
FAN1, a DNA Repair Nuclease, as a Modifier of Repeat Expansion Disorders
Abstract
FAN1 encodes a DNA repair nuclease. Genetic deficiencies, copy number variants, and single nucleotide variants of FAN1 have been linked to karyomegalic interstitial nephritis, 15q13.3 microdeletion/microduplication syndrome (autism, schizophrenia, and epilepsy), cancer, and most recently repeat expansion diseases. For seven CAG repeat expansion diseases (Huntington’s disease (HD) and certain spinocerebellar ataxias), modification of age of onset is linked to variants of specific DNA repair proteins. FAN1 variants are the strongest modifiers. Non-coding disease-delaying FAN1 variants and coding disease-hastening variants (p.R507H and p.R377W) are known, where the former may lead to increased FAN1 levels and the latter have unknown effects upon FAN1 functions. Current thoughts are that ongoing repeat expansions in disease-vulnerable tissues, as individuals age, promote disease onset. Fan1 is required to suppress against high levels of ongoing somatic CAG and CGG repeat expansions in tissues of HD and FMR1 transgenic mice respectively, in addition to participating in DNA interstrand crosslink repair. FAN1 is also a modifier of autism, schizophrenia, and epilepsy. Coupled with the association of these diseases with repeat expansions, this suggests a common mechanism, by which FAN1 modifies repeat diseases. Yet how any of the FAN1 variants modify disease is unknown. Here, we review FAN1 variants, associated clinical effects, protein structure, and the enzyme’s attributed functional roles. We highlight how variants may alter its activities in DNA damage response and/or repeat instability. A thorough awareness of the FAN1 gene and FAN1 protein functions will reveal if and how it may be targeted for clinical benefit.
TABLE OF CONTENTS
| 1. | FAN1 GENIC REGION AND DISEASE-ASSOCIATED GENETIC VARIANTS | 96 |
| 1.1. | Genes | 96 |
| 1.2. | Transcripts | 98 |
| 1.3. | MTMR10 and TRPM1 | 98 |
| 1.4. | 15q13.3 micro-deletion/-duplication, and CNVs | 98 |
| 2. | FAN1 PROTEIN STRUCTURE, DOMAINS, AND PROTEIN INTERACTIONS | 98 |
| 3. | FAN1 AND DNA REPAIR-RELATED PROCESSES | 101 |
| 4. | FAN1 AND DNA REPLICATION STRESS | 104 |
| 5. | FAN1 DEFICIENCIES AND VARIANTS IN GENETIC DISEASES AND CANCER | 105 |
| 5.1. | Karyomegalic interstitial nephritis (KIN; OMIM#614817) | 105 |
| 5.2. | Cancer | 107 |
| 5.3. | Neurodevelopmental and neuropsychiatric diseases | 107 |
| 6. | GENETIC MODIFIERS OF CAG/POLYQ EXPANSION DISEASE ONSET | 108 |
| 6.1. | FAN1 variants in CAG/polyQ expansion diseases hasten or delay disease onset | 108 |
| 6.2. | Modifiers may alter expression levels of FAN1 mRNA | 109 |
| 6.3. | FAN1 coding variants, DNA binding, and nuclease activities | 109 |
| 7. | FAN1 IN REPEAT INSTABILITY | 111 |
| 7.1. | Candidate nucleases in repeat instability, a questionable involvement of Flap endonuclease 1 (FEN1) | 111 |
| 7.2. | FAN1 variants modify disease by modifying repeat expansions | 111 |
| 7.3. | Role of FAN1 in replication-associated repeat instability | 113 |
| 7.4. | Role of FAN1 in repair-associated repeat instability | 114 |
| 7.5. | Role of FAN1 in transcription-associated repeat instability | 114 |
| 8. | CONCLUSIONS AND FUTUREPERSPECTIVES | 114 |
Genome integrity is maintained by the concerted action of distinct DNA repair pathways. DNA nucleases resolve damaged DNA and unusual DNA structures that arise during cellular processes such as replication, recombination, and transcription. DNA repair nucleases usually recognize aberrant DNA conformations of damaged and/or potentially mutagenic DNA intermediates, rather than DNA sequences; hence, they are referred to as ‘structure-specific nucleases’. DNA repair machinery corrects errors incurred during replication in mitotic cells, and repairs damaged DNA in non-mitotic cells [1]. Processing of aberrant DNA structures, particularly steps involving nucleolytic cleavage, represents a committed and tightly controlled biological ‘decision’ in genome maintenance pathways. If not kept in check, structure-specific nucleases can cause extensive DNA degradation and wreak havoc on the stability of the genome [2]. On the other hand, the essential nature of structure-specific nucleases is underscored by the fact that homozygous null mutations in genes encoding these enzymes are often embryonic lethal or lead to infertility; single site mutations may predispose to cancer or other diseases (for review, see [2]). This review focuses on FAN1, with particular attention to its potential roles as a modifier of disorders associated with genomic repeat expansions, including Huntington’s disease (HD), spinocerebellar ataxia type 1 (SCA1), SCA2, SCA3, SCA6, SCA7, SCA17, autism, schizophrenia, and others.
1FAN1 GENIC REGION AND DISEASE-ASSOCIATED GENETIC VARIANTS
1.1Genes
FAN1 (OMIM#613534) was initially identified as KIAA1018 in a human brain cDNA library and found to be ubiquitously expressed in the central nervous system and peripheral tissues [3]. The protein encoded by KIAA1018 interacts with the mismatch repair (MMR) proteins, PMS2 [4] and MLH1 [4], and FANCD2 [5–8]. Except where noted, we refer to the gene in question as FAN1. FAN1, at 15q13.3 (GRCh37/hg19 coordinates: chr15:31,196,076-31,235,31) contains 15 exons, of which 13 are coding. The gene’s cis-acting regions include promoter regions, CpG islands, transcription factor binding sites, and enhancers (Fig. 1b). It has three predicted promoters (as per EPD, ENSEMBL, and NCBI), two of which are located upstream of exon 1, while the third promoter resides in intron 8. A large CpG island of 918 base pairs (bp) spans exon1, promoter 1 and further upstream [9–11] (Fig. 1b). Various FAN1 variants and the FAN1 haplotypes associated with HD and SCAs are shown in Fig. 1 (summarized in 1d) and discussed in detail in Section 6.1 and 6.2.
Fig.1
Genomic region of human FAN1 at 15q13.3 (GRCh37/hg19). (a) Schematic organization of 15q13.3 microduplication/microdeletion region contains multiple genes including FAN1. (b) FAN1 and MTR10 are partially overlapped and include various coding and non-coding genetic variants identified in HD, various spinocerebellar ataxias (SCAs), autism (ASD), and schizophrenia (SZC). The murine homolog of p.S795P (mS798P, in between the TPR and the nuclease domain) was identified in a murine Rett syndrome modifier screen [86]. CAG/polyQ disease-hastening and disease-delaying variants are shown in red and green fonts, respectively. (c) FAN1 has various transcript variants, a limited set of full length and truncated FAN1 transcript variants are shown (NM: Transcript variant, NP: Isoforms of transcript variant). Information for making this figure is from the NCBI genome browser, UCSC genome browser, and Ensembl browser. For details of the 15q13.3 chromosomal rearrangements, see Supplementary Figure 1. (d) Table with HD modifier haplotypes in FAN1.
![Genomic region of human FAN1 at 15q13.3 (GRCh37/hg19). (a) Schematic organization of 15q13.3 microduplication/microdeletion region contains multiple genes including FAN1. (b) FAN1 and MTR10 are partially overlapped and include various coding and non-coding genetic variants identified in HD, various spinocerebellar ataxias (SCAs), autism (ASD), and schizophrenia (SZC). The murine homolog of p.S795P (mS798P, in between the TPR and the nuclease domain) was identified in a murine Rett syndrome modifier screen [86]. CAG/polyQ disease-hastening and disease-delaying variants are shown in red and green fonts, respectively. (c) FAN1 has various transcript variants, a limited set of full length and truncated FAN1 transcript variants are shown (NM: Transcript variant, NP: Isoforms of transcript variant). Information for making this figure is from the NCBI genome browser, UCSC genome browser, and Ensembl browser. For details of the 15q13.3 chromosomal rearrangements, see Supplementary Figure 1. (d) Table with HD modifier haplotypes in FAN1.](https://content.iospress.com:443/media/jhd/2021/10-1/jhd-10-1-jhd200448/jhd-10-jhd200448-g001.jpg)
1.2Transcripts
FAN1 transcripts are predicted to undergo alternative splicing, generating 28 potential transcript variants (Fig. 1c), which code for full-length or truncated proteins. Some transcripts are predicted to undergo nonsense-mediated decay, and some retain introns [11]. A FAN1 splicing effect on HD modifiers is unlikely, based upon analyses of data in non-disease expression databases [12]. FAN1/Fan1 circular RNA (circRNA) was identified in human and mouse heart, but is of unknown function [13]. The MTMR10 gene partially overlaps with FAN1 and is convergently transcribed (Fig. 1a, b). Disease onset-modifying variants in overlapping regions of these two genes have been identified (Fig. 1). MTMR10-tagging single nucleotide polymorphisms (SNPs) identified in individuals with HD or SCA2 may relate to FAN1 expression levels [14–16]. MTMR10 transcripts could hypothetically act in an antisense manner to regulate FAN1 mRNA stability and translatability. The same might be true where a FAN1 transcript regulates the MTMR10 mRNA. Evidence of such antisense regulation is not known for these genes but could provide insight into targeting for therapeutic purposes.
1.3MTMR10 and TRPM1
The 15q13.3 region with adjacent genes, FAN1, MTMR10, and TRPM1 (Transient Receptor Potential Melastatin Family member 1) (Fig. 1a), appears to be enriched with variants that modify HD onset. FAN1 has been referred to as MTMR15; however no data reveal any relation of FAN1 to myotubularin-related proteins (MTMs) [7]. In contrast, MTMR10 is an MTMR family member. Various tagSNPs associated with HD age of onset are within or proximal to the two overlapping convergently transcribed genes, MTMR10 and FAN1 (Fig. 1b, d). A protein coding variant in TRPM1 (rs3784588; p.V1434I) is associated with hastening HD onset [12]. The roles of MTMR10 or TRPM1, if any, in HD or any of the 15q13.3-related diseases, are unknown. TRPM1 and MTMR10 have been argued to not be HD modifiers, based upon genetic data [14] and the absence of an effect of the disease-delaying variant haplotype (15AM2) upon expression levels of either TRPM1 or MTMR10 in non-HD databases [12, 14]. Moreover, genetic ablation of Mtmr10 does not affect CAG instability in HD mice, whereas ablation of Fan1 does [18]. The effect of TRPM1/Trpm1 on CAG instability in human or mouse models has not been addressed.
1.415q13.3 micro-deletion/-duplication, and CNVs
Genetic variants of FAN1 include copy number variations (CNVs), inversions, and single nucleotide variants (SNVs). FAN1 and MTMR10 lie within a 2 Mb region, 15q13.3, which can present copy number variation of zero, one, two, three, or four copies [19, 20]. This 15q13.3 region contains at least seven genes [21–23], with at least five distinct human-specific structural configurations, including a common inversion [24] (Fig. 1 and Supplementary Figure 1). The genes include ARHGAP11B, FAN1, MTMR10, TRPM1, miR211, KLF13, OTUD7A, and CHRNA7 (Fig. 1). Microdeletions of 15q13.3 can be homozygous, heterozygous or compound heterozygous. Homozygous and compound heterozygous 15q13.3 microdeletions are associated with severe phenotypes, which may include intellectual disability, dysmorphic features, epilepsy, neuropsychiatric disturbances with or without cognitive impairment, schizophrenia and autism [21, 25]. In contrast, individuals with heterozygous 15q13.3 microdeletions may show a mild phenotype or complete absence of anomalies [21–23]. Duplications of 15q13.3 are less commonly observed than deletions, and less often associated with intellectual disability [19, 20]. CNVs of the 15q13.3 region could increase or decrease the levels of the FAN1, MTMR10, and TRPM1 gene products, which may affect phenotypes. However, the mechanism by which CNVs of any of these genes relate to clinical presentations remains unclear. Of the various genomic configurations of the 15q13.3 region (Supplementary Figure 1), it is not known which occur with the HD-modifying FAN1 and MTMR10 variants. It would be interesting to learn whether some 15q13.3 structural configurations are associated with modifying the presentation of HD, SCAs, autism, or schizophrenia.
2FAN1 PROTEIN STRUCTURE, DOMAINS, AND PROTEIN INTERACTIONS
In 2010, four groups independently identified KIAA1018 as a structure-specific nuclease required for DNA interstrand crosslink (ICL) repair; they collectively renamed the protein FAN1 (for FANCD2-FANCI-associated nuclease 1) due to its physical association with FANCD2— a central component of the Fanconi Anemia (FA) pathway [5–8]. The fundamental importance of FAN1 in cell physiology is underscored by its domain organization, which is highly conserved among eukaryotes and also present in some bacteria [5–8] (Fig. 2a, b). All FAN1 orthologs contain a C-terminal PDXn(D/E)XK motif located within an atypical virus-type replication-repair nuclease (VRR_NUC) domain [26, 27]. FAN1 is the only known eukaryotic nuclease with a VRR_NUC module, which is otherwise common in bacteriophages and bacteria as a stand-alone domain. FAN1 preferentially incises 5’-flap branched structures [5–7, 28] (Fig. 3). However, FAN1’s substrate specificity appears to be broad; its endonuclease activity can efficiently cleave different DNA intermediates and probably deal with several types of DNA lesions. Besides its ability to cleave DNA substrates containing a 5’-flap, FAN1 displays a 5’-3’ exo-nuclease activity that requires gapped, nicked, or recessed DNA as an entry point [5–7] (Fig. 3) and can cleave every 3rd nucleotide in small flap non-repeat DNA [29]. FAN1 can also cleave D-loop structures that form during homologous recombination repair (HRR) [6] (Fig. 3). DNA damage induced by exogenous or endogenous chemicals, including ICLs and oxidative DNA lesions, have also been considered as targets of FAN1 (detailed in Section 4).
Fig.2
Protein domains of FAN1. (a) FAN1 is composed of an unstructured N-terminal region containing the Ubiquitin Binding Zinc (UBZ) finger domain, a central region containing a SAP domain to mediate DNA binding and a TPR domain required for protein-protein interactions, and a C-terminal region embedding a VRR_NUC domain that harbors the catalytic site. A non-canonical PCNA-interacting peptide (PIP) motif and a KEN-box motif, which are required for interaction with PCNA and Cdh1, respectively, are located at the N-terminal region. Key residues of each domain are highlighted in red. Mutations associated with pancreatic cancer and early onset of HD are highlighted with red and black circles, respectively. (b) Crystal structure of FAN1 revealed that two FAN1 molecules can dimerize to form a head-to-tail dimer in the presence of DNA. Assembly of FAN1 dimer occurs via the TPR and NUC domains of the first FAN1 molecule interacting with the SAP domain of a second FAN1 [41]. (c) FAN1 can form different subcomplexes with MLH1, ubiquitylated FANCD2, ubiquitylated PCNA, or can exist as homodimer in solution.
![Protein domains of FAN1. (a) FAN1 is composed of an unstructured N-terminal region containing the Ubiquitin Binding Zinc (UBZ) finger domain, a central region containing a SAP domain to mediate DNA binding and a TPR domain required for protein-protein interactions, and a C-terminal region embedding a VRR_NUC domain that harbors the catalytic site. A non-canonical PCNA-interacting peptide (PIP) motif and a KEN-box motif, which are required for interaction with PCNA and Cdh1, respectively, are located at the N-terminal region. Key residues of each domain are highlighted in red. Mutations associated with pancreatic cancer and early onset of HD are highlighted with red and black circles, respectively. (b) Crystal structure of FAN1 revealed that two FAN1 molecules can dimerize to form a head-to-tail dimer in the presence of DNA. Assembly of FAN1 dimer occurs via the TPR and NUC domains of the first FAN1 molecule interacting with the SAP domain of a second FAN1 [41]. (c) FAN1 can form different subcomplexes with MLH1, ubiquitylated FANCD2, ubiquitylated PCNA, or can exist as homodimer in solution.](https://content.iospress.com:443/media/jhd/2021/10-1/jhd-10-1-jhd200448/jhd-10-jhd200448-g002.jpg)
Fig.3
FAN1 can process various unusual DNA structures, including interstrand cross-linked DNA (ICL), dsDNA (supercoiled, covalently closed circular, linear DNA), nicked DNA, flap-DNA, R-loop DNA, and repeat containing DNA (unknown). Purple and red arrows denote exo-nuclease and endo-nucleolytic cleavage, respectively.
