
A Double-Pronged Sword: XJB-5-131 Is a Suppressor of Somatic Instability and Toxicity in Huntington’s Disease
Abstract
Due to large increases in the elderly populations across the world, age-related diseases are expected to expand dramatically in the coming years. Among these, neurodegenerative diseases will be among the most devastating in terms of their emotional and economic impact on patients, their families, and associated subsidized health costs. There is no currently available cure or rescue for dying brain cells. Viable therapeutics for any of these disorders would be a breakthrough and provide relief for the large number of affected patients and their families. Neurodegeneration is accompanied by elevated oxidative damage and inflammation. While natural antioxidants have largely failed in clinical trials, preclinical phenotyping of the unnatural, mitochondrial targeted nitroxide, XJB-5-131, bodes well for further translational development in advanced animal models or in humans. Here we consider the usefulness of synthetic antioxidants for the treatment of Huntington’s disease. The mitochondrial targeting properties of XJB-5-131 have great promise. It is both an electron scavenger and an antioxidant, reducing both somatic expansion and toxicity simultaneously through the same redox mechanism. By quenching reactive oxygen species, XJB-5-131 breaks the cycle between the rise in oxidative damage during disease progression and the somatic growth of the CAG repeat which depends on oxidation.
INTRODUCTION
In the next decade, at least 2% of Americans will be afflicted with some form of Alzheimer’s disease (AD) (4,000,000) [1], Parkinson’s disease (PD) (1,500,000) [2], or Huntington’s disease (HD) (250,000) [3], among others. Each of these disorders can affect patients for decades with no hope of a cure [4]. Because the number of affected individuals will grow dramatically, the gap between the problem’s size and our capabilities for treatment will widen. Viable therapeutics for any of these disorders would be a breakthrough and provide relief for affected patients and their families. “Biologics” have been developed as therapeutics to offset both AD and HD. For example, the amyloid-β-directed monoclonal antibody aducanumab was recently approved for the treatment of AD [5], but its long-term efficacy is, as yet, unknown. For HD, there was hope that use of anti-sense oligonucleotide (ASO) therapy would reduce the expression level of the toxic mutant huntington protein, thereby rescuing disease. However, Roche halted a phase III study of its ASO drug, Tominersen (Ionis Pharmaceuticals), for lack of efficacy, and Wave Life Sciences discontinued the development of two of its HD ASOs that were in phase I/II clinical trials [6, 7]. These outcomes have underscored the need to continue the search for small molecule drugs for neurodegeneration [8–10]. In the past, antioxidants held promise as efficient inhibitors of damaging reactive oxygen species (ROS) in aging and age-related neurodegenerative diseases [11–13]. Yet, natural antioxidants such as Coenzyme Q10, (CoQ10) also failed in clinical trials for HD and PD [14–17], casting doubt as to the efficacy of a ROS inhibitor approach to drug development [18]. These failures, however, sparked interest in developing unnatural (synthetic) antioxidants, which increased their effective concentration by targeting mitochondria (MT) directly and reducing ROS at its source. Here, we consider the benefits of the synthetic, mitochondrial-targeted antioxidant, XJB-5-131, as a potential clinical candidate for HD.
CHECKERED HISTORY OF ANTIOXIDANT THERAPY
Considering the damage caused by ROS, it seemed obvious that antioxidants might be a good therapeutic strategy to offset at least some pathology that develops in HD patients (Fig. 1). Indeed, there is significant evidence that MT are hyperactive at early stages of disease [19–21], particularly in astrocytes [19–22], which express inflammatory mediators when they become reactive [20–22]. In affected striatal astrocytes, MT reprogram their fuel use from glucose to fatty acids under disease conditions, increasing β-oxidation and ROS production [20]. The mitochondrial bioenergetics for neurons and astrocytes in various HD models are discussed [23]. The naturally occurring oxidants were obvious candidates to offset the impact of elevated ROS. Vitamin E and CoQ10 have been a significant focus of antioxidants therapeutics in PD and HD in both rodent models [24–27] and in human clinical trials [14–17]. However, the triumphs and tribulations of the dietary supplementation with natural antioxidants has led to uncertainty as to the benefits of such an approach.
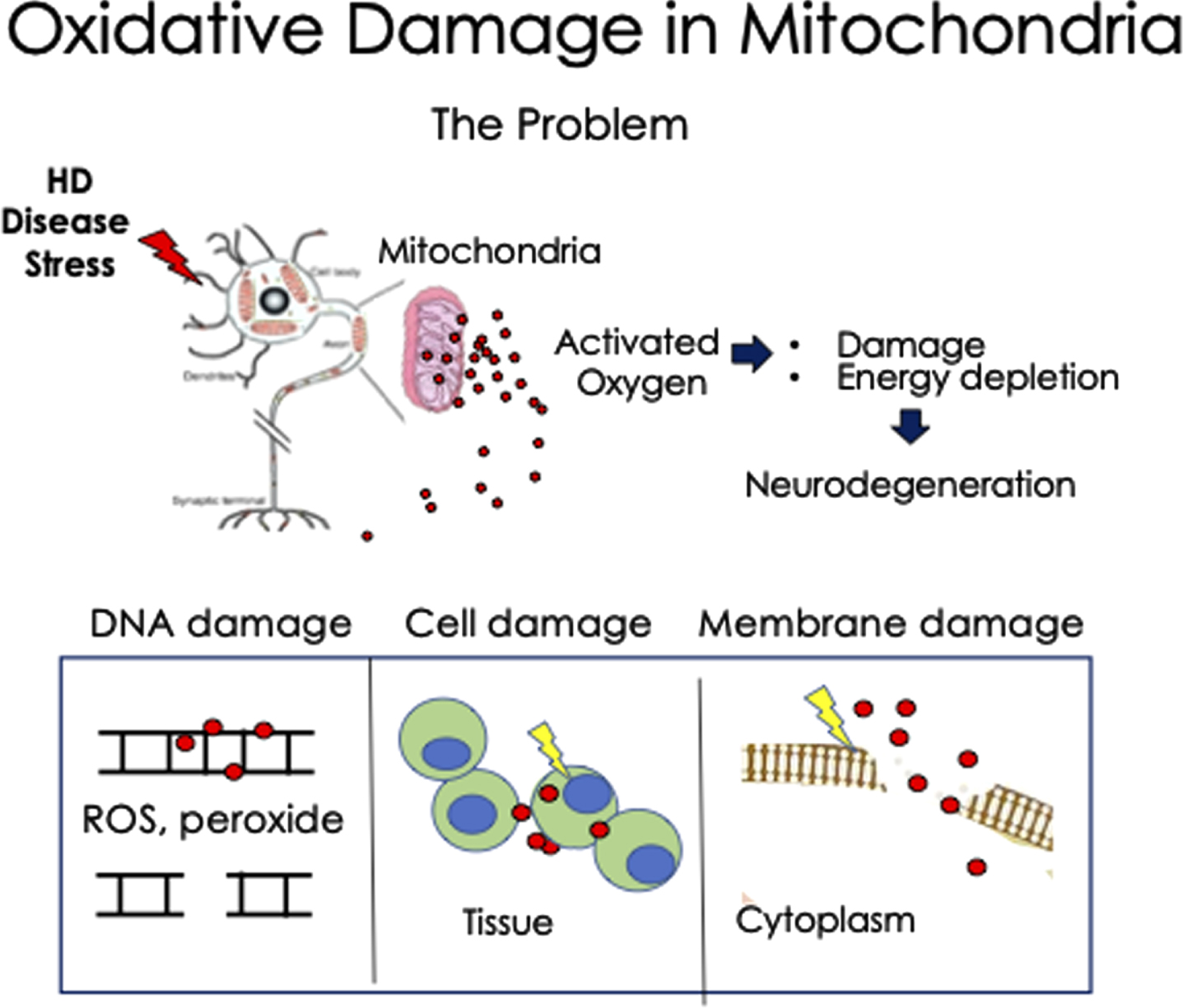
Fig. 1
Schematic diagram of the deleterious effects of oxidative damage in Huntington’s disease neurons. Under stress, MT release high levels of ROS, which can act at short distance as radicals and as peroxide at longer distance. The MT are the source and major target of oxidation. The activated ROS interacts with DNA, RNA, cell membranes, and cytoplasmic proteins, as examples. Additionally, damaged MT eventually diminish their ATP production leading to energy depletion and cell death in the HD neurons. This mechanism may apply to other neurodegenerative diseases as well.
CoQ10 (ubiquinone) is an essential biological cofactor of the electron transport chain (ETC) (Fig. 2A) and functions as an important endogenous antioxidant in mitochondrial and lipid membranes (Fig. 2B) [28, 29]. Primary CoQ10 deficiency can affect any part of the body, but particularly the brain, muscle, and kidney tissues, as a consequence of their high energy demands [30, 31]. CoQ10 deficiency in brain tissue can cause ataxia, together with a range of other neurological manifestations, suggesting that an increase might offset brain toxicity [32–35]. Thus, the lack of efficacy in clinical trials for CoQ10 for both PD and HD patients was puzzling [14–17].
