
Circulating Endocannabinoids in Huntington’s Disease: An Exploratory Cross-Sectional Study
Abstract
Huntington’s disease (HD) is an inherited neurodegenerative disease characterized by motor, cognitive and behavioral deficits. Some evidence suggests that the endocannabinoid system participates in the pathophysiology of HD. We conducted a cross-sectional study comparing plasma levels of anandamide and 2-arachidonoylglycerol in manifest HD gene-expansion carriers (HDGEC) and healthy controls, finding no difference in endocannabinoid levels between the groups. Correlations between endocannabinoid levels and clinical scales (Mini-Mental State Examination, Hospital Anxiety and Depression Scale, Unified Huntington Disease Rating Scale) were non-significant. We found a significant association between body mass index and anandamide levels in healthy controls but not in HDGEC.
INTRODUCTION
Huntington’s disease (HD) is an inherited neurodegenerative disease characterized by motor, cognitive, and behavioral deficits often manifesting between 30 and 50 years of age [1]. The causative mutation is a CAG triplet repeat expansion in the huntingtin gene located on the short arm of chromosome 4. It is considered pathogenic when there are more than 35 CAG repeats [2]. HD gene expansion carriers (HDGEC) are clinically diagnosed with manifest HD when unequivocal clinical signs are present [3].
The endocannabinoid system may play a role in the pathophysiology of HD [2, 4]. Anandamide (AEA) and 2-arachidonoylglycerol (2AG) are the two main modulators of the endocannabinoid system, and there are two specific G-protein coupled cannabinoid receptors: type 1 (CB1) and type 2 (CB2) [5]. CB1 receptors are expressed primarily on the presynaptic terminals of excitatory and inhibitory neurons. CB2 receptors are found predominantly in the peripheral immune system but may also be present in the microglia [5]. The endocannabinoid system plays an essential role in modulating basal ganglia, and CB1 receptors are highly expressed in these nuclei. In HDGEC and animal models of HD, there is a marked decrease of these receptors in the brain [6–8]. The down-regulation of CB1 expression seems to occur early in the disease and significantly affects the striatal medium spiny neurons [9]. These abnormalities seem to have a role in the pathophysiology of the disease. Furthermore, neuroprotective effects of some cannabinoids in HD models reinforce the association between the endocannabinoid system and HD [4, 10, 11].
HD is known to have systemic features, such as weight loss, altered glucose homeostasis, skeletal-muscle atrophy, and blood cell abnormalities [12, 13, 24]. The endocannabinoid system is also widely distributed throughout the human body and is expressed in several organs and systems with pleiotropic function, including regulation of metabolism and the immune system [14]. A previous study reported that fatty acid amide hydrolase activity was dramatically reduced in the peripheral lymphocytes of HDGEC, leading to increased levels of AEA, since it is metabolized by fatty acid amide hydrolase [15]. These observations may indicate that the peripheral endocannabinoid system could also be compromised in HDGECs. To our knowledge, no studies have assessed endocannabinoid levels in the plasma or serum of HDGECs. This study aimed to evaluate possible abnormalities of peripheral circulating endocannabinoids in HDGECs.
METHODS
We performed a cross-sectional study comparing plasma levels of AEA and 2AG in HDGECs and healthy controls. We invited all consecutive HDGECs clinically and genetically diagnosed with HD who were treated at the Movement Disorders Clinic of the Ribeirão Preto Medical School Hospital, São Paulo, Brazil between November 2016 and April 2018 to participate. We recruited healthy controls from the local community through local media announcements. The healthy controls were not genotyped for CAG expansion but were asked about their family history of neurological diseases. We performed a physical examination to rule out neurological abnormalities. The exclusion criteria for all participants were: any significant or decompensated systemic diseases (e.g., diabetes), diagnosis of other neurological diseases, active infectious or inflammatory disease, pregnancy, or history of drug abuse. We excluded healthy controls who were using antiepileptics, antidepressants, or antipsychotics, as well as those with any psychiatric disturbance (psychosis, anxiety, or depression).
