. Y.N., and Hir.N. analysed data; and A.B., His.N., and M.N. wrote the paper.
References
[1] | R. Warren B. Walker Jr. and V.R. Nathan, Environmental factors influencing public health and medicine: Policy implications, J Natl Med Assoc 94: (4) ((2002) ), 185–193. |
[2] | A. Prüss-Üstün and C. Corvalán, Preventing Disease Through Healthy Environments: Towards an Estimate of the Environmental Burden of Disease, World Health Organization, Geneva, (2006) . |
[3] | S. Valdivia, A. Patrone, M. Reynaldo and M. Perello, Acute high fat diet consumption activates the mesolimbic circuit and requires orexin signaling in a mouse model, PLoS ONE 9: (1) ((2014) ), e87478. doi:10.1371/journal.pone.0087478. |
[4] | E. Egecioglu, K.P. Skibicka, C. Hansson, M. Alvarez-Crespo, P.A. Friberg, E. Jerlhag, J.A. Engel and S.L. Dickson, Hedonic and incentive signals for body weight control, Rev Endocr Metab Disord 12: (3) ((2011) ), 141–151. doi:10.1007/s11154-011-9166-4. |
[5] | J. Legler, T. Hamer, M. van der Sluijs-van de Bor, G. Schoeters, L. van der Ven, M. Eggesbo, J. Koppe, M. Feinberg and T. Trnovec, The OBELIX project: Early life exposure to endocrine disruptors and obesity, Am J Clin Nutr 94: ((2011) ), 1933S–1938S. |
[6] | P.T. Tai, M. Nishijo, N.T. Anh, S. Maruzeni, H. Nakagawa, H. Van Luong, T.H. Anh, R. Honda, T. Kido and H. Nishijo, Dioxin exposure in breast milk and infant neurodevelopment in Vietnam, Occup Environ Med 70: ((2013) ), 656–662. doi:10.1136/oemed-2012-101021. |
[7] | P.T. Tai, M. Nishijo, T.N. Nghi, H. Nakagawa, H. Van Luong, T.H. Anh and H. Nishijo, Effects of perinatal dioxin exposure on development of children during the first 3 years of life, J Pediatr 175: ((2016) ), 159–166. doi:10.1016/j.jpeds.2016.04.064. |
[8] | M. Nishijo, P.T. Tai, T.N.A. Nguyen, T.N. Nghi, H. Nakagawa, H.V. Luong, A.H. Tran, Y. Morikawa, H.D. Manh, T. Kido, M.N. Nui, N.M. Hai and H. Nishijo, 2,3,7,8-tetrachlorodibenzo-p-dioxin in breast milk increases autistic traits of 3-year-old children in Vietnam, Mol Psychiatry 19: ((2014) ), 1220–1226. doi:10.1038/mp.2014.18. |
[9] | M. Nishijo, N.N. Tran, H. Nakagawa, K. Torii, T. Kondoh and H. Nishijo, Effects of perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on development of taste preference in rat offspring, J Addict Res Ther 5: ((2014) ), 173. |
[10] | T.A. Mably, R.W. Moore and R.E. Peterson, In utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin. 1. Androgenic status, Toxicol Appl Pharmacol 114: ((1992) ), 97–107. doi:10.1016/0041-008X(92)90101-W. |
[11] | D.J. Bjerke and R.E. Peterson, Reproductive toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in male rats: Different effects of in utero versus lactational exposure, Toxicol Appl Pharmacol 127: ((1994) ), 241–249. doi:10.1006/taap.1994.1158. |
[12] | M. Nishijo, J. Kuriwaki, E. Hori, K. Tawara, H. Nakagawa and H. Nishijo, Effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on fetal brain growth and motor and behavioral development in offspring rats, Toxicol Lett 173: (1) ((2007) ), 41–47. doi:10.1016/j.toxlet.2007.06.007. |
[13] | J. Lindén, S. Lensu, J. Tuomisto and R. Pohjanvirta, Dioxins, the aryl hydrocarbon receptor and the central regulation of energy balance, Front Neuroendocrinol 31: (4) ((2010) ), 452–478. doi:10.1016/j.yfrne.2010.07.002. |
[14] | R. Pohjanvirta, L. Tuomisto and J. Tuomisto, The central nervous system may be involved in TCDD toxicity, Toxicology 58: (2) ((1989) ), 167–174. doi:10.1016/0300-483X(89)90006-1. |