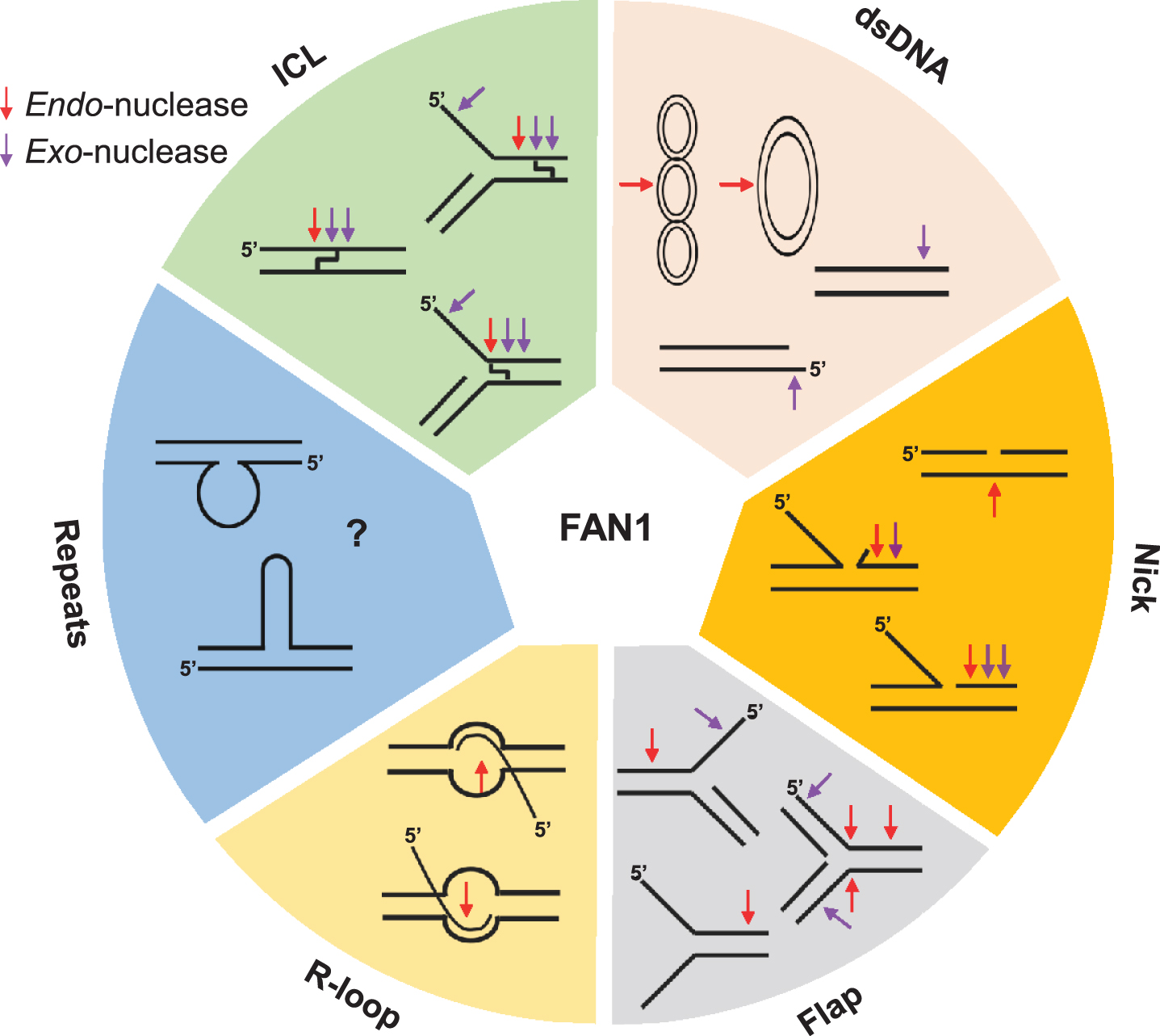
FAN1’s N-terminal region is largely unstructured, except for a RAD18-like ubiquitin-binding zinc finger (UBZ) domain, which is required for FAN1 localization to ICLs and stalled replication forks, possibly through binding to ubiquitylated FANCD2 [5–8, 30]. Consistently, a FAN1 zinc-finger mutant (C44A/C47A, Fig. 2a) fails to co-localize with FANCD2 in DNA damage-induced nuclear foci [5–8]. However, the UBZ mutant is competent in rescuing from mitomycin C (MMC) sensitivity of FAN1 knockout cells, suggesting that ubiquitin recognition is not essential for ICL repair [30–33]. In agreement with this, the FAN1 p.M50R cancer-associated mutant— located in the UBZ domain (Fig. 2a)— equally abolishes FAN1 foci but restores MMC resistance in FAN1-deficient cells [30], indicating that other FAN1 domains can mediate recruitment to ICLs. Indeed, FAN1 harbours a non-canonical proliferating cell nuclear antigen (PCNA)-interacting peptide (PIP) box motif that precedes and acts in concert with the UBZ domain in binding to ubiquitylated PCNA (ub-PCNA) [34] (Fig. 2a). A systematic and comparative analysis measuring the recruitment kinetics of DNA repair proteins at complex DNA lesions indicated two separate modes of FAN1 recruitment: an early step clustering with PCNA and a later step overlapping with FANCD2 [35]. Moreover, the PIP- and UBZ-dependent interaction of FAN1 with ubiquitylated PCNA is reminiscent of that for DNA polymerase η, acting in translesion DNA synthesis (TLS) [36–38], and for Spartan, a metallo-protease acting in DNA-protein crosslink repair [39]. Remarkably, both the PIP box and the UBZ are well conserved in FAN1 of multicellular eukaryotes including C. elegans, but absent in S. pombe, suggesting an evolutionary expansion of FAN1 interaction networks.
FAN1 contains highly conserved SAP (SAF-A/B, Acinus and PIAS) and TPR (tetratricopeptide repeat) domains implicated in DNA binding and FAN1 dimerization [28, 40] (Fig. 2a, b). FAN1 is monomeric in solution, but dimerizes in the presence of DNA [28]. Human FAN1 forms an asymmetrical ‘head-to-tail’ FAN1 dimer on DNA (Fig. 2b) [41]. Dimerization relies on a region of the SAP domain of one FAN1 molecule interacting with the TPR and NUC domains of a second FAN1 molecule [41]. The first SAP domain defines the DNA substrate orientation, whereas the TPR and NUC domains of the second FAN1 facilitate the nucleolytic reaction by engaging the cognate DNA in the catalytic site [41] (Fig. 2b). While FAN1 dimerization endows the protein with its specific endo-nuclease activity, dimerization is not absolutely required for its exo-nucleolytic function [41]. The FAN1 dimer-DNA crystal structure displays three different DNA binding modes (’substrate-scanning’, ‘substrate-latching’, and ‘substrate-unwinding’), which determine distinct mechanisms for DNA cleavage [41]. Along this line, FAN1 homodimers process long (40 bp) but not short flap (1-5 bp) DNA substrates [42]. Besides their role in tethering and orienting the DNA substrate for subsequent cleavage, these homodimers possibly evolved to bind DNA in a conformation that simultaneously allows interactions with PCNA, FANCD2-FANCI and other proteins (Fig. 2c). It would be interesting to elucidate the contribution of the endo- rather than the exo-nuclease activity in the processing of damaged DNA, by employing dimer-defective FAN1 variants (K525E/R526E/K528E triple mutant or a 510-518 deletion) [41, 42]. Curiously, FAN1 dimerization is unique to higher eukaryotes [28, 41].
FAN1 was initially detected in a proteomic analysis of MLH1- and PMS2-interacting proteins, undertaken to gain novel insights into the function of MMR [4]. Individual interactions between FAN1 and MutL homologs were subsequently validated using reciprocal immunoprecipitations and size-exclusion chromatography [5, 7]. It is currently unknown whether complex formation between FAN1 and MutLα, MutLβ, or MutLγ heterodimers is mediated via their common MLH1 subunit or via a common interaction motif in PMS2, PMS1 and MLH3. MutL-binding region(s) in FAN1 have not yet been mapped. MLH1 is not required for nuclease activities of FAN1 in vitro [6]. The FAN1-MLH1 interaction may play a structural role, which potentially determines DNA cleavage specificity. Finally, it has been proposed that FAN1 interacts with MSH2 following treatment with alkylating agents [43], but this finding has not been independently confirmed and direct interaction of FAN1 with MSH2 (or its partners MSH3 or MSH6) was not demonstrated.
3FAN1 AND DNA REPAIR-RELATED PROCESSES
Loss of FAN1 results in sensitivity to drugs that induce DNA crosslinks (e.g., MMC, chlorambucil, and platinum-based drugs) [5–8, 44], albeit far less (by at least 30-fold) than with deficiencies of either FANCA, FANCJ, or FANCQ (XPF/ERCC4) [6, 45, 46]. Moreover, FAN1 depletion leads to only a very mild (<5-fold) increase in spontaneous and MMC-induced chromoso-mal aberrations (chromatid breaks, exchanges, chro-mosome-type aberrations, multi-radials), compared to those in FA cells (up to 100-fold) [6, 45]. ICLs can be processed either in S-phase by replication-coupled pathways that require the NEIL glycosylase [47] or FA proteins [48, 49], or in a replication-independent manner involving TLS or MMR pathways [50, 51]. Both replication-dependent and replication-independent ICL repair pathways share several enzy-matic steps, which rely on the activity of structure-specific nucleases.
FAN1 can cleave ICL-stalled replication-like structures [29, 52] (Figs. 3 and 4a), indicating its role in S-phase-dependent ICL repair. There has been considerable effort to define the function of FAN1 in processing of DNA modified from endogenous or exogenous sources, but its precise role in ICL repair is still unclear. ICLs induced by exogenous compounds— such as cyclophosphamide, cisplatin, melphalan, psoralens, diepoxybutane, or MMC— have been used as proxies for the repair of endogenous ICLs caused by common metabolites, such as formaldehyde and acetaldehyde (including that derived from ingested alcohol), and oxidized lipid species [7, 53, 54]. FAN1 can resolve a subset of ICL-containing structures, including those where the replication forks remain some distance from the damage [52] (Figs. 3 and 4a), thereby generating a suitable substrate for subsequent nicking by a 3’ flap endo-nuclease (such as XPF-ERCC1) and DNA synthesis by TLS polymerases (Fig. 4b). Biochemical evidence suggests that the primary function of FAN1 in ICL repair lies in its ability to ‘unhook’ the DNA lesion [55]. Although FAN1’s nuclease activity is well suited for ICL incision in vitro [29], its depletion in Xenopus egg extracts does not lead to any incision defects [56]. This discrepancy might reflect a difference in the chemical nature of the lesions studied. In the replication-dependent ICL repair system of Xenopus, unhooking only occurs when replication forks are in very close proximity (at least 5 nucleotides) to the crosslink: an unfavorable scenario for FAN1 [52, 56]. Alternatively, immune depletion of FAN1 in Xenopus egg extracts may not cause discernable defects in ICL incision because of redundancy with other structure-specific nucleases (for review, see [57]). Thus, in mice, Fan1 is non-epistatic to Slx1 [58]— a nuclease that can process crosslinks by promoting the endo-nucleolytic cleavage of a wide range of branched structures, including 5’-flaps (Fig. 3). However, while Slx1 processes ICL-containing structures at the DNA branch point [59], FAN1 can incise both the single-stranded DNA (ssDNA) flap, and double-stranded DNA (dsDNA) region downstream [7, 28, 41]. FAN1 and SLX1 may cleave distinct ICL-containing DNA sequences, or they may operate at different steps of ICL repair, with SLX1 playing a more prominent role in the unhooking step. FAN1-mediated cleavage of ICL-containing substrates in vitro is reminiscent of the activity of SNM1A, the human homolog of Pso2, a structure-specific nuclease involved in ICL repair in S. pombe [60]. To date, SNM1A and FAN1 are the only established nucleases that can traverse crosslinks and exhibit both 5’-flap endo-nuclease and 5’-3’ exo-nuclease activities [53, 61]. Human SNM1A complements the ICL sensitivity in yeast that lack Pso2 [60], but its deficiency confers very mild crosslink sensitivity in mammalian cells [32], probably because of the redundancy with FAN1. Accordingly, yeast Fan1 and Pso2 are non-epistatic in terms of cisplatin sensitivity [40] and Snm1a partially compensates Fan1 loss in mouse cells [32].
Fig.4
FAN1 at the cross-road of genome maintenance pathways. (a) FAN1 promotes FA pathway-independent repair of ICLs. FAN1 catalyzes ICL unhooking by endo-nucleolytic incision followed by exo-nucleolytic digestion of DNA past the lesion, thereby generating a substrate for TLS polymerases (ICLs are indicated by red box). (b) FAN1 interacts with MutLα and may function as a compensatory nuclease in MMR in the absence of EXO1. (c) FAN1 promotes PCNA mono-ubiquitylation on the K164 residue. Further modification of the same residue via K63-linked polyubiquitination is then required for the recruitment of the ZRANB3 translocase and fork reversal, an error-free damage tolerance pathway. (d) FAN1 binds ubiquitylated PCNA on K164 and activates a feed forward loop that enhances PCNA ubiquitylation and possibly TLS (aberrant DNA lesions are indicated by open triangles).
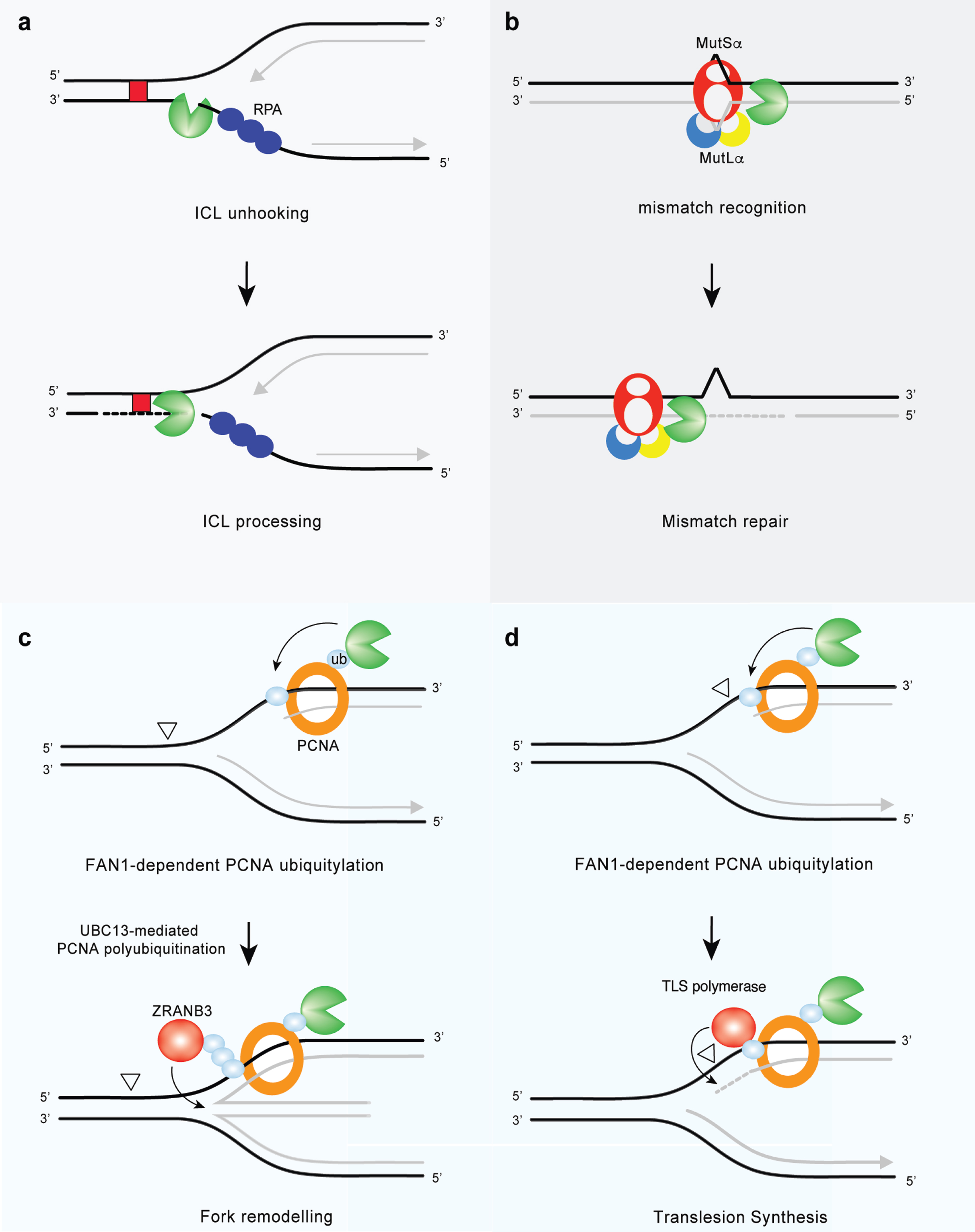
In addition to a direct role in ICL unhooking, it has been speculated that FAN1 may act on HRR intermediates [6, 7] that form during the last step of the ICL repair process (for review, see [62]). HRR requires the formation of a DNA double-strand break (DSB), followed by DNA end resection by the MRN-RPA-BLM-DNA2-EXO1 machinery [63]. Then, the BRCA1-PALB2-BRCA2 complex [64] allows assembly of the RAD51 nucleofilament in order to promote a homology search in the intact sister chromatid. While FAN1 seems not to be required for DSB resection [7], its deficiency leads to a delay in the disappearance of RAD51 foci [6, 7], indicating a role in the late stage of HRR. Alternatively, ICLs can result in fork collapse, generating one-ended DSBs that require break-induced replication (BIR) (a sub-pathway of HRR) for fork restart (for review, see [62]). Thus, the lagging strand 5’ to the ICL provides a potential substrate for FAN1 endo-nuclease activity, which can create a nick and further promote the trimming of the nascent DNA. This generates a 3’ ssDNA overhang, required for resumption of DNA synthesis [41, 52]. While FAN1 can cleave ICLs in vitro, its role in the cellular response to MMC or other crosslinking agents remains unknown. Therefore, it is of fundamental importance to functionally delineate FAN1-mediated ICL processing in cells and in vivo.
