
Mono- and Biallelic Inactivation of Huntingtin Gene in Patient-Specific Induced Pluripotent Stem Cells Reveal HTT Roles in Striatal Development and Neuronal Functions
Abstract
Background:
Mutations in the Huntingtin (HTT) gene cause Huntington’s disease (HD), a neurodegenerative disorder. As a scaffold protein, HTT is involved in numerous cellular functions, but its normal and pathogenic functions during human forebrain development are poorly understood.
Objective:
To investigate the developmental component of HD, with a specific emphasis on understanding the functions of wild-type and mutant HTT alleles during forebrain neuron development in individuals carrying HD mutations.
Methods:
We used CRISPR/Cas9 gene-editing technology to disrupt the ATG region of the HTT gene via non-homologous end joining to produce mono- or biallelic HTT knock-out human induced pluripotent stem cell (iPSC) clones.
Results:
We showed that the loss of wild-type, mutant, or both HTT isoforms does not affect the pluripotency of iPSCs or their transition into neural cells. However, we observed that HTT loss causes division impairments in forebrain neuro-epithelial cells and alters maturation of striatal projection neurons (SPNs) particularly in the acquisition of DARPP32 expression, a key functional marker of SPNs. Finally, young post-mitotic neurons derived from HTT-/- human iPSCs display cellular dysfunctions observed in adult HD neurons.
Conclusions:
We described a novel collection of isogenic clones with mono- and biallelic HTT inactivation that complement existing HD-hiPSC isogenic series to explore HTT functions and test therapeutic strategies in particular HTT-lowering drugs. Characterizing neural and neuronal derivatives from human iPSCs of this collection, we show evidence that HTT loss or mutation has impacts on neuro-epithelial and striatal neurons maturation, and on basal DNA damage and BDNF axonal transport in post-mitotic neurons.
INTRODUCTION
The human huntingtin (HTT) gene encodes a large multifunctional scaffold protein that is expressed at variable levels in all cells from the fertilized egg to adulthood [1–3]. Abnormal expansion of a CAG repeat tract in exon 1 of the HTT gene results in the elongation of a poly-glutamine (≥40Q) in the N-terminal of mutant HTT proteins (mut-HTT) and causes Huntington’s disease (HD) [4]. This neurodegenerative disorder is dominantly inherited and the neuropathological hallmark of HD is the progressive and massive loss of neurons, particularly striatal projection neurons (SPN) [5, 6]. Despite the mode of inheritance of HD that supports a toxic gain of function by HD mutations, the partial loss of wild type HTT (wt-HTT) may as well contribute to the neuropathology of HD [7–9].
While typically diagnosed in adulthood, a growing body of literature describes a significant developmental component to HD [10–18]. Wild-type HTT is involved in multiple developmental processes including gastrulation [19–21], spermatogenesis [7], epithelial morphogenesis and epithelial-to-mesenchymal transition [13, 22, 23]. In the developing brain, HTT is implicated in neural tube formation and is essential for the differentiation and migration of neuroblasts in mice [10, 17]. HTT is also linked to forebrain developmental defects in human fetuses [11, 24] confirming previous data in the cortex and striatum of HD animals and in human induced pluripotent stem cell (iPSC) models [13, 14, 25, 26]. Moreover, severe reduction of HTT protein causes an extremely rare, yet dramatic, neurodevelopmental disorder [27, 28]. The impacts of complete loss of HTT, HTT haplo-insufficiency and HTT mutations on early steps of human neural development and later on forebrain cells maturation and functions, the brain region most affected in HD, remain poorly understood.
In this study, we have tackled this question employing patient–specific iPSCs and present the generation of a collection of isogenic clones. These clones encompass mono- or biallelic inactivation of HTT and originate from an iPSC line derived from an HD-patient carrying 109 CAGs. We show that neither wild-type nor mutant HTT monoallelic or biallelic inactivation overtly compromises iPSC pluripotency. Furthermore, our investigations into neural differentiation of HTT-/- and -/mut clones reveal impairments in the self-organization of forebrain neuroepithelial cells into rosette structures, as well as alterations in the maturation of SPNs. Lastly, we provide evidence that dysfunctions related to DNA damage repair and BDNF axonal transport, which are well documented in adult HD neurons, are already displayed in young post-mitotic SPNs or cortical neurons derived from HTT-/- clones.
MATERIALS AND METHODS
Human iPSC and hESC cultures
The HD-hiPSC “109Q” line ND42222 (XX, 109 CAG, passage 42) was obtained from Coriell repository. This line is heterozygous for HTT p.Gln18[109] and thus has 109 CAG repeats in one of the two alleles of HTT. Human iPSC amplification, neuronal cell generation, and terminal differentiation were performed as previously described [29]. 109Q iPS cells were maintained on vitronectin-coated (Life Technology) plates in mTeSRplus medium (STEMCELL Technologies). Cultures were fed every other day and passaged via manual dissociation using 0.02% EDTA (0.25 mM; pH 7.2; Merck Sigma-Aldrich) every 4 to 5 days. The human embryonic stem cells lines were cultured as previously described for each line (WT-hiPSC: i90cl16 XX, passage 44 and WT-hESC: RC9 (WT, XY, passage 20–60, RoslinCells) [30]; HD-hESC: SIVF018 (XX, 46 CAG, passages 18–30, Sydney IVF Stem Cells) and SI187 (XY, 51 CAG, passages 12–25, Stemride, USA) [31]; HD-iPSC: HD-71Q (ND42228), XX 71 CAG, passages 30–35, Coriell repository) [32].
Generation of an isogenic series from 109Q-iPSCs
We used CRISPR/Cas9 technology to generate one series of isogenic hiPSC lines with different HTT proteins dosages (HTTwt/-; HTT-/mut and HTT-/-). The ND42222_109Q line (Coriell) was used as a parental line. To create this series, CRISPR/Cas9 was used to alter the ATG in the first exon of HTT, ensuring that no protein would be produced in the targeted allele [33]. Dissociated hiPS cells were electroporated with a mix of 30 nM of sgRNA (by duplexing RNA oligos: crRNA and tracrRNA, ordered from Integrated DNA Technologies (IDT)), 30 nM of SpCas9 purified protein (TACGENE: MNHN-CNRS UMR 7196/INSERM U1154 from Anne de Cian, Jean-Paul Concordet), according to the protocol from IDT. We identified 16 homozygous knockout clones (HTT-/-) and 16 hemizygous clones either expressing only the wild type allele (8 clones HTTwt/-) or only the mutant allele (8 clones HTT-/mut) out of 54 isolated clones. The genomic integrity of the clones was verified by M-Fish karyotyping and single nucleotide polymorphism (SNP) genotyping.
Neural and neuronal differentiations
For neural differentiation, hiPSC colonies were treated (DIV0) as previously described [30] in N2B27 medium consisting of 50% DMEMF-12 Glutamax, 50% Neurobasal medium, 2% B27 supplement 50×minus vitamin A, 1% N2 supplement and 50μM β-mercaptoethanol (Thermo Fisher Scientific) supplemented with SB431542 (20μM; Tocris), LDN-193189 (100 nM; Sigma-Aldrich), XAV-939 (1μM; Tocris), and 10μM ROCK inhibitor (Y27632, Calbiochem). For medium spiny neurons (MSN) and cortical neurons differentiation, hiPSC or hESC colonies were treated (DIV0) as previously described in [30, 34] and [29], respectively.