Fig. 2
Schematic diagram of Coenzyme Q10 in the electron transport chain of the mitochondrial inner membrane. A) The structure of Coenzyme Q10, a lipid soluble component of the mitochondrial inner membrane. B) Mechanisms of oxidative stress involving mitochondria. The mitochondrial ETC reoxidizes reduced cofactors (NADH and FADH2) using molecular oxygen as the final electron acceptor, and the energy released in this process is captured in the form of ATP. The electron transport chain (ETC) depicting the position of Coenzyme Q10 (red) in the inner membrane. Coenzyme Q10 is critical for electron transport in the mitochondrial respiratory chain. The enzyme carries electrons from complexes I and II to complex III, thus participating in ATP production. I, II, III, IV, and V indicate protein components of the ETC. ‘e’ indicates electrons. and F1 are subunits of ATP synthase complex V (Complex V). ATP and ADP, adenosine triphosphate and diphosphate, respectively; CoQ 10, coenzyme Q10; FAD, flavin adenine dinucleotide; FADH, reduced flavin adenine dinucleotide. Image published with permission from Dove Medical Press, Rodick et al., Nutr Diet Supplem. 2017;10:1-11 [107]. C) Several components of the ETC chain (most often CI and CIII) generate O2•–. (left) The radical is converted into H2O2 by mitochondrial SOD. Through the Fenton reaction, H2O2 is converted into •OH, a molecule that produces oxidative cell injury through DNA damage, carboxylation of proteins, and lipid peroxidation. (right) •NO is produced by the activity of intracellular NOS. •NO can be combined with O2•– to produce peroxynitrite (ONOO–), a molecule that acts as a strong oxidant and can damage many cellular structures and alter their function. •NO, nitric oxide; •OH, hydroxyl radical; H2O2, hydrogen peroxide; O2•–, superoxide anion; ONOO–, peroxynitrite.
![Schematic diagram of Coenzyme Q10 in the electron transport chain of the mitochondrial inner membrane. A) The structure of Coenzyme Q10, a lipid soluble component of the mitochondrial inner membrane. B) Mechanisms of oxidative stress involving mitochondria. The mitochondrial ETC reoxidizes reduced cofactors (NADH and FADH2) using molecular oxygen as the final electron acceptor, and the energy released in this process is captured in the form of ATP. The electron transport chain (ETC) depicting the position of Coenzyme Q10 (red) in the inner membrane. Coenzyme Q10 is critical for electron transport in the mitochondrial respiratory chain. The enzyme carries electrons from complexes I and II to complex III, thus participating in ATP production. I, II, III, IV, and V indicate protein components of the ETC. ‘e’ indicates electrons. and F1 are subunits of ATP synthase complex V (Complex V). ATP and ADP, adenosine triphosphate and diphosphate, respectively; CoQ 10, coenzyme Q10; FAD, flavin adenine dinucleotide; FADH, reduced flavin adenine dinucleotide. Image published with permission from Dove Medical Press, Rodick et al., Nutr Diet Supplem. 2017;10:1-11 [107]. C) Several components of the ETC chain (most often CI and CIII) generate O2•–. (left) The radical is converted into H2O2 by mitochondrial SOD. Through the Fenton reaction, H2O2 is converted into •OH, a molecule that produces oxidative cell injury through DNA damage, carboxylation of proteins, and lipid peroxidation. (right) •NO is produced by the activity of intracellular NOS. •NO can be combined with O2•– to produce peroxynitrite (ONOO–), a molecule that acts as a strong oxidant and can damage many cellular structures and alter their function. •NO, nitric oxide; •OH, hydroxyl radical; H2O2, hydrogen peroxide; O2•–, superoxide anion; ONOO–, peroxynitrite.](https://content.iospress.com:443/media/jhd/2022/11-1/jhd-11-1-jhd210510/jhd-11-jhd210510-g002.jpg)
The sources of the failures of CoQ10 in HD in clinical trials are unclear. The compound is well tolerated in humans, and often used as a general nutritional supplement and efforts to improve CoQ10 efficacy continue [36]. However, when administered as a dietary supplement, CoQ10 tends to be retained in cell membranes, is inefficient in entering the MT [37–39], and has low permeability across to the blood-brain barrier (BBB) [39–41]. Moreover, there is uncertainty as to whether plasma CoQ10 status, which is the result of both dietary intake and hepatic synthesis, reflects that of the brain [39–41]. The poor outcome of CoQ10 in humans is thought, at least in part, to occur due to feedback control, i.e., CoQ10 is the expressed product of an endogenous gene, and the cell compensates for a dietary increase by down-regulating CoQ10 synthesis [39, 41, 42]. Thus, even if plasma concentrations rise during treatment, it remains controversial whether dietary supplementation of CoQ10 will significantly enhance its steady-state level or reach a pharmacological effective in vivo concentration in the brain. Indeed, in vivo, the effects of CoQ10 treatment have been marginal, variable, or tissue specific [39, 42–46]. For example, MitoQ, a mitochondrial-targeted CoQ10 derivative, failed to offset neurodegeneration in PD clinical trials, but has shown unexpected promise in the periphery as a therapeutic in treating non-alcoholic fatty liver disease [47, 48].
A second consideration is the charge of CoQ10 and its derivatives, which can also diminish their efficacy. Loss of the charge gradient is a major feature of MT in dying neurons, yet mitochondrial entry of cationic antioxidants such as MitoQ requires the charge gradient [49]. Thus, uptake of these potential-driven antioxidants is self-limiting [50] as there is inevitable depolarization of MT with disease progression. Indeed, MitoQ and a mitochondrial targeted Vitamin E (MitoVitE) protect cultured fibroblasts from Friedrich’s ataxia (FRDA) patients; yet, their enhanced potency is abolished in cells pretreated with carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), an uncoupling agent that dissipates the mitochondrial membrane potential [51]. The diminution of efficacy in FRDA cells raises the issue as to whether MitoQ will have any benefit if treatment starts after disease onset. Treatment time is important, and many compounds have protective effects in offsetting toxicity in animals if treatment begins early. However, for most compounds, efficacy in counteracting toxicity after disease onset is typically untested but may be a better predictor of clinical success.
XJB-5-131: A PLATFORM TECHNOLOGY FOR DIRECT TARGETING OF MITOCHONDRIA
The failure of natural antioxidants to offset the feature of HD has prompted the search for unnatural (synthetic) analogs and new approaches to improve antioxidant specificity. Improvements have taken the form of increasing the steady state level of compounds, increasing the antioxidant metabolic stability or by modifying target specificity. In this regard, biological studies suggested that a synthetic antioxidant, XJB-5-131, was paradigm-shifting in that it provided a platform for targeting MT, leading to increased compound bioavailability and improved antioxidant efficacy (Fig. 3) [52–54]. XJB-5-131 is a bi-functional molecule comprising a peptide delivery component (Fig. 3A, black box), which directly targets the mitochondrial membrane and delivers an antioxidant nitroxide, 2,2,6,6-tetramethyl piperidine-1-oxyl (TEMPO) (Fig. 3A, red) to neutralize reactive species such as radicals, electrons, and oxidants [53, 54]. XJB-5-131 was bioinspired by the activity of naturally occurring antibiotic gramicidin S (GS) (Fig. 3B). The C2-symmetric GS (cyclo(Leu-D-Phe-Pro-Val-Orn)2) is well-known for its type II’ β-turn secondary structure backbone, which buries the polar amide functions in the interior space of the reverse turn, presents the lipophilic and charged side chains above and below the plane of the cyclopeptide backbone, and supports ambiphilic properties that convey a high affinity to microbial membranes.