FAN1-mediated processing of damaged DNA relies on its accurate recruitment to the site of the lesion. Although FAN1 structure might be crucial for its substrate preference, scaffold proteins are required to secure its timely localization. Initially, FAN1 has been associated with the FA pathway because of its binding to ub-FANCD2— a central component of the FA machinery. The FAN1-FANCD2 interaction mediates FAN1 recruitment to sites of ICLs and has been thought to be crucial for ICL repair. This long-held belief is now challenged by the evidence that cells expressing the UBZ-mutated form of FAN1 are not hypersensitive to MMC [30, 32]. Thus, the contribution of the UBZ domain in FAN1 to ICL repair, if any, remains largely unknown. It may bypass the requirement of the FA proteins and become crucial for ICL repair in a context where the FA pathway is inactive. Along this line, FAN1 deficiency enhances ICL-induced genomic instability in FANCD2 null cells and further sensitizes FANCA-deleted cells to MMC [32]. Chicken DT40 cells depleted of both FAN1 and FANCC are more sensitive than wild-type and single-knockout cells to cisplatin [44]. In addition, kinetics of FAN1 foci formation is largely unaffected in PD20 cells that lack FANCD2 [32]. Together, these findings suggest a role for FAN1 in ICL repair that is outside of the FA pathway, explaining why FAN1 mutations have not been identified in unclassified FA patients. The chemical nature of ICLs, the resulting DNA distortion, and the location of the damage will influence the mode of its repair. Thus, a role for FAN1 might be in the formation of a one-ended DSB, only for the subset of forks that do not physically encounter the cross-link and do not activate the FA core complex. Intriguingly, FAN1 appears earlier than the FA pathway on the evolutionary scale. A FAN1 homolog lacking a UBZ domain is indeed implicated in the repair of ICLs in unicellular eukaryotes, such as fission yeast (S. pombe), that do not possess the FA machinery [40]. We thus speculate that the FAN1-FANCD2 interaction is not directly related to the repair of DNA crosslinks, and that an as-yet unidentified factor is responsible for mobilizing FAN1 to sites of ICL in a UBZ-independent manner. Localization of FAN1 at sites of ICL damage is biphasic, with an initial rapid recruitment of the protein followed by a slower but steady accumulation, which coincides with FANCD2 binding [32]. While the retention of FAN1 at ICLs depends on its UBZ domain and is not associated with ICL sensitivity, the first, very fast phase of this process apparently confers resistance to ICL-inducing drugs and relies on the FAN1 SAP domain [32]. However, it is very unlikely that the direct interaction of FAN1 with DNA can specifically drive the protein to ICL sites in a timely manner. Since the SAP domain is also involved in FAN1 dimerization, mutations in this module can perhaps perturb FAN1 dimer structure and rewire interaction networks, ultimately affecting FAN1 recruitment.
A direct role for FAN1 in post-replicative MMR seems apparent, based on its interaction with MutL homologs MLH1 and PMS2, together forming the MutLα heterodimer [4] (Fig. 2c). The MMR system detects and removes non-Watson-Crick base pairs in both mitotic and non-mitotic cells. The absence of MMR in states of high proliferation leads to high levels of mutations [65]. Thus, functional MMR is essential to ensure the fidelity of DNA replication but also for genome integrity in non-mitotic cells. Cells treated with the alkylating agent, N-methyl N-nitrosourea (MNU), accumulate FAN1 nuclear foci in an MLH1-dependent manner [43]. Authors of this study propose that FAN1 exo-nuclease generates ssDNA to activate a DNA damage response, ultimately triggering apoptosis [43]. To formally prove a role of FAN1 in MMR, one should monitor the rate of G:C-to-A:T transversion mutations in a FAN1-deficient background following MNU treatment. Independent studies reported that FAN1 inactivation causes no evident defect in MMR [5, 66] in normal settings, but that FAN1 may be one of several redundant MMR nucleases, which may include (but may not be limited to) EXO1, MRE11, Artemis, FEN1, and XPF [66]. Thus, FAN1-deficiency may contribute to a partial MMR defect only in certain genetic backgrounds.
MMR complexes are implicated in recognition and processing of ICLs and other forms of chemically damaged DNA, in addition to mismatched bases. In particular, MutSα (MSH2-MSH6) binds to ICL-containing plasmids and promotes recruitment of MutLα [51], whereas MutSβ— which has higher specificity for larger insertion-deletion loops (IDLs)— associates with psoralen-induced ICLs [67]. Recent reports suggest a role for the MMR machinery upstream of the FA pathway in the mono-ubiquitination and chromatin localization of FANCD2 [68]. Thus, it is conceivable that the FAN1-MLH1 complex might be recruited to sites of ICLs in S-phase at an early step that precedes FANCD2 localization. Future experiments should address whether FAN1-MLH1 interaction is required for the FAN1-dependent ICL repair, analogously to what has been proposed for the DNA helicase FANCJ [69, 70]. In this respect, it will be crucial to test whether MutLα can act as a bridge allowing FAN1 nuclease to pair with the FANCJ helicase during ICLprocessing.
4FAN1 AND DNA REPLICATION STRESS
Replication stress is defined as perturbations in replication dynamics that alter fork progression. A number of DNA regions in the genome are intrinsically difficult to replicate and might represent a hurdle for the DNA replication machinery. For example, repetitive DNA sequences— such as ribosomal DNA, telomeres, GC-rich regions and expanded trinucleotide repeats— can assemble into secondary DNA structures that are thought to impede fork progression or cause replication slippage. Other DNA regions with low density of replication origins, such as common fragile sites, are prone to form gaps and stretches of ssDNA; thus, they are tied to replication stress. Heavily transcribed regions display elevated levels of stress because of increased probability of a clash between replication and transcription machineries. In addition to these endogenous sources, cells may experience various forms of exogenous DNA damage that perturb replication fork progression. Beyond its role in ICL repair, FAN1 contributes to DNA replication and is associated with replication stress responses (Fig. 4c, d). The first evidence linking FAN1 to replication fork processing implicates its nuclease activity in the restart of forks stalled by aphidicolin (APH), a specific inhibitor of B-family DNA polymerases [71]. At sites of APH-stalled replication forks, FAN1 associates with FANCD2 and the BLM helicase in a trimeric complex to promote fork recovery and suppresses new origin firing. In this context, the UBZ domain of FAN1 appears to be dispensable, whereas its chromatin localization requires non-ubiquitylated FANCD2 [71]. Upon fork stalling, FANCD2 targets the replisome by establishing a transient interaction with the replicative MCM helicase, independently of its mono-ubiquitylation by the FA core complex [72]. Non-ubiquitylated FANCD2 may grant FAN1 access to the replication machinery and permit fork restart in a timely manner. In the absence of FANCD2, FAN1 can be still mobilized to stalled forks and drive extensive nucleolytic processing of nascent DNA strands [71]. Thus, the major role of the FAN1-BLM-FANCD2 trimeric complex may be to restrain FAN1 nuclease activity and ensure appropriate processing of stalled forks. In particular, FANCD2 might limit FAN1 activity at stalled forks by stabilizing RAD51-DNA filaments [73]. The role ascribed to FAN1 in fork metabolism is reminiscent of that attributed to DNA2— another nuclease involved in fork restart [74], which can mediate over-resection of nascent DNA strands if not kept in check [74, 75]. FAN1 localization at APH-stalled forks also relies on MRE11’s 3’-5’ exo-nuclease activity [71], which can reshape the fork into a suitable substrate for FAN1. Besides its role in fork restart, FAN1 nuclease activity is required to restrain replisome progression and preserve genomic integrity, under conditions of dNTP shortage due to hydroxyurea treatment [30]. Surprisingly, the UBZ domain, which is dispensable for FAN1-mediated fork restart, appears to be crucial for limiting fork progression and chromosomal instability following replication stress [30]. Likewise, the FAN1 interaction with ubiquitylated-PCNA is required to limit the rate of DNA synthesis under replicative stress conditions [34]. Further investigation is needed to establish how FAN1 can, at the same time, limit fork progression and promote fork restart. Like FAN1, the HLTF translocase [76] or the RAD51 recombinase [77] can restrain DNA synthesis during replication stress by promoting fork remodeling— a process that converts replication forks into four-way structures and back. Replication fork reversal allows the replisome to bypass UV-damaged templates [78] and represents a pathway that allows DNA replication to pause and resume once the damage is repaired (for review, see [54]). Thus, it is tempting to speculate that FAN1-dependent processing of stalled forks might operate at the initial step of fork reversal by promoting localization of the SNF2 family DNA translocases— such as ZRANB3, HLTF and/or SMARCAL1— whose activity has been involved in fork regression [79]. In particular, ZRANB3 fulfills its functions by binding poly-ubiquitylated PCNA. In response to replication stress, FAN1 activates a feed-forward loop able to enhance levels of mono-ubiquitylated PCNA, providing potential substrates for poly-ubiquitin chain extension. The latter will ultimately help ZRANB3 to engage replication factories efficiently and promote regression of the stalled fork. Future research should aim to prove this model and determine whether FAN1 accomplishes its replicative function by promoting fork reversal (Fig. 4c). On the other hand, given that the formation of reversed forks places the DNA lesion back into the context of dsDNA, we cannot rule out a role for FAN1 in the incision of the region flanking the damage, analogous to its putative function in ICL repair.
Alternatively, FAN1 can channel stalled forks into the TLS pathway, allowing replication through a damaged template with low processivity and fidelity (Fig. 4d). FAN1 might indeed process forked DNA in a way that generates RPA-coated ssDNA: a pre-requisite for the recruitment of the E3 ligase, RAD18, and subsequent PCNA ubiquitylation [80]. Ubiquitylated -PCNA can enable mobilization of TLS DNA polymerases, which bypass bulky lesions in a post-replicative manner (Fig. 4d). The latter may account for the limited fork speed observed in FAN1-proficient cells upon replication stress induction. On the other hand, permissive replication detected in FAN1-deficient cells might be associated with extensive repriming and/or unscheduled origin firing. Both of these processes lead to an excess of ssDNA gaps that, if left unreplicated, might overwhelm the post-replicative repair mechanisms in G2, generating DSBs and chromosomal instability. Thus, a key objective for future research will be to investigate whether unrestrained replication in FAN1-deficient cells generates S1-nuclease-sensitive tracts, which would indicate ssDNA regions. Moreover, it will be important to explore the role of FAN1 in replisome surveillance in cells lacking either PrimPol— which has emerged as a key factor in re-priming [81]— or Cdc7 kinase activity, which otherwise stimulates dormant origin firing [82]. Such studies will help to define whether FAN1 may serve as a molecular switch to balance fork reversal, TLS, or re-priming events during replication stress.
5FAN1 DEFICIENCIES AND VARIANTS IN GENETIC DISEASES AND CANCER
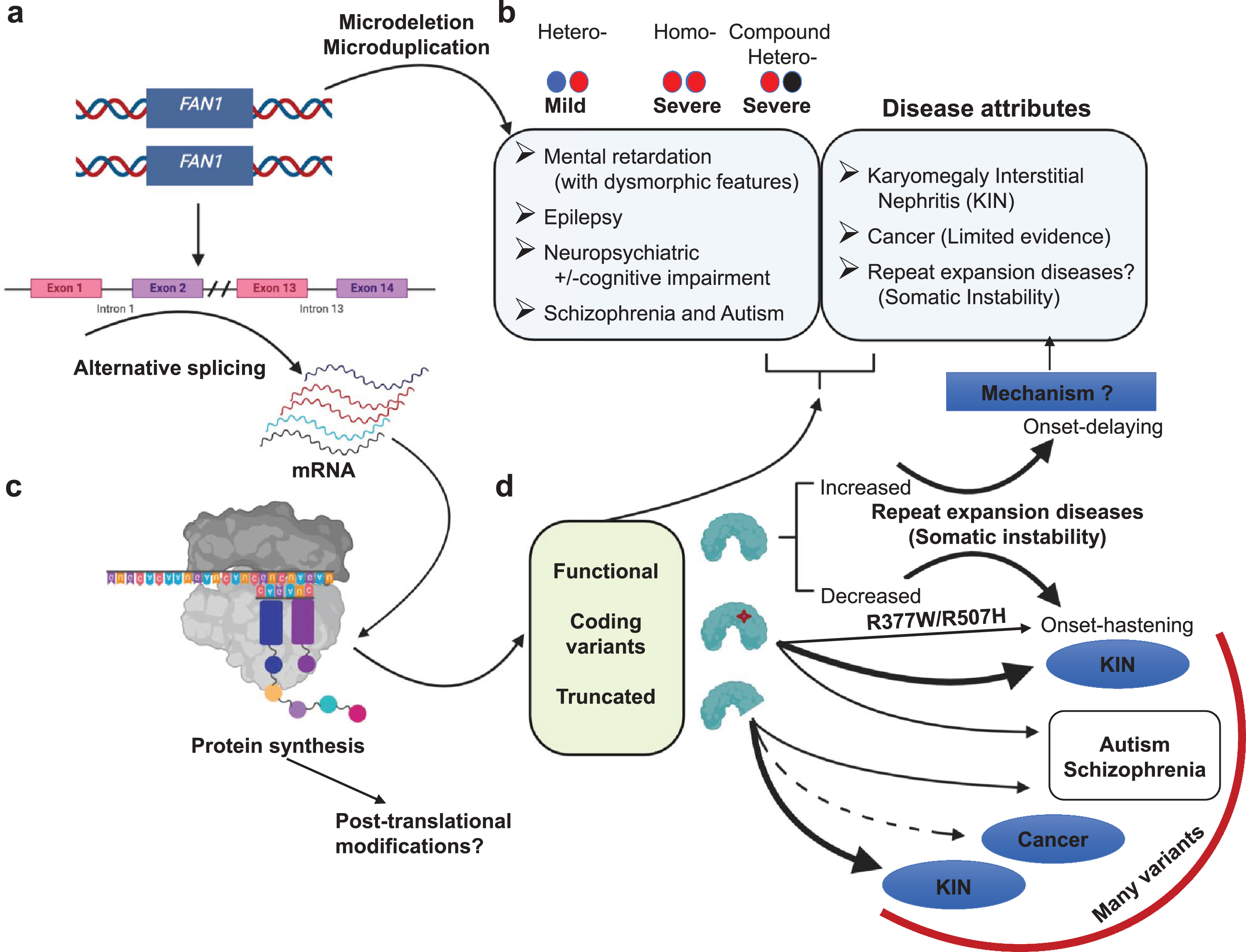
Many FAN1 variants have been associated with various diseases, including CAG/polyQ-diseases, karyomegalic interstitial nephritis (KIN), cancer, autism spectrum disorder, schizophrenia, epilepsy and Rett syndrome [14, 25, 31, 83–86] (Fig. 5). The relative contribution of FAN1 variants to each of the associated phenotypes is unknown. The connection of FAN1 to KIN is by far the strongest, possibly followed by the modifier association with HD. Although ranking the strength of the other disease associations with FAN1 is difficult, we have approximated them by the size of the arrows in Fig. 5. The weight of the data supporting these associations varies, as discussed below. These variants and associated clinical presentations are summarized in Table S1, with variants pertinent to this review highlighted in Fig. 1b and d. The mechanism by which any of these variants contribute to the various clinical presentations is unclear but discussed in the sections that follow.
Fig.5
Overview of FAN1 genetics and its associated diseases. (a) Heterozygous microdeletion in the FAN1 gene is associated with a mild form of 15q13.3 microdeletion syndrome. Whereas homozygous microdeletion or compound heterozygous microdeletion is associated with increased severity. Microduplications can be associated with various symptoms. (b) Defects in FAN1 identified in various diseases (c). These defects may be due to alternative splicing. It may generate aberrant transcript, which further on translation produces either functional protein, coding variants, or truncated FAN1 protein. (d) Various forms of FAN1 proteins are identified in multiple diseases such as truncated protein and coding variants are identified in various cancers, KIN, schizophrenia, autism, and some disease hastening coding variants identified in HD and SCAs. The relative strength of these associations varies widely and are discussed in the text. Arrows that are bold, medium and dotted denotes strong, moderate and limited evidence for the disease association, respectively. The mechanism associated FAN1 role with all of these diseases is unknown.
5.1Karyomegalic interstitial nephritis (KIN; OMIM#614817)
Bona fide autosomal recessive mutations in FAN1 lead to KIN (Fig. 5), a rare hereditary genetic disorder that leads to chronic interstitial nephropathy, first described more than four decades ago [87–89]. To date, more than 50 cases have been reported (reviewed by Isnard et al. [90]). KIN usually presents as a slowly progressing chronic kidney disease leading to end stage renal dysfunction in early adulthood [91]. The analysis of renal biopsy specimens usually shows non-specific but severe chronic interstitial fibrosis and tubular changes, associated with non-specific glomerulosclerosis and vascular lesions. Genetic deficiencies of FAN1 have been identified in individuals affected by KIN [31]. Parenchymatous organs, such as liver, and kidney, neuronal tissue, and reproductive tissues show high levels of FAN1 expression [31], thus explaining why FAN1 variants might cause KIN (Supplementary Table 1). The disorder is characterized by fibrosis of renal tubules; it differs from other forms of chronic kidney disease by virtue of the pronounced enlarged polyploid nuclei in the renal epithelium, rarely observed in other organs [92–95]. The source of the organ specificity of KIN is obscure. Kidney cells from KIN individuals showed > 84% spontaneous hyperploidy; most were hyper-tetraploid 8-16C, but some had as many as 81 genomic equivalents [91, 96]. Consistently, Fan1-deficient mouse models recapitulate hallmarks of KIN pathology and also display liver function abnormalities, with hepatocytes showing increased ploidy [32, 58, 97]. Zhou et al. studied ten families with KIN [31] and identified 12 different mutations of FAN1 in 9 of the families (Supplementary Table 1). Eight of the 12 mutations truncate the nuclease domain and three missense mutations (p.Q929P, p.G937D and p.D960N) are in evolutionarily conserved amino acid residues of the FAN1 nuclease domain [31]. They found none of these mutations in 96 healthy individuals. Consistently, mice expressing a truncated form of Fan1, devoid of its nuclease activity, develop a mild form of KIN [58]. Analysis of medical records showed that the p.R507H variant of FAN1 associated with KIN in population cohorts, contributing in unknown ways [98]. It is notable that multiple individuals with homozygous 15q13.3 microdeletions encompassing FAN1 do not show signs of KIN; rather they present with global developmental delay or intellectual disability, and severe hypotonia [23, 45, 99–106]. A major open question in the pathophysiology of KIN concerns the nature of the lesions repaired by FAN1. Polyploidy (up to 8C) observed in Fan1-deficient mice increases during development or in response to ICL-inducing agents, but not to hydroxyurea [32, 58], suggesting that increased ploidy arises by a defect in ICL repair or in the processing of ICL-stalled replication forks. The increased ploidy might be a direct consequence of re-duplication of the whole genome or might reflect the presence of portions of the genome that are under-replicated. Either way, the appearance of polyploidy implicates replication cycles not followed by accurate mitosis. The ICL-induced polyploidy is independent of FANCD2 [58].
