
What is the Pathogenic CAG Expansion Length in Huntington’s Disease?
Abstract
Huntington’s disease (HD) (OMIM 143100) is caused by an expanded CAG repeat tract in the HTT gene. The inherited CAG length is known to expand further in somatic and germline cells in HD subjects. Age at onset of the disease is inversely correlated with the inherited CAG length, but is further modulated by a series of genetic modifiers which are most likely to act on the CAG repeat in HTT that permit it to further expand. Longer repeats are more prone to expansions, and this expansion is age dependent and tissue-specific. Given that the inherited tract expands through life and most subjects develop disease in mid-life, this implies that in cells that degenerate, the CAG length is likely to be longer than the inherited length. These findings suggest two thresholds— the inherited CAG length which permits further expansion, and the intracellular pathogenic threshold, above which cells become dysfunctional and die. This two-step mechanism has been previously proposed and modelled mathematically to give an intracellular pathogenic threshold at a tract length of 115 CAG (95% confidence intervals 70– 165 CAG). Empirically, the intracellular pathogenic threshold is difficult to determine. Clues from studies of people and models of HD, and from other diseases caused by expanded repeat tracts, place this threshold between 60– 100 CAG, most likely towards the upper part of that range. We assess this evidence and discuss how the intracellular pathogenic threshold in manifest disease might be better determined. Knowing the cellular pathogenic threshold would be informative for both understanding the mechanism in HD and deploying treatments.
BACKGROUND
Huntington’s disease (HD) is one of > 50 diseases caused by expanded short tandem repeats [1, 2]. In those diseases where the repeat is coding, as in HD, the repeat unit is usually CAG and this is translated to a homopolymeric glutamine tract in the encoded protein. There are nine such diseases, often referred to collectively as the polyglutamine diseases. The sections of these proteins containing expanded glutamine form cellular aggregates [3]. The polyglutamine diseases have disease-causing expansion lengths that are much shorter than those in diseases where the repeats causing the expansion are not translated [4, 5], implying a possible constraint on length at the level of the protein. While somatic expansion is critical in reaching the intracellular pathogenic CAG length threshold, the subsequent events leading to cell dysfunction and death have not been conclusively defined (Fig. 1). Much attention has focused on the expanded glutamine tract in the protein but it has never been conclusively proven that this elicits toxicity in cells in human disease, and the genetic evidence implicates CAG length rather than polyglutamine length as critical in HD pathogenesis [6– 8]. Other potential pathogenic mechanisms that cannot be precluded include RNA-based toxicity as in myotonic dystrophy (OMIM 160900) [9], RAN translation [10] and aberrant exon 1 splicing [11]: all of these mechanisms would also be exacerbated by somatic expansion of the repeat in individual cells. Recent evidence of neurodevelopmental effects in HD [12], and early phenotypes in peripheral blood mononuclear cells [13, 14], may indicate other pathways impacted by the unexpanded CAG length, but the genetic evidence in HD subjects very clearly points to somatic expansion as likely to be important in disease manifestation.
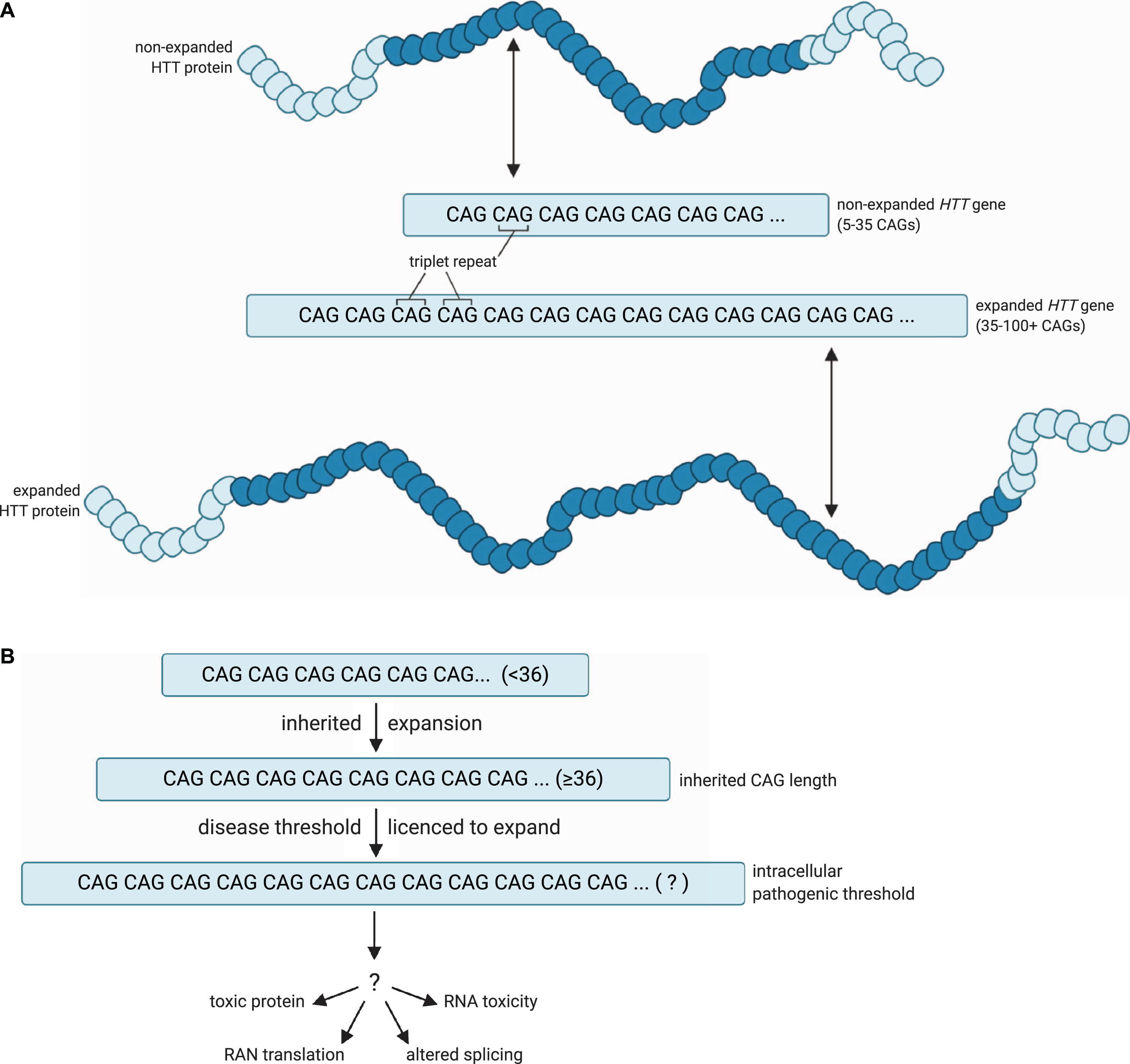
Fig. 1
A model for the pathogenic threshold in HD. A) HD pathogenesis is largely determined by an expanded cytosine-adenine-guanine (CAG) trinucleotide repeat within exon 1 of the huntingtin (HTT) gene, which is translated into an expanded polyglutamine tract in the corresponding HTT protein. Wild-type HTT possesses 5– 35 CAG repeats (non-expanded HTT gene), and can undergo expansion into the disease range in the germline to create apparent de novo HD subjects, but ≥36 + repeats is associated with a significantly increased risk of developing HD (expanded HTT gene). B) An expanded HTT allele with 36 or more repeats is unstable and licenced to further expand in cells over the lifespan of the HD at-risk individual. HD symptoms would manifest and progress as increasing numbers of disease-relevant cells undergo somatic expansion beyond an unknown intracellular pathogenic threshold that renders the gene toxic in those cells. Figures created using BioRender.com. Figure 1A adapted from a figure by National Institute of General Medical Sciences, National Institutes of Health.
In HD, age at onset of disease is largely determined by the length of the CAG tract [15– 17]. More recently, however, age at onset has been shown to be modulated by a series of genetic modifiers whose discovery has revolutionised the way we think about HD pathogenesis [7, 18– 20]. Two types of genetic modifier revealed so far have provided evidence that has made us rethink our notions of HD pathogenesis. First, some of the encoded proteins translated from these modifiers act directly on DNA and are most likely to exert their effect at the level of the mutated expanded DNA, through modulating the length of the CAG tract in both somatic and germline cells [18– 21], and indeed, this has been shown experimentally in cell culture [22, 23]. Second, the exact sequence at the HTT CAG locus exerts a strong influence on age at onset of disease [6– 8]. The length of the uninterrupted CAG tract correlates most closely with age at onset of disease and any interrupting bases that disrupt the continuous repeated sequence delay age at onset. Not all of the difference in onset age appears to be determined by uninterrupted CAG length at the HTT locus, in particular, having no CAA interruptions appears to hasten onset, while the effect of having more than one CAA at the 3’ end of the repeat tract is unclear [6, 7]. Together, these findings refocus attention on the causative repeat expansion at the level of the DNA, rather than the expanded glutamine tract in the mutant huntingtin protein that is translated from it.