[15] | K.E. Houlahan, S.D. Prokopec, I.D. Moffat, J. Lindén, S. Lensu, A.B. Okey, R. Pohjanvirta and P.C. Boutros, Transcriptional profiling of rat hypothalamus response to 2,3,7,8-tetrachlorodibenzo-ρ-dioxin, Toxicology 328: ((2015) ), 93–101. doi:10.1016/j.tox.2014.12.016. |
[16] | K. Funa and M. Sasahara, The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system, J Neuroimmune Pharmacol 9: (2) ((2014) ), 168–181. doi:10.1007/s11481-013-9479-z. |
[17] | C.H. Heldin, A. Ostman and L. Ronnstrand, Signal transduction via platelet-derived growth factor receptors, Biochimica et biophysica Acta 1378: ((1998) ), 79–113. |
[18] | A. Smits, M. Kato, B. Westermark, M. Nister, C.F. Heldin and K. Funa, Neurotrophic activity of platelet-derived growth factor (PDGF): Rat neuronal cells possess functional PDGF-beta type receptors and respond to PDGF, Proc Natl Acad Sci USA 88: ((1991) ), 8159–8163. doi:10.1073/pnas.88.18.8159. |
[19] | K. Sasaki, Y. Oomura, K. Suzuki, T. Muto, K. Hanai, I. Tooyama, H. Kimura and N. Yanaihara, Effects of fibroblast growth factors and platelet-derived growth factor on food intake in rats, Brain Res Bull 27: (3–4) ((1991) ), 327–332. doi:10.1016/0361-9230(91)90120-9. |
[20] | M. Meireles, C. Marques, S. Norberto, I. Fernandes, N. Mateus, C. Rendeiro, J.P. Spencer, A. Faria and C. Calhau, The impact of chronic blackberry intake on the neuroinflammatory status of rats fed a standard or high-fat diet, J Nutr Biochem 26: (11) ((2015) ), 1166–1173. doi:10.1016/j.jnutbio.2015.05.008. |
[21] | C. Pang, Z. Gao, J. Yin, J. Zhang, W. Jia and J. Ye, Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity, Am J Physiol Endocrinol Metab 295: (2) ((2008) ), E313–E322. doi:10.1152/ajpendo.90296.2008. |
[22] | M. Kakeyama and C. Tohyama, Developmental neurotoxicity of dioxin and its related compounds, Ind Health 41: (3) ((2003) ), 215–230. doi:10.2486/indhealth.41.215. |
[23] | J. Tuomisto, W. Andrzejewski, M. Unkila, R. Pohjanvirta, J. Linden, T. Vartiainen and L. Tuomisto, Modulation of TCDD-induced wasting syndrome by portocaval anastomosis and vagotomy in long-evans and Han/Wistar rats, Eur J Pharmacol 292: (3–4) ((1995) ), 277–285. |
[24] | J.T. Tuomisto, M. Viluksela, R. Pohjanvirta and J. Tuomisto, Changes in food intake and food selection in rats after 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure, Pharmacol Biochem Behav 65: (3) ((2000) ), 381–387. doi:10.1016/S0091-3057(99)00209-9. |
[25] | A.T. Nguyen, M. Nishijo, E. Hori, N.M. Nguyen, T.T. Pham, K. Fukunaga, H. Nakagawa, A.H. Tran and H. Nishijo, Influence of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on socioemotional behaviors in offspring rats, Environ Health Insights 7: ((2013) ), 1–14. doi:10.4137/EHI.S10346. |
[26] | M.N. Nguyen, M. Nishijo, A.T. Nguyen, A. Bor, T. Nakamura, E. Hori, H. Nakagawa, T. Ono and H. Nishijo, Effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on parvalbumin- and calbindin-immunoreactive neurons in the limbic system and superior colliculus in rat offspring, Toxicology 314: (1) ((2013) ), 125–134. doi:10.1016/j.tox.2013.09.005. |
[27] | H. Masuyama and Y. Hiramatsu, Additive effects of maternal high fat diet during lactation on mouse offspring, PLoS One 9: (3) ((2014) ), e92805. doi:10.1371/journal.pone.0092805. |