5.2Cancer
FAN1 deficiency or variants are associated with cancer (Supplementary Table 2), however, FAN1 is not a FA gene per se. Individuals carrying FAN1-deficiencies do not present with the clinical attributes typical of FA patients (i.e., anemia, bone marrow failure, skin pigmentation anomalies, skeletal abnormalities of the upper limbs, or spontaneous leukemia) [45]. Aged mice carrying a defective Fan1 nuclease domain do eventually develop pulmonary carcinomas and hepatic lymphomas [30]. Fan1-deficient mice do not display discernable defects in bone marrow histology nor attrition of the hematopoietic stem cell compartment [30]. The p.M50R of FAN1 increases risk of colorectal cancer (CRC) [107] and occurs as germline mutation in human hereditary pancreatic cancers [33]. Heterozygous germline FAN1 mutations in MMR-proficient families suggest that it may be a CRC susceptibility gene [108]. A meta-analysis suggested that the contribution of FAN1 to CRC is doubtful, as the evidence of segregation is weak (p = 0.125), with no somatic mutations of FAN1 in tumors from any of the FAN1 mutation carriers [108, 109]. Consistent with this, another meta-analysis raises serious concerns about the role of FAN1 in cancer progression and predisposition [85]. Among > 5000 cancer patients clinically suspected as having genetic predisposition to colorectal, ovarian, breast, and other cancers, monoallelic pathogenic FAN1 variants were identified in 0.35% . However, the expected frequency of heterozygous mutations in patients and the general population is also not significantly different [85]. No CRC was reported in the first- or second-degree relatives of any of the 16 index cases with pathogenic FAN1 variants. No cancer was identified in close blood relatives of four index cases with KIN carrying biallelic germline mutations of FAN1 nor in patients with homozygous 15q13.3 microdeletions encompassing FAN1 [85, 104]. Larger CRC series could provide more information about the prevalence of FAN1 pathogenic variants. A thorough analysis of genetic and genomic alterations found in FAN1-associated tumors will be important to clarify the underlying repair defects accumulated by these cells and mechanism of action of FAN1 in colorectal carcinogenesis. Interestingly, FAN1 may acquire a key role in cancer cells lacking BRCA2 [34]. BRCA2 is an important factor of HRR and acts as reservoir of the RAD51 recombinase [64, 110–113]. Deposition of RAD51 by BRCA2 not only facilitates HRR but has a fork-protective function by preventing replication stress in a manner distinct from HRR [114]. FAN1 limits replication stress and genome instability in BRCA2-deficient cells [34] and may provide a protective mechanism in cancer cells lacking BRCA2, ultimately affecting cell survival and treatment responses. In light of these data and resolution of the FAN1 crystal structure, FAN1 inhibitors could be developed for treatment of BRCA2-deficient tumors. Finally, increased FAN1 expression correlates with poor prognosis of breast and ovarian cancers that are refractory to therapy [115]. Therefore, FAN1 might be a novel promising target for cancer therapy or be used as potential biomarker to predict the insurgence of drug resistance in breast and ovarian cancer.
5.3Neurodevelopmental and neuropsychiatric diseases
FAN1 copy number variations (CNVs), via deletions and duplications of the 15q13.3 region, are associated with autism, schizophrenia, and epilepsy [21, 25, 83, 84] (Fig. 5). These neurodevelopmental, psychiatric, and neurological disorders, respectively, share genetic predisposition factors [21, 25, 83, 116, 117]. Whole exome sequencing of autism patients and whole genome sequencing of schizophrenia patients identified various variants including FAN1 missense variants associated with both autism and schizophrenia [25, 83, 84] (Supplementary Table 2). Likewise, recurrent FAN1 CNVs predispose to an array of psychiatric and neurodevelopmental disorders [116, 117]. A homozygous knock-out mouse model involving the ortholog to a 1.5 Mb region of chromosome 15q.13.3 spanning Fan1 and seven adjacent genes displays strong phenotypes related to autism, schizophrenia, and epilepsy [21]. However, the degree to which these phenotypes are related to CNVs of FAN1, as opposed to any of the adjacent genes, is unknown.
Soon after the discovery that myotonic dystrophy and HD are caused by expanded tandem DNA repeat sequences, it was hypothesized that autism spectrum disorder (ASD), schizophrenia (SCZ), and epilepsy might also be caused by repeat expansions [118–120]. Indeed, various forms of epilepsy, SCZ, and— most recently— ASD are associated with repeat expansions [121–126]. Strikingly, Yuen and colleagues recently broadened, by leaps and bounds, the awareness of both the sequence motifs and the number of repeat loci that can be associated with a neurological phenotype [122]. FMR1 CGG expansions cause a spectrum of clinical presentations, including fragile X syndrome (FXS), autism, fragile X-associated ataxia, premature ovarian failure/insufficiency, attention-deficit disorder, learning disabilities, as well as psychological, endocrine, autoimmune, and metabolic disorders [127]. Other fragile sites caused by CGG expansions have also been associated with ASD. Ionita-Laza et al. reported numerous variants in FAN1 as modifiers of ASD and SCZ [25], including p.R377W and p.R507H, which were subsequently identified as modifiers of onset in HD [14, 15]. The role of FAN1 in any of these disorders and the underlying mechanisms by which it might mediate pathological effects need further investigation.
A coding variant of the murine Fan1 is a modifier in Rett syndrome mice [86]— a neurodevelopmental autism-like disorder that occurs primarily in females. We feel that this finding is worth some attention for future studies in humans. Rett syndrome is caused predominantly by mutations in the Methyl-CpG-binding protein 2 (MECP2) gene; “atypical” Rett syndrome can result from variants in FOXG1 or CDKL5 [128–130]. A recent study screened for modifiers that improve subjective phenotypes in Mecp2/Y mice after mutagenesis with N-ethyl-N-nitrosourea [86]. Phenotypes assessed included limb clasping, tremors, body weight, activity, and longevity. Amongst several pathways, many of the identified modifier variants were in DNA damage response genes (Birc6/BRUCE, Brca1, Brca2, Fan1, Mre11a, Rad50, Rbbp8/Ctip, Tet1, and Spin1) [86] providing the first instance by which this mechanism may contribute to Rett syndrome. Absence of MECP2 is associated with the accumulation of DNA damage [131]. The Rett-modifier in murine Fan1 corresponds to a S798P change in human FAN1 (Fig. 1), which is in between a highly conserved TPR and nuclease domain. How this Fan1 variant modifies the Mecp2-deficient phenotypes, and whether such modifications extend to humans with Rett syndrome, requires furtherinvestigation.
6GENETIC MODIFIERS OF CAG/POLYQ EXPANSION DISEASE ONSET
Certain neurodegenerative diseases are caused by the expansion of gene-specific CAG repeat sequences that encode polyglutamine (polyQ) tracts. Such diseases include HD (OMIM#143100), spinocerebellar ataxia type 1 (SCA1 OMIM#164400), SCA2 (OMIM#183090), SCA3 (OMIM#109150), SCA6 (OMIM#183086), SCA7 (OMIM#164500), SCA17 (OMIM#607136), and others. The age of onset of symptoms correlates with the length of the inherited CAG tract at the disease locus [132]. For the same inherited CAG size a broad range of onset age can occur, but less so within a given family [133], suggesting genetic modifiers of disease onset, independent of CAG tract size, must exist. Furthermore, Wexler et al. showed that 40% of the variance in age at onset in HD not attributable to CAG length, was heritable and therefore genetic [134].
The most robust evidence of DNA repair gene variants— including in FAN1— as modifiers of disease is for HD, where genome-wide analyses involve the greatest number of patients (HD cohorts of ∼4000–9000), with strong clinical assessments (reviewed in [135]). The HD GWAS generated a statistical phenotype model, based upon HD subjects with 40–55 CAG repeats in the HTT gene in their blood DNA, to calculate the influence of the repeat length on age at onset of motor signs [15]. That study generated for each subject a “residual age at motor onset” value, which represents the difference in years between actual age at onset and that expected based upon the repeat size. These residual phenotype values were used to search the individuals genomes for genetic variations associated with HD age at onset, identifying tagSNPs that define the modifierhaplotype.
6.1FAN1 variants in CAG/polyQ expansion diseases hasten or delay disease onset
DNA repair genes, MLH1, PMS1, PMS2, MSH3, LIG1, RRM2B, and FAN1 act as modifiers of HD [14]. The strongest by far is FAN1, with both disease-hastening and disease-delaying tagSNP haplotypes [14,15] (Fig. 1b, d). Details about these HD modifier haplotypes are outlined here. For simplicity, we generally refer only to specific SNPs (rs#s), rather than to the haplotypes (please see Fig. 1d, and the review by Hong et al. [135]). Assessment of other diseases with CAG/polyQ expansion (SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17) also identified variants in FAN1 as the strongest modifiers [136–138]. Those that act as modifiers of age of onset, the haplotype size, tagSNPs, genic location, and expected and documented effects, are summarized in Fig. 1d. Notably, some FAN1 variants hastened or delayed disease onset by as much as ∼6 years from that expected for the inherited CAG expansion size.
6.2Modifiers may alter expression levels of FAN1 mRNA
The tagSNPs of FAN1 associated with modifying HD onset may be associated with altered expression of FAN1 mRNA, whereby increased or decreased FAN1 levels are beneficial or detrimental, respectively [12, 14, 15, 139]. Both expression quantitative trait loci (eQTL) and transcriptome-wide association studies [140] suggest that the SNPs are associated with changes of FAN1 mRNA expression levels in specific brain regions but have no apparent effect on the expression of proximal genes, including MTMR10 or TRPM1 [12, 141]. In some individuals with HD, the onset-delaying FAN1 modifier overrides the effect of the onset-hastening haplotype on the other FAN1 allele [12]. These data support the concept that increased expression of FAN1 protein, and/or its functions, correlates with delayed onset and slower disease progression. For example, the tagSNPs rs35811129 and rs34017474 both correspond to cis-eQTLs for increased FAN1 expression in cortex [12, 14, 141] and are associated with delayed age of onset. However, the 15AM4 haplotype associated with tagSNP rs34017474 does not capture single modifier effects; rather, a combination of the effects of the 15AM1, 15AM2, 15AM3 and 15AM5haplotypes.
Expression studies provide a guide to how specific genetic variants might affect disease onset and progression but must be interpreted with caution. While the effects of FAN1 variants upon its own mRNA expression in the cerebral cortex appear to correlate consistently with suspected increased or decreased levels of FAN1 protein, other factors must be considered. Notably, these FAN1 expression studies use database (GTEx, CommonMind Consortium, and ROSMAP) from tissues of genetically defined individuals that were not affected with a CAG/polyQ disease [12, 14, 137, 141]. Individuals with HD have transcriptome-wide alterations, including in DNA repair genes, which must be accounted for [142, 143]. These dysregulated transcripts are evident in cell lines and patient tissues, and in many instances (but not all) are matched at the protein level [142, 143]. The size of the HTT repeat expansion can affect the degree of DNA repair gene dysregulation. For example, isogenic HD human pluripotent stem cells (hPSCs) with CAG lengths of 30, 45, 65, or 81 repeats, show increased expression of the DNA repair genes, FAN1, PMS2 and MSH3 at longer CAG repeat lengths [139]. Levels of the FAN1, PMS2, and MSH3 proteins are significantly lower in HD brains compared to age-matched controls [139]. Such expression level variations may change as disease progresses. Tissue-specific expression effects can be important variables. For example, in different tissues, the rs3512 FAN1 variant can have increasing or decreasing effects upon FAN1 mRNA expression levels [137]. In FXS mice, expression levels of Fan1 mRNA does not correlate to levels of Fan1-linked suppression of CGG expansions between the tissues [144]. Changes in mRNA expression levels do not always parallel changes in protein levels. None of the initial FAN1 expression studies assessed FAN1 protein levels in any tissue [12, 14, 137, 141]. However, in human brains, FAN1 is decreased in the cortex of individuals with HD, while no significant difference is observed in the striatum [139]. Thus, haplotype-specific transcription level variations from non-HD databases are potent predictors of mechanism, but the expression level differences must be validated in the appropriate patient cells/tissues or experimental systems. Complexities of FAN1 expression on CAG instability are discussed further in Section 7.2.
Limited data from HD patients support the concept that altered levels of FAN1 correlate with disease onset. As noted above, CNVs of the 15q13.3 region could increase or decrease the levels of FAN1 and adjacent genes, which may affect disease. Some individuals with HD who carry three complete copies of FAN1 (due to duplication of chr15:30900000-31500000) have significantly delayed onset compared to those with two allelic copies [12]. This finding supports the concept that increased FAN1 levels can be protective for HD; however, one cannot exclude the possible contribution of increased levels of other genes in the triplicated region, nor the presence or absence of any associated variants. It is important to distinguish the possible contribution of CNVs relative to any identified disease-modifying single nucleotide variants. This is particularly important in regions prone to CNVs, such as 15q13.3, which encompasses FAN1. CNVs are not responsible for the effects of individual single modifiers in the region [12].
6.3FAN1 coding variants, DNA binding, and nuclease activities
Understanding the mechanism(s) by which the FAN1 coding variants may alter disease is of great interest. Current thinking is that FAN1 may modulate repeat instability [12, 141]: a concept supported by the hyper-instability of the expanded CGG repeat in somatic tissues of Fan1-deficient FXS mice [144]; however, biochemical evidence supporting this role is limited. So far there is no direct biochemical evidence of FAN1’s role in the binding and processing of DNA repeats. FAN1 can recognize various secondary structures in vitro, such as chemically crosslinked DNA and DNA flaps [5–7, 29, 52] (Fig. 3). Two variants of FAN1 that result in amino acid changes— p.R377W (rs151322829) and p.R507H (rs150393409)— are associated with hastening HD age of onset by 3.2 and 5 years, respectively [12, 14] (Fig. 1d). The p.R377W variant is proximal to Y374, which— along with Y436— contacts the DNA single-strand-duplex junction; these are required for appropriate DNA substrate orientation [29] but not DNA binding [41]. The missense mutation, Y374F, when coupled with Y436F, significantly impairs endo-nuclease activity on a 5’ flap [41]. The HD onset-hastening p.R507H variant is located within the SAP-containing domain implicated in DNA binding [41]. One might speculate that the FAN1 coding variants might affect the biochemical activities (DNA binding, protein dimerization, nuclease activity, etc.) of the FAN1 protein. The p.R377W and p.R507H FAN1 variants have decreased DNA binding to flapped-DNAs relative to that of the wild-type (wt) FAN1 [12]. Notably, this study used an indirect streptavidin-biotin-oligo pull-down method, with a crude nuclear extract from FAN1 knockout HEK293T cells over-expressing transfected FAN1 [12]. Binding constants were not calculated. The reduced DNA binding by the variants was evident whether the substrate contained a (CAG)10 loop-out or was devoid of repeats [12]. Binding activity for the FAN1 variants was assessed for only CAG (not CTG) slip-outs, and nuclease activity was not assessed. It is notable, that several nuclease-dead FAN1 mutants have DNA binding activities that do not reflect their absent nucleases activities. For example, the nuclease-dead mutant FAN1p . D960A, retains DNA binding capacity comparable to FAN1p . wt. In contrast, the FAN1p . K525E/R526E/K528E shows undetectable DNA binding, undetectable endo-nuclease activity, but exo-nuclease activity comparable to wildtype FAN1 [6–8, 41].
FAN1 does not preferentially interact with the expanded CAG tract of the endogenous mutant HTT gene, as measured with chromatin immunoprecipitation (ChIP) [141]. No difference in FAN1 binding could be detected by ChIP between FAN1 wt/wt or p.R507H/wt HD cells. FAN1 binding assessed by ChIP at different genes with CAG, CTG, or CGG repeat tracts at other disease related genes (TBP, ATXN3, DMPK, or FMR1) is also not enriched at the expanded HTT CAG repeat nor any of the other non-expanded repeat genes. It is possible that ChIP is unable to detect subtle differences in DNA substrate preference, or between FAN1wt/wt and p.R507H/wt.
Nucleases can be classified as sequence-specific or structure-specific. DNA nucleases cleave the phosphodiester bond, yielding one cleavage product containing a 5′-terminal phosphate and one containing a 3′-terminal hydroxyl group. Nucleases can be further classified as endo-nucleases— which hydrolyse internal phosphodiester bonds— or exo-nucleases, which hydrolyse from the 5′ or 3′ ends of nucleic acids. Some nucleases are either endo- or exo- or can have both endo- and exo-nucleolytic activity. FAN1 is a structure-specific DNA nuclease with both endo- and 5′ → 3′ exo-nuclease activity.
The nuclease activity of FAN1 was reported as not necessary for FAN1’s effect upon CAG/CTG instability [141]. To examine FAN1 function in repeat instability, FAN1 knockout U2OS cells with a stably integrated (CAG)118 construct, were reconstituted with FAN1p . wt, nuclease-dead FAN1p . D960A, or FAN1p . R507H [141]. Repeat expansion was significantly slower in all FAN1 reconstituted lines compared to that in FAN1 knockout cells. There was no difference in CAG expansion rates between cells expressing the FAN1p . wt and those with either the FAN1p . R507H or the nuclease-dead FAN1p . D960A mutant. Thus, neither the R507H nor the nuclease activity had any effect on CAG instability. Expression of the FAN1p . D960A mutation on a p.R507H background did not affect CAG expansion rate relative to that of the catalytically active FAN1p . R507H. These results led those authors to conclude that FAN1 “nuclease activity is not required for protection against CAG repeat expansion ... FAN1 affects somatic expansion of the CAG repeat through a nuclease-independent mechanism.” [141]. This is a counter-intuitive finding, as all FAN1 biological functions characterized to date depend on its nuclease activity [29, 32, 52, 97]. The exogenous repeat assay system and/or the induced overexpression of exogenous reconstituting FAN1 genes may not have been sensitive enough to “reveal’ a subtle contribution from the nuclease activity. It is possible that the proficient DNA binding activity of the nuclease-dead FAN1p . D960A [41] may, with its over-expression, in some way suppress CAG instability, as opposed to the expected enhanced CAG expansion observed in cells lacking FAN1. Therefore, FAN1 nuclease activity could be more directly assessed via targeted mutation of the endogenous FAN1 nuclease domain in cells or an animal model.