Protein extraction and western blotting
Protein extracts (5–10μg) were loaded on a 3–8% (NuPage Tris-Acetate gels, Invitrogen®) or 10% (NuPage Bis–Tris gels, Invitrogen®) gels and transferred onto Gel Transfer Stacks Nitrocellulose membranes (Invitrogen®) using the iBlot2 Dry Blotting System (Invitrogen®). Antibody binding was quantified using a LiCor Odyssee CLx imager and Image Studio Lite 5.2 software. All antibodies used are listed in the Supplementary Material.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde (PFA) with 4% sucrose and further permeabilized with 0.1% Triton X-100 (Sigma) and 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS, Sigma). Primary antibodies were then added and the samples incubated at 4°C overnight in PBS with 2% BSA and 0.1% Triton. Species-specific secondary antibodies coupled to Alexa 350, 488, 555, and 647 (1/1000, Invitrogen) and DAPI counterstain were applied for 1 h at room temperature. Primary Antibodies: CALB (Origene; TA318675; 1/500); CTIP2 (abcam; ab18465; 1/500); DARPP32 (Abcam; ab40801; 1/500); FOXP1 (Abcam; ab16645; 1/800); MAP2 (Biolegend; 822501; 1/1500); NANOG (Abcam; ab62734; 1/500); OCT4 (Cell signaling; 28405; 1/500); Pericentrin (Abcam; ab28144; 1/800); SSEA3 (Biolegend; 330312; 1/500); TBR1 (Abcam; ab31940; 1/500); γ-H2AX (Millipore; 05-636; 1/500).
DNA damage analyses
DNA damage was analyzed in undifferentiated iPSCs, striatal neurons, and cortical neurons stained with γ-H2AX and DAPI to visualize double-strand DNA breaks and nuclei. Twenty images were taken per experiment and processed using the HCS CellInsight CX7 Platform (Thermo Fisher Scientific). The software analyzed each image to determine the number of cells (DAPI positive nuclei) and the number of γ-H2AX foci in each nucleus. The number of foci per cell was calculated and normalized to the number of foci in HTT wt/mut cells.
Spindle orientation quantification, lumen size determination, and mitosis count
Spindle angle in metaphase cells were stained for pericentrin and DAPI to visualize the spindle poles, the lumen outer limit and chromatin, was calculated using ImageJ software (http://rsb.info.nih.gov/ij/, NIH, USA). The images were captured with a Leica DMI6000 confocal optical microscope (TCS SPE) equipped with a 63x oil-immersion objective controlled by LAS X software. Z-stack steps were of 0.64μm. For hiPSCs, the angle between the pole-pole axis and the substratum plane was calculated. Using imageJ software, a line crossing both spindle poles was drawn on the Z projection pictures and repositioned along the Z-axis using the stack of Z-sections. For R-NSCs, one line crossing both spindle poles and the tangent of the lumen outer limit were drawn on the Z projection pictures to determine the angle. Lumen area was calculated using Z-projection images. The outer boundary of the lumen was manually traced using pericentrin staining, and the perimeter was measured using ImageJ software. For mitosis counting, Z-projection images were employed to count the number of round DAPI-positive nuclei and cells in M-phase, characterized by condensed DAPI + chromosomes within rosette structures.
BDNF transport
Cortical progenitors were infected with BDNF-mCherry lentivirus upon seeding. At DIV17, we used an inverted microscope (Axio Observer, Zeiss) coupled to a spinning-disk confocal system (CSU-W1-T3, Yokogawa) connected to a wide-field electron-multiplying CCD camera (ProEM + 1024, Princeton Instrument) and maintained at 37°C and 5% CO2. We took images every 200 ms for 30 s BDNF-mCherry trafficking (×63 oil-immersion objective, 1.46 NA). Images were analyzed with the KymoToolBox plugin for ImageJ [35–37].
Statistical analysis
GraphPad Prism (GraphPad Software, Inc.) software was used for statistical analysis. All experiments were conducted blindly and consisted of at least three independent replicates. Data are expressed as the median. The criterion for statistical significance was set to p < 0.05.
RESULTS
In order to model the loss of wt-HTT, mut-HTT, or both isoforms during human neural and striatal development, we used CRISPR-Cas9 technology to inactivate the HTT gene in the human iPSC line (ND42222) generated from an HD patient carrying a mutant allele with 109 CAG repeats. We produced a collection of isogenic clones with mono- or biallelic inactivation of the HTT alleles mediated by Cas9 and a sgRNA targeting a sequence close to the ATG of the gene (Fig. 1A). We screened gene-edited clones based on wt and/or mut-HTT protein expression measured by western blot (Fig. 1B, C; Supplementary Figure 1A). Quantification of the protein level of total-HTT, wt-HTT, and mut-HTT isoforms of our selection of clones confirmed the corresponding complete inactivation of HTT alleles (Fig. 1C). In hemizygous clones, the level of the isoform encoded by the non-edited allele was unaffected (Fig. 1C). Sequencing of HTT alleles at the site of editing in selected clones by the guide RNA revealed indels, resulting in frameshifts and early stop codons, thereby preventing the synthesis of the expanded CAG track (Supplementary Figure 2). Undifferentiated iPSC cultures displayed no alteration of proliferation or pluripotency parameters analyzed for each clone and genotype by immunocytochemistry and RNAseq looking at canonical pluripotency master regulators (OCT4, NANOG), membrane bound pluripotency markers (SSEA3, TRA1-81) or proliferation genes (KI67 and PCNA) (Supplementary Figure 1B–D; Supplementary Figure 3). We concluded that partial or complete loss of wt or mut-HTT does not alter the basic properties of human iPSCs.
Fig. 1
Generation of isogenic clones of 109Q-iPSC with monoallelic or biallelic HTT inactivation and alteration of neural rosette derived from these clones. A) Schematic diagram of the HTT gene locus at the transcription start site (TSS) showing the guide RNA (sgHTT: in blue) targeting a sequence close to the translation start codon ATG in exon 1 (in green). B) Representative western blot and (C) quantification of HTT (D7F7) or mutant HTT (P1874) in HTT-/-, HTTwt/-, HTT-/mut iPSC clones. Individual data points, mean and SEM are shown as fold change (FC) compared to HTTwt/mut cells, n = 4/genotype. *p < 0.05, **p < 0.01, one-way ANOVA test with Tukey’s multiple comparison post-test. All the isogenic clones generated maintain pluripotency as shown by (D). Positive immunostaining for the pluripotency marker NANOG. (Two clones from each genotype were fully characterized; scale bar: 100μm). E) Principal component analyses (PCA) of RNAseq data of undifferentiated iPSCs of all 4 genotypes (n = 3–4 edited clones per genotype). All the isogenic clones were able to differentiate into neural cells forming rosette shaped structures (R-NSC). F) Immunostaining of pericentrin (green) identifying rosette lumens and DNA staining (DAPI, blue) at DIV7. G) Picture of one cell in division in a R-NSC illustrating the measurement of the spindle angle α. H) Schematic representation of the three different types of division observed in R-NSCs. I) Quantification of α angles relative to the tangent of the lumen, the mean angle of division of cells in R-NSCs from HTT-/mut clones is higher than that of R-NSCs derived from HTTwt/- clones. (Individual data points, mean and SEM are shown. n = 52-76 from three independent experiments are analyzed. ***p < 0.001, one-way ANOVA test with Tukey’s multiple comparison post-test).