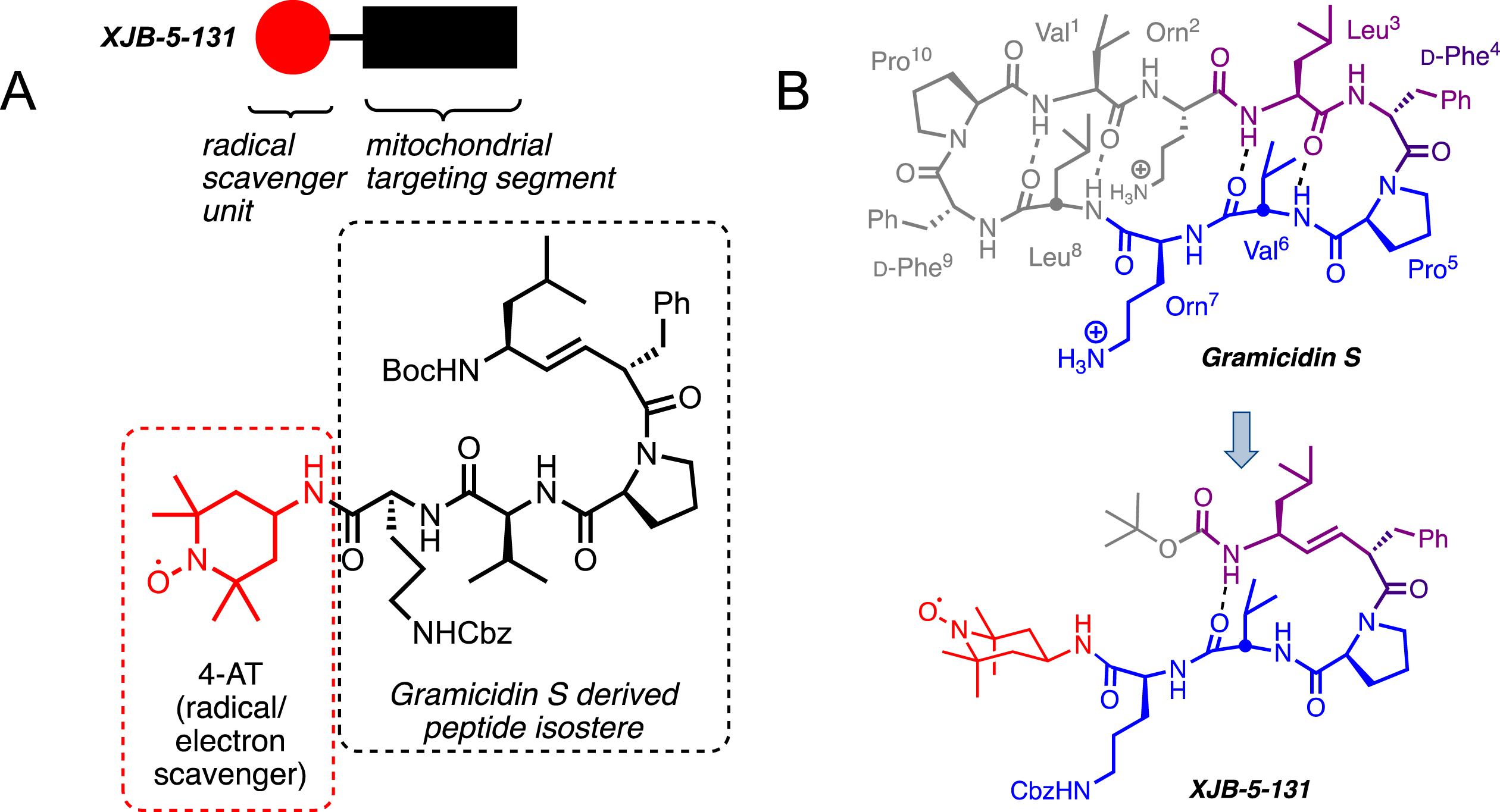
Fig. 3
A) The structure of XJB-5-131. XJB-5-131 is a bifunctional molecule with a peptide isostere for delivery (black box and structure in dashed box) of the antioxidant and a radical scavenger, 4-amino-TEMPO (red circle and structure in red dashed box). B) XJB-5-131 was “bioinspired” by the cyclopeptide antibiotic Gramicidin S (see blue and purple portions of Gramicidin S). Structure, sequence, and hairpin folding of Gramicidin S (GS; top) and the GS-analog XJB-5-131 (bottom). Regions of close structural analogy are highlighted in purple, identical sequence is marked in blue; unrelated moieties are grayed. The purple dipeptide segment has a regular amide bond in GS, which is replaced by an alkene peptide bond isostere in XJB-5-131. The red payload only present in XJB-5-131 is the ROS scavenging 4-amino-Tempo (4-AT) nitroxide. Boc, tert-butoxycarbonyl; Cbz, benzyloxycarbonyl.
While targeted antioxidants such as MitoQ are often cationic, XJB-5-131 has no charge at physiological pH, which enhances its permeability for lipid membrane entry. In contrast to GS, the δ-NH2 of Orn of XJB-5-131 is capped by a benzyl carbamate to avoid a positive charge that could lead to membrane disruption. These features of XJB-5-131’s targeting not only enhance penetration of the pentapeptide mimetic but also do so without destabilizing the mitochondrial membranes. XJB-5-131 crosses the BBB, and both electron paramagnetic resonance (EPR) studies and mass spectrometry confirmed a 600-fold accumulation of XJB-5-131 in MT [55] (Fig. 4A). XJB-5-131 targeting was tested in primary striatal neurons and synaptosomes [56, 57] from HdHQ(150/150) animal model for HD [56]. HdhQ(150/150) mice harbor a disease-length 150 CAG tract “knocked-into” both alleles of the mouse HD gene homologue [58]. Neurons isolated from these animals were treated with BODIPY-FL-XJB-5-131, a derivative labeled with a fluorescent (FL) boron-dipyrromethene (BODIPY) dye (Fig. 4B) [56]. Within one hour of incubation, BODIPY-FL-XJB-5-131 crosses the plasma membrane and stains MT, as verified by co-staining with MitoTracker Deep Red (Fig. 4C) [56]. Thus, XJB-5-131 has high affinity for its target, the MT.
Fig. 4
Distribution and mitochondrial accumulation of XJB-5-131 in vivo and in vitro. A) Design and results of in vivo EPR detection of XJB-5-131 distribution in rats. A) Top: Computed tomography (CT) scan of a rat and CT contour drawing; Bottom: EPR detection of XJB-5-131 nitroxide (green, center) indicating its distribution in the brain 5 min and 25 min after administration, together with two EPR standards at the tips of two cannulae (green, top left and right). Taken with permission from [55]. B) Schematic of XJB-5-131-BODIPY conjugate. The derivative is analogous to XJB-5-131 as shown in Fig. 3 but adds the BODIPY fluorescent marker (black dashed box). Taken with permission from [56]. C) Co-localization of XJB-5-131-BODIPY (right, blue) and MitoTracker (left, red) establishing the accumulation of XJB-5-131 in MT in a primary neuron from the striatum of an HdhQ(150/150) animal. Taken with permission from [56].
![Distribution and mitochondrial accumulation of XJB-5-131 in vivo and in vitro. A) Design and results of in vivo EPR detection of XJB-5-131 distribution in rats. A) Top: Computed tomography (CT) scan of a rat and CT contour drawing; Bottom: EPR detection of XJB-5-131 nitroxide (green, center) indicating its distribution in the brain 5 min and 25 min after administration, together with two EPR standards at the tips of two cannulae (green, top left and right). Taken with permission from [55]. B) Schematic of XJB-5-131-BODIPY conjugate. The derivative is analogous to XJB-5-131 as shown in Fig. 3 but adds the BODIPY fluorescent marker (black dashed box). Taken with permission from [56]. C) Co-localization of XJB-5-131-BODIPY (right, blue) and MitoTracker (left, red) establishing the accumulation of XJB-5-131 in MT in a primary neuron from the striatum of an HdhQ(150/150) animal. Taken with permission from [56].](https://content.iospress.com:443/media/jhd/2022/11-1/jhd-11-1-jhd210510/jhd-11-jhd210510-g004.jpg)
XJB-5-131 mechanisms of action
The centerpiece of the XJB-5-131 efficacy is targeting the active nitroxide moiety as cargo directly to MT (Fig. 3A and 4C). ROS are highly reactive, transient species but are rapidly converted to hydrogen peroxide by superoxide dismutase (SOD) under conditions of normal redox homeostasis [59–61] (Fig. 2C and Fig. 5A). The TEMPO moiety of XJB-5-131 regulates the amount of ROS by supporting redox homeostasis, and, similar to SOD, undergoes one- or two-electron transfer processes to hydroxylamines or oxoammonium cations, respectively [54, 59] (Fig. 5A,B). The nitroxide moiety in XJB-5-131 can serve as a superoxide dismutase-mimic and participates in a similar reduction-oxidation (redox) mechanism (Fig. 5B) [60, 61]. This electron scavenging activity prevents superoxide radical anion formation, neutralizes ROS when it is formed, and prevents the occurrence of organic peroxides and subsequent mitochondrial DNA (mtDNA) damage. Peroxides can also diffuse away from MT to other cell components. Mechanistically, to avoid the formation of superoxide radical anion, the redox cycling ability of XJB-5-131 prevents the leakage of electrons during the reduction of O2 to H2O at cytochrome c oxidase. Additionally, in the hydroxylamine state, XJB-5-131 can reduce a radical species by hydrogen atom donation to regenerate the nitroxide state [54, 59–61] (Fig. 5B). Thus, XJB-5-131 is both an electron scavenger and an antioxidant [54]. When located in the membrane, the nitroxide or hydroxylamine moiety returns the electrons to the ETC after the reduction of water, and therefore XJB-5-131 can exert antioxidant properties without significantly diminishing ATP production (Fig. 2) [54].