One HD individual with a heterozygous rare mutation truncating the FAN1 nuclease domain (rs184745027; p.R952*), had disease onset 11.7 years earlier than expected from CAG repeat length [12]. Biochemical experiments of FAN1 acting on repeat DNAs, additional cellular, and in vivo model system data are needed to determine the roles of the known FAN1 activities in modulating repeats and disease. It is worth noting that Exo1, like Fan1, suppresses CGG expansions, where Exo1 acts mainly in the gonads and Fan1 affects expansions in somatic tissues, but less so in the germline. Exo-/- did not affect CAG instability in brain, liver, kidney, or heart, but did in intestine. In contrast, Fan1-/- affected both CGG and CAG instability in each of those organs, but not in the germline transmitted lengths. Interestingly, the Exo1p . D173A mutant, which abolishes its nuclease activity but retains its native conformation and DNA binding activity, shows a partial suppression of transmitted CGG expansions, intermediate between Exo1 +/+ and Exo1 -/- animals [144]. Thus, in the mouse germline, this supports suppression of CGG expansions by Exo1 via both nuclease-dependent and nuclease-independent pathways.
7FAN1 IN REPEAT INSTABILITY
7.1Candidate nucleases in repeat instability; questionable involvement of Flap endonuclease 1 (FEN1)
There must be a DNA nuclease involved in the process of dynamic disease-associated repeat expansions and contractions; however, it is unclear which one is key. Candidates that have been tested include both endo- and exo-nucleases that recognize unusual DNA structures [145]. These include Rad27/Fen1/FEN1, DNA2, RecJ, Exo1/EXO1, SbcCD/Rad50/Mre11, Mus81-Eme1, APE1, GEN1, ERCC1-XPF, XPG, and WRN, which have been tested for a possible role in repeat processing or instability; in vitro assays and models range from bacteria, yeast, fruit flies, and mice, to human cells [146–193]. Considerable effort has been invested into FEN1, the Flap endo-nuclease 1 and its possible role in disease-associated repeat instability. In yeast, deficiencies of Rad27— the yeast FEN1 homolog— increase CAG repeat expansions and contractions [149]. Yeast show a CAG contraction bias, in contrast to the expansion bias in humans. Transgenic mice and humans reveal no involvement of FEN1 in CAG repeat instability [167, 188, 194]. Moreover, genetic variants of FEN1, known to be involved in cancer, show no disease association in families with either HD or Fuchs endothelial corneal dystrophy (FECD) (both CAG/CTG expansion diseases) [188, 194]. While it seems intriguing that FEN1 can act upon flap-DNAs free of repeats, but its nuclease activity is inhibited by the presence of CAG/CTG hairpins, the relevance of this to repeat instability is unclear [160–162]. Until recently, there had been no human genetic evidence implicating involvement of any particular exo-nuclease. FAN1 was the first endo-/exo-nuclease implicated in humans, as FAN1 is the strongest modifier in genome-wide association studies searching for modifiers of age of onset for HD [14, 15].
7.2FAN1 variants modify disease by modifying repeat expansions
Attempts to decipher the role of FAN1 and its variants in repeat instability have yielded mixed findings. One mechanism by which the coding FAN1 variants might hasten onset of CAG/polyQ disease is through enhancing high levels of somatic repeat expansions. Initially, variants were tested for their impact on ICL repair/sensitivity. The effects of two coding FAN1 HD-modifying variants, p.R377W and p.R507H, upon the function of FAN1 are limited, and somewhat contradictory. A knockout of FAN1 in HEK293T cells causes mildly increased sensitivity to MMC, which is rescued by transient expression of wt FAN1, but not by either variant form [12]. In contrast, the wt or the p.R507H FAN1 rescued to comparable levels the increased MMC sensitivity in FAN1-deficient U2OS cells [141]. Lymphoblasts from HD individuals with p.R507H/wt FAN1 show increased MMC sensitivity, compared to those with wt FAN1 [12]; however, another report shows no such difference [141]. The contradictory results may reflect other genetic variations between individuals, the numbers of HD lines studied, or different model cell systems, using different experimental conditions. The link (or lack thereof) of FAN1’s roles in repeat instability and any of its many known functions, including ICL processing,is unknown.
There are other instances of seemingly contradictory findings for the effect of the FAN1 variants. For example, the non-coding rs3512 FAN1 variant is one of the strongest modifiers delaying onset for HD, SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 [15, 136–138], and would be predicted to be associated with decreased somatic CAG expansions. However, while rs3512 is associated with increased FAN1 expression in blood [137], increased CAG expansion rates were detected in blood cells of HD individuals with this variant [137]. The association of rs3512 with increased somatic CAG expansions is inconsistent with the suspected decrease in somatic CAG expansions by which this variant would mediate a later age of onset. However, it is possible that this variant acts upon repeat instability differently in brain than in blood.
Deciphering the mechanism by which any one DNA repair gene variant may act on HD age of onset and/or CAG instability, is likely to be complex. For example, the FAN1 rs3512 variant was associated with delayed onset for HD, SCA2, and some, but not all SCA3 populations [138, 195]. The rs3512 variant in a cohort of Chinese SCA3 patients was not significant, a discrepancy attributed to the diverse allele frequencies between ethnicities [196]. Moreover, the effect of the rs3512 variant in another cohort of SCA3 affected individuals was modulated by the size of non-diseased CAG lengths of the ATXN2 CAG repeat, whose repeat when expanded causes SCA2 [138]. In fact, mounting evidence suggests that the presence of intermediate non-diseased sized CAG tract lengths at other disease repeat expansion genes can significantly modify the age of onset for a repeat expansion at another disease loci in the same individual [138]. The age of onset in HD individuals with HTT CAG expansions is modulated by CAG repeat sizes in the normal range of three other repeat expansion disease loci, ATXN3, (SCA3) CACNA1A (SCA6) and AR (SBMA) [197]. These associations suggest an interaction of these genes/gene products, possibly in a shared pathway. Notably, mutant HTT protein was recently shown to interact with the ataxin-3 protein, encoded by ATXN3, in a DNA repair complex [198, 199]. Asides from the ATXN2-SCA3-FAN1 rs3512 association, whether these non-disease CAG tract lengths affect other disease modifiers is unknown. Also, an onset-delaying FAN1 modifier can effectively be cancelled by other mutations. For example, one HD individual homozygous for the onset-delaying FAN1 modifier rs35811129, expected to over-express FAN1 also had a rare mutation truncating the nuclease domain (rs184745027; p.R952*) in one FAN1 allele [12]. Rather than delayed disease onset, this individual had onset 11.7 years earlier than expected by their inherited CAG repeat length. Thus, the heterozygous loss of FAN1 nuclease activity produced an onset-hastening effect, over-riding the effect homozygous onset-delaying modifier. Thus, modifiers within the same modifier gene, or in separate modifier genes may synergize or cancel each other out.
In FAN1-knock-out U2OS cells, CAG expansions in an HTT (CAG)118 construct were greater than in cells that had wt FAN1 added [141]. However, as noted in Section 6.2, that system was unable to distinguish between the effect of wt FAN1 and that of p.R507H, or the nuclease-dead, p.D960A, even with the latter on a p.R507H background [141]. Thus, the effect of either the HD-hastening variant, R507H, or the nuclease domain could not be discerned relative to CAG instability. In iPSCs derived from an HD individual with (CAG)120 in the endogenous HTT gene, the repeat tract expanded by ∼1 CAG unit every 12.4 days, over ∼60 days [141]. ShRNA-mediated knock-down of FAN1 significantly increased this to ∼1 CAG unit every 9 days [141]. Similar effects were observed when the iPSCs were differentiated to post-mitotic medium spiny neurons: the vulnerable cell type in HD. Similarly, knocking out FAN1 in HD-derived iPSC cells with (CAG)72, using CRISPR/Cas9, lead to gradual increases in the modal repeat size by ∼1-5 units over six months of cell culture [12]. These results support a role of FAN1 in slowing, but not arresting, the rate of spontaneous CAG expansions of the endogenous HTT expanded CAG repeat. Since FAN1 can affect both CGG and CAG repeat instability, this now poses the question as to whether FAN1 might act upon any of the more recently discovered repeat expansion loci, and upon how many different repeat sequence motifs FAN1 might act? [121, 122,200, 201].
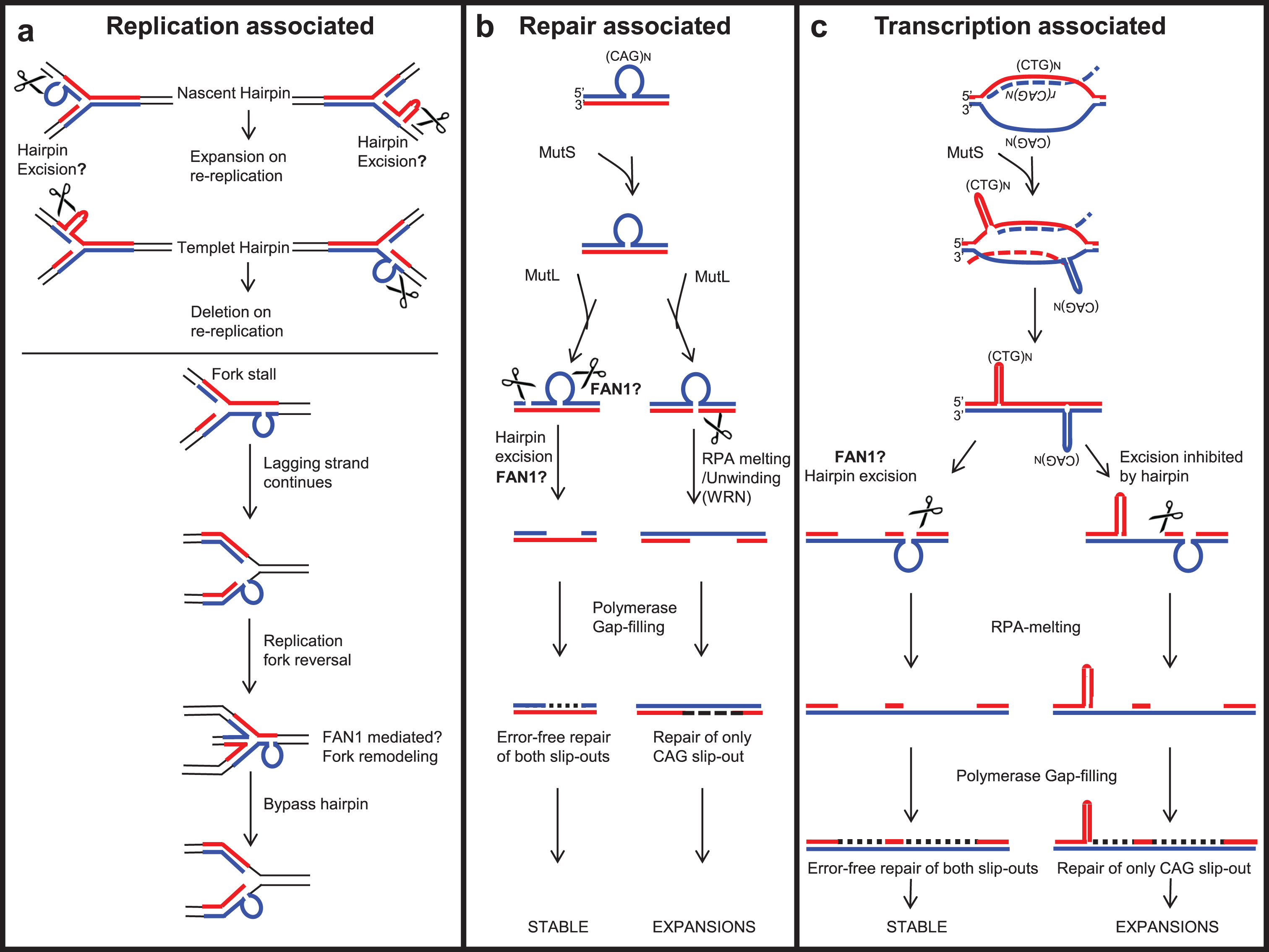
The mechanisms by which FAN1 may suppress CAG or CGG repeat instability are unclear. Models proposed [202, 203] include a) replication-associated repeat instability, b) repair-associated repeat instability, and c) transcription-associated (R-loop) repeat instability (Fig. 6). That FAN1 can affect CAG instability in post mitotic cells argues against a role of FAN1 in instability at replication forks. The common ground for all these models is aberrant DNA structures formed by the expanded sequence, called slipped-DNAs, as intermediates of the expanded mutations [202, 204, 205]. Unusual non-B-DNA structures can form in vitro and in vivo at each of the unstable repeats (reviewed in [205–210]). In addition, all models of repeat instability require nucleases to process the slipped-DNA and thus protect the repeat tract from further expansion. Various DNA repair endo- and exo-nucleases have been studied with respect to repeat instability, such as FEN1, EXO1, MutLα and MutLγ [186, 202, 211–213]. Nonetheless, FAN1 is genetically implicated as a critical nuclease involved in repeat instability, yet the exact role of FAN1 nuclease in processing slipped-DNAs formed by repeat tracts is still unexplored (see Section 4.4 for details).
Fig.6
Possible roles of FAN1 in processing repeat-containing DNA, protecting against repeat instability. (a) Replication-associated repeat instability; FAN1 may process slip-outs formed on a template or nascent strand during replication and may stabilize repeat length or induce contraction. (b) A slip-out formed by repeat-containing DNA can be processed by a non-mitotic DNA repair associated mechanism. FAN1 may act as one of the critical nucleases to process repeat slip-outs to stabilize the repeat tract against length variations. (c) In non-replicating cells, repeat instability is associated with active transcription across the expanded repeat. FAN1 may process the slip-outs formed following unidirectional or bidirectional transcription across the expanded repeat, to stabilize repeat tract against length variations.
7.3Role of FAN1 in replication-associated repeat instability
The replication-associated repeat instability model [202, 214–216] suggests that during replication of repeat-containing DNA, slip-out may form on nascent or template DNA [202]. The slip-out on nascent DNA may lead to expansions by re-repli-cation, whereas that on template DNA leads to contraction [217, 218] (Fig. 6a). Possibly an unknown nuclease processes the slip-out formed on nascent DNA, protecting it from expansion. With both endo- and exo-nuclease activity on structured DNA, FAN1 could perhaps fill this role.
The slip-out formed on the template strand may cause replication machinery to pause and allow dissociation of the replisome from the replication fork [202, 219, 220]. However, the lagging strand continuously synthesizes DNA, resulting in fork reversal (Fig. 6a). FAN1 may be involved in the TLS pathway and may process stalled replication forks (discussed in Section 3), protecting it from repeat instability. The recruitment of FAN1 to slipped-DNA following fork stalling has yet to be proven.
7.4Role of FAN1 in repair-associated repeat instability
In HD patients, tissue-specific expansion-biased repeat instability is observed mainly in the brain and liver [154, 221]. However, brain neuronal cells do not undergo active replication, suggesting that somatic repeat instability is not limited to DNA replication, but may be induced by DNA repair mechanisms. Expression of FAN1 is high in brain cells, supporting its possible role in protecting against repeat instability. It is an interacting partner of MMR proteins, notably MLH1 [4, 5, 7, 34, 43], suggesting that it may be involved in MMR. Knowing how disruption of the FAN1-MLH1 interaction affects the severity of repeat expansion could be relevant to prediction of the onset of neurological disorders.
The requirement of MMR complexes— including MutSα (MSH2-MSH3), MutSβ (MSH2-MSH6), MutLα (MLH1-PMS2) and MutLγ (MLH1-MLH3)— in CAG/CTG, CGG/CCG, and GAA/TTC repeat instability has been extensively studied [185, 210, 213, 222–229]. MustSβ (MSH2-MSH3), through its ability to bind slip-DNA structures, may facilitate their formation [210]. MustSβ recruits MutL complexes (Fig. 6b). MutLα has endo-nuclease activity on both the slipped-out and non-slipped-out strands, in unnicked DNA [186, 230]. In contrast, MutLγ endo-nuclease cleaves only the non-slipped-out strand [186]. MutLα or MutLγ generate a nick in the slip-out, potentially serving as an entry point for exo-nucleases like EXO1 or FAN1, both known to interact with MLH1 [231–233]. Any of these proteins may subsequently process the slip-out and stabilize the repeat expansion. In contrast, cleavage on the non-slip-out strand by MutLγ may lead to WRN-RPA-mediated melting of hairpin DNA [234]. Resulting gaps in the DNA may be filled by DNA polymerase β, which may lead to repeat expansion [166, 186, 234, 235] (Fig. 6b). FAN1 co-localizes with RPA at stalled replication forks in cells [236] and the nuclease activity of FAN1 is enhanced by RPA [237], but a direct interaction was not tested.
7.5Role of FAN1 in transcription-associated repeat instability
In non-replicating cells, repeat instability can be driven by active transcription through the expanded repeat. Transcription of repeat-containing DNA leads to the formation of DNA:RNA hybrids, commonly referred to as ‘R-loops’ (Fig. 6c). Slip-out structures may then form on the non-transcribed strand. Bidirectional transcription may lead to the formation of slip-outs in both strands [203, 238–241], which are recognized by MutSβ and activate MutL complexes. As with repair-associated repeat stability, the relative contribution of MutLα or MutLγ will determine the fate of the slip-out, giving rise to either expansions, contractions, or stability. Repair of only non-transcribing slip-outs may result in repeat expansions, whereas repair of both slip-outs may lead to repeat stability [203, 210, 238–241] (Fig. 6c). However, for repair to happen, the slipped-DNA is processed by DNA nucleases: an aspect that needs to be further explored. In summary, it is possible that FAN1, through its endo- and/or exo-nuclease activity, is involved in the processing of these slip-outs, thereby protecting against further repeat expansion.