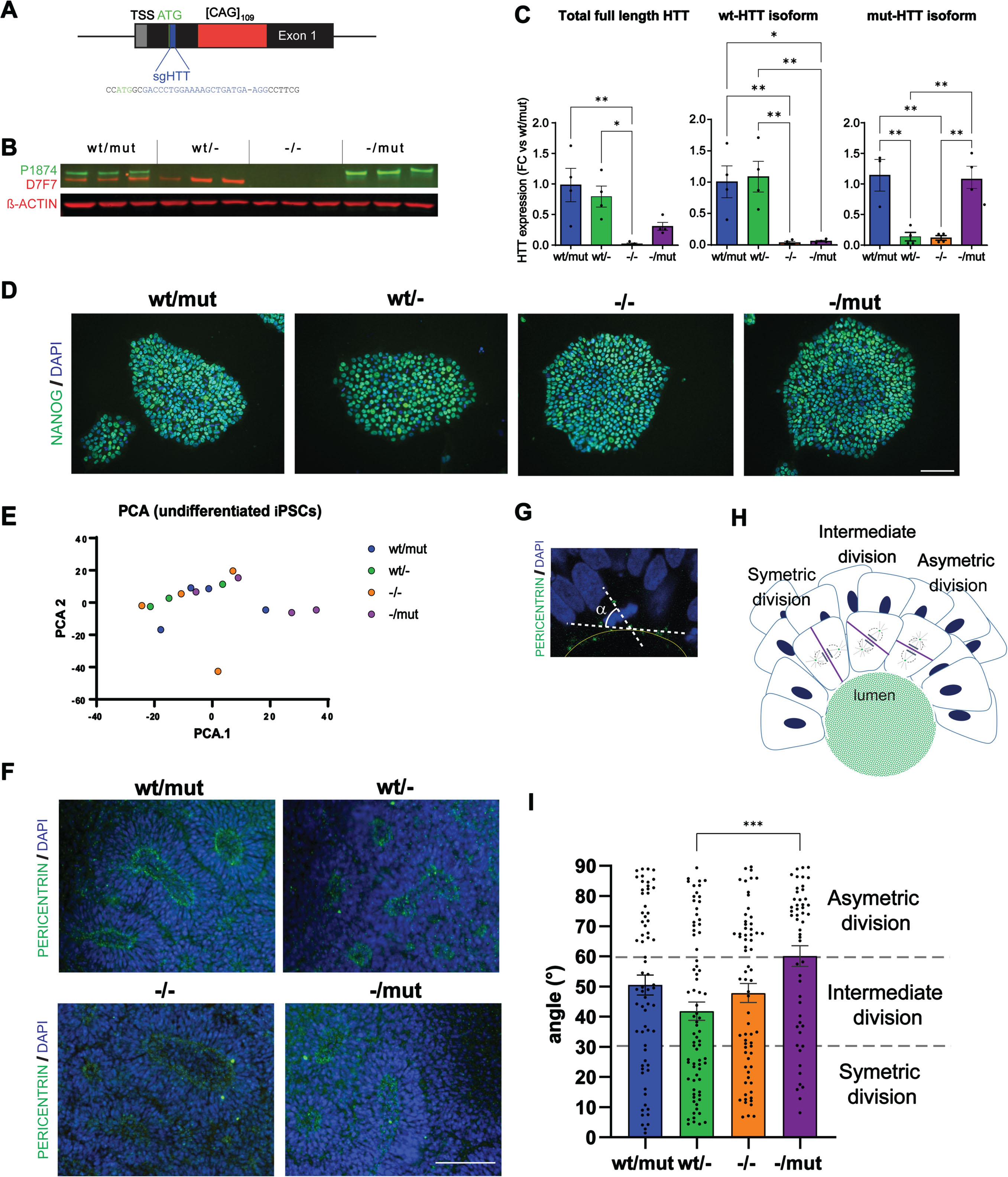
We then used our clones to record the effect of wt and/or mut-HTT loss on human neural induction and neuro-epithelial cells formation and organization. We did not record any change in the transition from pluripotency to neuroectodermal fate when monitoring OCT4 and PAX6 levels between DIV0 to 8 (Supplementary Figure 4A). Investigating the next stage when neuro-epithelial cells progressively emerge into rosette-like structures (R-NSC), the lumen size of DIV7 rosettes remained unchanged across genotypes (Fig. 1I; Supplementary Figure 4B). In contrast, we detected statistically higher cell division in HTT-/- rosettes compared with HTT-/mut (Supplementary Figure 4C) as well as changes in the orientation of cell division of cells adjacent to R-NSC lumen. Spindle orientation drives the self-organization of R-NSCs as symmetric division gives rise to two daughter neuroepithelial cells and permits the expansion of rosette size while asymmetric division (alpha spindle angle 0–30°) generates a daughter more likely to mature into post-mitotic neurons [26]. Ruzo and collaborators reported that wt human embryonic stem cells (hESC) derived R-NSCs (DIV28) show a bias toward symmetric division that is significantly reduced in rosettes derived from HD-hESCs [26]. In our R-NSC cultures, we observed that HTT-/mut R-NSCs are biased towards asymmetric divisions while HTTwt/- cells are biased towards symmetric division (Fig. 1F–H; Supplementary Figure 4D). These conclusions are consistent with our previous observations of HD-hESC derived neural stem cells (DIV > 50) treated with RNAi silencing both HTT alleles. Interestingly, the spindle orientation of undifferentiated iPSCs remained unchanged in all genotypes (Supplementary Figure 4E–G). Overall, our data suggest that HTT loss or mutation has significant impacts on cell division, polarity, and self-organization of developing structures by human neuro-epithelial cells but may have less or no impact on cell division of non-polarized cells during development.
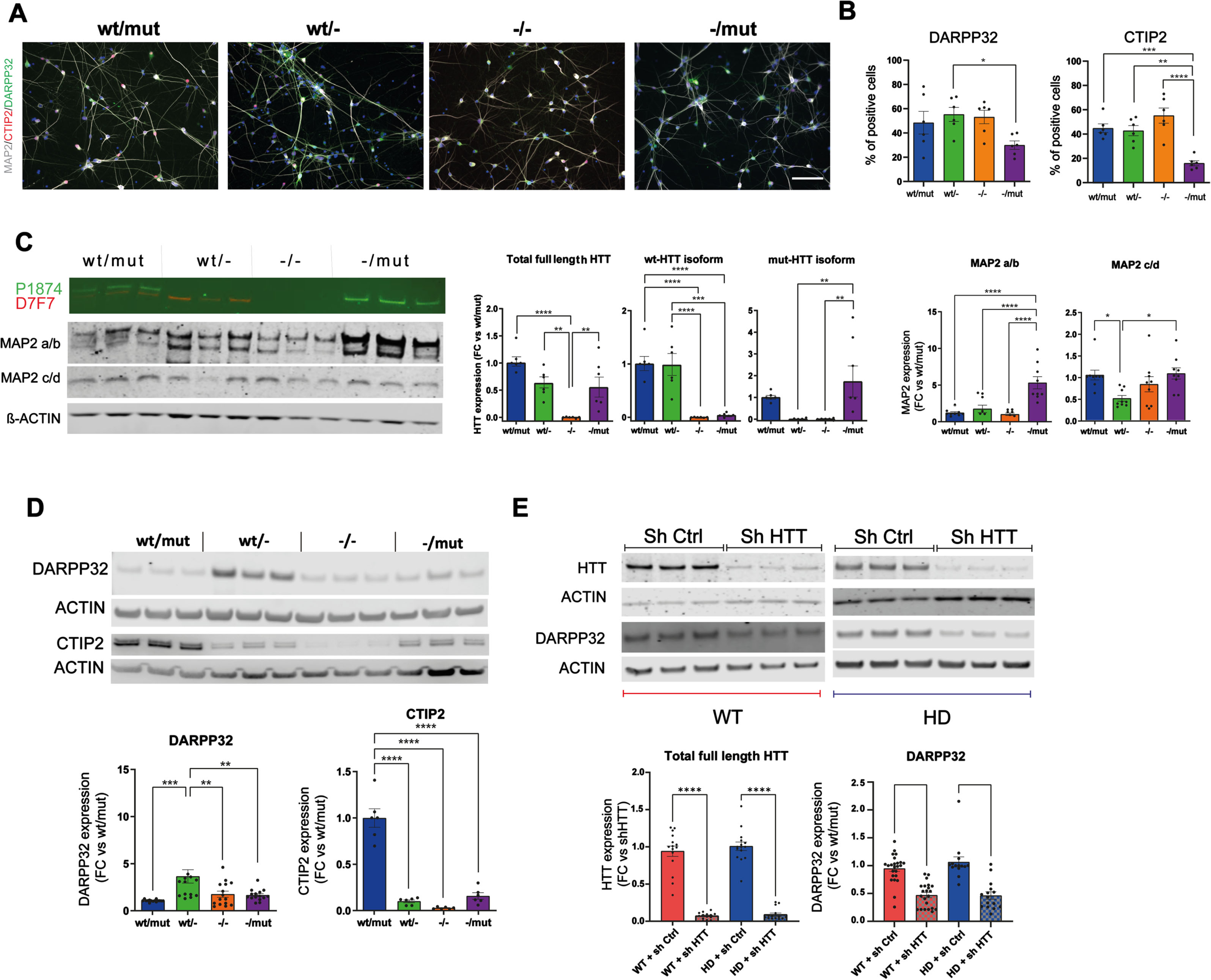
Considering that the alteration in rosette self-organization might influence their differentiation into post-mitotic neurons, we investigated the maturation of R-NSCs from all genotypes (HTTwt/mut, wt/-, -/mut, -/-) into striatal neurons, the subtype of neurons most affected in HD. We assessed the cultures at DIV55 by immunocytochemistry (Fig. 2A, B; Supplementary Figure 5A, B), western blot (Fig. 2C, D), and RNAseq (Supplementary Figure 6) for markers of post-mitotic neurons (MAP2), and striatal projecting GABAergic neurons (SPN: DARPP32/PPP1R1B, CTIP2/BCL11B). Overall, all cultures produced neuronal populations enriched in striatal cells. Proportion of positive cells and/or the protein level of DARPP32 and CTIP2 (BCL11B) cells, a transcription factor essential for the formation of DARPP32 + SPN, was significantly higher in HTTwt/- than in HTT-/mut. (Fig. 2A, B). To determine if the loss of wt- or wt/mut-HTT would impair DARPP32 expression in post-mitotic striatal neurons expressing this SPN marker, we used lentiviruses expressing short hairpin RNA (shRNA) targeting HTT mRNA. We differentiated three HD and two WT iPSC or hESC lines into striatal neurons transduced with either shHTT or control shRNA lentivirus at DIV35, a stage at which DARPP32 + SPNs become detectable (Supplementary Figure 5D, E). Western blot analysis at DIV55 shows that the level of DARPP32 was significantly reduced in all striatal cultures derived from WT or HD lines transduced with shHTT viruses (Fig. 2E). Overall, our data suggest that wt-HTT is involved in DARPP32 protein homeostasis in human SPNs.
Fig. 2
Loss of HTT impairs striatal differentiation of human iPSCs. A) Representative immunostaining of neuronal marker (MAP2) and SPN marker (DARPP32, and CTIP2) at DIV55 (scale bar: 100μm). B) Percentage of cells expressing DARPP32 and CTIP2 (1 clone per genotype, n = 6–12; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). C, D) Representative western blot and quantification of HTT isoforms, MAP2 isoforms (C) and DARPP32 and CTIP2 (D) protein level for each genotype. Individual data points, mean and SEM are shown, (n = 3 independent maturations of 2 independent differentiations per clones; 1–2 clones per genotype; *p < 0.05, **p < 0.01; ***p < 0.001; ****p < 0.0001, one-way ANOVA test with Tukey’s multiple comparison post-test). (E) Representative western blot and quantification of HTT (D7F7) and DARPP32 protein level in striatal cultures derived from 2 WT and 3 HD hPSC lines at DIV48, 15 days after transduction with shCTRL or shHTT lentiviruses. Individual data points, mean and SEM are shown, n = 3–9 per hPSC lines. ****p < 0.0001, one-way ANOVA test with Tukey’s multiple comparison post-test.