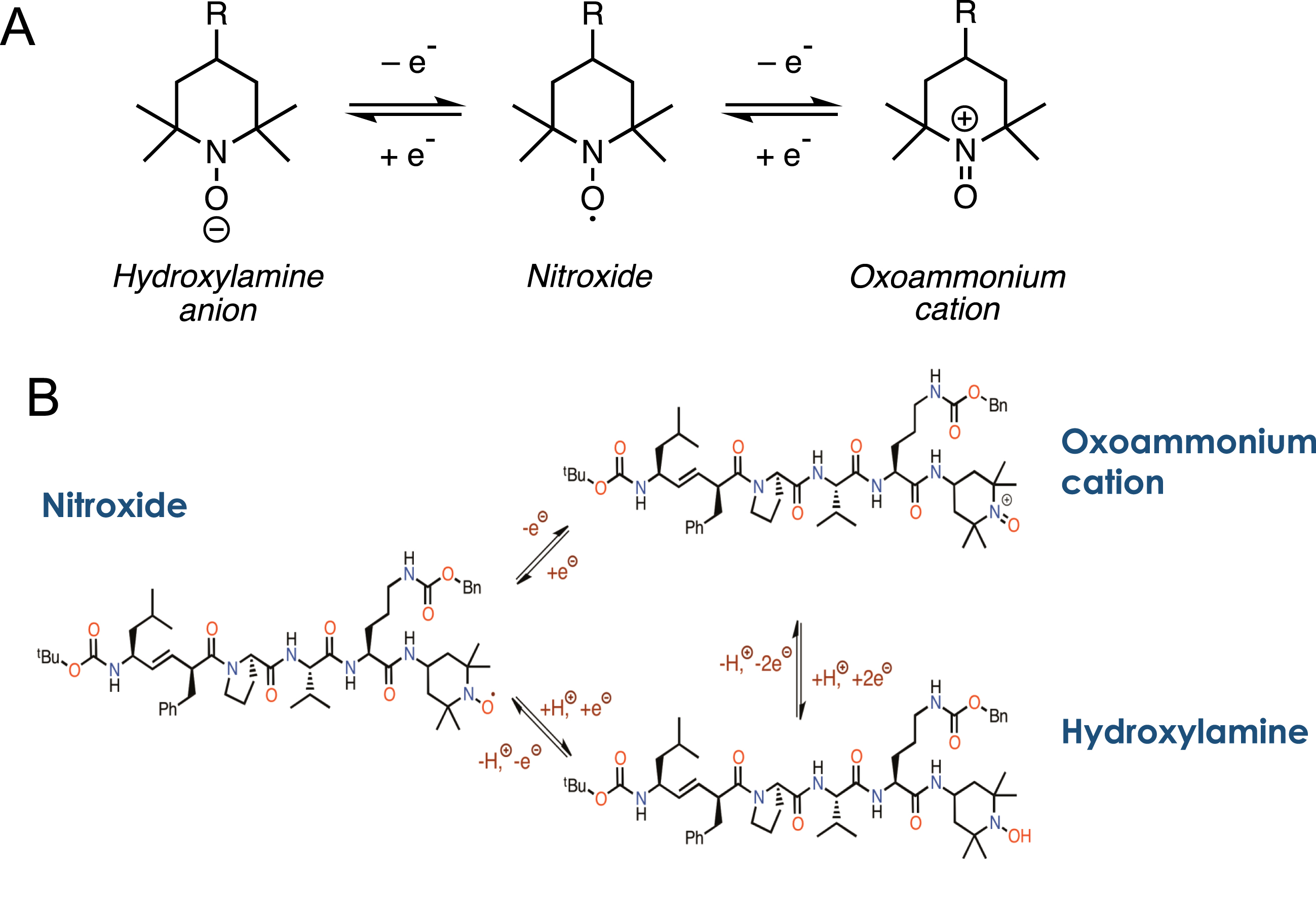
Fig. 5
Schematic of Redox chemistry in XJB-5-131. A) The basic chemistry of a hydroxylamine-nitroxide-oxoammonium redox cycle as described in the text. B) The structure of the three redox forms of XJB-5-131.
XJB-5-131 rescues features of toxicity in HD models for disease
The antioxidant properties of XJB-5-131 as therapeutic for HD were tested initially in high quality preparations of synaptosomes [56] from the HdHQ(150/150) mice. Synaptosomes are “pinched off” nerve terminals that are generated during brain homogenization [57]. They harbor intact MT and represent a simple and robust system to test mitochondrial function within a physiological milieu [56, 57]. In synaptosomes from untreated HdHQ(150/150) animals, oxidation of mtDNA was elevated and mtDNA copy number was diminished in aging animals [56]. However, treatment with XJB-5-131 restored the copy number of MT genomes [56]. XJB-5-131 also improved survival of cultured control neurons, suggesting the favorable effects of the compound were not limited to a specific HD pathology trigger for mitochondrial dysfunction [56]. These findings imply that the beneficial effects of specific targeting of this synthetic antioxidant to MT in vitro and in vivo arose from its function as an antioxidant and regulator of redox homeostasis. While XJB-5-131 has minimal effects on mitochondrial metabolism in the resting state, XJB-5-131 neutralizes the rise in ROS under stress and sustains the bioenergetics of MT in HD animals.
The antioxidant properties of XJB-5-131 may also reduce aggregation, which has relevance to both HD and AD. For AD, there is great interest in the pharmaceutical industry to identify molecules that can degrade (NPT002 from Neurophage) or prevent proteolytic formation (various BACE inhibitors) of amyloid-β plaques [62]. Oxidative damage to protein and to membranes of MT lowers the rates of clearance and enhances the gradual accumulation of toxic partially degraded peptides (Fig. 1). A mitochondrial-targeted antioxidant with electron scavenging properties is predicted to decrease aggregation, to reduce the level of small, toxic peptides, and to enhance their clearance. In AD the accumulation of oxidized lipid species is observed in plaques, which are hallmarks of the disease [1]. Similarly, the elevated oxidized lipid species, which are observed in HD animals [20, 63, 64], are likely to contribute to inclusion formation. Indeed, XJB-5-131 is effective in reversing the accumulation of oxidized lipids in older HdHQ(150/150) animals concomitantly with rescue of pathology [20].
Efficacy of XJB-5-131 as a suppressor of toxicity in HD models
Due to its strong electron scavenging and antioxidant properties, XJB-5-131 has been tested in several diseases or adverse conditions where elevated ROS is thought to contribute significantly to pathology [20, 65–71]. The most detailed analysis of XJB-5-131 was performed using late and early onset mouse models for HD [20, 56, 65]. To test efficacy in vivo, HdHh(150/150) mice were intraperitoneally injected with XJB-5-131 at 1 mg/kg dosing three times a week for up to 57 weeks, and tested for improvement of disease phenotypes. XJB-5-131 had striking in vivo effects [20, 56, 65]. Chronic treatment of HdhQ(150/150) mice with XJB-5-131 suppressed weight loss, a feature that is commonly observed in HD patients [1, 65]. Genetically matched BL6 control animals increased in weight to an average of 44±5 g at 52 weeks. Age and gender-matched HdhQ(150/150) mouse (32±3 g) were smaller than control mice; treatment of HdhQ(150/150) littermates with XJB-5-131 suppressed weight loss and resulted in an 18% increase in the average body mass. Early signs of HD pathophysiology manifest as motor abnormalities [56, 65]. The effect of XJB-5-131’s efficacy on motor function has also been evaluated [56, 65]. Standard tests of rotarod performance and grip strength were measured at 9, 28, and 57 weeks of XJB-5-131 treatment. All animals at 9 weeks displayed robust and equivalent performance on the rotarod [56]. However, by 28 weeks, motor function in HdhQ(150/150) mice declined, and was substantially diminished by 57 weeks. Remarkably, little decline in rotarod performance was observed between 9 and 57 weeks [56] in XJB-5-131-treated HdhQ(150/150) littermates, whose performance was similar to WT animals.
Long term treatment of HD mice administered by intraperitoneal (IP) administration with 1 mg/kg for 1 year, and 2 mg/kg XJB-5-131 for 2.5 years had no adverse effects in controls at these concentrations [20, 56, 65]. Thus, the phenotypic improvement depended on the compound treatment. Beneficial effects were not limited to HD models. XJB-5-131 treatment improved mitigated lethal hemorrhagic shock in a rat model [69], resulted in a reduction in oxidative damage, decreased radiation damage [68], reduced the ischemia-reperfusion injury during brain trauma [55], and offset aging-related intervertebral disc degeneration [70], among others [66]. An important finding in HD animals was that XJB-5-131 treatment was not only effective in blocking the progression of neurodegenerative disease if treatment was early but was also effective in blocking disease progression if treatment began after disease onset [65]. A derivative of XJB-5-131 with enhanced metabolic stability in humans would be a promising candidate for clinical testing. XJB-5-131 also improved pathological features of HD in R6/2 mice [71], a severe, early onset model of HD. Chronic treatment of R6/2 mice with XJB-5-1-131 reduced weight loss and improved the motor functions, especially in male mice [71]. Low doses of XJB-5-131 that were used in the study had no effect on the lifespan of R6/2 mice but the analysis has left open the possibility that higher doses might do so [71].
XJB-5-131, an efficient inhibitor of somatic mutation in HD animal models
The importance of oxidative DNA damage in promoting trinucleotide expansion led to the testing of XJB-5-131 as a suppressor of expansion in mice [67, 71]. Indeed, XJB-5-131 effectively suppressed expansion [67, 71]. To resolve the mechanism, we proposed over fifteen years ago that base excision repair (BER) and mismatch repair (MMR) cooperated in a “toxic oxidation cycle” to cause somatic mutation (Fig. 6A) [72]. The cycle was proposed based on wide-spread findings that loss of components of the MMR machinery (reviewed recently, [73]), MSH2 and MSH3 [74–82], PMS2, MulLα MutLγ) [83–89] and loss of the BER machinery (8-oxo-guanine glycosylase (OGG1) [72], NEILS 1 [90], and Polymerase β [91]) suppressed CAG expansion. Conversely, interaction of MSH2-MSH3 and Polβ promoted expansion [92–94]. The involvement of the MMR and BER pathways was consistent with genome wide association studies in HD patients, which confirmed that components of the MMR machinery and the factors regulating the metabolism of MT influenced onset of HD pathology in humans [95–98]. Thus, the toxic oxidation model provided a plausible mechanism for expansion that integrated both repair pathways in causing the mutation.
Fig. 6
A) The step schematic of the toxic-oxidation cycle, image modified with permission from [72], as described in the text. B) XJB-5-131 blocks somatic expansion in R6/2 animals as they age [71]. Blue is the number of repeats in the CAG tract of the tail DNA at birth, while the red is the number of repeats in the repeat tract at 8 or 12 weeks in brain tissue from the striatum. Vehicle treated R6/2 animals expand their repeats tract over time, while somatic expansion is suppressed in XJB-5-131-treated animals. The repeats tracts are illustrated by Genescan plots, which display the size distribution of the most prevalent repeat tract lengths as peaks in the distribution. The results are taken from Polyzos et al., PLoS One. 2018;13(4):e0194580 [71], which is freely available under the Creative Commons CC0 public domain dedication.