We collected demographic and clinical data from all participants. All participants were assessed with the Mini-Mental State Examination (MMSE) and the healthy controls were also assessed with the Mini International Neuropsychiatric Interview to exclude psychiatric disorders. We evaluated HDGECs with the Hospital Anxiety and Depression Scale (HADS) and the Unified Huntington Disease Rating Scale (UHDRS).
We collected peripheral blood samples by venous puncture (approximately 15 mL per individual), be-tween 8 am and 10 am, since the endocannabinoid system may be affected by circadian rhythm [14]. The blood was immediately centrifuged, and the plasma was frozen at -70°C. We asked all participants to fast for at least 8 h prior to blood collection, as well as to avoid exercise and alcohol on the day before collection.
The plasma levels of AEA and 2AG were measured using a previously validated column switching method with bi-dimensional chromatography-tan-dem mass spectrometry [16]. Blood analysis was performed blindly, and each sample was measured in triplicate. We also examined C-reactive protein and glucose, total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides, and hepatic transaminase levels.
We used the nonparametric Mann-Whitney test for statistical analysis. We tested for sex differences between the groups using the Z-score for two population proportions. Correlations were analyzed with Spearman’s correlation coefficient, and the Shapiro-Wilk test was used to assess data distribution.
We estimated the sample size according to the number of manifest HDGEC patients regularly seen in our clinic (approximately 50). The local ethics committee approved the study (HCRP-USP, number 1.828.036, November 2016/CAAE 59355416.3.0000.5440). All participants provided written in-formed consent.
RESULTS
We invited 50 manifest HDGECs to participate in the study, but 6 declined. We excluded 2 because it was impossible to collect complete clinical data or blood samples. We evaluated 32 healthy controls and excluded 2 due to significant depressive symptoms. Table 1 describes the demographic and clinical features of all participants. Most variables were non-normally distributed.
Table 1
Demographic and clinical features of HD gene expansion carriers (HDGEC) and healthy controls (HC). Values are represented as median (IQR), absolute numbers (n), or percentage (%)
| HDGEC | HC | p | |
| Number (n) | 42 | 30 | |
| Female (%) | 59.5% | 56.6% | 0.66a |
| Age (y) | 48.5 (43–58) | 47.0 (39–54) | 0.12b |
| Education (y) | 4.5 (4–10) | 11.0 (11–15) | <0.001*,b |
| Weight (kg) | 61.0 (54.5–70) | 69.5 (65.1–77.5) | 0.002*,b |
| Height (cm) | 163 (155–168) | 166 (160–170) | 0.136b |
| Body mass index (kg/m2) | 22.7 (20.3–25.7) | 24.9 (23.2–28.2) | 0.024*,b |
| MMSE | 20.5 (16–23) | 29.5 (29–30) | <0.001*,b |
| MMSE (z score) | –2.0 (–3; –0.6) | 0.8 (0.4;1.2) | <0.001*,b |
| HADS-A | 3.0 (1–6) | – | – |
| HADS-D | 4.0 (1–11) | – | – |
| Antidepressant use (n) | 32 | 0 | – |
| Antipsychotic use (n) | 34 | 0 | – |
| Disease duration (y) | 7.0 (4–10.4) | – | – |
| UHDRS motor score (0–124 pts) | 37.5 (30.5–71) | – | – |
| Independence scale (10–100%) | 70% (60–85) | – | – |
HADS, Hospital Anxiety and Depression Scale; HADS-A, anxiety score; HADS-D, depression score; IQR, interquartile range; MMSE, Mini-Mental State Examination; UHDRS, Unified Huntington Disease Rating Scale; *p < 0.05; aZ-score test; bMann-Whitney test
The final sample included 42 manifest HDGECs and 30 healthy controls. The groups were matched for age and sex, and women predominated in both groups. Regarding disease stage, 19% of the HDGECs were Shoulson-Fahn stage I, 29% were stage II, 19% were stage III, and 33% were in the late stages (IV–V). The HDGEC group had a significantly lower education, weight, body mass index (BMI), and MMSE scores than the control group. Median disease duration in the HDGEC group was 7 years, and most used antidepressants and/or antipsychotics.