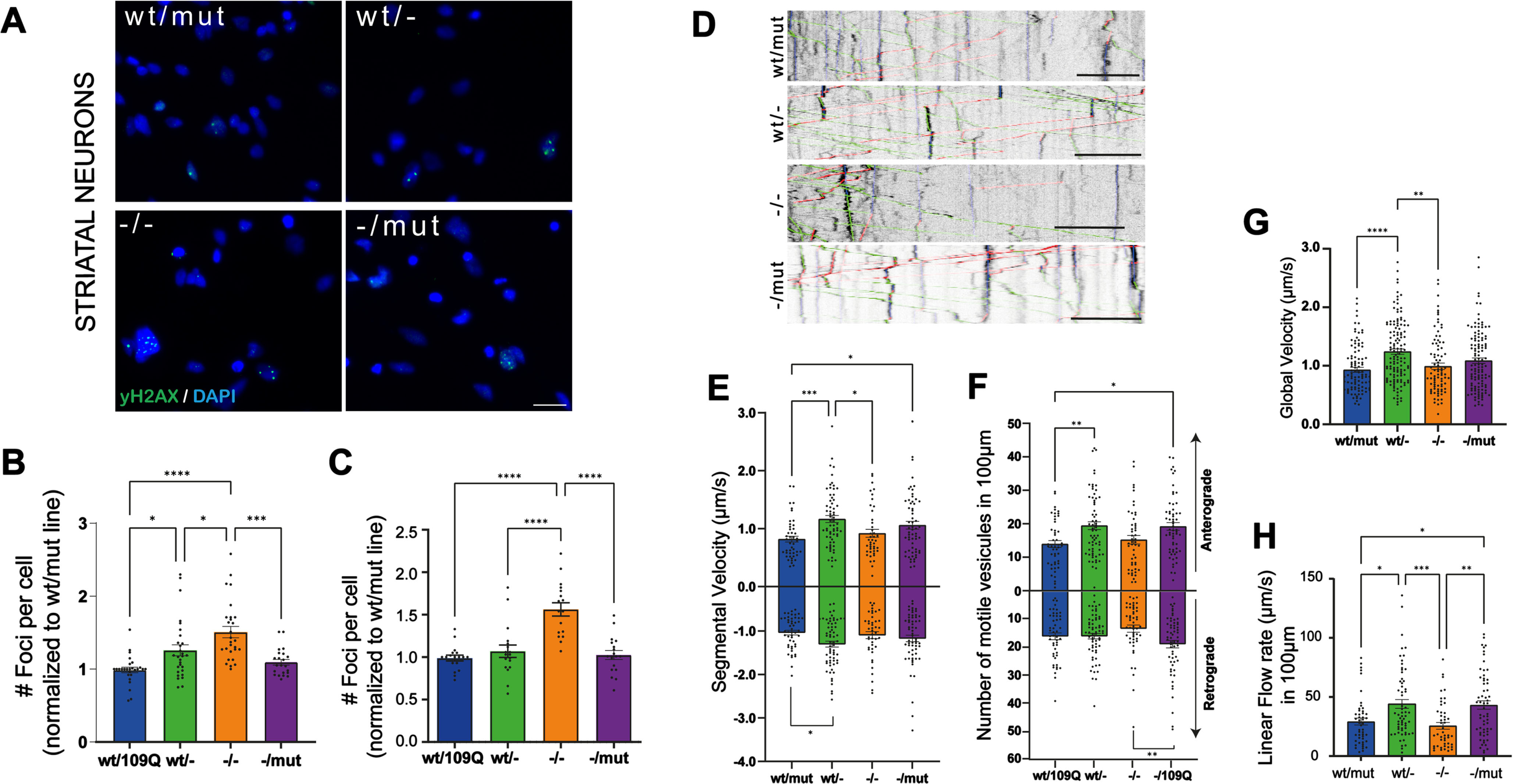
While impaired functions of striatal and cortical neurons are well described in adult HD mice this is far less the case in patient-derived iPSC neuronal derivatives. We explored two functions: DNA damage and BDNF transport in neurons in the derivatives of our iPSC clones. Increased levels of DNA damage are found in HD neural progenitor cells derived from a panel of isogenic, allelic human ESCs [38]. In order to assess whether the loss of wt- or wt/mut-HTT could modulate this phenotype, we quantified nuclear foci in striatal (Fig. 3A, B; Supplementary Figure 7D) and cortical neurons (Fig. 3C; Supplementary Figure 7A–C) at DIV55 with γ-H2AX, a double-stranded DNA breaks marker. We identified a significant increase in basal DNA damage in both cortical and striatal neurons derived from HTT-/- clones while no change in DNA damage marks were observed in undifferentiated iPSCs of different genotypes (Supplementary Figure 7E, F). These results suggest that DNA damage phenotype in HD might be resulting from a loss of wt-HTT function in HD neurons. Impaired vesicular transport of BDNF along axons of cortical neurons projecting onto striatal SPN has been described in HD mice and in immature neurons derived from HD-hESCs [39]. As impaired transport and release of BDNF have profound consequences on the survival of the cortico-striatal pathway in HD, we examined the dynamics of BDNF-mCherry containing vesicles recording their trafficking (Fig. 1D). We analyzed the segmental velocity (the speed of a given vesicle without pauses and according to their direction), number of vesicles migrating in a given direction (anterograde or retrograde), global velocity (speed, including pausing and static vesicles), and linear flow. Linear flow accounts for the velocity and number of motile vesicles, providing an estimated numerical value of the overall transport within the dendrites (Fig. 3E–H). The majority of BDNF transport parameters were improved in cortical neurons derived from HTTwt/- clones relative to neurons from parental line. Together, these results support the dominant-negative activity of mutant HTT on BDNF transport in HD neurons. Considering the relative immaturity of human iPSC-derived neurons and because increased γH2AX staining may result from increased DNA damage and/or reduced DNA damage repair processes, our results suggest that alterations of BDNF transport and DNA damage and/or DNA damage repair already occur in young forebrain neurons in the developing fetus carrying the HD mutation.
Fig. 3
Mutation or loss of HTT isoforms impair DNA damage response and BDNF transport in human neurons. A) Immunostaining of yH2AX marker (green) to identify double-stranded DNA breaks in striatal neurons at DIV 55 (scale bar 20μm). Number of foci per cell normalized to HTTwt/mut line in striatal neurons (B) and cortical neurons (C). (Individual data points (mean of 20 pictures/well), mean and SEM are shown; n indicates the total number of neurons per condition in at least four independent experiments; n = 31,251 wt/mut; n = 33,513 wt/-; n = 24,597 -/-; n = 27,809 -/mut; *p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA test with Tukey’s multiple comparison post-test.). D) Representative kymographs showing BDNF-mCherry axonal trafficking for each genotype in cortical neurons (DIV 37–38; anterograde movement in green, retrograde movement in red and posing time in blue; scale bar: 10μm). E) Anterograde and retrograde segmental velocities of BDNF-mCherry-containing vesicles (μm/s). F) Number of anterograde and retrograde vesicles trafficking along 100μm of axon. G) Global velocity of BDNF-mCherry-containing vesicles (μm/s). H) Linear flow rate (μm/s). E–H) Individual data points, mean and SEM are shown, n indicates the number of axons per condition in at least three independent experiments; n = 49 wt/mut; n = 65 wt/-; n = 50 -/-; n = 61 -/mut; *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA test with Tukey’s multiple comparison post-test.
DISCUSSION