![A) The step schematic of the toxic-oxidation cycle, image modified with permission from [72], as described in the text. B) XJB-5-131 blocks somatic expansion in R6/2 animals as they age [71]. Blue is the number of repeats in the CAG tract of the tail DNA at birth, while the red is the number of repeats in the repeat tract at 8 or 12 weeks in brain tissue from the striatum. Vehicle treated R6/2 animals expand their repeats tract over time, while somatic expansion is suppressed in XJB-5-131-treated animals. The repeats tracts are illustrated by Genescan plots, which display the size distribution of the most prevalent repeat tract lengths as peaks in the distribution. The results are taken from Polyzos et al., PLoS One. 2018;13(4):e0194580 [71], which is freely available under the Creative Commons CC0 public domain dedication.](https://content.iospress.com:443/media/jhd/2022/11-1/jhd-11-1-jhd210510/jhd-11-jhd210510-g006.jpg)
The most common oxidative damage product in DNA is 8-oxoguanine. In the toxic oxidation cycle, the damaged base is enzymatically removed by OGG1, leaving an abasic site with an unpaired cytosine (Fig. 6A). This site is nicked by apoendonuclease I (APE1), leaving a transient single strand break (SSB) as an intermediate and a 3’-OH end for extension by gap-filling polymerases. MSH2-MSH3 stimulates polymerase β to traverse the repeat tract and complete gap filling synthesis [92]. In the process, the MSH2-MSH3-polβ complex displaces the CAG flap [92, 93], which folds back into a stable hairpin. MSH2-MSH3 binds to the mismatched hairpin stem at the end of gap filling synthesis and facilitates ligation [94]. An “in trans” endonuclease clips across from the hairpin strand. Extrusion of the DNA hairpin loop and its incorporation into the duplex DNA results in expansion (Fig. 6A). Loss of MutL homologues 1 (MLH1) and MLH3 [85–89], which are partners with MSH2-MSH3 in MMR, stops expansion in mice, suggesting that MLH-related endonucleases constitute at least one of the “in trans” endonucleases that process the loop. Since repair of the oxygen modification constituted at least one important pathway for initiating the somatic expansion, it made sense that XJB-5-131, if it reduced ROS, should also block expansion. Indeed, in R6/2 animals, treatment with XJB-5-131 attenuated somatic expansion at 12 weeks of treatment and reduced the density of inclusions (Fig. 6B) [71]. XJB-5-131 also suppressed the expansion mutation in HdhQ(150/150) animals compared to WT animals at older ages [67]. The ability for XJB-5-131 to reduce expansion is perhaps the most direct evidence that expansion arises from oxidative DNA damage and that targeted neutralization of oxidation in MT can offset HD onset and progression. (Some models for somatic expansion have been recently reviewed in a special issue of the Journal of Huntington’s Disease, 2021). Toxicity and expansion are directly related by oxidation and form a feedback loop in which the two processes exacerbate each other. By reducing reactive oxygen species, XJB-5-131 breaks the cycle between the rise in oxidative damage during disease progression and the somatic growth of the CAG repeat, which is a response to the oxidation.
SUMMARY: THE BENEFICIAL PROPERTIES OF XJB-5-131
In conclusion, the use of XJB-5-131 in neurodegenerative pathologies has three main advantages that overcome at least some potential sources of past clinical failures of antioxidant therapies (Fig. 7). Since XJB-5-131 is a synthetic antioxidant, it is not modulated by gene expression and is capable of achieving and maintaining pharmacologically effective concentrations in cells. XJB-5-131 crosses the BBB and is readily visualized in the brains of rats by in vivo EPR imaging [55]. Furthermore, XJB-5-131 is charge neutral and does not influence the mitochondrial membrane potential nor does it depend on the electrostatic gradient to enter MT (Fig. 7). These properties of XJB-5-131 provide a mechanism for improving mitochondrial function even after membrane depolarization. Mitochondrial targeting delivers the antioxidant directly to the specific site needed for neutralization, increases the lifetime of the antioxidant, and protects the mtDNA, membranes and proteins against damage (Fig. 7). The use of synthetic derivatives avoids the inevitable downregulation of endogenous natural antioxidants production that limits the efficacy of vitamin Q10 supplements, for example. Taken together, direct targeting of the synthetic antioxidant to MT is poised to provide a level of specificity and efficacy that enables a viable therapeutic strategy. Because mitochondrial decline is a common and central feature of toxicity in the brain, targeted nitroxides such as XJB-5-131 are expected to be useful in a wide spectrum of neurodegenerative diseases, or in other deleterious ROS-induced conditions and mitochondrial malfunctions. Indeed, recent studies have also established XJB-5-131’s efficacy in renal injury [99], progressive optic neuropathy [100] and in worm models for PD [101]. Collectively, XJB-5-131 and its derivatives are promising clinical candidates, and this class of compounds warrants further investigation for their efficacy.
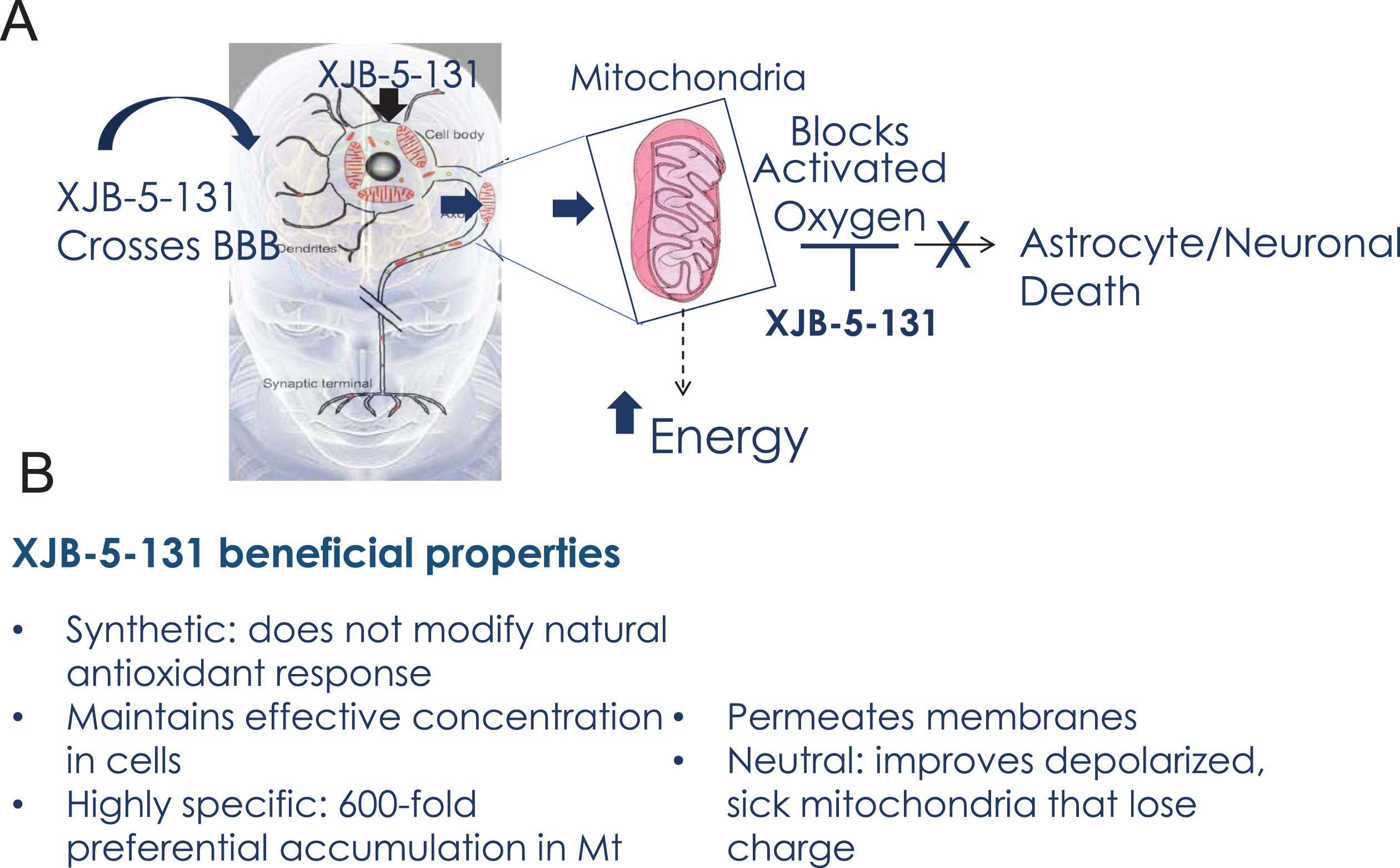
Fig. 7
Beneficial properties of XJB-5-131. A) Schematic diagram of XJB-5-131 entry into brain MT and the mechanism of neuronal protection. B) Summary of the beneficial properties of XJB-5-131.