There was no difference between the groups regarding C-reactive protein, glucose, total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides, and aminotransferase plasma levels. Figure 1 shows plasma endocannabinoid levels in the HD and control groups.
Fig. 1
Plasma endocannabinoid levels (anandamide –AEA 2-arachidonoyl-glycerol –2AG) of HD gene expansion carriers (HDGEC) and healthy controls (HC). Values are represented in ng/ml. AEA p = 0.11, 2AG p = 0.14.
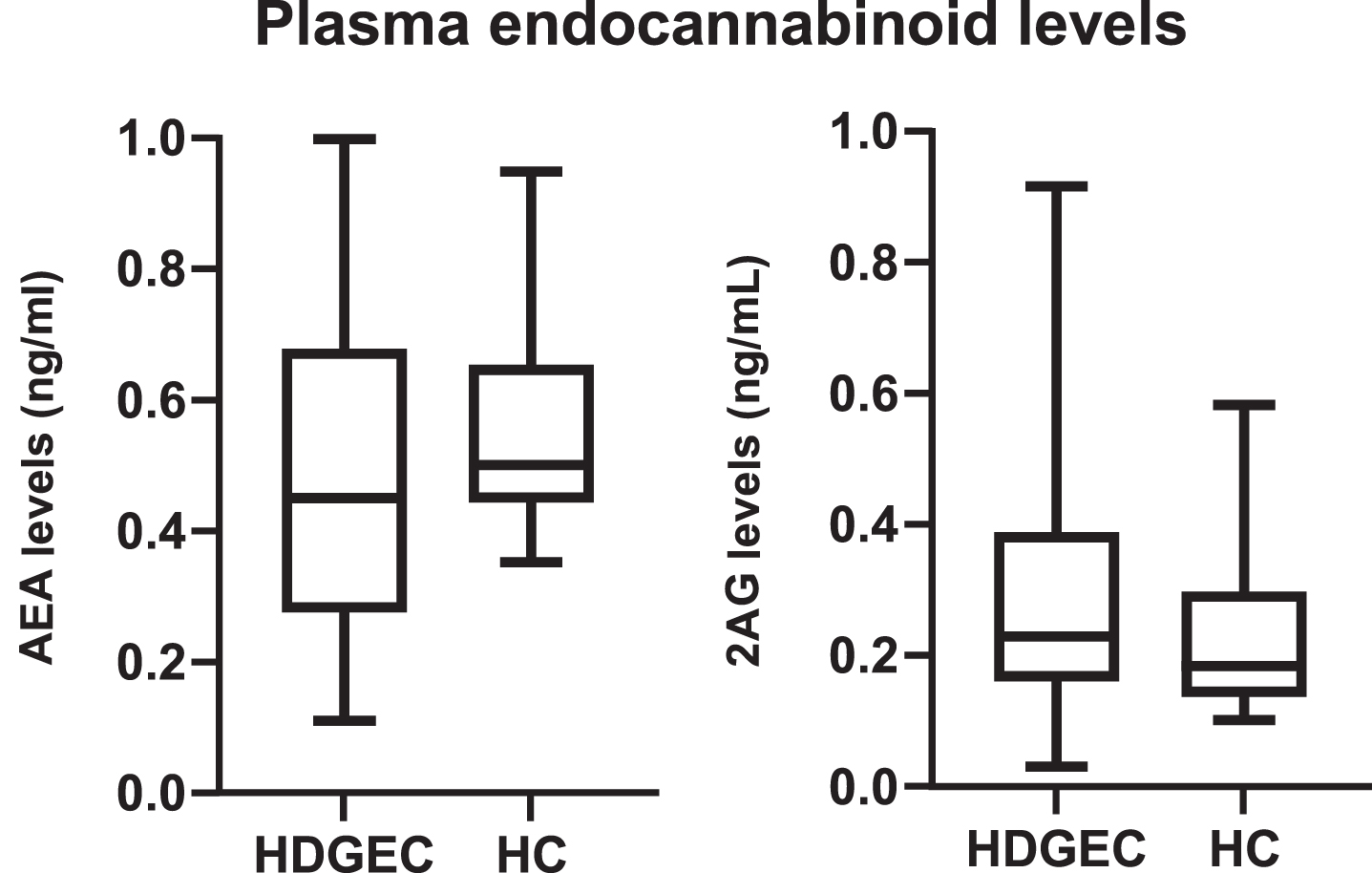
As presented in Fig. 1, there was no significant difference in endocannabinoid levels between the groups (AEA p = 0.11; 2AG p = 0.14). We found no difference between endocannabinoid levels in the HDGEC group, regardless of antidepressant or antipsychotic use (antidepressants vs. AEA p = 0.85; antidepressants vs. 2AG p = 0.54; antipsychotics vs. AEA p = 0.39; antipsychotics vs. 2AG p = 0.76). There was also no significant correlation between endocannabinoid levels and HADS scores (HADS vs. AEA p = 0.64, r = 0.07; HADS vs. 2AG p = 0.26, r = 0.17), endocannabinoid levels and MMSE scores (MMSE vs. AEA p = 0.98, r = 0.003; MMSE vs. 2AG p = 0.99, r = –0.002), or endocannabinoid levels and UHDRS motor scores (UHDRS vs. AEA p = 0.56, r = –0.08; UHDRS vs. 2AG p = 0.24, r = 0.17)
We found a significant correlation between BMI and AEA levels in healthy controls (p = 0.02), although the correlation coefficient was weak-to-moderate (r = 0.42). Other associations between BMI and endocannabinoid levels were non-significant (healthy control BMI vs. 2AG p = 0.83, r = 0.03; HD BMI vs. AEA p = 0.99, r = 0.0004; HD BMI vs. 2AG p = 0.28, r = 0.16).
DISCUSSION