To date, although extensive research has been conducted on HTT inactivation [40] or lowering [41] as a potential strategy to improve HD pathogenesis, the translation into the clinic of this obvious approach for HD has not yet been substantiated. The largest clinical trial thus far (NCT03761849) by Roche examined non-allele selective antisense oligonucleotide (ASO) targeting HTT mRNA (Tominersen) and reported outcomes less favorable than those of a placebo [42]. While the neuroprotective role of wt-HTT protein is increasingly described in animal models, loss of function mutations in wt-HTT have only recently been described as causing important neurodevelopmental defects in humans [27]. Consistently with these observations, our study contributes evidence supporting the involvement of HTT protein in distinct stages of human neurodevelopment emphasizing developmental aspect of HD. This complements existing research that bolsters the hypothesis of a developmental component in HD [10–18].
We created a novel collection of isogenic mono- or biallelic HTT knock-out iPSC clones to investigate the functions of the HTT gene and molecular determinants of HD. Interestingly, among the iPSC clones with confirmed HTT protein disruption we generated, we observed a relatively even distribution: 50% homozygous clones (HTT-/-), 25% wild-type hemizygous clones (HTTwt/-), and 25% mutant hemizygous clones (HTT-/mut). Likewise, we observed equal expression of key pluripotency markers, normal growth rate and no alteration of spindle orientation during division across all genotypes in human iPSCs. Overall, our observation of no alterations of cell division or pluripotency in iPSCs with partial or complete loss of wt or mut-HTT is consistent with those in mouse ES null and hemizygous HTT line (Hdh -/- & -/+) and in HTT-/- human embryonic stem cells [19, 26]. Moreover, we show that selectively targeting the mutant HTT isoform in human iPSCs and neurons did not affect the expression of the wild-type HTT allele, which has implications for allele-selective clinical approaches aimed at lowering mutant HTT in HD patients. This result is consistent with observations on HD cell lines where the mutant allele was specifically targeted using Zinc finger proteins [43]. Our editing strategy did not specifically target the [CAG]n repeat expansion within the expanded HTT allele, nor did it significantly alter the expression level of HTT mRNA in the isoclone-derived iPSCs or neuronal cultures. Consequently, our collection of clones is not suitable for investigating cellular impairments triggered by repeat-associated non-ATG (RAN) translation proteins produced from the expanded HTT allele. Similarly, mutant HTT can induce persistent epigenetic modifications [44], which might persist even after gene editing eliminates mutant HTT protein in HTT-/- and HTTwt/- clones and subsequent iPSCs amplification and differentiation. Therefore, our collection may not be able to detect phenotypic changes caused by mutant HTT-mediated epigenetic modifications, even when comparing different isogenic clones.
Characterizing neural and neuronal derivatives of human iPSCs of this collection, we show evidence that HTT loss or mutation has impacts on neuro-epithelial and striatal neurons maturation in line with the growing body of literature in support of a role for HTT protein in specific steps of human neurodevelopment and for a developmental component to HD. A case of hypomorphic wt-HTT caused by a t(4;12) balanced chromosome translocation, reducing by half the expression of HTT, was not associated with any detectable abnormal phenotype in the translocation carriers at least up to 46 years old [45]. Likewise, heterozygote carriers of two types of single HTT variant causing approximately 15% (HTT c.8157T > A) and 40% (HTT c.4469 + 1 G > A) reduction in HTT level produce no phenotype in adult [27]. However, the severe HTT lowering below 10% of normal level caused by the compound heterozygote mutations of both variants in three of their children underlie a dramatic neurodevelopmental disorder (LOMARS; OMIM #617435), not resembling HD [27, 28]. These authors although observed that HTT loss of function variant suffer negative selection in the human population based on the observation in gnomAD database of much fewer than expected damaging loss-of-function mutation while, in contrast, HTT missense variants are only slightly underrepresented [27]. In HD patients, the non-coding single nucleotide polymorphism (SNP) rs13102260:G > A, in the promoter of the wt-HTT allele is a cis-regulatory variant that reduces wt-HTT transcription and is associated with earlier age of onset of HD mutation carrier [46]. Overall, these reports indicate that the loss of wt-HTT may be more consequential than expected and that if significantly beyond 50% of normal level, it can result in neurodevelopmental defect in humans.