A TWO-STAGE HYPOTHESIS OF DISEASE PATHOGENESIS
We know that further expansion of the HTT CAG tract occurs in the brain cells of HD subjects [24– 26], that longer CAG tracts are more likely to expand [27, 28], and that the greater the expansion in brain the earlier the onset of disease [29]. These observations in humans validate previous findings in mice and provide a link between genetic data from human subjects and disease models of repeat instability [18, 20, 30– 32]. These genetic data imply that at the point at which the CAG tract elicits toxicity in susceptible cells, which we refer to as the intracellular pathogenic CAG length, it is likely to be longer than the diagnostic disease-causing length of 36 CAG or more measured in the subjects’ blood. It is plausible that the inherited disease-causing threshold of 36 CAG in HD is the threshold beyond which further CAG repeat expansions are permissible in susceptible somatic cells (Fig. 1). It is also possible that there is a vicious cycle where alterations in DNA damage and repair exacerbate somatic expansion of the repeat [33]. So how can we define the intracellular pathogenic repeat length in such cells? Or at least the length that is pathogenic in a high enough proportion of the appropriate cell types to cause disease symptoms? This important information is lacking: defining such a threshold would be useful in clinical prognosis and clinical trials. A version of this interesting idea has been proposed previously by Kaplan and colleagues [34]. They postulated a mechanism where onset and progression of the disease are determined by the rate of expansion of the repeat tract in specific cells in the subject, and that the disease manifests only when the repeat tract has expanded beyond a certain threshold in a sufficient number of these cells. Progression is then determined by the rate at which more cells cross the pathogenic threshold for repeat length. The expansion rate and pathogenic threshold are estimated from clinical data (onset and repeat length of the inherited allele). The model assumes that the expansion rate is linear with respect to repeat length above an initial threshold, determined from clinical data (37 for HD). The assumption of linear expansion rate has not been confirmed experimentally, although Kaplan et al. [34] found that the onset predictions given by their model appeared to fit observed data for a number of repeat disorders, including HD, the polyglutamine tract mediated spinocerebellar ataxias (SCAs), myotonic dystrophy 1 (DM1, OMIM 160900) and Friedreich’s ataxia (FA, OMIM 229300). This model also predicts that earlier onset and longer CAG are correlated with faster progression, consistent with recent clinical observations [19, 35]. It may also explain why pre-clinical symptoms are evident up to 15 years prior to disease onset [36– 39]. This premanifest period reflects a proportion of cells that have somatic expansion which induces neuronal dysfunction, but too few to manifest overt clinical symptoms (Fig. 2). It is also likely that with a slow degeneration of neurons during this period, there is functional compensation in the CNS [40, 41]. The new genetic evidence makes this a compelling hypothesis in the pathogenesis of HD and other repeat disorders. This prescient paper has notably only been cited eleven times in 13 years [34].
Fig. 2
Potential relationship of CAG tract expansion and clinical Huntington’s disease events. The premanifest period of the disease may reflect the presence of a proportion of disease-relevant cells with sufficient somatic expansion to induce neuronal dysfunction, but too few to manifest overt clinical symptoms. Premanifest HD includes a presymptomatic period where no signs or symptoms are present, and prodromal HD, characterised by the onset of subtle signs and symptoms, which may be the result of the HTT CAG length expanding beyond an unknown pathogenic threshold in increasing numbers of disease-relevant cells. Manifest HD— characterised by chorea and gradual worsening of motor and cognitive difficulties— may then arise once a significant number of disease-relevant cells have passed this threshold. Somatic expansion in susceptible cell populations is likely to be occurring throughout the premanifest and prodromal stages of disease as indicated by the hypothetical dashed line, although the actual trajectory of this expansion will depend on the inherited repeat length and is likely to differ in different cell types. Therefore, the relationship between the trajectory of somatic expansion and clinical phenotypes is currently hypothetical. Figure adapted from Ross et al. [45] and Bates et al. [3] and created using BioRender.com.
![Potential relationship of CAG tract expansion and clinical Huntington’s disease events. The premanifest period of the disease may reflect the presence of a proportion of disease-relevant cells with sufficient somatic expansion to induce neuronal dysfunction, but too few to manifest overt clinical symptoms. Premanifest HD includes a presymptomatic period where no signs or symptoms are present, and prodromal HD, characterised by the onset of subtle signs and symptoms, which may be the result of the HTT CAG length expanding beyond an unknown pathogenic threshold in increasing numbers of disease-relevant cells. Manifest HD— characterised by chorea and gradual worsening of motor and cognitive difficulties— may then arise once a significant number of disease-relevant cells have passed this threshold. Somatic expansion in susceptible cell populations is likely to be occurring throughout the premanifest and prodromal stages of disease as indicated by the hypothetical dashed line, although the actual trajectory of this expansion will depend on the inherited repeat length and is likely to differ in different cell types. Therefore, the relationship between the trajectory of somatic expansion and clinical phenotypes is currently hypothetical. Figure adapted from Ross et al. [45] and Bates et al. [3] and created using BioRender.com.](https://content.iospress.com:443/media/jhd/2021/10-1/jhd-10-1-jhd200445/jhd-10-jhd200445-g002.jpg)
The in silico modelling of Kaplan et al. [34] predicted that the intracellular pathogenic CAG length threshold in HD was 115 CAG. However, the confidence intervals for this threshold are wide (95% CI 93– 170 CAG), possibly due to the model being fitted to a relatively small sample (n = 336). Do we have any evidence that would allow us to determine whether 115 CAG is a reasonable estimate of the threshold, or to refine that wide confidence interval? This is critical, as under this scenario, the period of an HD at-risk subject’s life before such a threshold is reached is a window of opportunity for therapies that address the expansion of the repeat (Fig. 2). It is potentially a large window as expansion is likely to occur throughout life [26, 42– 44] and at-risk subjects remain largely indistinguishable from their non-HD at-risk peers for a substantial period of that time [39].
There are some potential clues to the intracellular pathogenic threshold. We might be able to improve the definition of the edge of the pathogenic thresholds using data from mouse models. In mouse models the repeat is normally expanded to 100 CAG or more in order to induce a disease-like phenotype in the short-lived mouse [46, 47]. Even in the presence of tracts of over 100 CAG in their Htt gene, mice may only develop subtle phenotypes late in their lifespan. Other diseases where a CAG tract is translated to a polyglutamine tract may also offer some clues about thresholds for intracellular CAG toxicity [4, 48– 50]. Possible inferences from these sources of evidence are discussed below.
EVIDENCE FROM HD ANIMAL MODELS
There are many animal models of HD generated in a number of different ways (Table 1). They can be divided into those expressing transgenes with a truncated section of human HTT carrying the CAG tract, or full length human HTT, and those with long CAG tracts replacing mouse Htt in one way or another [3, 46, 47, 51]. Instability of the repeat sequence has been seen in many of the mouse models and was noted in the first HD models ever reported, the R6 series [52, 53].