In this study, we did not find altered levels of circulating endocannabinoids in HDGECs. In contrast, a previous study reported high levels of AEA in the peripheral lymphocytes of HDGECs due to reduced fatty acid amide hydrolase activity [15]. Multiple factors can affect circulating endocannabinoid levels, such as stress, exercise, inflammation, chronic pain, drug abuse, circadian rhythm, and food presentation and consumption [14]. AEA can also originate from multiple sources, such as blood cells, adipose tissue, muscles, the gastrointestinal tract, and the brain [14]. Several factors can contribute to changes in the blood levels of endocannabinoids, such as metabolic system dysregulation, immune system dysfunction, or altered production in the central nervous system (CNS). For this reason, circulating endocannabinoids may be less predictable and more subject to variation than measurements taken in the CNS or the immune system, which explains our findings.
We investigated whether antidepressants and depression could have modulated the plasma endocannabinoid levels of our patients but found no relationship between depressive symptoms or antidepressant use and endocannabinoid levels in HDGECs [23, 29].
Experimental studies in animal models of HD have shown low AEA and 2AG levels in basal ganglia and high AEA levels in the cerebral cortex, suggesting that endocannabinoid levels in the CNS could be altered in HD [21, 22]. Although altered endocannabinoid production in the CNS could reflect peripherally, the correlation between central and peripheral endocannabinoid levels is not predictable. A study that evaluated endocannabinoid levels in cannabis users and controls found no association between cerebrospinal fluid and serum levels of AEA or 2AG [25].