[28] | S. Ojiha, V. Saroha, M.E. Symonds and H. Budge, Excess nutrient supply in early life and its later metabolic consequences, Clin Exp Pharmacol Physiol 40: (11) ((2013) ), 817–823. doi:10.1111/1440-1681.12061. |
[29] | U.S. Pettersson, T.B. Waldén, P.O. Carlsson, L. Jansson and M. Phillipson, Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue, PLoS ONE 7: (9) ((2012) ), e46057. doi:10.1371/journal.pone.0046057. |
[30] | M. Laplante and D.M. Sabatini, mTOR signaling in growth control and disease, Cell 149: (2) ((2012) ), 274–293. doi:10.1016/j.cell.2012.03.017. |
[31] | H. Zhang, N. Bajraszewski, E. Wu, H. Wang, A.P. Moseman, S.L. Dabora, J.D. Griffin and D.J. Kwiatkowski, PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR, J Clin Invest 117: (3) ((2007) ), 730–738. doi:10.1172/JCI28984. |
[32] | S. Bernales, M.M. Soto and E. McCullagh, Unfolded protein stress in the endoplasmic reticulum and mitochondria: A role in neurodegeneration, Front Aging Neurosci 4: ((2012) ), 5. |
[33] | Z. Duan, J. Zhao, X. Fan, C. Tang, L. Liang, X. Nie, J. Liu, Q. Wu and G. Xu, The PERK-eIF2α signaling pathway is involved in TCDD-induced ER stress in PC12 cells, Neurotoxicology 44: ((2014) ), 149–159. doi:10.1016/j.neuro.2014.06.005. |
[34] | S. Amin, R.W. Moore, R.E. Peterson and S.L. Schantz, Gestational and lactational exposure to TCDD or coplanar PCBs alters adult expression of saccharin preference behavior in female rats, Neurotoxicol Teratol 22: ((2000) ), 675–682. doi:10.1016/S0892-0362(00)00094-5. |
[35] | H. Nishijo, T. Uwano, R. Tamura and T. Ono, Gustatory and multimodal neuronal responses in the amygdala during licking and discrimination of sensory stimuli in awake rats, J Neurophysiol 79: (1) ((1998) ), 21–36. |
[36] | S.E. Rhodes and E.A. Murray, Differential effects of amygdala, orbital prefrontal cortex, and prelimbic cortex lesions on goal-directed behavior in rhesus macaques, J Neurosci 33: (8) ((2013) ), 3380–3389. doi:10.1523/JNEUROSCI.4374-12.2013. |
[37] | K.D. Niswender, C.D. Morrison, D.J. Clegg, R. Olson, D.G. Baskin, M.G. Myers Jr., R.J. Seeley and M.W. Schwartz, Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: A key mediator of insulin-induced anorexia, Diabetes 52: (2) ((2003) ), 227–231. doi:10.2337/diabetes.52.2.227. |
[38] | B.F. Belgardt and J.C. Brüning, CNS leptin and insulin action in the control of energy homeostasis, Ann N Y Acad Sci 1212: ((2000) ), 97–113. doi:10.1111/j.1749-6632.2010.05799.x. |
[39] | G.C. Castro, M.F. Areias, L. Weissmann, P.G. Quaresma, C.K. Katashima, M.J. Saad and P.O. Prada, Diet-induced obesity induces endoplasmic reticulum stress and insulin resistance in the amygdala of rats, FEBS Open Bio 3: ((2013) ), 443–449. doi:10.1016/j.fob.2013.09.002. |
[40] | M.F. Areias and P.O. Prada, Mechanisms of insulin resistance in the amygdala: Influences on food intake, Behav Brain Res 282: ((2015) ), 209–217. doi:10.1016/j.bbr.2015.01.003. |
[41] | L. Zheng, Y. Ishii, A. Tokunaga, T. Hamashima, J. Shen, Q.L. Zhao, S. Ishizawa, T. Fujimori, Y. Nabeshima, H. Mori, T. Kondo and M. Sasahara, Neuroprotective effects of PDGF against oxidative stress and the signaling pathway involved, J Neurosci Res 88: (6) ((2010) ), 1273–1284. |
[42] | F. Lin, J. Zhu, M.G. Tonnesen, B.R. Taira, S.A. McClain, A.J. Singer and R.A. Clark, Fibronectin peptides that bind PDGF-BB enhance survival of cells and tissue under stress, J Invest Dermatol 134: (4) ((2014) ), 1119–1127. doi:10.1038/jid.2013.420. |