Table 1
Animal models of Huntington’s disease with up to 100 CAG repeats
| Animal model | Construct | Promoter | PolyQ repeat length | Pure repeat length/interruptions | Phenotype | Somatic expansion? | References |
| YAC46 (mouse) | FL human HTT gene within a YAC | HTT | Q46 | 46 mixed CAA/CAG | Increased NMDA-induced Ca + response, but no behavioural or cognitive phenotype | Not reported | [54– 56] |
| HD46 | N-terminal 3 kb of human HTT cDNA | Rat NSE | Q46 | (CAG)n(CAA)(CAG) | Increased incidence of clasping, abnormal gait and abnormal activity- though time-points are not clear. HTT inclusions in cortex and striatum. | Not reported | [57] |
| HD48 | FL human HTT | CMV | Q48 | (CAG)n(CAA)(CAG) | Limb clasping from 8– 24 weeks, hyperactivity from 20 weeks, reduced exploration from 24 weeks. Neuronal loss and fibrillary astrocytosis in the striata. HTT aggregates. | Not reported | [58] |
| HdhQ50 | Chimeric human exon 1/ mouse Htt | Htt | Q50 | (CAG)48(CAA)(CAG) | No behavioural or neuropathological changes observed at 6 months of age. | Not reported | [59, 60] |
| HDQ50 | CAG-only tract knocked into mouse exon 1 | Htt | Q52 | (CAG)50(CAA)(CAG) | No behavioural or pathological changes observed. | Not reported but germline expansion is present. The allelic line HDQ150 has somatic expansion. | [25, 61, 62] |
| Tg51 (rat) | 1962 bp rat Htt cDNA fragment | Htt | Q51 | Not clear | Reduced anxiety from 2 months, impaired coordination from 10 months, nuclear inclusions from 12 months. | Not reported | [63] |
| CAG71 | Chimeric human exon 1/ mouse Htt | Htt | Q71 | (CAG)71 (R42) Arginine residue at position 42 | No behavioural abnormalities. | Not reported | [64, 65] |
| YAC72 (mouse) | Full length human HTT gene within a yeast artificial chromosome | HTT | Q72 | (CAG)n(CAA)(CAG) | Circling and foot-clasping from 9 months, hyperactivity at 7 months, HTT aggregates and striatal degeneration from 12 months. | Not reported | [54, 56, 66] |
| Hdh6/Q72 | Chimeric human exon 1/ mouse Htt | Htt | Q72 | (CAG)n(CAA)(CAG) | Hyperaggressive behaviour from 3 months. No neuropathological changes. | Yes. Multiple tissues. Expansion bias in striatum where > 80% of cells showed expansions greater than+5 CAG. | [67] |
| Htt-Q79 | Chimeric human exon 1/ mouse Htt | Htt | Q77 | (CAG)n(CAA)(CAG) | Aggressive behaviour. Reactive gliosis from 40 weeks, nuclear inclusions in the striatum from 80 weeks. | Yes. Multiple tissues (brain, liver, kidney and stomach). | [68, 69] |
| Hdh4/Q80 | Chimeric human exon 1/ mouse Htt | Htt | Q80 | (CAG)n(CAA)(CAG) | Hyperaggressive behaviour from 3 months. Diffuse nuclear staining in the striatum from 17 weeks, HTT aggregates from 48 weeks. | Yes, high levels observed in the striatum at 24 months. Expansions were also observed in the cortex, cerebellum, hippocampus, hindbrain, spinal cord, olfactory bulb, kidney and eye. | [67, 70– 72] |
| N171-82Q | N-terminal 171 amino acids of human HTT cDNA | Prp | Q82 | (CAG)n(CAA)(CAG) | Motor deficit from 5 months, HTT nuclear inclusions. | Not reported | [73] |
| N586-82Q | N-terminal 586 amino acids of human HTT cDNA | Prp | Q82 | (CAG)n(CAA)(CAG) | Rotarod deficit from 4 months, hyperactivity from 5 months, HTT aggregates and cognitive impairment from 8 months. | Not reported | [74] |
| HD89 | FL human HTT | CMV | Q89 | (CAG)n(CAA)(CAG) | Limb clasping from 8 weeks, hyperactivity from 20 weeks, less exploration from 24 weeks. Neuronal loss and fibrillary astrocytosis in the striata. | Not reported | [58] |
| R6/1-89Q | Human HTT exon 1 | HTT | Q89 | (CAG)n(CAA)(CAG) | Clasping behaviour from 24 weeks, diffuse nuclear staining in cerebral cortex and hippocampus from 11 weeks, body weight loss from 28 weeks. | Yes. Expansions in motor cortex and hippocampus from 9 weeks. | [75] |
| HdhQ92 | Chimeric human exon 1/mouse Htt | Htt | Q92 | (CAG)n(CAA)(CAG) | Cognitive deficits from 4 months, mild motor deficit and HTT aggregates from 6 months, striatal cell loss from 8 months. | Yes, in striatum and liver | [60] |
| CAG94 | Chimeric human exon 1/mouse Htt | Htt | Q94 | (CAG)94 (R42) Arginine residue at position 42 | Increased sensitivity to NMDA from 7 weeks. Increased rearing from 9 weeks, decreased motor and exploratory activity from 18 weeks. | Not reported | [64, 65] |
| Hu97/18 (mouse) | Two full human HTT alleles, one mutant and one wild type | HTT | Q97 | (CAG)n(CAA)(CAG) | Learning and motor deficit from 2 months, progressive cognitive deficits from 6 months, reduced cortical and striatal volume from 12 months. | Not reported | [76, 77] |
| BAC HD (mouse) | FL human HTT | HTT | Q97 | 97 mixed CAA/CAG | Motor deficit at 2 months significant by 6 months, no HTT aggregates. | No | [78– 80] |
| BAC HD (rat) | FL human HTT | HTT | Q97 | 97 mixed CAA/CAG | Motor deficit from 2 months, hypoactivity from 4 months, learning deficit from 6 months. | No | [81– 83] |
| HD100 | N-terminal 3 kb of human HTT cDNA | Rat NSE | Q100 | (CAG)98(CAA)(CAG) | Rotarod impairment, clasping, abnormal gait from 13– 78 weeks. HTT inclusions from 13 weeks. | Not reported | [57] |
| HDQ50 | CAG tract knocked into mouse exon 1 | Htt | Q100 | (CAG)100(CAA)(CAG) | No behavioural or pathological changes observed. | Not reported but germline expansion is present. The allelic line HDQ150 has somatic expansion. | [25, 61, 62] |
NSE, neuron-specific enolase; CMV, cytomegalovirus; PrP, prion gene promoter. Not reported means no data were available. No means somatic expansion was investigated and not seen.
Although there are multiple rodent models which have been deployed to help us understand the biology of HD and begin the search for therapies, many are limited in their ability to inform us of the effects of genetic modifiers of disease, as they often present with repeats well above the presumed intracellular pathogenic threshold and a severe phenotype. The most useful are those with relatively short repeats (Table 1) though they have differences in their genetic manipulations that make straightforward inferences about the threshold for intracellular pathogenesis complex. They all have either full length human HTT transgenes or human exon 1 replacing mouse exon 1. This may lead to differences in transcription and translation compared with humans. Often several copies of a transgene are present in a genome and RNA and protein expression levels are variable. While the human disease is completely dominant and expression levels of HTT seem irrelevant to disease manifestation and course [84], that is not true in all animal models where expression levels do appear to influence phenotype, as discussed below. Perhaps most challenging is that in many of these models a human HTT gene has been used with a long pure CAG tract. This will almost invariably lead to somatic expansion thus the intracellular pathogenic threshold we are interested in is a moving target— it will have expanded in individual cells from the inherited or engineered repeat length but by an unknown amount. Finally, somatic expansion itself can be transcriptionally mediated [85– 87], meaning that alterations in transcription across an expansion prone-repeat may themselves alter levels of expansion.
An added complication is that very long repeats appear less pathogenic than shorter disease-causing repeats, and more prone to contraction than expansion [88], though it is not clear why [89, 90]. The earliest onset and most deleterious phenotypes are seen around 150 CAG with longer CAG tracts giving later phenotypic changes [89– 91] though it should be noted that in mice with an inherited ∼150 CAG there is also somatic expansion and the repeat length in the susceptible cells is likely to be longer than 150 CAG. Very long repeat tracts form unusual DNA structures [2] that can inhibit transcription or translation of HTT, though there is evidence that the somatically expanded CAG tracts in DNA are transcribed and translated into expanded polyglutamine-containing mutant HTT (mHTT) [92]. Such very long repeat tracts might prevent downstream events that promote pathogenesis such as production of exon 1 fragments [11, 93, 94] or nucleo-cytoplasmic shuttling [95]. Landles et al. [96] demonstrated that a version of the R6/2 mouse with 90 CAG, R6/2(CAG)90, showed earlier mHTT nuclear aggregation than an R6/2 line with 200 CAG, R6/2(CAG)200, but later phenotype onset. The R6/2(CAG)90 brains contained nuclear aggregates that had a diffuse punctate appearance and remained partly detergent soluble, which correlated with the onset of transcriptional changes, whereas the R6/2(CAG)200 brains had cytoplasmic aggregates that gave larger inclusion bodies which correlated with behavioural changes. Both lines of mice showed somatic expansion of the CAG tract, therefore the exact CAG tract length giving rise to these different molecular sequelae remains unknown. Further detailed studies in animals with less extreme repeat lengths such as this might well yield more insights into the pathogenic mechanism and threshold.
A number of models, still encoding glutamine but using a mixed CAACAG rather than a pure CAG tract, can help to establish a window for a pathogenic repeat length. The mixed CAACAG stabilises the repeat tract [78], preventing germline and somatic expansion.
The BAC HD model with 97 glutamines encoded by a mixed CAACAG tract fulfils this criteria— the mixed CAACAG tract prevents both germline and somatic expansion in mice but is still pathogenic (Table 1) [78]. These mice have 5 copies of the transgene integrated into their genome and express the BAC HD HTT at higher than endogenous levels, estimated at three-fold the level of transcript and 1.5– 2-fold the level of protein. They also notably show functional deficits, but no HTT-positive inclusions, and the translated mHTT protein is largely full-length and mainly located in the cytoplasm [78]. Other sequelae of the mixed tract may exert effects unrelated to CAG expansion: the CAACAG tract is likely to form different DNA structures to pure CAG tracts and this may well affect transcription at the locus. Differential codon usage may further affect translation efficiency of HTT: CAG is used to encode glutamine three times as frequently as CAA in brain tissue [97].