Peripheral mechanisms should also be considered when determining plasma endocannabinoid levels. One study found a strong positive association between adiposity and circulating AEA, but not with cerebrospinal fluid AEA levels [26].
We found a significant correlation between BMI and AEA levels in healthy controls but not in HDGECs. Nevertheless, it was a weak to moderate association (r = 0.42), and we cannot rule out a false-positive result. Nevertheless, this finding could indicate subtle endocannabinoid system impairment in HDGEC regarding the control of energy metabolism or other metabolic routes. Previous studies have reported positive, but not always straightforward, correlations between endocannabinoids and BMI, and these were mainly observed in obese individuals, which was not a characteristic of our study participants. One study found an association between BMI and 2AG, but not AEA [27], while another reported an association between BMI and AEA, but not 2AG [28].
Our findings neither exclude nor confirm compartmentalized changes in endocannabinoid levels in some peripheral tissues, such as in the immune system [14]. The presented data is insufficient to draw definitive conclusions regarding the possible role of peripheral endocannabinoid system in the pathophysiology of HD.
Our study had several limitations. The small sample size undermined our statistical power. The lack of an endocannabinoid analysis in the CNS (e.g., cerebrospinal fluid study) limited comparison with circulating endocannabinoid levels. The existence of factors that could influence peripheral endocannabinoid levels can complicate the interpretation of plasma endocannabinoid levels [14].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Ghosh R , Tabrizi SJ . Huntington disease. Handb Clin Neurol (2018) ;147: :255–78. |
[2] | Tabrizi SJ , Flower MD , Ross CA , Wild EJ . Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol (2020) ;16: (10):529–46. |
[3] | McColgan P , Tabrizi SJ . Huntington’s disease: a clinical review. Eur J Neurol (2018) ;25: (1):24–34. |
[4] | Lastres-Becker I , De Miguel R , Fernandez-Ruiz JJ . The endocannabinoid system and Huntington’s disease. Curr Drug Targets CNS Neurol Disord (2003) ;2: (5):335–47. |
[5] | Mechoulam R , Parker LA . The endocannabinoid system and the brain. Annu Rev Psychol (2013) ;64: :21–47. |
[6] | Blazquez C , Chiarlone A , Sagredo O , Aguado T , Pazos MR , Resel E , et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington’s disease. Brain. (2011) ;134: :119–36. |
[7] | Casteels C , Martinez E , Bormans G , Camon L , de Vera N , Baekelandt V , et al. Type 1 cannabinoid receptor mapping with [18F]MK-PET in the rat brain after quinolinic acid lesion: a comparison to dopamine receptors and glucose metabolism. Eur J Nucl Med Mol Imaging. (2010) ;37: (12):2354–63. |
[8] | Van Laere K , Casteels C , Dhollander I , Goffin K , Grachev I , Bormans G , et al. Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. J Nucl Med. (2010) ;51: (9):1413–7. |
[9] | Naydenov AV , Sepers MD , Swinney K , Raymond LA , Palmiter RD , Stella N , et al. Genetic rescue of CB1 receptors on medium spiny neurons prevents loss of excitatory striatal synapses but not motor impairment in HD mice. Neurobiol Dis. (2014) ;71: :140–50. |
[10] | Sepers MD , Raymond LA . Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov Today. (2014) ;19: (7):990–96. |
[11] | Laprairie RB , Bagher AM , Rourke JL , Zrein A , Cairns EA , Kelly MEM , et al. Positive allosteric modulation of the type 1 cannabinoid receptor reduces the signs and symptoms of Huntington’s disease in the R6/2 mouse model. Neuropharmacology. (2019) ;151: :1–12. |