8CONCLUSIONS AND FUTURE PERSPECTIVES
FAN1 was initially thought to be associated with Fanconi anemia (FA); however, rather than FA, homozygous loss of FAN1 causes karyomegalic interstitial nephritis (KIN) [31, 45]. Most KIN mutations ablate nuclease activity of the enzyme, indicating that this activity protects normal kidney function [31]. Nuclease-defective FAN1 causes mild hypersensitivity to DNA crosslinking agents in cells and in mice, suggesting defects in ICL processing [31]. FAN1 can process ICLs in an FA-independent manner [29]. Its function in ICL repair and replication fork-protection might be responsible for safeguarding cells from endogenous and exogenous insults, including platinum-based and mitomycin C-based anti-cancer therapies. Loss of FAN1 function does not increase the burden of human cancers [85]. Considering that some cytotoxic anticancer agents act by inducing ICLs and/or replication stress, FAN1 nuclease activity is likely a good target for sensitization of cancer cells to killing by conventional chemotherapy. However, this might not be applicable to HD subjects who display lower cancer incidence than age- and sex-matched controls [242].
FAN1 is the strongest modifier in HD [14, 15]. Increased FAN1 expression and DNA binding may protect against somatic repeat expansion of trinucleotide repeats [12, 14, 141]. However, the full set of FAN1 functions— including the biochemistry of its DNA binding activity, dimerization, nuclease activities, various interaction partners, and roles in DNA replication and repair— need further investigation, particularly with regard to repeat DNAs. How this diverse set of DNA metabolizing functions may contribute to FAN1’s involvement in multiple diseases is now a key focus. Variants in FAN1 contribute to numerous diseases (including autism, schizophrenia, and epilepsy), some of which are caused by expanded repeat DNAs [20, 25, 122, 200, 201]. Current genetic analysis supports a role of ongoing somatic repeat expansions, play a role in the age-of-onset and progression rate of many such diseases. Regulation of CAG/CTG and CGG/CCG repeats by FAN1 opens the possibility that it may be involved with other unstable repeat motifs associated with disorders such as autism or schizophrenia [121, 122, 200, 201]. A focus on elucidating the mechanistic role of FAN1 in repeat instability is warranted, to learn if and how it modifies disease by modulating somatic repeatinstability.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We acknowledge the input of Peixiang Wang, and thank Dr. Lesley Jones for discussions regarding the HD haplotypes and Mahreen Khan and Janet Buchanan for proof reading. This work was partially funded by grants from the Canadian Institutes of Health Research (CIHR FRN148910 to C.E.P.; CIHR FDN-388879 to J.Y.M.), the Hereditary Disease Foundation (to A.L.D.), the Swiss National Science Foundation (grant number: 31003A_176161 to A.A.S.), and the Swiss Cancer Research Foundation (grant number: KFS-4702-02-2019 to A.A.S.). C.E.P. holds a Tier 1 Canada Research Chair in Disease-Associated Genome Instability. J.Y.M holds a Tier 1 Canada Research Chair in DNA Repair and Cancer Therapeutics. C.E.P. and A.L.D. conceived, designed, and coordinated the review.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JHD-200448.
REFERENCES
[1] | Iyama T , Wilson DM . DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). (2013) ;12: (8):620–36. |
[2] | Dehé PM , Gaillard PHL . Control of structure-specific endonucleases to maintain genome stability. Nat Rev Mol Cell Biol. (2017) ;18: (5):315–30. |
[3] | Nagase T , Ishikawa KI , Suyama M , Kikuno R , Hirosawa M , Miyajima N , et al. Prediction of the coding sequences of unidentified human genes. XII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. (1998) ;5: (6):355–64. |
[4] | Cannavo E , Gerrits B , Marra G , Schlapbach R , Jiricny J . Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J Biol Chem. (2007) ;282: (5):2976–86. |
[5] | Smogorzewska A , Desetty R , Saito TT , Schlabach M , Lach FP , Sowa ME , et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell. (2010) ;39: (1):36–47. |
[6] | Kratz K , Schöpf B , Kaden S , Sendoel A , Eberhard R , Lademann C , et al. Deficiency of FANCD2-associated nuclease KIAA/FAN1 sensitizes cells to interstrand crosslinking agents. Cell. (2010) ;142: (1):77–88. |
[7] | MacKay C , Déclais AC , Lundin C , Agostinho A , Deans AJ , MacArtney TJ , et al. Identification of KIAA/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. (2010) ;142: (1):65–76. |
[8] | Liu T , Ghosal G , Yuan J , Chen J , Huang J . FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science. (2010) ;329: (5992):693–6. |
[9] | Barski A , Cuddapah S , Cui K , Roh TY , Schones DE , Wang Z , et al. High-resolution profiling of histone methylations in the human genome. Cell. (2007) ;129: (4):823–37. |
[10] | Eukaryotic Promoter Database. https://epd.epfl.ch//index.php |
[11] | Ensembl Genome Browser. https://uswest.ensembl.org/index.html |
[12] | Kim KH , Hong EP , Shin JW , Chao MJ , Loupe J , Gillis T , et al. Genetic and functional analyses point to fan1 as the source of multiple Huntington disease modifier effects. Am J Hum Genet. (2020) ;107: (1):96–110. |
[13] | Aufiero S , van Den Hoogenhof MMG , Reckman YJ , Beqqali A , van der Made I , Kluin J , et al. Cardiac circRNAs arise mainly from constitutive exons rather than alternatively spliced exons. RNA. (2018) ;24: (6):815–27. |
[14] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900.e14. |
[15] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. |
[16] | Liu J , Ciarochi J , Calhoun VD , Paulsen JS , Jeremy Bockholt H , Johnson HJ , et al. Genetics modulate gray matter variation beyond disease burden in prodromal Huntington’s disease. Front Neurol. (2018) ;29;9: :190. |
[17] | Alonso A , Sasin J , Bottini N , Friedberg I , Friedberg I , Osterman A , et al. Protein tyrosine phosphatases in the human genome. Cell. (2004) ;117: (6):699–711. |
[18] | Loupe JM , Pinto RM , Kim KH , Gillis T , Mysore JS , Andrew MA , et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet. (2020) ;29: (18):3044–53. doi: 10.1093/hmg/ddaa196 |
[19] | Gillentine MA , Schaaf CP . The human clinical phenotypes of altered CHRNA7 copy number. Biochem Pharmacol. (2015) ;97: (4):352–62. |
[20] | Szafranski P , Schaaf CP , Person RE , Gibson IB , Xia Z , Mahadevan S , et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA Benign or pathological? Hum Mutat. (2010) ;31: (7):840–50. |
[21] | Forsingdal A , Fejgin K , Nielsen V , Werge T , Nielsen J . 15q13.3 homozygous knockout mouse model display epilepsy-, autism-and schizophrenia-related phenotypes. Transl Psychiatry. (2016) ;6: (7):e860. |
[22] | Hoppman-Chaney N , Wain K , Seger PR , Superneau DW , Hodge JC . Identification of single gene deletions at 15q13. Further evidence that CHRNA7 causes the 15q13.3 microdeletion syndrome phenotype. Clin Genet. (2013) ;83: (4):345–51. |
[23] | LePichon JB , Bittel DC , Graf WD , Yu S . A 15q13.3 homozygous microdeletion associated with a severe neurodevelopmental disorder suggests putative functions of the TRPM1, CHRNA7, and other homozygously deleted genes. Am J Med Genet Part A. (2010) ;152: (5):1300–4. |
[24] | Antonacci F , Dennis MY , Huddleston J , Sudmant PH , Steinberg KM , Rosenfeld JA , et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet. (2014) ;46: (12):1293–302. |
[25] | Ionita-Laza I , Xu B , Makarov V , Buxbaum JD , Roos JL , Gogos JA , et al. Scan statistic-based analysis of exome sequencing data identifies FAN1 at 15q13.3 as a susceptibility gene for schizophrenia and autism. Proc Natl Acad Sci U S A. (2014) ;111: (1):343–8. |
[26] | Gwon GH , Jo A , Baek K , Jin KS , Fu Y , Lee JB , et al. Crystal structures of the structure-selective nuclease Mus81-Eme1 bound to flap DNA substrates. EMBO J. (2014) ;33: (9):1061–72. |
[27] | Newman M , Murray-Rust J , Lally J , Rudolf J , Fadden A , Knowles PP , et al. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. (2005) ;24: (5):895–905. |
[28] | Pennell S , Déclais AC , Li J , Haire LF , Berg W , Saldanha J , et al. FAN1 activity on asymmetric repair intermediates is mediated by an atypical monomeric virus-type replication-repair nuclease domain. Cell Rep. (2014) ;8: (1):84–93. |
[29] | Wang R , Persky NS , Yoo B , Ouerfelli O , Smogorzewska A , Elledge SJ , et al. Mechanism of DNA interstrand cross-link processing by repair nuclease FAN1. Science. (2014) ;346: (6213):1127–30. |
[30] | Lachaud C , Moreno A , Marchesi F , Toth R , Blow JJ , Rouse J . Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science. (2016) ;351: (6275):846–9. |
[31] | Zhou W , Otto EA , Cluckey A , Airik R , Hurd TW , Chaki M , et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. (2012) ;44: (8):910–5. |
[32] | Thongthip S , Bellani M , Gregg SQ , Sridhar S , Conti BA , Chen Y , et al. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev. (2016) ;30: (6):645–59. |
[33] | Smith AL , Alirezaie N , Connor A , Chan-Seng-Yue M , Grant R , Selander I , et al. Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett. (2016) ;370: (2):302–12. |
[34] | Porro A , Berti M , Pizzolato J , Bologna S , Kaden S , Saxer A , et al. FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat Commun. (2017) ;8: (1):1073. |
[35] | Aleksandrov R , Dotchev A , Poser I , Krastev D , Georgiev G , Panova G , et al. Protein dynamics in complex DNA lesions. Mol Cell. (2018) ;69: (6):1046–61.e5. |
[36] | Haracska L , Johnson RE , Unk I , Phillips B , Hurwitz J , Prakash L , et al. Physical and functional interactions of human DNA polymerase η with PCNA. Mol Cell Biol. (2001) ;21: (21):7199–206. |
[37] | Hishiki A , Hashimoto H , Hanafusa T , Kamei K , Ohashi E , Shimizu T , et al. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J Biol Chem. (2009) ;284: (16):10552–60. |
[38] | Bienko M , Green CM , Crosetto N , Rudolf F , Zapart G , Coull B , et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. (2005) ;310: (5755):1821–4. |
[39] | Centore RC , Yazinski SA , Tse A , Zou L . Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Mol Cell. (2012) ;46: (5):625–35. |
[40] | Fontebasso Y , Etheridge TJ , Oliver AW , Murray JM , Carr AM . The conserved Fanconi anemia nuclease Fan1 and the SUMO E3 ligase Pli1 act in two novel Pso2-independent pathways of DNA interstrand crosslink repair in yeast. DNA Repair (Amst). (2013) ;12: (12):1011–23. |
[41] | Zhao Q , Xue X , Longerich S , Sung P , Xiong Y . Structural insights into 5 € 2 flap DNA unwinding and incision by the human FAN1 dimer. Nat Commun. (2014) ;5: :5726. |
[42] | Rao T , Longerich S , Zhao W , Aihara H , Sung P , Xiong Y . Importance of homo-dimerization of Fanconi-associated nuclease 1 in DNA flap cleavage. DNA Repair (Amst). (2018) ;64: :53–8. |
[43] | Rikitake M , Fujikane R , Obayashi Y , Oka K , Ozaki M , Hidaka M . MLH1-mediated recruitment of FAN1 to chromatin for the induction of apoptosis triggered by O6-methylguanine. Genes Cells. (2020) ;25: (3):175–86. |
[44] | Yoshikiyo K , Kratz K , Hirota K , Nishihara K , Takata M , Kurumizaka H , et al. KIAA/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci U S A. (2010) ;107: (50):21553–7. |
[45] | Trujillo JP , Mina LB , Pujol R , Bogliolo M , Andrieux J , Holder M , et al. On the role of FAN1 in Fanconi anemia. Blood. (2012) ;120: (1):86–9. |
[46] | Rageul J , Kim H . Fanconi anemia and the underlying causes of genomic instability. Environ. Mol. Mutagen. (2020) ;61: (7):693–708. |
[47] | Budzowska M , Zhang J , Budzowska M , Drohat AC , Walter JC . Replication-dependent unhooking of dna interstrand cross-links by the NEIL3 glycosylase. Cell. (2016) ;167: (2):498–511.e14. |
[48] | Räschle M , Knipsheer P , Enoiu M , Angelov T , Sun J , Griffith JD , et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. (2008) ;134: (6):969–80. |
[49] | Knipscheer P , Räschle M , Smogorzewska A , Enoiu M , Ho TV , Schärer OD , et al. The fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. (2009) ;326: (5960):1698–701. |
[50] | Williams HL , Gottesman ME , Gautier J . Replication-Independent repair of DNA interstrand crosslinks. Mol Cell. (2012) ;47: (1):140–7. |
[51] | Kato N , Kawasoe Y , Williams H , Coates E , Roy U , Shi Y , et al. Sensing and processing of DNA interstrand crosslinks by the mismatch repair pathway. Cell Rep. (2017) ;21: (5):1375–85. |
[52] | Pizzolato J , Mukherjee S , Schärer OD , Jiricny J . FANCD2-associated nuclease 1, but not exonuclease 1 or flap endonuclease 1, is able to unhook DNA interstrand cross-links in vitro . J Biol Chem. (2015) ;290: (37):22602–11. |
[53] | Wang AT , Sengerová B , Cattell E , Inagawa T , Hartley JM , Kiakos K , et al. Human SNM1a and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. (2011) ;25: (17):1859–70. |
[54] | Neelsen KJ , Lopes M . Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat Rev Mol Cell Biol. (2015) ;16: (4):207–20. |
[55] | Jin H , Cho Y . Structural and functional relationships of FAN1. DNA Repair (Amst). (2017) ;56: :135–43. |
[56] | Klein Douwel D , Boonen RACM , Long DT , Szypowska AA , Räschle M , Walter JC , et al. XPF-ERCC1 acts in unhooking DNA Interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. (2014) ;54: (3):460–71. |
[57] | Zhang J , Walter JC . Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst). (2014) ;19: :135–42. |
[58] | Lachaud C , Slean M , Marchesi F , Lock C , Odell E , Castor D , et al. Karyomegalic interstitial nephritis and DNA damageinduced polyploidy in fan1 nuclease-defective knock-in mice. Genes Dev. (2016) ;30: (6):639–44. |
[59] | Fricke WM , Brill SJ . Slx1 - Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1 - Top3. Genes Dev. (2003) ;17: (14):1768–78. |
[60] | Hazrati A , Ramis-Castelltort M , Sarkar S , Barber LJ , Schofield CJ , Hartley JA , et al. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair (Amst). (2008) ;7: (2):230–8. |
[61] | Buzon B , Grainger R , Huang S , Rzadki C , Junop MS . Structure-specific endonuclease activity of SNM1A enables processing of a DNA interstrand crosslink. Nucleic Acids Res. (2018) ;46: (17):9057–66. |
[62] | Deans AJ , West SC . DNA interstrand crosslink repair and cancer. Nat Rev Cancer. (2011) ;11: (7):467–80. |
[63] | Ronato DA , Mersaoui SY , Busatto FF , Affar EB , Richard S , Masson JY . Limiting the DNA double-strand break resectosome for genome protection. Trends Biochem Sci. (2020) ;45: (9):779–93. |
[64] | Ducy M , Sesma-Sanz L , Guitton-Sert L , Lashgari A , Gao Y , Brahiti N , et al. The tumor suppressor PALB Inside out. Trends Biochem. Sci. (2019) ;44: (3):226–40. |
[65] | Shlien A , Campbell BB , De Borja R , Alexandrov LB , Merico D , Wedge D , et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet. (2015) ;47: (3):257–62. |
[66] | Desai A , Gerson S . Exo1 independent DNA mismatch repair involves multiple compensatory nucleases. DNA Repair (Amst). (2014) ;21: :55–64. |
[67] | Zhang N , Lu X , Zhang X , Peterson CA , Legerski RJ . hMutSβ is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol. (2002) ;22: (7):2388–97. |
[68] | Williams SA , Wilson JB , Clark AP , Mitson-Salazar A , Tomashevski A , Ananth S , et al. Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum Mol Genet. (2011) ;20: (22):4395–410. |
[69] | Peng M , Xie J , Ucher A , Stavnezer J , Cantor SB . Crosstalk between BRCA - Fanconi anemia and mismatch repair pathways prevents MSH 2-dependent aberrant DNA damage responses. EMBO J. (2014) ;33: (15):1698–712. |
[70] | Peng M , Litman R , Xie J , Sharma S , Brosh RM , Cantor SB . The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. (2007) ;26: (13):3238–49. |
[71] | Chaudhury I , Stroik DR , Sobeck A . FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol Cell Biol. (2014) ;34: (21):3939–54. |
[72] | Lossaint G , Larroque M , Ribeyre C , Bec N , Larroque C , Décaillet C , et al. FANCD2 binds MCM proteins and controls replisome function upon activation of S phase checkpoint signaling. Mol Cell. (2013) ;51: (5):678–90. |
[73] | Sato K , Shimomuki M , Katsuki Y , Takahashi D , Kobayashi W , Ishiai M , et al. FANCI-FANCD2 stabilizes the RAD51-DNA complex by binding RAD51 and protects the 5’-DNA end. Nucleic Acids Res. (2016) ;44: (22):10758–71. |