Given the BAC HD line with a stable tract of 97 glutamine-encoding codons has a phenotype [78] this sets an upper bound to the likely intracellular pathogenic length (Table 1). The HdhQ92 mouse with a human exon 1 pure CAG tract knocked into mouse Htt has a late behavioural phenotype and mHTT inclusions. This is one of the minority of HD mouse models that has been assessed systematically for the presence of somatic instability, which is seen especially in brain and liver [60, 98, 99]. This means that the CAG tract length associated with the intracellular pathogenic threshold in these mice is unknown. Similarly the HdhQ72 and HdhQ80 models show some mild phenotypes but also show substantial somatic instability, again with high levels of instability in the striatum, consistent with the HdhQ92 mice [28, 67, 72]. Other rodent lines with CAG tracts of less than 100 repeats might provide further clarification. However, in most of these somatic expansion is likely to occur, though it has not been reported, as a human HTT transgene with a long pure CAG tract was used to generate these mice (Table 1) [54, 58, 63, 73, 74, 100]. If expansion is occurring, in the germline or somatically, it is likely this will provoke changes in phenotype within a cohort. More interesting are the models reported by Levine et al. (1999) with 71 and 94 CAGs. Both have a (coding) interruption in the middle of the CAG tract [64]. The 71 CAG mice have no reported abnormal behaviour and the 94 CAG mice only some minor changes [64]. Given the interruption in the CAG array of the transgene it is likely that somatic expansion is attenuated in these mouse models. These animal models may well be either side of a threshold that defines pathogenesis, so it would be useful to investigate whether they display somatic expansion.
There are additional limitations in extrapolation from mice and other animal models to people [101]. Expression levels of the gene and protein are not necessarily at endogenous levels. Genetically the most accurate animal models are those with long CAGs knocked into their mouse Htt gene (Table1). Most contain a human exon1/mouse Htt chimeric sequence which has the disadvantage of not being a gene that appears in nature at all. There is an allelic series where only the mouse CAGCAA sequence encoding 7 glutamines is replaced in mouse exon 1 [102] (Table 1), but this may not show appropriate human-relevant downstream pathogenic events. Animals are not humans and have inherent limitations such as short lifespans and differences in underlying biology, seen in DNA repair systems and oxidative damage [103], that may well be important in determining the pathogenic threshold in particular models.
While both people with HD and the animal models of disease have development of phenotypic changes over time, animals do not have an age at onset of manifest disease, as at clinical diagnosis in humans. In both people and models the changes seen depend on what phenotypes are examined and how they are measured [38, 104, 105]. The differences in disease manifestation in people are not reflected in mice, because laboratory mice are much less genetically diverse and live in a more uniform environment. Genetic variation in HD subjects influences the presentation of many non-motor symptoms for instance [106]. Most HD mouse models, despite possession of a repeat length that would give juvenile HD with its different clinical presentation, show a similar motor phenotype (though this may be an artefact of how this is measured) (Table 1) [47]. They also display very little frank neurodegeneration, though they often have smaller and lighter brains than their wildtype counterparts [46]. A series of matched knock-in lines with identical glutamine encoding stretches in Htt have been generated: one with pure CAG tracts and a parallel line with CAACAG alternating tracts [51]. These lines encode 45, 80 and 105 glutamines and should reveal the pathogenic threshold in mice provided expression levels are similar in the parallel lines, though the processing of m Htt may still be different in the mouse gene from the human gene. If extrapolating the pathogenic repeat threshold from mouse models is difficult, is it then possible to garner more relevant information from other human diseases caused by similar repeat expansions?
EVIDENCE FROM OTHER DISEASES
Repeat sequences are common in the genome and biologically functional [107] and there is a growing list of diseases caused by expanded repeated sequences in DNA [1, 2, 108]. A series of neurodegenerative diseases are caused by expanded CAG sections in their coding sequence, invariably translated to a polyglutamine tract [109]. These diseases have some striking similarities: the repeat threshold at which disease is caused is in most cases a similar length [4, 48], they show a strong relationship of repeat length with age at onset of disease, many show somatic and germline expansion of their causative repeat [31, 48, 110– 112] and they have similar genetic modifiers of their ages at onset [113] (Table 2). This implies that the underlying events leading to expansion of the CAG tracts in these diseases might have common mechanisms that can be used to inform all of these diseases, though the molecular pathogenic events downstream of the CAG tract may be specific to each disease.
Table 2
Evidence from human CAG-repeat disorders
| Disease | Normal CAG range | Disease CAG range | Pure repeat length/interruptions | Phenotype | Somatic expansion observed? | References |
| HD | 6– 35 | 36– 200+ | (CAG)n(CAA)(CAG) CAG tract usually ends CAACAG. Alleles without CAA or with multiple CAAs observed. | Age at onset 2– 80 years (n = 40 years). Pure CAG alleles give earlier onset, multiple CAA interruptions later onset. | Yes, in neural and peripheral tissues. More CAG expansion in brain, especially striatum, globus pallidus and cerebral cortex, than peripheral tissues (except the cerebellar cortex). Repeat expansion is greater in neurons than glial cells. | [7, 8, 24, 26– 29, 122] |
| SCA1 | 6– 35 | 39+(no CAT) 45– 81+ | (CAG)n(CAT)(CAG)n OR (CAG)n(CAT)(CAG)(CAT)(CAG)n CAG tract is usually interrupted by CAT or CATCAGCAT. Alleles without interruptions or with additional CATs observed. | Age at onset 4– 74 years (n = 40 years). Pure CAG alleles give earlier onset, CAT interruptions later onset. | Yes, in neural and peripheral tissues. More CAG expansion in brain than the peripheral tissues (except the cerebellar cortex). | [116, 123– 136] |
| SCA2 | 14– 31 | 32– 270+ | (CAG)n(CAA)(CAG)n OR (CAG)n(CAA)(CAG)4(CAA)(CAG)n OR (CAG)n(CAA)(CAG)4(CAA)(CAG)4 (CAA)(CAG)n CAG tract is usually interrupted by 1– 3 CAAs separated by 4x CAGs. Alleles with no CAA, or with no CAAs and a CCG or CCGCCG interruption have also been observed. | Age at onset 1– 66 years (n = 32 years). Pure CAG alleles – ataxic phenotype, CAA interruptions – Parkinsonism (SCA2-P) or ALS phenotype. Lack of CAA plus a CCG or CCGCCG exclusively observed in disease alleles | Yes, in blood. No evidence of somatic expansion has been found in alleles with interrupted repeats involved in SCA2-P phenotype. | [116, 125, 137– 150] |
| SCA3 | 12– 44 | 52– 86+ | (CAG)2(CAA)(AAG)(CAG)(CAA)(CAG)n(CGG) OR (CAG)2(CAA)(AAG)(CAG)(CAA)(CAG)n(GGG) CAG tract ends with either CGG or GGG and is usually interrupted by two CAAs and one AAG. Alleles with one less CAA have been observed. | Age at onset 4– 70 years (n = 36 years). No relationship between repeat interruptions and phenotype. 3’ flank CGG rather than GGG is associated with disease | Yes, in neural and peripheral tissues. More CAG expansion in brain than the peripheral tissues (except the cerebellar cortex). | [116, 131– 134, 151– 155] |
| SCA6 | 7– 16 | 21– 28+ | (CAG)n No interruptions in disease or normal allele. | Age at onset 19– 71 years (n = 48 years). | No | [115– 117] |
| SCA7 | 7– 35 | 34– 460+ | (CAG)n No interruptions in disease or normal allele. | Age at onset 1– 45 years (n = 20 years). | Yes, in blood, kidney, and skeletal muscle (limited studies) | [116, 156– 162] |
| SCA12 | 7– 32 | 55– 78+ | (CAG)n No interruptions in disease or normal allele. | Age at onset 8– 55 years (n = 33 years). | Yes, in blood (limited studies) | [116, 163– 165] |
| SCA17 | 25– 44 | 47– 63+ | (CAG)3(CAA)3(CAG)n(CAA)(CAG)(CAA) (CAG)n(CAA)(CAG) CAG tract typically begins with (CAG)3(CAA)3, ends with CAACAG and has a central CAACAGCAA interruption. Alleles without CAACAGCAA have been observed. | Age at onset 3– 75 years (n = 35 years). Alleles without CAACAGCAA are associated with disease phenotype | Yes, in blood (limited studies). Mosaicism and therefore mutation frequency in patients with pure CAG repeats (without the central CAACAGCAA) is 2-3 fold of those with CAA interruptions. | [116, 166, 167] |
| DRPLA | 6– 35 | 48– 83+ | (CAG)n No interruptions in disease or normal allele. | Age at onset 1– 69 years (n = 30 years). | Yes, in neural and peripheral tissues. More CAG expansion in the brain than the peripheral tissues (except the cerebellar cortex). Repeat expansion is greater in glial cells than neurons. In cerebellum Purkinje cells and cerebral neuronal cells show more expansion than cerebellar granule cells. | [133, 153, 168– 173] |
| SBMA | 9– 36 | 38– 62+ | (CAG)n No interruptions in disease or normal allele. | Age at onset 14– 75 years (n = 43 years). | Yes, in neural and peripheral tissues. Skeletal and cardiac muscle showed the most expansion whereas central nervous system tissues, liver and spleen showed the least. | [122, 174– 178] |
Table 2 shows the diseases caused by expanded CAG tracts where the repeat is definitely or likely to be translated to a polyglutamine tract in the cognate protein— it is perhaps of relevance that most of the polyglutamine protein products have a role in DNA repair [33, 109, 114]. Only spinocerebellar ataxia 6 (SCA6, OMIM 183086) shows no evidence of somatic expansion of the CAG tract, though there is genetic anticipation in families, implicating germline expansion [115– 117]. SCA6 may therefore be an exception, not requiring intracellular somatic expansion to elicit pathogenesis. The CAG tract disease range is shorter than in the other diseases, and the repeat occurs in CACNA1A, encoding a calcium channel. Nuclear inclusions are seldom observed [118– 120], and as in SCA2 (OMIM 183090), are mainly cytoplasmic, thus it is likely that the cell toxicity in this disease is mediated through other, protein-based, mechanisms [121]. SCA7 (OMIM 164500), SCA12 (OMIM 604326), dentatorubral-pallidoluysian atrophy (DRPLA, OMIM 125370) and spinal and bulbar muscular atrophy (SBMA, OMIM 313200) show no repeat tract interruptions and all have at least some evidence of somatic expansion (Table 2). Interruptions are present in the CAG tracts of the causative genes for SCA1 (OMIM 164400), SCA2 (OMIM 183090), SCA3 (also known as Machado-Joseph’s disease, OMIM 109150), and SCA17 (OMIM 607136), though loss of interruptions is associated with disease-causing alleles in SCA2. These diseases are perhaps most informative in our quest to define the pathogenic CAG length range, as interruptions are known to stabilise expanded tracts, such that the inherited allele repeat length is likely to be the maintained in most cells. Information on the extent to which interruptions reduce the rate of expansion and delay age at onset could be used to modify the Kaplan model and thus improve the estimate of the pathogenic threshold.