[12] | Turner C , Cooper JM , Schapira AH . Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov Disord. (2007) ;22: (12):1715–21. |
[13] | Lalic NM , Maric J , Svetel M , Jotic A , Stefanova E , Lalic K , et al. Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol. (2008) ;65: (4):476–80. |
[14] | Hillard CJ . Circulating endocannabinoids: from whence do they come and where are they going? Neuropsychopharmacology (2018) ;43: (1):155–72. |
[15] | Battista N , Bari M , Tarditi A , Mariotti C , Bachoud-Levi AC , Zuccato C , et al. , Severe deficiency of the fatty acid amide hydrolase (FAAH) activity segregates with the Huntington’s disease mutation in peripheral lymphocytes. Neurobiol Dis. (2007) ;27: (1):108–16. |
[16] | Marchioni C , de Souza ID , Grecco CF , Crippa JA , Tumas V , Queiroz MAC . A column switching ultrahigh-performance liquid chromatography-tandem mass spectrometry method to determine anandamide and 2-arachidonoylglycerol in plasma samples. Anal Bioanal Chem. (2017) ;409: (14):3587–96. |
[17] | Li C , Jones PM , Persaud SJ . Role of the endocannabinoid system in food intake, energy homeostasis and regulation of the endocrine pancreas. Pharmacol Ther. (2011) ;129: (3):307–20. |
[18] | Sharma DS , Paddibhatla I , Raghuwanshi S , Malleswarapu M , Sangeeth A , Kovuru N , et al. Endocannabinoid system: Role in blood cell development, neuroimmune interactions and associated disorders. J Neuroimmunol. (2021) ;353: :577501. |
[19] | Watkins BA , Hutchins H , Li Y , Seifert MF . The endocannabinoid signaling system: a marriage of PUFA and musculoskeletal health. J Nutr Biochem. (2010) ;21: (12):1141–52. |
[20] | Simon V , Cota D . Mechanisms in Endocrinology: Endocannabinoids and metabolism: past, present and future. Eur J Endocrinol.R. (2017) ;176: (6):309–24. |
[21] | Bisogno T , Martire A , Petrosino S , Popoli P , Di Marzo V . Symptom-related changes of endocannabinoid and palmitoylethanolamide levels in brain areas of R6/2 mice, a transgenic model of Huntington’s disease. Neurochem Int. (2008) ;52: (1-2):307–13. |
[22] | Bari M , Battista N , Valenza M , Mastrangelo N , Malaponti M , Catanzaro G , et al. In vitro and in vivo models of Huntington’s disease show alterations in the endocannabinoid system. FEBS J. (2013) ;280: (14):3376–88. |
[23] | Colangeli R , Teskey GC , Di Giovanni G . Endocannabinoid-serotonin systems interaction in health and disease. Prog Brain Res. (2021) ;259: :83–134. |
[24] | Djoussé L , Knowlton B , Cupples LA , Marder K , Shoulson I , Myers RH . Weight loss in early stage of Huntington’s disease. Neurology. (2002) ;59: (9):1325–30. |
[25] | Morgan CJA , Cerebrospinal fluid anandamide levels, cannabis use and psychotic-like symptoms. Br J Psychiatry. (2013) ;202: (5):381–2. |
[26] | Jumpertz R , Guijarro A , Pratley RE , Piomelli D , Krakoff J . Central and peripheral endocannabinoids and cognate acylethanolamides in humans: association with race, adiposity, and energy expenditure. J Clin Endocrinol Metab. (2011) ;96: (3):787–91. |
[27] | Cote M , Matias I , Lemieux I , Petrosino S , Almeras N , Despres JP , Di Marzo V . Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int J Obes (Lond). (2007) ;31: (4):692–99. |
[28] | Di Marzo V , Verrijken A , Hakkarainen A , Petrosino S , Mertens I , Lundbom N , et al. Role of insulin as a negative regulator of plasma endocannabinoid levels in obese and nonobese subjects. Eur J Endocrinol. (2009) ;161: (5):715–22. |
[29] | Hill MN , Miller GE , Ho WSV , Gorzalka BB , Hillard CJ . Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry. (2008) ;41: (2):48–53. |