[74] | Thangavel S , Berti M , Levikova M , Pinto C , Gomathinayagam S , Vujanovic M , et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol. (2015) ;208: (5):545–62. |
[75] | Higgs MR , Reynolds JJ , Winczura A , Blackford AN , Borel V , Miller ES , et al. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol Cell. (2015) ;59: (3):462–77. |
[76] | Kile AC , Chavez DA , Bacal J , Eldirany S , Korzhnev DM , Bezsonova I , et al. HLTF’s ancient HIRAN domain binds 3’ dna ends to drive replication fork reversal. Mol Cell. (2015) ;58: (6):1090–100. |
[77] | Zellweger R , Dalcher D , Mutreja K , Berti M , Schmid JA , Herrador R , et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. (2015) ;208: (5):563–79. |
[78] | Higgins NP , Kato K , Strauss B . A model for replication repair in mammalian cells. J Mol Biol. (1976) ;101: (3):417–25. |
[79] | Poole LA , Cortez D . Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit Rev Biochem Mol Biol. (2017) ;52: (6):696–714. |
[80] | Davies AA , Huttner D , Daigaku Y , Chen S , Ulrich HD . Activation of ubiquitin-dependent dna damage bypass is mediated by replication protein A. Mol Cell. (2008) ;29: (5):625–36. |
[81] | Mourón S , Rodriguez-Acebes S , Martínez-Jiménez MI , García-Gómez S , Chocrón S , Blanco L , et al. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol. (2013) ;20: (12):1383–9. |
[82] | Bousset K , Diffley JFX . The Cdc7 protein kinase is required for origin firing during S phase. Genes Dev. (1998) ;12: (4):480–90. |
[83] | Chen J , Calhoun VD , Perrone-Bizzozero NI , Pearlson GD , Sui J , Du Y , et al. A pilot study on commonality and specificity of copy number variants in schizophrenia and bipolar disorder. Transl Psychiatry. (2016) ;6: (5):e824. |
[84] | Jian X , Chen J , Li Z , Fahira A , Shao W , Zhou J , et al. Common variants in FAN1, located in 15q13.3, confer risk for schizophrenia and bipolar disorder in Han Chinese. Prog Neuropsychopharmacology Biol Psychiatry. (2020) ;103: :109973. doi:10.1016/j.pnpbp.2020.109973 |
[85] | Fievet A , Mouret-Fourme E , Colas C , de Pauw A , Stoppa-Lyonnet D , Buecher B . Prevalence of pathogenic variants of FAN1 in more than patients assessed for genetic predisposition to colorectal, breast, ovarian, or other cancers. Gastroenterology. (2019) ;156: (6):1919–20. |
[86] | Enikanolaiye A , Ruston J , Zeng R , Taylor C , Schrock M , Buchovecky CM , et al. Suppressor mutations in Mecp2-null mice implicate the DNA damage response in Rett syndrome pathology. Genome Res. (2020) ;30: (4):540–52. |
[87] | Sclare G . A case of unexplained karyomegaly. Beitr Pathol. (1976) ;157: (3):301–6. |
[88] | Mihatsch MJ , Gudat F , Zollinger HU , Heierli C , Thölen H , Reutter FW . Systemic karyomegaly associated with chronic interstitial nephritis. A new disease entity? Clin Nephrol. (1979) ;12: (2):54–62. |
[89] | Burry AF . Extreme dysplasia in renal epithelium of a young woman dying from hepatocarcinoma. J Pathol. (1974) ;113: (3):147–50. |
[90] | Isnard P , Rabant M , Labaye J , Antignac C , Knebelmann B , Zaidan M . Karyomegalic Interstitial Nephritis: A case report and review of the literature. Medicine (Baltimore). (2016) ;95: (20):e3349. |
[91] | Bhandari S Kalowski S , Collett P , Cooke BE , Kerr P , Newland R , et al. Karyomegalic nephropathy: An uncommon cause of progressive renal failure. Nephrol Dial Transplant. (2002) ;17: (11):1914–20. |
[92] | Spoendlin M , Moch H , Brunner F , Brunner W , Burger HR , Kiss D , et al. Karyomegalic interstitial nephritis: Further support for a distinct entity and evidence for a genetic defect. Am J Kidney Dis. (1995) ;25: (2):242–52. |
[93] | Akyürek LM , Hussein A , Nicholson AG , Mauritz NJ , Mölne J . Pulmonary manifestations of systemic karyomegaly. Respir Med Case Reports. (2020) ;29: :101032. |
[94] | Vadiaka M , Sotsiou F , Koufos C . A case of systemic karyomegaly associated with interstitial nephritis. Ann Med Interne (Paris). (1998) ;149: (5):291–4. |
[95] | Moch H , Spondlin M , Schmassmann A , Mihatsch MJ . Systemic karyomegaly with chronic interstitial nephritis. Discussion of the disease picture based on an autopsy case. Pathologe. (1994) ;15: (1):44–8. |
[96] | Tagliente DJ , Voss JS , Peters SG , Aubry MC , Cornell LD , Maleszewski JJ . Systemic karyomegaly with primary pulmonary presentation. Hum Pathol. (2014) ;45: (1):180–4. |
[97] | Airik R , Schueler M , Airik M , Cho J , Porath JD , Mukherjee E , et al. A FANCD2/FANCI-associated nuclease 1-knockout model develops karyomegalic interstitial nephritis. J Am Soc Nephrol. (2016) ;27: (12):3552–9. |
[98] | Bastarache L , Hughey JJ , Hebbring S , Marlo J , Zhao W , Ho WT , et al. Phenotype risk scores identify patients with unrecognized mendelian disease patterns. Science. (2018) ;359: (6381):1233–9. |
[99] | Simon J , Stoll K , Fick R , Mott J , Lawson-Yuen A . Homozygous 15q13. 3 microdeletion in a child with hypotonia and impaired vision: A new report and review of the literature. Clin Case Reports. (2019) ;7: (12):2311–5. |
[100] | Liao J , Deward SJ , Madan-Khetarpal S , Surti U , Hu J . A small homozygous microdeletion of 15q13.3 including the CHRNA7 gene in a girl with a spectrum of severe neurodevelopmental features. Am J Med Genet Part A. (2011) ;155: (11):2795–800. |
[101] | Spielmann M , Reichelt G , Hertzberg C , Trimborn M , Mundlos S , Horn D , et al. Homozygous deletion of chromosome 15q13.3 including CHRNA7 causes severe mental retardation, seizures, muscular hypotonia, and the loss of KLF13 and TRPM1 potentially cause macrocytosis and congenital retinal dysfunction in siblings. Eur J Med Genet. (2011) ;54: (4):e441–5. doi: 10.1016/j.ejmg.2011.04.004 |
[102] | Prasun P , Hankerd M , Kristofice M , Scussel L , Sivaswamy L , Ebrahim S . Compound heterozygous microdeletion of chromosome 15q13.3 region in a child with hypotonia, impaired vision, and global developmental delay. Am J Med Genet Part A. (2014) ;164: (7):1815–20. |
[103] | Masurel-Paulet A , Andrieux J , Callier P , Cuisset JM , Le Caignec C , Holder M , et al. Delineation of 15q13.3 microdeletions. Clin Genet. (2010) ;78: (2):149–61. |
[104] | Lowther C , Costain G , Stavropoulos DJ , Melvin R , Silversides CK , Andrade DM , et al. Delineating the 15q13.3 microdeletion phenotype: A case series and comprehensive review of the literature. Genet Med. Nature Publishing Group. (2015) ;17: (2):149–57. |
[105] | Masurel-Paulet A , Drumare I , Holder M , Cuisset JM , Vallée L , Defoort S , et al. Further delineation of eye manifestations in homozygous 15q13.3 microdeletions including TRPM A differential diagnosis of ceroid lipofuscinosis. Am J Med Genet Part A. (2014) ;164A: (6):537–44. |
[106] | Le Pichon JB , Yu S , Kibiryeva N , Graf WD , Bittel DC . Genome-wide gene expression in a patient with 15q13.3 homozygous microdeletion syndrome. Eur J Hum Genet. (2013) ;21: (10):1093–9. |
[107] | Belhadj S , Terradas M , Munoz-Torres PM , Aiza G , Navarro M , Capellá G , et al. Candidate genes for hereditary colorectal cancer: Mutational screening and systematic review. Hum Mutat. (2020) ;41: (9):1563–76. |
[108] | Seguí N , Mina LB , Lázaro C , Sanz-Pamplona R , Pons T , Navarro M , et al. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology. (2015) ;149: (3):563–6. |
[109] | Broderick P , Dobbins SE , Chubb D , Kinnersley B , Dunlop MG , Tomlinson I , et al. Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients— a systematic review. Gastroenterology. (2017) ;152: (1):75–77.e4. |
[110] | Moynahan ME , Pierce AJ , Jasin M . BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. (2001) ;7: (2):263–72. |
[111] | Jensen RB , Carreira A , Kowalczykowski SC . Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. (2010) ;467: (7316):678–83. doi:10.1038/nature09399 |
[112] | Shivji MKK , Davies OR , Savill JM , Bates DL , Pellegrini L , Venkitaraman AR . A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucleic Acids Res. (2006) ;34: (14):4000–11. |
[113] | Felicio PS , Grasel RS , Campacci N , de Paula AE , Galvão HCR , Torrezan GT , et al. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum Mutat. 2020. doi:10.1002/humu.24158 |
[114] | Schlacher K , Christ N , Siaud N , Egashira A , Wu H , Jasin M . Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. (2011) ;145: (4):529–42. |
[115] | Santarpia L , Iwamoto T , Di Leo A , Hayashi N , Bottai G , Stampfer M , et al. DNA repair gene patterns as prognostic and predictive factors in molecular breast cancer subtypes. Oncologist. (2013) ;18: (10):1063–73. |
[116] | Green EK , Rees E , Walters JTR , Smith KG , Forty L , Grozeva D , et al. Copy number variation in bipolar disorder. Mol Psychiatry. (2016) ;21: (1):89–93. |
[117] | Kirov G . The role of copy number variation in schizophrenia. Expert Rev Neurother. (2010) ;10: (1):25–32. |
[118] | Ross CA , McInnis MG , Margolis RL , Li SH . Genes with triplet repeats: Candidate mediators of neuropsychiatric disorders. Trends Neurosci. (1993) ;16: (7):254–60. |
[119] | McInnis MG , McMahon FJ , Chase GA , Simpson SG , Ross CA , DePaulo JR . Anticipation in bipolar affective disorder. Am J Hum Genet. (1993) ;53: (2):385–90. |
[120] | Margolis RL , McInnis MG , Rosenblatt A , Ross CA . Trinucleotide repeat expansion and neuropsychiatric disease. Arch Gen Psychiatry. (1999) ;56: (11):1019–31. |
[121] | Song JHT , Lowe CB , Kingsley DM . Characterization of a human-specific tandem repeat associated with bipolar disorder and schizophrenia. Am J Hum Genet. (2018) ;103: (3):421–30. |
[122] | Trost B , Engchuan W , Nguyen CM , Thiruvahindrapuram B , Dolzhenko E , Backstrom I , et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature. (2020) ;586: (7827):80–6. |
[123] | Lafrenière RG , Rochefort DL , Chrétien N , Rommens JM , Cochius JI , Kälviäinen R , et al. Unstable insertion in the 5’ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat Genet. (1997) ;15: (3):298–302. |
[124] | Virtaneva K , D’Amato E , Miao J , Koskiniemi M , Norio R , Avanzini G , et al. Unstable minisatellite expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat Genet. (1997) ;15: (4):393–6. |
[125] | Lalioti MD , Scott HS , Buresi C , Rossier C , Bottani A , Morris MA , et al. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. (1997) ;386: (6627):847–51. |
[126] | Ishiura H , Tsuji S . Advances in repeat expansion diseases and a new concept of repeat motif-phenotype correlation. Curr Opin Genet Dev. (2020) ;65: :176–85. |
[127] | Hagerman RJ , Protic D , Rajaratnam A , Salcedo-Arellano MJ , Aydin EY , Schneider A . Fragile X-associated neuropsychiatric disorders (FXAND). Front Psychiatry. (2018) ;9: :564. |
[128] | Cogliati F , Giorgini V , Masciadri M , Bonati MT , Marchi M , Cracco I , et al. Pathogenic variants in STXBP1 and in genes for GABAa receptor subunities cause atypical rett/rett-like phenotypes. Int J Mol Sci. (2019) ;20: :3621. |
[129] | Guerrini R , Parrini E . Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia. (2012) ;53: (12):2067–78. |
[130] | Ehrhart F , Sangani NB , Curfs LMG . Current developments in the genetics of Rett and Rett-like syndrome. Curr Opin Psychiatry. (2018) ;31: (2):103–8. |
[131] | Squillaro T , Alessio N , Cipollaro M , Renieri A , Giordano A , Galderisi U . Partial silencing of methyl cytosine protein binding 2 (MECP2) in mesenchymal stem cells induces senescence with an increase in damaged DNA. FASEB J. (2010) ;24: (5):1593–603. |
[132] | Donaldson J , Powell S , Rickards N , Holmans P JL . What is the pathogenic CAG expansion length in Huntington’s disease? J Huntingtons Dis. 2020. doi:10.3233/JHD-200445 |
[133] | Rosenblatt A , Brinkman RR , Liang KY , Almqvist EW , Margolis RL , Huang CY , et al. Familial influence on age of onset among siblings with Huntington disease. Am J Med Genet - Neuropsychiatr Genet. (2001) ;105: (5):399–403. |
[134] | Wexler NS , Lorimer J , Porter J , Gomez F , Moskowitz C , Shackell E , et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. (2004) ;101: (10):3498–503. |
[135] | Hong EP , MacDonald ME , Wheeler VC , Jones L , Holmans P , Orthe M et al. Huntington’s disease pathogenesis: Two sequential components. J Huntingtons Dis. 2020. doi:10.3233/JHD-200427 |
[136] | Bettencourt C , Hensman-Moss D , Flower M , Wiethoff S , Brice A , Goizet C , et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. (2016) ;79: (6):983–90. |
[137] | Ciosi M , Maxwell A , Cumming SA , Hensman Moss DJ , Alshammari AM , Flower MD , et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. (2019) ;48: :568–80. |
[138] | Mergener R , Furtado GV , de Mattos EP , Leotti VB , Jardim LB , Saraiva-Pereira ML . Variation in DNA repair system gene as an additional modifier of age at onset in spinocerebellar ataxia type 3/Machado-Joseph disease. Neuromolecular Med. (2020) ;22: (1):133–8. |
[139] | Wright GEB , Caron NS , Ng B , Casal L , Casazza W , Xu X , et al. Gene expression profiles complement the analysis of genomic modifiers of the clinical onset of Huntington disease. Hum Mol Genet. (2020) ;29: (16):2788–802. |
[140] | Gusev A , Ko A , Shi H , Bhatia G , Chung W , Penninx BWJH , et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. (2016) ;48: (3):245–52. |
[141] | Goold R , Flower M , Moss DH , Medway C , Wood-Kaczmar A , Andre R , et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. (2019) ;28: (4):650–61. |
[142] | Ament SA , Pearl JR , Cantle JP , Bragg RM , Skene PJ , Coffey SR , et al. Transcriptional regulatory networks underlying gene expression changes in Huntington’s disease. Mol Syst Biol. (2018) ;14: (3):e7435. |
[143] | Świtońska K , Szlachcic WJ , Handschuh L , Wojciechowski P , Marczak Ł , Stelmaszczuk M , et al. Identification of altered developmental pathways in human juvenile HD iPSC with 71Q and 109Q using transcriptome profiling. Front Cell Neurosci. (2019) ;12: :528. |
[144] | Zhao XN , Usdin K . FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst). (2018) ;69: :1–5. |
[145] | Castel AL , Cleary JD , Pearson CE . Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. (2010) ;11: (3):165–70. |
[146] | Nguyen JHG , Viterbo D , Anand RP , Verra L , Sloan L , Richard GF , et al. Differential requirement of Srs2 helicase and Rad51 displacement activities in replication of hairpin-forming CAG/CTG repeats. Nucleic Acids Res. (2017) ;45: (8):4519–31. |
[147] | Ye Y , Kirkham-McCarthy L , Lahue RS . The Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex promotes trinucleotide repeat expansions independently of homologous recombination. DNA Repair (Amst). (2016) ;43: :1–8. |
[148] | Marcadier JL , Pearson CE . Fidelity of primate cell repair of a double-strand break within a (CTG)·(CAG) tract: Effect of slipped DNA structures. J Biol Chem. (2003) ;278: (36):33848–56. |
[149] | Freudenreich CH , Kantrow SM , Zakian VA . Expansion and length-dependent fragility of CTG repeats in yeast. Science. (1998) ;279: (5352):853–6. |
[150] | Tarantino ME , Bilotti K , Huang J , Delaney S . Rate-determining step of flap endonuclease 1 (FEN1) reflects a kinetic bias against long flaps and trinucleotide repeat sequences. J Biol Chem. (2015) ;290: (34):21154–62. |
[151] | Crespan E , Hübscher U , Maga G . Expansion of CAG triplet repeats by human DNA polymerasesλ and β in vitro, is regulated by flap endonuclease 1 and DNA ligase 1. DNA Repair (Amst). (2015) ;29: :101–11. |
[152] | Xu M , Lai Y , Torner J , Zhang Y , Zhang Z , Liu Y . Base excision repair of oxidative DNA damage coupled with removal of a CAG repeat hairpin attenuates trinucleotide repeat expansion. Nucleic Acids Res. (2014) ;42: (6):3675–91. |
[153] | Ma L , Chen K , Clarke DJ , Nortcliffe CP , Wilson GG , Edwardson JM , et al. Restriction endonuclease TseI cleaves A:A and T:T mismatches in CAG and CTG repeats. Nucleic Acids Res. (2013) ;41: (9):4999–5009. |
[154] | Goula AV , Pearson CE , Della Maria J , Trottier Y , Tomkinson AE , Wilson DM , et al. The nucleotide sequence, DNA damage location, and protein stoichiometry influence the base excision repair outcome at CAG/CTG repeats. Biochemistry. (2012) ;51: (18):3919–32. |
[155] | Li M , Völker J , Breslauer KJ , Wilson DM . APE1 incision activity at abasic sites in tandem repeat sequences. J Mol Biol. (2014) ;426: ()11:2183–98. |