[43] | E.L. Whiteman, J.J. Chen and M.J. Birnbaum, Platelet-derived growth factor (PDGF) stimulates glucose transport in 3T3-L1 adipocytes overexpressing PDGF receptor by a pathway independent of insulin receptor substrates, Endocrinology 144: (9) ((2003) ), 3811–3820. doi:10.1210/en.2003-0480. |
[44] | F. Nyberg, Growth hormone in the brain: Characteristics of specific brain targets for the hormone and their functional significance, Front Neuroendocrinol 21: (4) ((2000) ), 330–348. doi:10.1006/frne.2000.0200. |
[45] | J.B. Martin, Plasma growth hormone (GH) response to hypothalamic or extrahypothalamic electric stimulation, Endocrinology 91: (1) ((1971) ), 107–115. doi:10.1210/endo-91-1-107. |
[46] | D.P. Figlewicz and S.C. Benoit, Insulin, leptin, and food reward: Update, Am J Physiol Regul Integr Comp Physiol 296: (1) ((2008) ), R9–R19. doi:10.1152/ajpregu.90725.2008. |
[47] | S. Fulton, P. Pissios, R.P. Manchon, L. Stiles, L. Frank, E.N. Pothos, E. Maratos-Flier and J.S. Flier, Leptin regulation of the mesoaccumbens dopamine pathway, Neuron 51: (6) ((2006) ), 811–822. doi:10.1016/j.neuron.2006.09.006. |
[48] | J.N. Oak, N. Lavine and H.H. Van Tol, Dopamine D(4) and D(2L) receptor stimulation of the mitogen-activated protein kinase pathway is dependent on trans-activation of the platelet-derived growth factor receptor, Mol Pharmacol 60: (1) ((2001) ), 92–103. |
[49] | S.A. Kotecha, J.N. Oak, M.F. Jackson, Y. Perez, B.A. Orser, H.H. Van Tol and J.F. MacDonald, A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission, Neuron 35: (6) ((2002) ), 1111–1122. doi:10.1016/S0896-6273(02)00859-0. |
[50] | Y. Ohi, Y. Ishii, A. Haji, S. Noguchi, T. Sasaoka, T. Fujimori, Y. Nabeshima, M. Sasahara and Y. Hattori, Platelet-derived growth factor (PDGF)-BB inhibits AMPA receptor-mediated synaptic transmission via PDGF receptor-beta in murine nucleus tractus solitarius, Brain Res 1159: ((2007) ), 77–85. doi:10.1016/j.brainres.2007.05.037. |
[51] | M.A. Beazely, A. Lim, H. Li, C. Trepanier, X. Chen, B. Sidhu and J.F. Macdonald, Platelet-derived growth factor selectively inhibits NR2B-containing N-methyl-D-aspartate receptors in CA1 hippocampal neurons, J. Biol. Chem. 284: (12) ((2009) ), 8054–8063. doi:10.1074/jbc.M805384200. |
[52] | C.F. Valenzuela, A. Kazlauskas, S.J. Brozowski, J.L. Weiner, K.A. Demali, B.J. McDonald, S.J. Moss, T.V. Dunwiddie and R.A. Harris, Platelet-derived growth factor receptor is a novel modulator of type Aγ-aminobutyric acid-gated ion channels, Mol Pharmacol 48: ((1995) ), 1099–1107. |
[53] | F. Peng, H. Yao, X. Bai, X. Zhu, B.C. Reiner, M. Beazely, K. Funa, H. Xiong and S. Buch, Platelet-derived growth factor-mediated induction of the synaptic plasticity gene Arc/Arg3.1, J. Biol. Chem. 285: (28) ((2010) ), 21615–21624. doi:10.1074/jbc.M110.107003. |
[54] | P.T. Nguyen, T. Nakamura, E. Hori, S. Urakawa, T. Uwano, J. Zhao, R. Li, N.D. Bac, T. Hamashima, Y. Ishii, T. Matsushima, T. Ono, M. Sasahara and H. Nishijo, Cognitive and socio-emotional deficits in platelet-derived growth factor receptor-β gene knockout mice, PLoS One 6: (3) ((2011) ), e18004. doi:10.1371/journal.pone.0018004. |
[55] | N. Shioda, S. Moriguchi, T. Oya, Y. Ishii, J. Shen, T. Matsushima, H. Nishijo, M. Sasahara and K. Fukunaga, Aberrant hippocampal spine morphology and impaired memory formation in neuronal platelet-derived growth factor β-receptor lacking mice, Hippocampus 22: (6) ((2012) ), 1371–13718. doi:10.1002/hipo.20973. |