XJB-5-131 may be effective alone or as an adjuvant. Indeed, several hybrid compounds of tacrine and structural moieties derived from natural sources such as flavonoids [quercetin, rutin, coumarin, gallamine, resveratrol, scutellarin, anisidine, hesperetin, (-)-epicatechin], melatonin, and trolox have also been applied as multitarget-directed ligands that reduce oxidation in cells [102–106]. Co-treatment with XJB-5-131 might improve efficacy. By either approach (alone or as an adjuvant), the properties of synthetic XJB-5-131 show considerable promise and overcome at least some sources of past failures.
LIABILITIES AND FUNCTIONAL IMPROVEMENTS
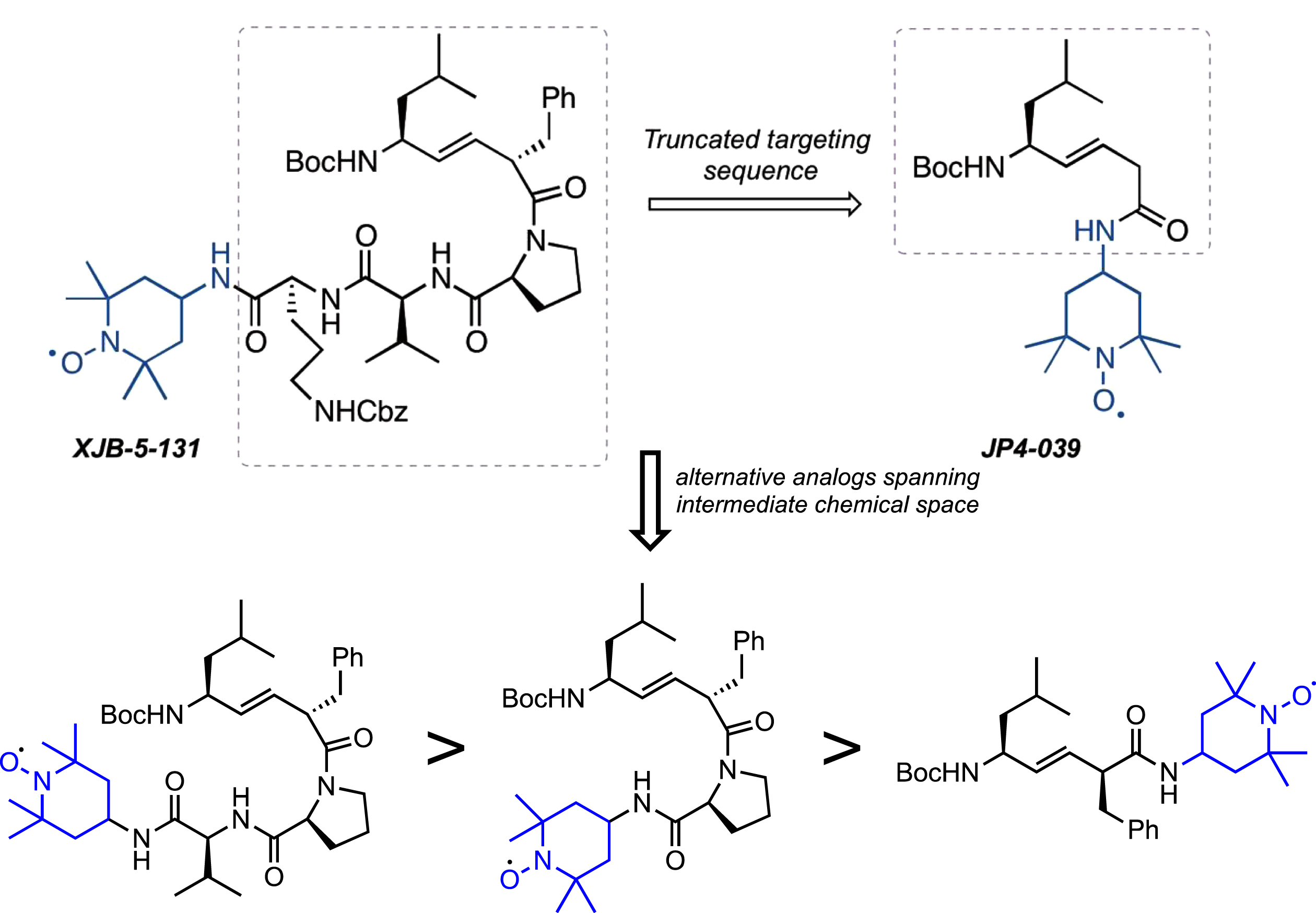
The first-generation molecule, XJB-5-131, is enriched in mitochondria 600-fold over the cytosol [55]. However, the compound has a high molecular weight, and low water solubility [53, 54]. Although it is typically a detriment for small molecules, in the case of XJB-5-131, the low solubility appears to play a beneficial role in that it drives the compounds from the cytosol into the MT, preventing rapid clearance at its target. However, the cost of this is that animals must be treated continuously with the drug to achieve pharmacological activity (by intraperitoneal injection). Additionally, the synthesis of XJB-5-131 requires a minimum of 17 steps, which is challenging for scale-up [54]. A simplified second-generation analog, JP4-039, has substantially improved the physical properties of XJB-5-131 [66] (Fig. 8). This molecule benefits from lower MW, and higher solubility, and an easily scalable synthesis. In preliminary testing, JP4-039 has attractive biological properties while maintaining significant mitochondrial localization and ROS-scavenging abilities. The structural complexity from XJB-5-131 to JP4-039 retains a significant amount of structural diversity and provides structural variations and conformations that are yet to be explored. While it is realistic to anticipate that future analogs of XJB-5-131 are likely to have even better properties, clinical development would require a complete set of preclinical studies in multiple animal models to establish merit.
Fig. 8
Analogs of XJB-5-131. Upper: Schematic diagram of JP4-039 and alternative analogs. The tripeptide moiety in XJB-5-131 is deleted in the truncated JP4-039, which only retains the dipeptide alkene isostere and nitroxide components. Lower: Structures of 3 hypothetical XJB-5-131analogs with decreasing structural complexity from left to right and increasing similarity to JP4-039.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants NS060115 (to CTM) and GM119161 (to CTM). HDTRA1-16-1-0041 and U19-AI1068021 (to PW).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Alzheimer’s disease facts and figures. Alzheimers Dement (2021) ;17: (3):327–406. |
[2] | Marras C , Beck JC , Bower JH , Roberts E , Ritz B , Ross GW , et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. (2018) ;4: :21–8. |
[3] | McColgan P , Tabrizi SJ . Huntington’s disease: A clinical review. Eur J Neurol. (2017) ;25: (1):24–34. |
[4] | Grady PA . Advancing the health of our aging population: A lead role for nursing science. Nurs Outlook. (2011) ;59: (4):207–9. |
[5] | Yang P , Sun F . Aducanumab: The first targeted Alzheimer’s therapy. Drug Discov Ther. (2021) ;15: (3):166–8. |
[6] | Kingwell K . Double setback for ASO trials in Huntington disease. Nat Rev Drug Discov. (2021) ;20: (6):412–3. |
[7] | KwonD . Failure of genetic therapies for Huntington’s devastates community. Nature. (2021) ;593: (7858):180. |
[8] | Nakamori M , Mochizuki H . Targeting expanded repeats by small molecules in repeat expansion disorders. Mov Disord. (2020) ;36: (2):298–305. |
[9] | Oliver DMA , Reddy PH . Small molecules as therapeutic drugs for Alzheimer’s disease. Mol Cell Neurosci. (2019) ;96: :47–62. |
[10] | Wiggins R , Feigin A . Emerging therapeutics in Huntington’s disease. Exp Opin Emerging Drugs. (2021) ;26: (3):295–302. |
[11] | Chandran R , Mehta SL , Vemuganti R . Antioxidant combo therapy protects white matter after traumatic brain injury. Neuromol Med. (2021) ;23: (3):344–7. |
[12] | Konno T , Melo EP , Chambers JE , Avezov E . Intracellular sources of ROS/H(2)O(2) in health and neurodegeneration: Spotlight on endoplasmic reticulum. Cells. (2021) ;10: (2):233. |
[13] | Tauffenberger A , Magistretti PJ . Reactive oxygen species: Beyond their reactive behavior. Neurochem Res. (2021) ;46: (1):77–87. |
[14] | Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology. (2001) ;57: (3):397–404. |
[15] | NINDS NET-PD Investigators. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. (2007) ;68: (1):20–8. |
[16] | McGarry A , McDermott M , Kieburtz K , de Blieck EA , Beal F , Marder K , et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology. (2017) ;88: (2):152–9. |
[17] | Parkinson Study Group QE3 Investigators, Beal MF , Oakes D , Shoulson I , Henchcliffe C , Galpern WR , et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. (2014) ;71: (5):543–52. |
[18] | Costas C , Faro LRF . Do naturally occurring antioxidants protect against neurodegeneration of the dopaminergic system? A systematic revision in animal models of Parkinson’s disease. Curr Neuropharmacol. 2021; doi: 10.2174/1570159X19666210421092725 |
[19] | Gu Y , Cheng X , Huang X , Yuan Y , Qin S , Tan Z , et al. Conditional ablation of reactive astrocytes to dissect their roles in spinal cord injury and repair. Brain Behav Immun. (2019) ;80: :394–405. |
[20] | Polyzos AA , Lee DY , Datta R , Hauser M , Budworth H , Holt A , et al. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in Huntington mice. Cell Metab. (1258) ;29: (6):1258–73.e11. |
[21] | Weydt P , Pineda VV , Torrence AE , Libby RT , Satterfield TF , Lazarowski Eduardo R , et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. (2006) ;4: (5):349–62. |
[22] | Assefa BT , Gebre AK , Altaye BM . Reactive astrocytes as drug target in Alzheimer’s disease. Biomed Res Int. 2018: :4160247. |
[23] | Polyzos AA , McMurray CT . The chicken or the egg: Mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech Age Dev. (2017) ;161: (Pt A):181–97. |
[24] | Hickey MA , Zhu C , Medvedeva V , Franich NR , Levine MS , Chesselet M-F . Evidence for behavioral benefits of early dietary supplementation with CoEnzymeQ10 in a slowly progressing mouse model of Huntington’s disease. Mol Cell Neurosci. (2012) ;49: (2):149–57. |
[25] | Menalled LB , Patry M , Ragland N , Lowden PAS , Goodman J , Minnich J , et al. Comprehensive behavioral testing in the R6/2 mouse model of Huntington’s disease shows no benefit from CoQ10 or minocycline. PLoS One. (2010) ;5: (3)::e9793–e. |
[26] | Schilling G , Coonfield ML , Ross CA , Borchelt DR . Coenzyme Q10 andremacemide hydrochloride ameliorate motor deficits in a Huntington’sdisease transgenic mouse model. Neurosci Lett. (2001) ;315: (3):149–53. |
[27] | Yang L , Calingasan NY , Wille EJ , Cormier K , Smith K , Ferrante RJ , et al. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J Neurochem. (2009) ;109: (5):1427–39. |
[28] | Aaseth J , Alexander J , Alehagen U . Coenzyme Q10 supplementation –In ageing and disease. Mech Age Dev. (2021) ;197: :111521. |