SCA1 disease-causing expanded CAG tracts are 39 CAGs or more with no interruption, or 45– 81 with interruptions. Lack of interruptions gives earlier disease onset [135] and in uninterrupted alleles there is a strong length correlation with age at onset [127]. The interruptions are CAT, encoding histidine rather than glutamine, and the later onset of disease was assumed to be mediated by the resulting change in the protein [126], but it appears more likely to be mediated at the level of DNA by the somatic expansion widely seen in this disease [123, 124, 129, 131– 133, 136, 179]. The pathology of SCA1 is concentrated in the cerebellum with a characteristic early and severe degeneration of the Purkinje cells [4] although recent evidence shows that subjects have widespread degeneration in deep cerebellar structures and the brainstem as well as cerebral pathology [180]. In postmortem SCA1 human brain, the highest levels of somatic expansion are not seen in the cerebellar regions and the Purkinje cells most affected in the disease [129], though at the end stage of disease the earliest affected cells may have been lost. Additionally, Purkinje cells are low in number compared with other cerebellar neurons [181], and thus rare, large expansions in these cells are likely to be underestimated when looking at whole cerebellar tissue. However, elegant work in mice has shown that it is likely to be protein interactions, particularly with capicua, that drive cell-specific intracellular pathogenesis in the Purkinje cells [182, 183]. Nevertheless, somatic expansion may drive other pathogenic events in SCA1: a similar genetic modification signal was seen in SCA1 as in HD, implying that age at onset is at least partly modulated by similar events in both diseases [113].
SCA2 is more complicated. Most CAG tract alleles have CAA interruptions, but may also be interrupted by CCG, encoding glycine. Pure CAG tracts over 34 CAG cause the ataxic phenotype of SCA2 [5, 50, 145], but interrupted alleles in what would normally be considered the long normal or low SCA2 range (see Table 2), give a Parkinsonian or amyotrophic lateral sclerosis phenotype [138, 141, 148, 184]. No evidence of somatic expansion has been seen in the phenotypes associated with interruptions [149, 150] but it is seen in SCA2 [140].
SCA3 is perhaps the most interesting and informative of the SCAs with respect to the CAG length pathogenic threshold. Normal alleles may have repeat lengths up to 44 CAG, whereas disease-associated alleles range from 52– 75 CAG, with most disease alleles harbouring repeat lengths of over 60 CAG (Table 2) [48]. There is a window where no repeat lengths have been reported between the normal and disease ranges in SCA3 as in DRPLA, SCA12 and SCA17. The CAG tract is usually interrupted by two CAAs and there does not seem to be an association between the presence of interruptions and phenotype. Notably the somatic mosaicism observed is of the order of a few repeats even in the presence of CAG tracts of 70– 80, and expansions are more prevalent in peripheral tissues than in nervous tissue [153, 154]. Though these analyses are in relatively few brains and do not use techniques that would reveal individual large expansions, nevertheless this appears to be a more stable CAG repeat tract than in HD or SCA1 for instance, especially given the CAG tract length. This provides a repeat tract length for neurodegeneration of a minimum of 60 CAG in SCA3.
SCA17 is caused by an expanded mixed CAA/CAG tract in TBP. The normal repeat number in TBP is up to 40 CAG/CAA, reduced penetrance alleles have 41– 48 repeats, and full penetrance alleles carry more than 48 CAG/CAA repeats. These interrupted repeats have a complex structure [185, 186] (Table 2). SCA17 predominantly occurs in subjects who do not carry a central CAACAGCAA interrupting sequence. The instability of the expanded CAG repeat is dependent on repeat configuration, and CAA interruption is a limiting factor for further CAG repeat expansion [166]. There is some germline instability, raising the possibility that there is some somatic expansion also occurring despite the non-pure CAG tract.
There are limitations to extrapolating from other diseases. They have different pathologies and different susceptible cell types. Notably in most of these diseases regional pathology and somatic expansion are not correlated, but relatively few subjects have been analysed in anatomical detail and only one study conducted at the single cell level. This study, measuring somatic expansion in single cells in DRPLA, compared somatic mosaicism in cerebellar structures in early versus late onset patients [173]. Higher rates of expansion were more evident in late onset case than early onset cases, though this may well be a function of age [88, 187]. The frequency of expansions was highest in glial cells, with Purkinje cells lower and granular cells lower again. Relative levels of expression of the cognate genes in the most susceptible cells are not known, but are assumed to underlie differential spatial pathogenesis [121, 188], and transcription appears to be important in promoting somatic repeat tract length changes [85– 87, 189– 192]. Finally, surviving cells that are examined in post-mortem human brain may be resistant to the ongoing toxicity mechanisms and therefore uninformative about the intracellular pathogenic repeat length threshold.
MOUSE MODELS IN OTHER REPEAT DISORDERS
There are multiple mouse models of each non-HD polyglutamine repeat disorder, most of which have not had somatic expansion of the repeat surveyed systematically (Table 3). Most alleles were cloned from patients as transgenes or knocked into the endogenous mouse genes, and often required longer CAG repeat lengths than in humans to evoke a phenotype. As in HD animal models, transgenic mouse models of these diseases often demonstrate severe early-onset neuropathology and behavioural syndromes whilst knock-in mouse models tend to show milder late-onset phenotypes that perhaps parallel the disease more accurately, but are slower to produce phenotypes. Consistent with animal models of HD, animal models of other triplet repeat disorders tend to show increased disease phenotype as CAG repeat length increases, though this is influenced by the promoter used, transgene copy number and resultant transgene expression. Cemal et al. [193] generated a series of eight YAC SCA3 models and found that disease severity increased both with an increased CAG repeat tract length and an increased transgene copy number such that an animal with 72 CAG and one copy of the transgene developed symptoms later than an animal with 67 CAG repeats and two copies of the transgene.