Previously, we reported dominant-negative effects of HD mutations leading to alteration of the division of human ESC-derived neural cells [47]. These human neural stem cells present limited apico-basal polarization features oriented perpendicular to the substratum of culture. In the present study, we analyzed both undifferentiated iPSCs, which are not polarized cells, and DIV7 early neuro-epithelial cells that self-organized into rosette structures composed of highly polarized forebrain neural cells which apico-basal polarity oriented radially to the rosette structure [48]. While polarized neural cells present significant difference in the orientation of their cell division based on their HTT genotype, undifferentiated iPSCs from the four genotypes tested did not and divided uniformly perpendicular to their substratum. Although the rate of division of HTT-/- rosette cells was the highest, similar lumen areas were measured across genotypes unlike previous description of HD-hESC/iPSC derived rosettes [26, 49] and HTT-/- hESC-derived rosettes at later stage (DIV28) [26]. Overall, our data are consistent and expand results of studies that show mitotic angle and cell polarity defects in different epithelial neural, or mammary cells [15, 26, 47, 49] and suggest that HTT loss or mutation has significant impact on cell division, polarity and self-organization of developing structures by human epithelial cells but may have less or no impact on cell division of non-polarized cells during development.
The striatal projection neurons (SPN), which are the largest neuronal population in the striatum and most prominently affected in HD [51], arise from the Gsx2-positive progenitors in the lateral ganglionic eminence [52] and express DARPP32, a central mediator of dopamine signaling and other first messengers in these cells [53]. Pluripotent Hdh-/- cells injected in the mouse blastocyst mostly fail to colonize the striatum of chimeric mice [17]. Conversely, loss of HTT in the Gsx2 lineage leads to late-life neuronal loss in the striatum and is accompanied by reduced DARPP32 immunoreactivity [54]. Conditional deletion of HTT in the brain or specifically in SPNs produces similar results on genesis, differentiation, and long-term survival of SPNs [7, 55]. Likewise, striatal development in HD mice revealed defective SPN neurogenesis in the striatum [14]. Using our collection of HTT-edited human iPSC clones, we investigated whether the loss of wt- and/or mut-HTT similarly deregulates striatal development and SPN specification or survival in humans. We confirmed that HTT is not required for post-mitotic neuron generation nor for SPN specification. We observed that striatal neuronal cultures only expressing mutant HTT expressed more adult isoform of MAP2 than the other cultures and are thus likely more matured. This may be explained by a higher proportion of rosette progenitors that undergo, earlier, asymmetric divisions. Conversely, DARPP32 levels were highest in neuronal cultures that only expressed wt-HTT and were significantly reduced in already specified wt and HD human SPNs in which HTT expression was largely abolished by RNA interference. Overall, our data suggest both the loss of wild-type HTT and the presence of mutant HTT alter the development of the main neuronal population of the striatum.
Altered survival of neurons in the telencephalon of HD patients has been linked to several neuronal dysfunctions [56]. Disruptions of the homeostasis of BDNF and of the DNA damage repair machinery play key roles in the pathological cascades that lead to neuronal loss. BDNF gene expression, BDNF protein axonal transport efficiency and release in the striatum depend on HTT function and are altered in HD [36, 37, 57]. Robust DNA damage response is closely connected to aging [58]. In HD, damage to nuclear DNA in patient samples and mouse models, in the form of strand breaks and damaged bases, has been extensively reported [59–62], including in prodromal patients [63], and along with elevated ATM signaling (Ataxia telangiectasia mutated), a core component of the DNA repair system [64]. Equally, human genome-wide association studies identified polymorphisms in DNA handling factors as potential modifiers of the age at neurological onset in HD [65, 66]. We showed in cortical and striatal neurons an increased basal level of DNA damage in HTT-/- neurons. This result support the participation of wt-HTT in normal DNA damage repair machinery in human neurons. In contrast to the increased level of DNA damage described in neural progenitor cells (NPC) derived from isogenic 45Q and 81Q HD-hESCs [38], we could not record significantly different levels of DNA damage in the neurons derived from hemizygote clones (HTTwt/- or -/mut) or the parental line (HTTwt/mut). This might be caused by the proliferative nature of NPCs or to the media composition difference between NPCs and neurons. Interestingly, the DNA damage level of undifferentiated iPSCs was similar across genotypes although percentage of cells without any mark of DNA damage was significantly lower than that of neurons. Previously we reported the impact of allele-specific RNA interference of mut-HTT in HD-hESC derived neurons [39]. Our data supported a dominant-negative effect of mutant HTT on the normal function of wt-HTT on BDNF transport in human neurons. BDNF transport in cortical neurons derived from HTT-/- iPSCs was globally reduced compared to that of wt-hemizygote neurons but not significantly different from neurons derived from HTTwt/mut or HTT-/mut clones. This confirmed that only an allele-specific approach targeting the mut-HTT allele could revert the BDNF transport alteration in HD patients.
In summary, the newly generated collection of isogenic iPSC clones provides a valuable resource for studying HTT functions during human development and in adult cells. It complements existing collections of isogenic hESC and h-iPSC clones with different CAG length variations in exon 1 of HTT, facilitating a deeper understanding of molecular and cellular differences resulting from various HD mutations.
ACKNOWLEDGMENTS
We would like to thank Anne de Cian and Jean-Paul Concordet from TACGENE: MNHN-CNRS UMR 7196/INSERM U1154 for the SpCas9 purified protein.
FUNDING
This work benefited from support from European Commission H2020 Project Joint Programme –Neurodegenerative Disease Research (JPND) ModelPolyQ grant 643417), NeurATRlS ANR-11-INBS-0011, of the French lnvestissements d’Avenir Program run by the Agence Nationale pour la Recherche.”, the Laboratoire d’Excellence Revive (ANR-10-LABX-73), Parkington project ANR-17-CE18-0026, Neurolead project BPI France and Institut national de la santé et de la recherche médicale (Inserm) (A.L.P). I-Stem and CECS are supported by the Association Française contre les Myopathies (AFM).
CONFLICT OF INTEREST
Frédéric Saudou is an Editorial Board member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer review.
DATA AVAILABILITY
The whole-genome sequence data have been uploaded to the Sequence Read Archive at NCBI under Accession number GSE228254. Other data supporting the findings of this study are available on request from the corresponding author.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JHD-231509.