[29] | Fernández-Del-Río L , Rodríguez-López S , Gutiérrez-Casado E , González-Reyes JA , Clarke CF , Burón MI , et al. Regulation of hepatic coenzyme Q biosynthesis by dietaryomega-3 polyunsaturated fatty acids. Redox Biol. (2021) ;46: :102061. |
[30] | Gueguen N , Baris O , Lenaers G , Reynier P , Spinazzi M . Secondary coenzyme Q deficiency in neurological disorders. Free Radical Biol Med. (2021) ;165: :203–18. |
[31] | Rabanal-Ruiz Y , Llanos-González E , Alcain FJ . The use ofcoenzyme Q10 in cardiovascular diseases. Antioxidants. (2021) ;10: (5):755–74. |
[32] | Emmanuele V , López LC , Berardo A , Naini A , Tadesse S , Wen B , et al. Heterogeneity of coenzyme Q10 deficiency: Patient study and literature review. Arch Neurol. (2012) ;69: (8):978–83. |
[33] | Hargreaves I , Heaton RA , Mantle D . Disorders of human coenzyme Q10 metabolism: An overview. Int J Mol Sci. (2020) ;21: (18):6695–708. |
[34] | Manzar H , Abdulhussein D , Yap TE , Cordeiro MF . Cellular consequences of coenzyme Q10 deficiency in neurodegeneration of the retina and brain. Int J Mol Sci. (2020) ;21: (23):9299. |
[35] | Spindler M , Beal MF , Henchcliffe C . Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr Dis Treat. (2009) ;5: :597–610. |
[36] | Arenas-Jal M , Suñé-Negre JM , García-Montoya E . CoenzymeQ10 supplementation: Efficacy, safety, and formulation challenges. Comp Rev Food Sci Food Saf. (2020) ;19: (2):574–94. |
[37] | Barcelos IPd , Haas RH . CoQ10 and aging. Biology. (2019) ;8: (2):28–50. |
[38] | Montero R , Yubero D , Salgado MC , González MJ , Campistol J , O’Callaghan MDM , et al. Plasma coenzyme Q(10) status is impaired inselected genetic conditions. Sci Rep. (2019) ;9: (1):793–800. |
[39] | Turton N , Bowers N , Khajeh S , Hargreaves IP , Heaton RA . Coenzyme Q10 and the exclusive club of diseases that show a limited response to treatment. Exp Opin Orphan Drugs. (2021) ;9: (5):151–60. |
[40] | Niklowitz P , Sonnenschein A , Janetzky B , Andler W , Menke T . Enrichment of coenzyme Q10 in plasma and blood cells: Defense against oxidative damage. Int J Biol Sci. (2007) ;3: (4):257–62. |
[41] | Wainwright L , Hargreaves IP , Georgian AR , Turner C , Dalton RN , Abbott NJ , et al. CoQ(10) Deficient endothelial cell culture model for the investigation of CoQ(10) blood-brain barrier transport. J Clin Med. (2020) ;9: (10):3236–57. |
[42] | Kwong LK , Kamzalov S , Rebrin I , Bayne A-CV , Jana CK , Morris P , et al. Effects of coenzyme Q10 administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic Biol Med. (2002) ;33: (5):627–38. |
[43] | Orsucci D , Mancuso M , Ienco EC , LoGerfo A , Siciliano G . Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr Med Chem. (2011) ;18: (26):4053–64. |
[44] | Palamakula A , Khan MA . Evaluation of cytotoxicity of oils used in coenzyme Q10 Self-Emulsifying Drug Delivery Systems (SEDDS). Int J Pharm. (2004) ;273: (1-2):63–73. |
[45] | Sohal RS , Forster MJ . Coenzyme Q, oxidative stress and aging. Mitochondrion. (2007) ;7: (Suppl):S103–11. |
[46] | Sumien N , Heinrich KR , Shetty RA , Sohal RS , Forster MJ . Prolonged intake of coenzyme Q10 impairs cognitive functions in mice. J Nutr. (2009) ;139: (10):1926–32. |
[47] | Curcio A , Romano A , Cuozzo S , Nicola AD , Grassi O , Schiaroli D , et al. Silymarin in combination with vitamin C, vitamin E, coenzyme Q10 and selenomethionine to improve liver enzymes and blood lipid profile in NAFLD patients. Medicina. (2020) ;56: (10):544–52. |
[48] | Farhangi MA , Alipour B , Jafarvand E , Khoshbaten M . Oral coenzyme Q10 supplementation in patients with nonalcoholic fatty liver disease: Effects on serum vaspin, chemerin, pentraxin 3, insulin resistance and oxidative stress. Arch Med Res. (2014) ;45: (7):589–95. |
[49] | Oyewole AO , Birch-Machin MA . Mitochondria-targeted antioxidants. FASEB J. (2015) ;29: (12):4766–71. |
[50] | Zhang Y , Aberg F , Appelkvist EL , Dallner G , Ernster L . Uptake of dietary coenzyme Q supplement is limited in rats. J Nutr. (1995) ;125: (3):446–53. |
[51] | Jauslin ML , Meier T , Smith RAJ , Murphy PM . Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. (2003) ;17: (13):1–10. |
[52] | Fink MP , Macias CA , Xiao J , Tyurina YY , Jiang J , Belikova N , et al. Hemigramicidin–TEMPO conjugates: Novel mitochondria-targeted anti-oxidants. Biochem Pharmacol. (2007) ;74: (6):801–9. |
[53] | Hoye AT , Davoren JE , Wipf P , Fink MP , Kagan VE . Targeting mitochondria. Acc Chem Res. (2008) ;41: (1):87–97. |
[54] | Wipf P , Xiao J , Jiang J , Belikova NA , Tyurin VA , Fink MP , et al. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin–TEMPO conjugates. J Amer Chem Soc. (2005) ;127: (36):12460–71. |
[55] | Ji J , Kline AE , Amoscato A , Samhan-Arias AK , Sparvero LJ , Tyurin VA , et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. (2012) ;15: (10):1407–13. |
[56] | Xun Z , Rivera-Sánchez S , Ayala-Peña S , Lim J , Budworth H , Skoda EM , et al. Targeting of XJB-5-131 to mitochondria suppressesoxidative DNA damage and motor decline in a mouse model ofHuntington’s disease. Cell Rep. (2012) ;2: (5):1137–42. |
[57] | Choi SW , Gerencser AA , Nicholls DG . Bioenergetic analysis ofisolated cerebrocortical nerve terminals on a microgram scale: Sparerespiratory capacity and stochastic mitochondrial failure. J Neurochem. (2009) ;109: (4):1179–91. |
[58] | Lin CH . Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. (2001) ;10: (2):137–44. |
[59] | Sheng Y , Abreu IA , Cabelli DE , Maroney MJ , Miller A-F , Teixeira M , et al. Superoxide dismutases and superoxide reductases. Chem Rev. (2014) ;114: (7):3854–918. |
[60] | Luo W , Muller JG , Rachlin EM , Burrows CJ . Characterization of hydantoin products from one-electron oxidation of 8-Oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. (2001) ;14: (7):927–38. |
[61] | Parry JD , Pointon AV , Lutz U , Teichert F , Charlwood JK , Chan PH , et al. Pivotal role for two electron reduction in 2,3-Dimethoxy-1,4-naphthoquinone and 2-Methyl-1,4-naphthoquinone metabolism and kinetics in vivo that prevents liver redox stress. Chem Res Toxicology. (2009) ;22: (4):717–25. |
[62] | Cantuti-Castelvetri I , Gannon K , Veinbergs I , Rassoulpour A , Gannon K , Rockenstein E , et al. P1- NPT002 reduces beta-amyloidaggregates and improves cognitive performance in aged TGmice. Alzheimers Dement.P. (2012) ;8: (4S_Part_6):226. |
[63] | Carroll JB , Deik A , Fossale E , Weston RM , Guide JR , Arjomand J , et al. HdhQ111 Mice exhibit tissue specific metabolite profiles that include striatal lipid accumulation. PLoS One. (2015) ;10: (8):e0134465. |
[64] | Zheng S , Clabough EBD , Sarkar S , Futter M , Rubinsztein DC , Zeitlin SO . Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. (2010) ;6: (2):e1000838. |
[65] | Polyzos A , Holt A , Brown C , Cosme C , Wipf P , Gomez-Marin A , et al. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum Mol Genet. (2016) ;25: (9):1792–802. |
[66] | Bernard ME , Kim H , Berhane H , Epperly MW , Franicola D , Zhang X , et al. GS-nitroxide (JP4-039)-mediated radioprotection of human fanconi anemia cell lines. Radiat Res. (2011) ;176: (5):603–12. |
[67] | Budworth H , Harris FR , Williams P , Lee DY , Holt A , Pahnke J , et al. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. (2015) ;11: (8):e1005267. |
[68] | Gokhale A , Rwigema JC , Epperly MW , Glowacki J , Wang H , Wipf P , et al. Small molecule GS-nitroxide ameliorates ionizing irradiation-induced delay in bone wound healing in a novel murine model. In Vivo. (2010) ;24: (4):377–85. |