Table 3
Animal models in other repeat disorders
| Disease | Animal model | Construct | Promoter | PolyQ repeat length | Pure repeat length/interruptions | Phenotype | Somatic expansion observed? | References | |
| DRPLA | AT-FL-65Q | FL human ATN1 | Prp | Q65 | (CAG)n | Motor deficits from 9 weeks. NIIs from 19 weeks. | Not reported | [194, 195] | |
| ATN-1[Q76] | FL human ATN1 | ATN1 | Q76 | (CAG)n | Motor deficits from 8 weeks. DNS from 14 weeks. | Yes at 64 weeks. High levels of somatic expansion in cortex, liver, kidney. Low levels in cerebellum and heart. | [196– 198] | ||
| Q96 | FL human ATN1 | ATN1 | Q96 | (CAG)n | Motor deficits from 8 weeks. Decreased life span (54 weeks). | Not reported | [197] | ||
| Q113 | FL human ATN1 | ATN1 | Q113 | (CAG)n | Motor deficits from 7 weeks. Decreased life span from 21 weeks. | Not reported | [197] | ||
| Atro-118Q | FL human ATN1 | NSE | Q118 | (CAG)n | Motor deficits from 5 weeks. DNS and NIIs from 12 weeks. Decreased lifespan from 14 weeks. | Not reported | [199] | ||
| Tg(ATN1)Q129Stsu | FL human ATN1 | ATN1 | Q129 | (CAG)n | Motor deficits from 3 weeks. DNS from day 4. NIIs at 9 weeks. Decreased lifespan from 10 weeks. | Somatic instability not reported. Germline ATN1 instability reported. | [197, 198, 200] | ||
| SCA1 | Sca178Q | Chimeric human exon 8/ mouse Atxn1 | Atxn1 | Q78 | (CAG)n | Motor deficits from 9 months. No neuropathological changes up to 18 months. | Somatic instability not reported. Intergenerational repeat instability (+2 to – 6 CAG) | [201] | |
| PS-82 or B05 | FL human ATXN1 | Pcp2/L7 | Q82 | (CAG)n | Motor deficits from 5 weeks. NIIs in Purkinje cells at 6 weeks. Purkinje cell loss 24 weeks. | Not reported. Intergenerationally stable. | [202– 204] | ||
| PrP-tTA/TRE-SCA1 | FL human ATXN1 | Tet-Prp | Q100 | (CAG)n | Motor deficits from 5 weeks. Purkinje cell loss, dendritic degeneration and reactive gliosis from 20 weeks | Not reported | [205] | ||
| Sca1154Q | Chimeric human exon 8/ mouse Atxn1 | Atxn1 | Q154 | (CAG)n | Motor deficits from 8 weeks. NIIs in hippocampus and cerebral cortex from 7 weeks. NIIs in cerebellum at 40 weeks. | Yes, observed in most tissues at 40 weeks. Considerable expansions in striatum (>210 CAG), spinal cord and pons. | [206, 207] | ||
| SCA2 | Atxn2-CAG42-Knock-In | Expanded CAG in exon 2 of mouse Atxn2 | Atxn2 | Q42 | (CAG)n | Motor deficit from 18 months. DCS and CI in cerebellum at 13 months. | No somatic instability or intergenerational instability. | [208] | |
| 58Q-5B | FL human ATXN2 | Pcp2/L7 | Q58 | (CAG)n | Motor deficits from 16 weeks. CIs and decreased number of Purkinje cells at 24 weeks. | Not reported | [209, 210] | ||
| BAC-Q72 | FL human ATXN2 | Atxn2 | Q72 | (CAG)n | Motor deficits from 8 weeks. Morphological changes in Purkinje cells at 24 weeks. | Not reported | [211] | ||
| 75Q-SCA2 | FL human ATXN2 | Atxn2 | Q75 | (CAG)n | Motor deficits from 6 weeks. Purkinje cell degeneration from 52 weeks | Not reported. Intergenerationally stable. | [212] | ||
| Atxn2-CAG100-KIN | Expanded CAG in exon 2 of mouse Atxn2 | Atxn2 | Q100 | (CAG)n | Motor deficits from 20 weeks. Progressive brain atrophy and neuronal aggregation in multiple brain regions from 13 weeks. | Yes, expansions > 128 CAG observed. Somatic mosaicism observed in multiple mice. | [213] | ||
| ATXN2 | FL human ATXN2 | Pcp2/L7 | Q127 | (CAG)n | Motor deficits from 8 weeks. Extranuclear aggregates and DCS in the cerebellum from 4 weeks. Purkinje cell loss from 12 weeks. | Not reported | [214] | ||
| SCA3 | MJD 64.8 | FL human ATXN3 | ATXN3 | Q64 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)58 | Motor deficits from 3 weeks. Severe neuronal loss and gliosis. Widespread NIIs from 5 months. | Not reported | [193] | |
| MJD67.2 | FL human ATXN3 | ATXN3 | Q67 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)61 | Motor deficits from 4 weeks. NIIs in multiple brain regions. | Not reported | [193] | ||
| Pcp2/ATXN3_69CAG | c-terminus human ATXN3 | Pcp2/L7 | Q69 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)63 | Motor deficits from 3 weeks. Cerebellar atrophy from 3 weeks. DCS in cerebellum at day 5, NIIs at 7 weeks. | Not reported | [215] | ||
| 70.61 CAG | FL human ATXN3 | Prp | Q70 | Not reported | Motor deficits from 12 weeks. Widespread NIIs at 3-4 months | Not reported | [216, 217] | ||
| Q71B/C | FL human ATXN3 | Prp | Q71 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)65 | Motor deficits from 11 weeks. Widespread NIIs at 2-3 months | Not reported | [218] | ||
| MJD72.1 | FL human ATXN3 | ATXN3 | Q72 | (CAG)4(CAA)(CAG)67 | Motor deficits from 34 weeks. NIIs in 10% pontine and dentate neurons | Not reported | [193] | ||
| MJD76.1 (founder) | FL human ATXN3 | ATXN3 | Q76 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)70 | Motor deficits from 4 weeks. | Not reported | [193] | ||
| PrP/MJD77-het/hom | FL human ATXN3 | Tet-Prp | Q77 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)72 | Motor deficits from 9 weeks. NIIs in cerebral cortex at 4 weeks. | Not reported | [219] | ||
| HA-Q79 | FL human ATXN3 | Pcp2/L7 | Q79 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)n | No behavioural phenotype or neurological phenotype at 23 weeks | Not reported | [220] | ||
| HA-Q79 | Truncated ATXN3 | Pcp2/L7 | Q79 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)n | Ataxia from 4 weeks, failed to rear | Reported as stable on transmission. | [220] | ||
| Ataxin-3-Q79HA | FL human ATXN3 | Prp | Q79 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)n | Motor deficits from 20 weeks. Widespread NIIs at 43 weeks. | Not reported | [221] | ||
| Atxn3Q82/Q6 | Chimeric human exon 10/ mouse Atxn3 | ATXN3 | Q86 | (CAG)2(CAAAAG) (CAG)82 | No motor impairment at 52 weeks. NIIs in deep cerebellar nuclei at 10 weeks. Extranuclear inclusions in striatum, hippocampus. | Somatic instability not reported but intergenerational instability observed | [222] | ||
| MJD84.2 | FL human ATXN3 (two copies) | ATXN3 | Q84 | CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)78 | Motor deficits from 4 weeks. Widespread DNS (52 weeks). NIIs cerebellum and pons. Purkinje cell loss (80 weeks). | Not reported | [193, 223] | ||
| MJD22.1/84.1 | FL human ATXN3 | ATXN3 | Q84 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)12 (CAA)(CAG)65 | Motor deficits not reported, mild neuronal loss and gliosis. NIIs in pontine and dentate. | Not reported | [193] | ||
| Ki91 | Chimeric human exon 7– 11/ mouse Atxn3 | ATXN3 | Q91 | Not reported | Motor deficits from 90 weeks. Cerebellar degeneration and mild loss of Purkinje cells from 52 weeks. | Yes, age-dependent expansions prominent in the striatum, pons and testes at 40 weeks. Intergenerational instabilities also observed. | [224] | ||
| Hemi-CMVMJD94 | FL human ATXN3 | CMV | Q94 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)77 | Motor deficits from 16 weeks. Mild neuronal atrophy in pontine and dentate nuclei (16 weeks). No NIIs at 12 months. | Yes, age-dependent increase observed in multiple tissues. Pontine nuclei, substantia nigra, striatum and liver showed the highest rates. | [225] | ||
| CMVMJD135 | FL human ATXN3 | CMV | Q135 | (CAG)2(CAA)(AAG) (CAG)(CAA)(CAG)129 | Motor deficits from 10 weeks. NIIs in pons and spinal cord from 20 weeks. Neuronal loss in the pons from 60 weeks. | Somatic mosaicism observed. Intergenerational instability in 54– 79% of transmissions. | [226] | ||