REFERENCES
[1] | Dure LS , Landwehrmeyer GB , Golden J , McNeil SM , Ge P , Aizawa H , et al. IT15 gene expression in fetal human brain. Brain Res (1994) ;659: ;33–41. https://doi.org/10.1016/0006-8993(94)90860-5. |
[2] | Li SH , Schilling G , Young WS , 3rd Li XJ , Margolis RL , Stine OC , et al. Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron (1993) ;11: ;985–93. |
[3] | Strong TV , Tagle DA , Valdes JM , Elmer LW , Boehm K , Swaroop M , et al. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat Genet (1993) ;5: ;259–65. https://doi.org/10.1038/ng1193-259. |
[4] | Group THDCR A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research GrouCell (1993) ;72: ;971–83. |
[5] | Handley OJ , Naji JJ , Dunnett SB , Rosser AE Pharmaceutical, cellular and genetic therapies for Huntington’s disease. Clin Sci (Lond) (2006) ;110: ;73–88. |
[6] | Reiner A , Albin RL , Anderson KD , D’Amato CJ , Penney JB , Young AB Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. (1988) ;85: ;5733–7. https://doi.org/10.1073/pnas.85.15.5733. |
[7] | Dragatsis I , Levine MS , Zeitlin S Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet (2000) ;26: ;300–6. |
[8] | Van Raamsdonk JM , Pearson J , Rogers DA , Bissada N , Vogl AW , Hayden MR , et al. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet (2005) ;14: ;1379–92. https://doi.org/10.1093/hmg/ddi147. |
[9] | Zhang S , Feany MB , Saraswati S , Littleton JT , Perrimon N Inactivation of Drosophila Huntingtin affects long-term adult functioning and the pathogenesis of a Huntington’s disease model. Dis Model Mech (2009) ;2: ;247–66. https://doi.org/10.1242/dmm.000653. |
[10] | Barnat M , Le Friec J , Benstaali C , Humbert S Huntingtin-mediated multipolar-bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron (2017) ;93: ;99–114. https://doi.org/10.1016/j.neuron.2016.11.035. |
[11] | Barnat M , Capizzi M , Aparicio E , Boluda S , Wennagel D , Kacher R , et al. Huntington’s disease alters human neurodevelopment. Science (2020) ;369: ;787–93. https://doi.org/10.1126/science.aax3338. |
[12] | Capizzi M , Carpentier R , Denarier E , Adrait A , Kassem R , Mapelli M , et al. Developmental defects in Huntington’s disease show that axonal growth and microtubule reorganization require NUMA1. Neuron (2022) ;110: ;36–50.e5. https://doi.org/10.1016/j.neuron.2021.10.033. |
[13] | Lo Sardo V , Zuccato C , Gaudenzi G , Vitali B , Ramos C , Tartari M , et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci (2012) ;15: ;713–21. https://doi.org/10.1038/nn.3080. |
[14] | Molero AE , Gokhan S , Gonzalez S , Feig JL , Alexandre LC , Mehler MF Impairment of developmental stem cell-mediated striatal neurogenesis and pluripotency genes in a knock-in model of Huntington’s disease. Proc Natl Acad Sci U S A (2190) ;106: ;0–5. https://doi.org/10.1073/pnas.0912171106. |
[15] | Molina-Calavita M , Barnat M , Elias S , Aparicio E , Piel M , Humbert S Mutant huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J Neurosci (2014) ;34: ;10034–40. https://doi.org/10.1523/JNEUROSCI.0715-14.2014. |
[16] | Nopoulos PC , Aylward EH , Ross CA , Mills JA , Langbehn DR , Johnson HJ , et al. Smaller intracranial volume in prodromal Huntington’s disease: Evidence for abnormal neurodevelopment. Brain (2011) ;134: ;137–42. https://doi.org/10.1093/brain/awq280. |
[17] | Reiner A , Del Mar N , Meade CA , Yang H , Dragatsis I , Zeitlin S , et al. Neurons lacking huntingtin differentially colonize brain and survive in chimeric mice. J Neurosci (2001) ;21: ;7608–19. |
[18] | White JK , Auerbach W , Duyao MP , Vonsattel JP , Gusella JF , Joyner AL , et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet (1997) ;17: ;404–10. https://doi.org/10.1038/ng1297-404. |
[19] | Duyao MP , Auerbach AB , Ryan A , Persichetti F , Barnes GT , McNeil SM , et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science (1995) ;269: ;407–10. |
[20] | Nasir J , Floresco SB , O’Kusky JR , Diewert VM , Richman JM , Zeisler J , et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell (1995) ;81: ;811–23. |
[21] | Zeitlin S , Liu JP , Chapman DL , Papaioannou VE , Efstratiadis A Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet (1995) ;11: ;155–63. https://doi.org/10.1038/ng1095-155. |
[22] | Piccolo FM , Kastan NR , Haremaki T , Tian Q , Laundos TL , De Santis R , et al. Role of YAP in early ectodermal specification and a Huntington’s disease model of human neurulation. ELife. (2022) ;11: ;e73075. https://doi.org/10.7554/eLife.73075. |
[23] | Thion MS , McGuire JR , Sousa CM , Fuhrmann L , Fitamant J , Leboucher S , et al. Unraveling the role of huntingtin in breast cancer metastasis. J Natl Cancer Inst. (2015) ;107: ;djv208. https://doi.org/10.1093/jnci/djv208. |
[24] | Braz BY , Wennagel D , Ratié L , de Souza DAR , Deloulme JC , Barbier EL , et al. Treating early postnatal circuit defect delays Huntington’s disease onset and pathology in mice. Science (2022) ;377: ;eabq5011. https://doi.org/10.1126/science.abq5011. |
[25] | Haremaki T , Metzger JJ , Rito T , Ozair MZ , Etoc F , Brivanlou AH Self-organizing neuruloids model developmental aspects of Huntington’s disease in the ectodermal compartment. Nat Biotechnol (2019) ;37: ;1198–208. https://doi.org/10.1038/s41587-019-0237-5. |
[26] | Ruzo A , Croft GF , Metzger JJ , Galgoczi S , Gerber LJ , Pellegrini C , et al. Chromosomal instability during neurogenesis in Huntington’s disease. Development. (2018) ;145: ;dev156844. https://doi.org/10.1242/dev.156844. |
[27] | Jung R , Lee Y , Barker D , Correia K , Shin B , Loupe J , et al. Mutations causing Lopes-Maciel-Rodan syndrome are huntingtin hypomorphs. Hum Mol Genet (2021) ;30: ;135–48. https://doi.org/10.1093/hmg/ddaa283. |
[28] | Rodan LH , Cohen J , Fatemi A , Gillis T , Lucente D , Gusella J , et al. A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur J Hum Genet (2016) ;24: ;1826–7. https://doi.org/10.1038/ejhg.2016.74. |
[29] | Gribaudo S , Bousset L , Courte J , Melki R , Peyrin J-M , Perrier AL Propagation of distinct alpha-synuclein strains within human reconstructed neuronal network and associated neuronal dysfunctions. In: Cieplak AS, editor. Protein Aggregation: Methods and Protocols. Springer Science + Business Media, LLC; 2022, p. 357-78. |
[30] | Nicoleau C , Varela C , Bonnefond C , Maury Y , Bugi A , Aubry L , et al. Embryonic stem cells neural differentiation qualifies the role of Wnt/beta-catenin signals in human telencephalic specification and regionalization. Stem Cells (2013) ;31: ;1763–74. https://doi.org/10.1002/stem.1462. |
[31] | Feyeux M , Bourgois-Rocha F , Redfern A , Giles P , Lefort N , Aubert S , et al. Early transcriptional changes linked to naturally occurring Huntington’s disease mutations in neural derivatives of human embryonic stem cells. Hum Mol Genet (2012) ;21: (17):3883–95. https://doi.org/10.1093/hmg/dds216 |
[32] | Fjodorova M , Louessard M , Li Z , De La Fuente DC , Dyke E , Brooks SP , et al. CTIP2-regulated reduction in PKA-dependent DARPP32 phosphorylation in human medium spiny neurons: Implications for Huntington disease. Stem Cell Reports (2019) ;13: ;448–57. https://doi.org/10.1016/j.stemcr.2019.07.015. |