[69] | Macias CA , Chiao JW , Xiao J , Arora DS , Tyurina YY , Delude RL , et al. Treatment with a novel hemigramicidin-TEMPO conjugate prolongs survival in a rat model of lethal hemorrhagic shock. Ann Surg. (2007) ;245: (2):305–14. |
[70] | Nasto LA , Robinson AR , Ngo K , Clauson CL , Dong Q , St Croix C , et al. Mitochondrial-derived reactive oxygen species (ROS) play a causal role in aging-related intervertebral disc degeneration. J Orthop Res. (2013) ;31: (7):1150–7. |
[71] | Polyzos AA , Wood NI , Williams P , Wipf P , Morton AJ , McMurray CT . XJB-5-131-mediated improvement in physiology and behaviour of the R6/2 mouse model of Huntington’s disease is age- and sex- dependent. PLoS One. (2018) ;13: (4):e0194580. |
[72] | Kovtun IV , Liu Y , Bjoras M , Klungland A , Wilson SH , McMurray CT . OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. (2007) ;447: (7143):447–52. |
[73] | Iyer RR , Pluciennik A . DNA mismatch repair and its role in Huntington’s disease. J Huntingtons Dis. (2021) ;10: (1):75–94. |
[74] | Dragileva E , Hendricks A , Teed A , Gillis T , Lopez ET , Friedberg EC , et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. (2009) ;33: (1):37–47. |
[75] | Gomes-Pereira M . Pms2 is a genetic enhancer of trinucleotide CAG. CTG repeat somatic mosaicism: Implications for the mechanism of triplet repeat expansion. Hum Mol Genet. (2004) ;13: (16):1815–25. |
[76] | Lokanga RA , Zhao X-N , Usdin K . The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum Mutat. (2014) ;35: (1):129–36. |
[77] | Manley K , Shirley TL , Flaherty L , Messer A . Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. (1999) ;23: (4):471–3. |
[78] | Owen BAL , Yang Z , Lai M , Gajek M , Badger JD , Hayes JJ , et al. (CAG)n-hairpin DNA binds to Msh2–Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. (2005) ;12: (8):663–70. |
[79] | Savouret C , Brisson E , Essers J , Kanaar R , Pastink A , te Riele H , et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. (2003) ;22: (9):2264–73. |
[80] | Tomé S , Manley K , Simard JP , Clark GW , Slean MM , Swami M , et al. MSH3 polymorphisms and protein levels affect CAG repeat instabilityin Huntington’s disease mice. PLoS Genet. (2013) ;9: (2):e1003280. |
[81] | van den Broek WJAA . Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. (2002) ;11: (2):191–8. |
[82] | Wheeler VC . Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. (2000) ;9: (4):503–13. |
[83] | Bourn RL , De Biase I , Pinto RM , Sandi C , Al-Mahdawi S , Pook MA , et al. Pms2 suppresses large expansions of the (GAA·TTC)n sequence in neuronal tissues. PLoS One. (2012) ;7: (10):e47085. |
[84] | Ezzatizadeh V , Sandi C , Sandi M , Anjomani-Virmouni S , Al-Mahdawi S , Pook MA . MutLα heterodimers modify the molecular phenotype of Friedreich ataxia. PLoS One. (2014) ;9: (6):e100523. |
[85] | Kadyrova LY , Gujar V , Burdett V , Modrich PL , Kadyrov FA . Human MutLγ, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc Natl Acad Sci U S A. (2020) ;117: (7):3535–42. |
[86] | Pinto RM , Dragileva E , Kirby A , Lloret A , Lopez E , St Claire J , et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. (2013) ;9: (10):e1003930. |
[87] | Pluciennik A , Burdett V , Baitinger C , Iyer RR , Shi K , Modrich P . Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLα endonuclease activation. Proc Natl Acad Sci U S A. (2013) ;110: (30):12277–82. |
[88] | Roy JCL , Vitalo A , Andrew MA , Mota-Silva E , Kovalenko M , Burch Z , et al. Somatic CAG expansion in Huntington’s disease is dependent on the MLH3 endonuclease domain, which can be excluded via splice redirection. Nucleic Acids Res. (2021) ;49: (7):3907–18. |
[89] | Zhao X-N , Kumari D , Gupta S , Wu D , Evanitsky M , Yang W , et al. Mutsβ generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum Mol Genet. (2015) ;24: (24):7087–96. |
[90] | Møllersen L , Moldestad O , Rowe AD , Bjølgerud A , Holm I , Tveterås L , et al. Effects of anthocyanins on CAG repeat instability and behaviour in Huntington’s disease R6/1 mice. PLoS Curr. (2016) ;8: :ecurrents.hd.58d04209ab6d5de0844db7ef5628ff67. |
[91] | Lokanga RA , Senejani AG , Sweasy JB , Usdin K . Heterozygosity for a hypomorphic Polβ mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genet. (2015) ;11: (4):e1005181. |
[92] | Lai Y , Budworth H , Beaver JM , Chan NLS , Zhang Z , McMurray CT , et al. Crosstalk between MSH2-MSH3 and polβ promotes trinucleotide repeat expansion during base excision repair. Nat Comm. (2016) ;7: :12465–80. |
[93] | Guo J , Gu L , Leffak M , Li G-M . MutSβ promotes trinucleotide repeat expansion by recruiting DNA polymerase β to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res. (2016) ;26: (7):775–86. |
[94] | Liu Y , Prasad R , Beard WA , Hou EW , Horton JK , McMurray CT , et al. Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion. J Biol Chem. (2009) ;284: (41):28352–66. |
[95] | Genetic Modifiers of Huntington’s Disease Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. |
[96] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Electronic address: [email protected]; Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900 e14. |
[97] | Lee JM , Chao MJ , Harold D , Abu Elneel K , Gillis T , Holmans P , et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet. (2017) ;26: (19):3859–67. |
[98] | Moss DJH , Pardinas AF , Langbehn D , Lo K , Leavitt BR , Roos R , et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. (2017) ;16: (9):701–11. |
[99] | Zhao Z , Wu J , Xu H , Zhou C , Han B , Zhu H , et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. (2020) ;11: (8):629–44. |
[100] | Rao VR , Lautz JD , Kaja S , Foecking EM , Lukács E , Stubbs EB . Mitochondrial-targeted antioxidants attenuate TGF-β2signaling in human trabecular meshwork cells. Invest OpthalmolVisual Sci. (2019) ;60: (10):3613–24. |
[101] | Sammi SR , Foguth RM , Nieves CS , De Perre C , Wipf P , McMurray CT , et al. Perfluorooctane sulfonate (PFOS) produces dopaminergic neuropathology in Caenorhabditis elegans. Toxicol Sci. (2019) ;172: (2):417–34. |
[102] | Devi S , Kumar V , Singh SK , Dubey AK , Kim JJ . Flavonoids: Potential candidates for the treatment of neurodegenerative disorders. Biomedicines. (2021) ;9: (2):99–119. |
[103] | Fotoohi A , Moloudi MR , Hosseini S , Hassanzadeh K , Feligioni M , Izadpanah E . A novel pharmacological protective role for safranal in an animal model of Huntington’s disease. Neurochem Res. (2021) ;46: (6):1372–9. |
[104] | Lum PT , Sekar M , Gan SH , Bonam SR , Shaikh MF . Protective effect of natural products against Huntington’s disease: An overview of scientific evidence and understanding their mechanism of action. ACS Chem Neurosci. (2021) ;12: (3):391–418. |
[105] | Luo F , Sandhu AF , Rungratanawanich W , Williams GE , Akbar M , Zhou S , et al. Melatonin and autophagy in aging-related neurodegenerative diseases. Int J Mol Sci. (2020) ;21: (19):7174–204. |
[106] | Simunkova M , Alwasel SH , Alhazza IM , Jomova K , Kollar V , Rusko M , et al. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch Toxicol. (2019) ;93: (9):2491–513. |
[107] | Rodick TC , Seibels DR , Babu JR , Huggins KW , Ren G , Mathews ST . Potential role of coenzyme Q10 in health and disease conditions. Nutr Diet Supplem. (2017) ;10: :1–11. |