| 148.19 CAG | FL human ATXN3 | Prp | Q148 | Not clear | Motor deficits from 9 weeks. Widespread NIIs (3-4 months) | Not reported | [217] | ||
| HDPromMJD148 | FL human ATXN3 | Htt | Q148 | Not clear | Motor deficits from 57 weeks. NIIs in red nucleus, pons and the cerebellum from 78 weeks. | Intergenerational instability | [227] | ||
| SCA3-KI | Expanded CAG in exon 10 of mouse Atxn3 | ATXN3 | Q304 | (CAACAGCAG)n | Motor deficits and widespread NIIs from 18 months | Not reported | [228] | ||
| SCA6 | CT-longQ27PC | FL human CACNA1A | CMV/ ACTB | Q27 | (CAG)n | Motor deficit from 8 months. Purkinje cell degeneration. | Not reported | [229] | |
| Cav2.1 knock-in mice | Insertion of CAG into exon 47 | CACNA1A | Q28 | (CAG)n | Motor deficits from 6 months | Not reported | [230] | ||
| SCA6_30Q | Chimeric human exon 47/ mouse Cacna1a | CACNA1A | Q30 | (CAG)n | No motor impairment | Not reported | [231] | ||
| SCA6_84Q | Chimeric human exon 47/ mouse Cacna1a | CACNA1A | Q84 | (CAG)n | Motor deficits from 30 weeks. NIIs in Purkinje cells at 22 months. | Not reported | [231, 232] | ||
| MPI-118Q | Chimeric exon 47 with splicing site mutation | CACNA1A | Q118 | 4 CAA interruptions | Motor deficits from 6 weeks. Purkinje cell number decrease from 6 weeks and cytoplasmic inclusions from 7 weeks. | Not reported | [233] | ||
| SCA7 | ataxin-7-Q52 | FL human ATXN7 | PDGF-B | Q52 | (CAG)n | Motor deficits from 7 months. DNS at 39 weeks, NIIs in cerebellum from 43 weeks | Not reported | [234, 235] | |
| P7E | FL human ATXN7 | Pcp2 | Q90 | (CAG)n | Motor deficits from 48 weeks. DNS in Purkinje cells from 3 weeks and NIIs from 35 weeks | Not reported | [236] | ||
| R7E | FL human ATXN7 | Rhodopsin | Q90 | (CAG)n | No motor deficits reported. DNS in eye retina from 2 weeks and DCS from 4 weeks | Not reported | [236, 237] | ||
| PrP-SCA7-c92Q | FL human ATXN7 | Prp | Q92 | (CAG)n | Motor deficits from 8 weeks. NIIs in photoreceptors (11 weeks) and in the pons, hippocampus and medulla (19 weeks). | Yes, mild somatic mosaicism (100 CAGs). | [238– 240] | ||
| Gfa2-SCA7-92Q | FL human ATXN7 | GFAP (Gfa2) | Q92 | (CAG)n | Motor deficits from 34 weeks. Reactive gliosis, dendritic degeneration (39 weeks), DNS and DCS in cerebellum (52 weeks) | Not reported | [241] | ||
| PrP-floxed-SCA7-92Q BAC | FL human ATXN7 | Prp | Q92 | (CAG)n | Motor deficits from 21 weeks. Loss of cells in molecular layer and los of glial processes (40 weeks) | Not reported | [242] | ||
| Atxn7 100Q | Chimeric human exon 3/ mouse Atxn7 | Atxn7 | Q100 | (CAG)n | Cerebellar molecular layer atrophy at 8-9 months. | Not reported | [243] | ||
| B7E2 | FL human ATXN7 | PDGF-B | Q128 | (CAG)n | Motor deficits from 22 weeks. DCS in cerebellum and cerebral cortex (4 weeks), DNS (9 weeks) and NIIs (17 weeks). | Not reported | [244] | ||
| Sca7 266Q/5Q | Chimeric human exon 3/ mouse Atxn7 | Atxn7 | Q266 | (CAG)n | Motor deficits from 5 weeks. Widespread NIIs from 5 weeks. Brain volume decrease, apoptosis increase, cell loss from 12– 15 weeks. | Not reported. Intergenerational instability reported. | [245] | ||
| SCA17 | TBPQ64 (rat) | FL human TBP | Prp | Q64 | 64 mixed CAA/CAG | Motor deficits from 3.5 months. Degeneration of Purkinje cell layers at 9 months. | Not reported. Intergenerational stability across three generations | [246] | |
| TBP-71Q-27 | FL human TBP | Prp | Q71 | 71 mixed CAA/CAG | Motor deficits from 6 weeks. DNS in multiple brain regions from 15 weeks. Purkinje cell degeneration. | Not reported | [247] | ||
| TBP-105Q | FL human TBP | Prp | Q105 | 105 mixed CAA/CAG | Motor deficits from 6 weeks. NIIs in cerebellum from 10 weeks. | Not reported | [247] | ||
| L7-hTBP | FL human TBP | Pcp2/L7 | Q109 | 109 mixed CAA/CAG | Motor deficits from 9 weeks. Purkinje cell loss (4 weeks). DNS in brainstem, cerebellum, cerebral cortex and striatum (22 weeks). | Not reported | [248] | ||
| SBMA | Mx-AR | FL human AR | Mx | Q45 | (CAG)n | No behavioural phenotype. | Somatic instability not reported. No change in repeat across 4 generations. | [249] | |
| NSE-AR | FL human AR | NSE | Q45 | (CAG)n | No behavioural phenotype. | Somatic instability not reported. No change in repeat across 4 generations. | [249] | ||
| AR YAC CAG 45 | 450-kb YAC construct with 100-kb human AR gene | AR | Q45 | (CAG)n | No behavioural phenotype. | No somatic instability. Intergenerational repeat instability in 10% of transmissions. | [250] | ||
| AR48Q | Chimeric human exon 1/ mouse Ar | mAR | Q48 | (CAG)n | No behavioural phenotype at 23 months. Gene expression alteration in testes at 26 weeks. | Not reported | [251] | ||
| AR65 | FL human AR | CMV | Q65 | (CAG)n | Motor deficits from 16 weeks. | Not reported | [252] | ||
| AR-97Q | FL human AR | Chicken β-actin | 97 | (CAG)n | Motor deficits from 9 weeks. Widespread DNS and NIIs from 15 weeks | Not reported | [253] | ||
| AR100 | FL human AR | hAR | 100 | (CAG)n | Motor deficits in males at 48 weeks. NIIs and DNS in spinal cord and hypothalamus (61 weeks). | Not reported | [254] | ||
| 112Q | FL human AR | Prp | 112 | (CAG)n | Motor deficits from 6 weeks. NIIs in spinal cord (6 weeks) | Not reported | [255] | ||
| AR113Q | Chimeric human exon 1/ mouse Ar | mAR | 113 | (CAG)n | Motor deficits from 8 weeks. NIIs in skeletal muscle (10 weeks). Reduced fertility. | Not reported | [256, 257] | ||
| AR120 | FL human AR | CMV | 120Q | (CAG)n | Motor deficits from 3 weeks. Cell loss in the spinal cord from 13 weeks, brain volume decreases from 26 weeks | Not reported | [252] | ||
| AR239Q | FL human AR | hAR | 239Q | (CAG)n | Motor deficits from 4 weeks. Widespread NIIs (8 weeks). | Somatic instability not reported. Minor intergenerational instability. | [258] |
FL, full length; Prp, prion protein; NSE, neuron specific enolase; NIIs, neuronal intranuclear inclusions; DCS, diffuse cerebellar staining; DNS, diffuse nuclear staining; CI, cerebellar inclusions.
In some cases, allelic series have been ‘naturally’ generated through intergenerational expansions or contractions following extensive breeding [196– 198, 225]. These models allow us to explore the effect of CAG repeat length in a well-controlled system. One such system is a series of transgenic DRPLA mouse models carrying 76, 96, 113 and 129 CAG, whose motor deficits and cognition worsen with CAG repeat length and age. High levels of somatic expansion were observed in the cortex, liver and kidney of the Q76 mice [196], and although no behavioural phenotype was initially reported in the Q76 after 64 weeks, they showed reduced survival and body weight when compared with non-transgenic littermates [197] as well as neuronal intranuclear accumulation [196]. Again, repeat instability is likely to occur in all models, but was only examined in Q76 animals.
Genomic context is an important driver of repeat instability in these models. Early studies of independent transgenic mouse models of SBMA with 45 AR CAG repeats revealed no behavioural phenotype at this repeat length, but did highlight the importance of genomic context in mediating intergenerational repeat instability in mice possessing repeats at the lower end of the pathogenic range [249, 250]. Consistent with this, a knock-in mouse model of SBMA with 48 AR CAG repeats demonstrated no overt behavioural phenotype at 23 months but did show changes in gene expression. Genomic context also appears to govern intergenerational and somatic expansion in a mouse model of SCA7 [240].