[33] | Merienne N , Vachey G , de Longprez L , Meunier C , Zimmer V , Perriard G , et al. The self-inactivating KamiCas9 system for the editing of CNS disease genes. Cell Rep (2017) ;20: ;2980–91. https://doi.org/10.1016/j.celre2017.08.075. |
[34] | Arber C , Precious SV , Cambray S , Risner-Janiczek JR , Kelly C , Noakes Z , et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development (2015) ;142: ;1375–86. https://doi.org/10.1242/dev.117093. |
[35] | Zala D , Hinckelmann M-V , Yu H , Lyra da Cunha MM , Liot G , Cordelières FP , et al. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell (2013) ;152: ;479–91. https://doi.org/10.1016/j.cell.2012.12.029. |
[36] | Gauthier LR , Charrin BC , Borrell-Pages M , Dompierre JP , Rangone H , Cordelieres FP , et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell (2004) ;118: ;127–38. |
[37] | Virlogeux A , Moutaux E , Christaller W , Genoux A , Bruyère J , Fino E , et al. Reconstituting corticostriatal network on-a-chip reveals the contribution of the presynaptic compartment to Huntington’s disease. Cell Rep (2018) ;22: ;110–22. https://doi.org/10.1016/j.celre2017.12.013. |
[38] | Ooi J , Langley SR , Xu X , Utami KH , Sim B , Huang Y , et al. Unbiased profiling of isogenic huntington disease hPSC-derived CNS and peripheral cells reveals strong cell-type specificity of CAG length effects. Cell Rep (2508) ;26: ;2494–2508.e7. https://doi.org/10.1016/j.celre2019.02.008. |
[39] | Drouet V , Ruiz M , Zala D , Feyeux M , Auregan G , Cambon K , et al. Allele-specific silencing of mutant huntingtin in rodent brain and human stem cells. PloS One (2014) ;9: ;e99341. https://doi.org/10.1371/journal.pone.0099341. |
[40] | Yamamoto A , Lucas JJ , Hen R Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell (2000) ;101: ;57–66. https://doi.org/10.1016/S0092-8674(00)80623-6. |
[41] | Kaemmerer WF , Grondin RC The effects of huntingtin-lowering: What do we know so far? Degener Neurol Neuromuscul Dis (2019) ;9: ;3–17. https://doi.org/10.2147/DNND.S163808. |
[42] | Tabrizi SJ , Estevez-Fraga C , van Roon-Mom WMC , Flower MD , Scahill RI , Wild EJ , et al. Potential disease-modifying therapies for Huntington’s disease: Lessons learned and future opportunities. Lancet Neurol (2022) ;21: ;645–58. https://doi.org/10.1016/S1474-4422(22)00121-1. |
[43] | Zeitler B , Froelich S , Marlen K , Shivak DA , Yu Q , Li D , et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat Med (2019) ;25: ;1131–42. https://doi.org/10.1038/s41591-019-0478-3. |
[44] | Hyeon JW , Kim AH , Yano H Epigenetic regulation in Huntington’s disease. Neurochem Int (2021) ;148: ;105074. https://doi.org/10.1016/j.neuint.2021.105074. |
[45] | Ambrose CM , Duyao MP , Barnes G , Bates GP , Lin CS , Srinidhi J , et al. Structure and expression of the Huntington’s disease gene: Evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet (1994) ;20: ;27–38. https://doi.org/10.1007/BF02257483. |
[46] | Becanović K , Nørremølle A , Neal SJ , Kay C , Collins JA , Arenillas D , et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci (2015) ;18: ;807–16. https://doi.org/10.1038/nn.4014. |
[47] | Lopes C , Aubert S , Bourgois-Rocha F , Barnat M , Rego AC , Deglon N , et al. Dominant-negative effects of adult-onset huntingtin mutations alter the division of human embryonic stem cells-derived neural cells. PloS One. (2016) ;11: ;e0148680. https://doi.org/10.1371/journal.pone.0148680. |
[48] | Hríbková H , Grabiec M , Klemová D , Slaninová I , Sun Y-M Calcium signaling mediates five types of cell morphological changes to form neural rosettes. J Cell Sci (2018) ;131: ;jcs206896. https://doi.org/10.1242/jcs.206896. |
[49] | Xu X , Tay Y , Sim B , Yoon S-I , Huang Y , Ooi J , et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Reports (2017) ;8: ;619–33. https://doi.org/10.1016/j.stemcr.2017.01.022. |
[50] | Keryer G , Pineda JR , Liot G , Kim J , Dietrich P , Benstaali C , et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest (2011) ;121: ;4372–82. https://doi.org/10.1172/JCI57552. |
[51] | Vonsattel JP , Myers RH , Stevens TJ , Ferrante RJ , Bird ED , Richardson EP Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol (1985) ;44: ;559–77. https://doi.org/10.1097/00005072-198511000-00003. |
[52] | Corbin JG , Gaiano N , Machold RP , Langston A , Fishell G The Gsh2 homeodomain gene controls multiple aspects of telencephalic development. Development (2000) ;127: ;5007–20. |
[53] | Ouimet CC , Langley-Gullion KC , Greengard P Quantitative immunocytochemistry of DARPP-32-expressing neurons in the rat caudatoputamen. Brain Res (1998) ;808: ;8–12. https://doi.org/10.1016/s0006-8993(98)00724-0 |
[54] | Mehler MF , Petronglo JR , Arteaga-Bracho EE , Gulinello ME , Winchester ML , Pichamoorthy N , et al. Loss-of-huntingtin in medial and lateral ganglionic lineages differentially disrupts regional interneuron and projection neuron subtypes and promotes Huntington’s disease-associated behavioral, cellular, and pathological hallmarks. J Neurosci (2019) ;39: ;1892–909. https://doi.org/10.1523/JNEUROSCI.2443-18.2018 |
[55] | Burrus CJ , McKinstry SU , Kim N , Ozlu MI , Santoki AV , Fang FY , et al. Striatal projection neurons require huntingtin for synaptic connectivity and survival. Cell Rep (2020) ;30: ;642–657.e6. https://doi.org/10.1016/j.celre2019.12.069 |
[56] | Ross CA , Tabrizi SJ Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol (2011) ;10: ;83–98. https://doi.org/10.1016/S1474-4422(10)70245-3 |
[57] | Zuccato C , Tartari M , Crotti A , Goffredo D , Valenza M , Conti L , et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet (2003) ;35: ;76–83. https://doi.org/10.1038/ng1219 |
[58] | Madabhushi R , Pan L , Tsai L-H DNA damage and its links to neurodegeneration. Neuron (2014) ;83: ;266–82. https://doi.org/10.1016/j.neuron.2014.06.034 |
[59] | Bogdanov MB , Andreassen OA , Dedeoglu A , Ferrante RJ , Beal MF Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem (2001) ;79: ;1246–9. |
[60] | Browne SE , Bowling AC , MacGarvey U , Baik MJ , Berger SC , Muqit MM , et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann Neurol (1997) ;41: ;646–53. |
[61] | Enokido Y , Tamura T , Ito H , Arumughan A , Komuro A , Shiwaku H , et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol (2010) ;189: ;425–43. https://doi.org/10.1083/jcb.200905138 |
[62] | Illuzzi J , Yerkes S , Parekh-Olmedo H , Kmiec EB DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res (2009) ;87: ;733–47. https://doi.org/10.1002/jnr.21881 |
[63] | Castaldo I , De Rosa M , Romano A , Zuchegna C , Squitieri F , Mechelli R , et al. DNA damage signatures in peripheral blood cells as biomarkers in prodromal huntington disease. Ann Neurol (2019) ;85: ;296–301. https://doi.org/10.1002/ana.25393 |
[64] | Lu X-H , Mattis VB , Wang N , Al-Ramahi I , van den Berg N , Fratantoni SA , et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med (2014) ;6: ;268ra178. https://doi.org/10.1126/scitranslmed.3010523 |
[65] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell (2015) ;162: ;516–26. https://doi.org/10.1016/j.cell.2015.07.003 |
[66] | Moss DJH , Pardiñas AF , Langbehn D , Lo K , Leavitt BR , Roos R , et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol (2017) ;16: ;701–11. https://doi.org/10.1016/S1474-4422(17)30161-8 |