[156] | Liu Y , Wilson SH . DNA base excision repair: A mechanism of trinucleotide repeat expansion. Trends Biochem Sci. (2012) ;37: (4):162–72. |
[157] | Jackson A , Okely EA , Leach DRF . Expansion of CAG repeats in Escherichia coli is controlled by single-strand DNA exonucleases of both polarities. Genetics. (2014) ;198: (2):509–17. |
[158] | Andreoni F , Darmon E , Poon WCK , Leach DRF . Overexpression of the single-stranded DNA-binding protein (SSB) stabilises CAG•CTG triplet repeats in an orientation dependent manner. FEBS Lett. (2010) ;584: (1):153–8. |
[159] | Richard GF , Goellner GM , McMurray CT , Haber JE . Recombination-induced CAG trinucleotide repeat expansions in yeast involve the MRE11-RAD50-XRS2 complex. EMBO J. (2000) ;19: (10):2381–90. |
[160] | Spiro C , Pelletier R , Rolfsmeier ML , Dixon MJ , Lahue RS , Gupta G , et al. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol Cell. (1999) ;4: (6):1079–85. |
[161] | Singh P , Zheng L , Chavez V , Qiu J , Shen B . Concerted action of exonuclease and gap-dependent endonuclease activities of FEN-1 contributes to the resolution of triplet repeat sequences (CTG) n- and (GAA)n-derived secondary structures formed during maturation of Okazaki fragments. J Biol Chem. (2007) ;282: (6):3465–77. |
[162] | Henricksen LA , Tom S , Liu Y , Bambara RA . Inhibition of flap endonuclease 1 by flap secondary structure and relevance to repeat sequence expansion. J Biol Chem. (2000) ;275: (22):16420–7. |
[163] | Vallur AC , Maizels N . Complementary roles for exonuclease 1 and flap endonuclease 1 in maintenance of triplet repeats. J Biol Chem. (2010) ;285: (37):28514–9. |
[164] | Liu Y , Prasad R , Beard WA , Hou EW , Horton JK , McMurray CT , et al. Coordination between polymerase β and FEN1 can modulate CAG repeat expansion. J Biol Chem. (2009) ;284: (41):28352–66. |
[165] | Spiro C , McMurray CT . Nuclease-Deficient FEN-1 Blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability. Mol Cell Biol. (2003) ;23: (17):6063–74. |
[166] | Hou C , Chan NLS , Gu L , Li GM . Incision-dependent and error-free repair of (CAG)n/(CTG) n hairpins in human cell extracts. Nat Struct Mol Biol. (2009) ;16: (8):869–75. |
[167] | Moe SE , Sorbo JG , Holen T . Huntingtin triplet-repeat locus is stable under long-term Fen1 knockdown in human cells. J Neurosci Methods. (2008) ;171: (2):233–8. |
[168] | Yang M , Guo H , Wu C , He Y , Yu D , Zhou L , et al. Functional FEN1 polymorphisms are associated with DNA damage levels and lung cancer risk. Hum Mutat. (2009) ;30: (9):1320–8. |
[169] | Sundararajan R , Gellon L , Zunder RM , Freudenreich CH . Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics. (2010) ;184: (1):65–77. |
[170] | Lin S , Wang M , Liu X , Lu Y , Gong Z , Guo Y , et al. FEN1 gene variants confer reduced risk of breast cancer in chinese women: A case-control study. Oncotarget. (2016) ;7: (47):78110–8. |
[171] | Kwiatkowski D , Czarny P , Toma M , Korycinska A , Sowinska K , Galecki P , et al. Association between single-nucleotide polymorphisms of the hOGG1, NEIL1, APEX1, FEN1, LIG1, and LIG3 genes and Alzheimer’s disease risk. Neuropsychobiology. (2016) ;73: (2):98–107. |
[172] | Ying N , Wang S , Xu H , Wang Y . Association between FEN1 polymorphisms -69G>A and G>T with susceptibility in human disease: A meta-analysis. Iran J Public Health. (2015) ;44: (12):1574–9. |
[173] | Perry JJP , Yannone SM , Holden LG , Hitomi C , Asaithamby A , Han S , et al. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. (2006) ;13: (5):414–22. |
[174] | Dudášová Z , Chovanec M . Artemis, a novel guardian of the genome. Neoplasma. (2003) ;50: (5):311–8. |
[175] | Bassing CH , Swat W , Alt FW . The mechanism and regulation of chromosomal V(D)J recombination. Cell. (2002) ;109: (Suppl):S45–45. |
[176] | Arya R , Bassing CH . V(D)J Recombination exploits DNA damage responses to promote immunity. Trends Genet. (2017) ;33: (7):479–89. |
[177] | Schärer OD . XPG: Its products and biological roles. Adv Exp Med Biol. (2008) ;637: :83–92. |
[178] | Han C , Huang X , Hua R , Song S , Lyu L , Ta N , et al. The association between XPG polymorphisms and cancer susceptibility: Evidence from observational studies. Medicine (Baltimore). (2017) ;96: (32):e7467. |
[179] | Notarangelo LD , Kim MS , Walter JE , Lee YN . Human RAG mutations: Biochemistry and clinical implications. Nat Rev Immunol. (2016) ;16: (4):234–46. |
[180] | Mittelman D , Moye C , Morton J , Sykoudis K , Lin Y , Carroll D , et al. Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc Natl Acad Sci U S A. (2009) ;106: (24):9607–12. |
[181] | Syed A , Tainer JA . The MRE11-RAD50-NBS1 Complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. (2018) ;87: :263–94. |
[182] | Itan Y , Shang L , Boisson B , Patin E , Bolze A , Moncada-Vélez M , et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A. (2015) ;112: (44):13615–20. |
[183] | Bergstralh DT , Sekelsky J . Interstrand crosslink repair: Can XPF-ERCC1 be let off the hook? Trends Genet. (2008) ;24: (2):70–6. |
[184] | Kavanagh D , Spitzer D , Kothari PH , Shaikh A , Liszewski MK , Richards A , et al. New roles for the major human 3′-5′ exonuclease TREX1 in human disease. Cell Cycle. (2008) ;7: (12):1718–25. |
[185] | Park VS , Pursell ZF . POLE proofreading defects: Contributions to mutagenesis and cancer. DNA Repair (Amst). (2019) ;76: :50–9. |
[186] | Kadyrova LY , Gujar V , Burdett V , Modrich PL , Kadyrov FA . Human MutLγ, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc Natl Acad Sci U S A. (2020) ;117: (7):3535–42. |
[187] | Gomes-Pereira M , Fortune MT , Ingram L , McAbney JP , Monckton DG . Pms2 is a genetic enhancer of trinucleotide CAG-CTG repeat somatic mosaicism: Implications for the mechanism of triplet repeat expansion. Hum Mol Genet. (2004) ;13: (16):1815–25. |
[188] | Nanda GG , Kumar MV , Pradhan L , Padhy B , Sundaray S , Das S , et al. rs215 is targeted by hsa-miRto regulate FEN1 expression but is not associated with Fuchs’ endothelial corneal dystrophy. PLoS One. (2018) ;13: (9):e0204278. |
[189] | Zahra R , Blackwood JK , Sales J , Leach DRF . Proofreading and secondary structure processing determine the orientation dependence of CAG·CTG trinucleotide repeat instability in Escherichia coli. Genetics. (2007) ;176: :27–41. |
[190] | van den Broek WJAA , Nelen MR , van der Heijden GW , Wansink DG , Wieringa B . Fen1 does not control somatic hypermutability of the (CTG)n · (CAG)n repeat in a knock-in mouse model for DM1. FEBS Lett. (2006) ;580: (22):5208–14. |
[191] | Shen B , Singh P , Liu R , Qiu J , Zheng L , Finger LD , et al. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. BioEssays. (2005) ;27: (7):717–29. |
[192] | Liu Y , Zhang H , Veeraraghavan J , Bambara RA , Freudenreich CH . Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol Cell Biol. (2004) ;24: (9):4049–64. |
[193] | Callahan JL , Andrews KJ , Zakian VA , Freudenreich CH . Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Mol Cell Biol. (2003) ;23: (21):7849–60. |
[194] | Otto CJ , Almqvist E , Hayden M , Andrew SE . The “flap” endonuclease gene FEN1 is excluded as a candidate gene implicated in the CAG repeat expansion underlying Huntington disease. Clin Genet. (2001) ;59: (2):122–7. |
[195] | Du Montcel ST , Durr A , Bauer P , Figueroa KP , Ichikawa Y , Brussino A , et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain. (2014) ;137: (9):2444–55. |
[196] | Wang C , Chen Z , Peng H , Peng Y , Zhou X , Yang H , et al. Investigation on modulation of DNA repair pathways in Chinese MJD patients. Neurobiol Aging. (2018) ;71: :267e5–267e6. doi: 10.1016/j.neurobiolaging.2018.06.024 |
[197] | Stuitje G , Van Belzen MJ , Gardiner SL , Van Roon-Mom WMC , Boogaard MW , Tabrizi SJ , et al. Age of onset in Huntington’s disease is influenced by CAG repeat variations in other polyglutamine disease-associated genes. Brain. (2017) ;140: (7):e42. |
[198] | Massey TH , Jones L . The central role of DNA damage and repair in CAG repeat diseases. DMM Dis Model Mech. (2018) ;11: (1):dmm031930. doi: 10.1242/dmm.031930 |
[199] | Gao R , Chakraborty A , Geater C , Pradhan S , Gordon KL , Snowden J , et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife. (2019) ;8: :e42988. |
[200] | York N , Lat F . Repeat DNA expands our understanding of autism spectrum disorder. Nature. (2021) ;589: (7841):200–2. |
[201] | Mitra I , Huang B , Mousavi N , Ma N , Lamkin M , Yanicky R , et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature. (2021) ;589: (7841):246–50. doi: 10.1038/s41586-020-03078-7 |
[202] | Pearson CE , Edamura KN , Cleary JD . Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet. (2005) ;6: (10):729–42. |
[203] | Reddy K , Schmidt MHM , Geist JM , Thakkar NP , Panigrahi GB , Wang YH , et al. Processing of double-R-loops in (CAG)•(CTG) and C9orf72 (GGGGCC)•(GGCCCC) repeats causes instability. Nucleic Acids Res. (2014) ;42: (16):10473–87. |
[204] | Castel AL , Cleary JD , Pearson CE . Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. (2010) ;11: (3):165–70. |
[205] | Axford MM , Wang YH , Nakamori M , Zannis-Hadjopoulos M , Thornton CA , Pearson CE . detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type 1 disease locus in patient tissues. PLoS Genet. (2013) ;9: (12):e1003866. |
[206] | Brown RE , Freudenreich CH . Structure-forming repeats and their impact on genome stability. Curr Opin Genet Dev. (2021) ;67: :41–51. doi: 10.1016/j.gde.2020.10.006 |
[207] | Poggi L , Richard G . Alternative DNA structures in vivo: Molecular evidence and remaining questions. (2021) ;85: (1):1–24. |
[208] | Wright GEB , Black HF , Collins JA , Gall-Duncan T , Caron NS , Pearson CE , et al. Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. (2020) ;19: (11):930–9. doi: 10.1016/S1474-4422(20)30343-4 |
[209] | Khristich AN , Mirkin SM . On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J Biol Chem. (2020) ;295: (13):4134–70. |
[210] | Nakamori M , Panigrahi GB , Lanni S , Gall-Duncan T , Hayakawa H , Tanaka H , et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet. (2020) ;52: (2):146–59. |
[211] | Cleary JD , Tomé S , Castel AL , Panigrahi GB , Foiry L , Hagerman KA , et al. Tissue-and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type 1 locus. Nat Struct Mol Biol. (2010) ;17: (9):1079–87. |
[212] | Gazy I , Hayward B , Potapova S , Zhao X , Usdin K . Double-strand break repair plays a role in repeat instability in a fragile X mouse model. DNA Repair (Amst). (2019) ;74: :63–9. |
[213] | Zhao X , Zhang Y , Wilkins K , Edelmann W , Usdin K . MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. (2018) ;14: (10):e1007719. |
[214] | Cleary JD , Nichol K , Wang YH , Pearson CE . Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat Genet. (2002) ;31: (1):37–46. |
[215] | Yang Z , Lau R , Marcadier JL , Chitayat D , Pearson CE . Replication inhibitors modulate instability of an expanded trinucleotide repeat at the myotonic dystrophy type 1 disease locus in human cells. Am J Hum Genet. (2003) ;73: (5):1092–105. |
[216] | Freudenreich CH , Lahiri M . Structure-forming CAG/CTG repeat sequences are sensitive to breakage in the absence of Mrc1 checkpoint function and S-phase checkpoint signaling: Implications for trinucleotide repeat expansion diseases. Cell Cycle. (2004) ;3: (11):1370–4. |
[217] | Pearson CE , Nichol Edamura K , Cleary JD . Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet. (2005) ;6: (10):729–42. |
[218] | Cleary JD , Pearson CE . Replication fork dynamics and dynamic mutations: The fork-shift model of repeat instability. Trends Genet. (2005) ;21: (5):272–80. |
[219] | Kang S , Ohshima K , Shimizu M , Amirhaeri S , Wells RD . Pausing of DNA synthesis in vitro at specific loci in CTG and CGG triplet repeats from human hereditary disease genes. J Biol Chem. (1995) ;270: (45):27014–21. |
[220] | Samadashwily GM , Raca G , Mirkin SM . Trinucleotide repeats affect DNA replication in vivo . Nat Genet. (1997) ;17: (3):298–304. |
[221] | Mason AG , Tomé S , Simard JP , Libby RT , Bammler TK , Beyer RP , et al. Expression levels of DNA replication and repair genes predict regional somatic repeat instability in the brain but are not altered by polyglutamine disease protein expression or age. Hum Mol Genet. (2014) ;23: (6):1606–18. |
[222] | Wheeler V , Dion V . Modifiers of CAG repeat instability: Insights from model systems. J Huntingtons Dis. 2020. doi:10.3233/JHD-200426 |
[223] | Gannon AMM , Frizzell A , Healy E , Lahue RS . MutSβ and histone deacetylase complexes promote expansions of trinucleotide repeats in human cells. Nucleic Acids Res. (2012) ;40: (20):10324–33. |
[224] | Halabi A , Ditch S , Wang J , Grabczyk E . DNA mismatch repair complex MutSβ promotes GAA·TTC repeat expansion in human cells. J Biol Chem. (2012) ;287: (35):29958–67. |
[225] | Panigrahi GB , Slean MM , Simard JP , Gileadi O , Pearson CE . Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSβ, but clustered slip-outs are poorly repaired. Proc Natl Acad Sci U S A. (2010) ;107: (28):12593–8. |
[226] | Tomé S , Holt I , Edelmann W , Morris GE , Munnich A , Pearson CE , et al. MSH2 ATPase domain mutation affects CTG•CAG repeat instability in transgenic mice. PLoS Genet. (2009) ;5: (5):e1000482. 10.1371/journal.pgen.1000482 |
[227] | Maiuri T , Hung CLK , Suart CE , Begeja N , Barba-Bazan C , Peng Y , et al. DNA repair in neurodegeneration: Somatic expansion and alternative hypotheses. J Huntingtons Dis. 2020. doi:10.3233/JHD-200414 |
[228] | Benn C , Gibson K , Reynolds D . Drugging DNA damage repair pathways for trinucleotide repeat expansion diseases. J Huntingtons Dis. 2020. doi:10.3233/JHD-200421 |
[229] | Iyer R , Pluciennik A . DNA mismatch repair and its role in Huntington’s disease. J Huntingtons Dis. 2020. doi:10.3233/JHD-200438 |
[230] | Pluciennik A , Burdett V , Baitinger C , Iyer RR , Shi K , Modrich P . Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLa endonuclease activation. Proc Natl Acad Sci U S A. (2013) ;110: (30):12277–82. |
[231] | Dherin C , Gueneau E , Francin M , Nunez M , Miron S , Liberti SE , et al. Characterization of a highly conserved binding site of Mlh1 required for exonuclease 1-dependent mismatch repair. Mol Cell Biol. (2009) ;29: (3):907–18. |
[232] | Pedrazzi G , Perrera C , Blaser H , Kuster P , Marra G , Davies SL , et al. Direct association of Bloom’s syndrome gene product with the human mismatch repair protein MLH1. Nucleic Acids Res. (2001) ;29: (21):4378–86. |
[233] | Langland G , Kordich J , Creaney J , Goss KH , Lillard-Wetherell K , Bebenek K , et al. The Bloom’s syndrome protein (BLM) interacts with MLH1 but is not required for DNA mismatch repair. J Biol Chem. (2001) ;276: (32):30031–5. |
[234] | Chan NLS , Hou C , Zhang T , Yuan F , Machwe A , Huang J , et al. The Werner syndrome protein promotes CAG/CTG repeat stability by resolving large (CAG)n/(CTG)n hairpins. J Biol Chem. (2012) ;287: (36):30151–6. |
[235] | Guo J , Gu L , Leffak M , Li GM . MutSβ promotes trinucleotide repeat expansion by recruiting DNA polymerase β to nascent (CAG) n or (CTG) n hairpins for error-prone DNA synthesis. Cell Res. (2016) ;26: (7):775–86. |
[236] | Shereda RD , Machida Y , Machida YJ . Human KIAA/FAN1 localizes to stalled replication forks via its ubiquitin-binding domain. Cell Cycle. (2010) ;9: (19):3977–83. |
[237] | Takahashi D , Sato K , Hirayama E , Takata M , Kurumizaka H . Human FAN1 promotes strand incision in 5′-flapped DNA complexed with RPA. J Biochem. (2015) ;158: (3):263–70. |
[238] | Schmidt MHM , Pearson CE . Disease-associated repeat instability and mismatch repair. DNA Repair (Amst). (2016) ;38: :117–26. |
[239] | Lin Y , Dent SYR , Wilson JH , Wells RD , Napierala M . R loops stimulate genetic instability of CTG·CAG repeats. Proc Natl Acad Sci U S A. (2010) ;107: (2):692–7. |
[240] | Reddy K , Tam M , Bowater RP , Barber M , Tomlinson M , Nichol Edamura K , et al. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. (2011) ;39: (5):1749–62. |
[241] | Freudenreich CH . R-loops: Targets for nuclease cleavage and repeat instability. Curr Genet. (2018) ;64: (4):789–94. |
[242] | McNulty P , Pilcher R , Ramesh R , Necuiniate R , Hughes A , Farewell D , et al. Reduced cancer incidence in Huntington’s disease: Analysis in the registry study. J Huntingtons Dis. (2018) ;7: :209–22. |