Marked repeat instability has been observed in a transgenic mouse model of SCA3, CMVMJD94, which carries 94 CAG repeats [225]. Expansion was observed in multiple tissues, but within the brain mosaicism was most notable in the pontine nuclei, substantia nigra and striatum. Somatic instability correlated well with neuronal atrophy and gliosis in the pontine nuclei and substantia nigra, but pathological involvement was not seen in the striatum [225]. Another mouse model of SCA3, Ki91, and a mouse model of SCA1, Sca1154Q/2Q, also demonstrate similar tissue-specific patterns of repeat expansions, with notable expansions in the striatum [207, 224]. This extends to other repeats— the same tissue distribution of expansion is seen in models of myotonic dystrophy [187]. These data suggest that whilst repeat instability is not associated with cerebellar neuronal vulnerability in models of SCA, it is likely that repeat instability in areas other than the cerebellum might contribute to disease pathogenesis [259]. Intergenerational instability has been observed in numerous models of SCA3 despite the interrupted CAG tract; this could be due to the presence of a long uninterrupted stretch of CAG at the 3’ end of the tract [222, 224, 226, 227]. These findings suggest that somatic instability is occurring in these model systems.
Some models have allowed us to examine the substantial effect that inheriting only 1 or 2 CAG additional repeats may have on phenotype [225]. SCA3 mice with 83 CAG repeats did not demonstrate behavioural differences, yet SCA3 mice with 94 CAG repeats and similar expression levels demonstrated rotarod deficits and behavioural abnormalities from 16 weeks. It was concluded that the threshold for disease in this model was between 84– 94 CAG repeats. Analysis of data from two cohorts of Q94 also revealed an inverse correlation between the length of the CAG repeat tract and the time spent on the rotarod [225].
Whilst animal models have been invaluable in examining pathogenesis in these diseases, as in HD models, to date, it has been difficult to show directly that somatic expansions are causative to neuronal dysfunction, earlier age at onset and faster disease progression. Interpretation of results is difficult when repeat sequence and length are not clearly defined or have not been examined. Many of the issues that arise in the HD animal models also arise in animal models of other repeat disorders and for many of the same reasons. However, the conclusion from human CAG repeat disorders, and also the corresponding mouse models, would indicate that a repeat length of less than 100 CAGs is toxic to cells— at the shorter end of that estimated by Kaplan et al. [34]. Exactly where the intracellular pathogenic threshold falls remains unclear, but the evidence would place it at over 60 CAG. The question remains whether it is possible to define the intracellular pathogenic threshold more accurately.
WHAT EVIDENCE DO WE NEED TO REFINE OUR DEFINITION OF THE INTRACELLULAR PATHOGENIC THRESHOLD?
The parameters used to establish the CAG-length threshold for HD pathogenesis by Kaplan et al. [34] included the CAG size threshold for disease to arise, the subject’s inherited repeat length as measured in blood, and their current age: these data are available. However, they also require a measure of the cell group critical portion— of the most susceptible cell population(s), what proportion have died, or are dysfunctional, at onset of clinical disease? The final unknown, for HD and the other repeat diseases, is the basal expansion rate of the repeat over time. In HD, the cell group critical portion can be estimated from previous work that showed around half of the most susceptible D2R-expressing medium spiny neurons in the striatum have been lost at onset [260– 262]. This parameter could likely be estimated in living subjects from imaging data, as recent well-standardised structural imaging and clinical data has been collected in prospective studies in both manifest and premanifest subjects [38, 263].
The basal expansion rate of the repeat in the most susceptible cells is much more difficult to measure or to derive from existing data. Given the likely stochastic nature of the allele expansion process and the data available in human brain which indicates very long repeats in some cells [24], this will be hard to estimate. However, the very long repeats could be rare events and indeed, could be protective in those surviving cells, as such repeat lengths are seen to reduce phenotype severity and delay onset in mice [90]. The most useful data are likely to come from single cell approaches in a combination of human and mouse brain. It would be ideal if all the data we needed could be derived from human brain, but this is unlikely to be sufficient as human postmortem brain is at the end stage of disease, and the only cells that can be surveyed are those that have survived. These are likely not representative of those that died earlier, and they may well themselves have been dysfunctional at death. Nevertheless, given this is likely a stochastic process there might be surviving cells at different points in the pathological trajectory that could be used in single cell experiments to define the pathogenic CAG tract length threshold. There are methods to sequence and size the HTT CAG tract accurately [264, 265], which could potentially be applied to single cells, but these would have to be tied to the single cell RNA gene expression data— achievable, but technically challenging.
Mouse brain is likely to offer a clearer picture of the dynamics of the pathological process, as tissues can be taken across the lifespan of the mouse and can be processed immediately to generate high quality single cell data. One major disadvantage of most HD mouse models is that they show little frank neurodegeneration, and in this respect do not recapitulate the human disease, but rather display neuronal dysfunction. However, for some analyses this is an advantage. Current data indicates that HD cellular dysfunction can be measured by single cell RNA-seq [266– 268], though the disconnect between behavioural and gene expression changes observed by Landles et al. [96] may make this difficult to interpret. The barrier here is gaining a measure of HTT CAG tract length in individual cells and matching that up with the gene expression signature of the same cells— the same technical challenge as noted above in human brain but perhaps easier to overcome in mouse brain. Single cell studies in mouse brain would also be a way to answer a long-standing question in HD: do vulnerable neurons die because of intracellular pathological events induced by the CAG tract, as we argue here, or do they die because of aberrant intercellular events, or both [269– 271]? In addition, mice with a shorter repeat length than those currently widely used in HD research will be necessary to examine the intracellular pathogenic threshold as they will need to start with a repeat length below that threshold. Another advantage of mice is that blood and brain somatic instability can be directly correlated. If this relationship can be established then it may be possible to extrapolate to human subjects where only blood is available.
Using the age at onset genetic modifier data obtain-ed in people might help to establish the pathogenic threshold. The effect sizes and directions of the known modifiers can be used to construct a polygenic risk score, which here consists of the sum of all known modifier alleles, weighted by the effect of each allele on onset [272]. This score can be used to predict somatic expansion in individuals without requiring expansion to be measured directly, thereby greatly increasing sample size, and may be incorporated into the Kaplan model [34]. This assumes that age at onset is a surrogate for measuring somatic expansion: Ciosi et al. [6] showed that individuals with higher blood DNA HTT CAG expansion have earlier HD onset and that the level of expansion in blood was associated with variants in selected genome-wide significant DNA repair genes from the modifier GWAS [7]. The question then becomes what is the relationship between blood DNA HTT CAG expansion and brain DNA HTT CAG expansion? Again, mice will be the model best able to test this directly. It would also be useful to have a prospective study of somatic expansion in blood in manifest and premanifest carriers to investigate how expansion increases over time and with proximity to onset: this could be performed in TRACK and TRACK-ON [38, 273] though studies of longer duration might be needed to establish longitudinal trajectories of repeat length. Then, if the relationship between expansion in blood and brain can be established experimentally, these trajectories may help infer the pathogenic HTT CAG threshold in the most susceptible cell populations in brain, likely to be neurons [25]. As larger genetic studies are performed in HD the various polygenic risk scores will become more accurate and account for more phenotypic variation. In particular, risk scores estimating somatic expansion directly will be important in improving inferences about the pathogenic CAG length threshold via the Kaplan model. This will require genome-wide association studies of somatic expansion with large sample sizes.
Given the recent interest in targeting somatic expansion of the expanded CAG in HTT, it behoves us to understand just how much of that expansion we need to prevent in order to substantially delay onset and slow the progression of disease. Evidence from animal models of HD and from other diseases caused by expanded CAG tracts place the threshold for cellular dysfunction above 60 CAG but below 100 CAG. Modelling in blood gives an expansion parameter that predicts age at onset [6] and this may prove a useful biomarker especially in clinical trials that target somatic expansion. While the prediction works well at a population level it is not currently clinically useful for individuals. There are substantial differences between individuals that could well be improved by using other peripheral tissues to measure expansion or by improving the model using genetics. As a first step, the CAG length and sequence themselves need to be more accurately measured [20, 274]. It should further be borne in mind that there are likely to be different repeat length dynamics in different cell types and the nature of the rate of expansion in different cell types is unknown. Cellular toxicity may be driven by different mechanisms in different cell types and different diseases. Specifically, in HD, in order to improve power in clinical trials, it would be very helpful to know how the HTT CAG expansion measured in blood relates to expansions in specific vulnerable cell types in the brain, and what influences that relationship. This might allow us to increase the power and shorten clinical trials by using a model that includes blood or other peripheral tissue HTT CAG expansion data, genetic data and imaging data. This could speed up getting treatments to the clinic [275].
CONFLICT OF INTEREST
LJ is a member of the Scientific Advisory Boards of LoQus23 Therapeutics and Triplet Therapeutics. JD, SP, NR and PH have no conflicts of interest.
REFERENCES
[1] | Hannan AJ . Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet. (2018) ;19: (5):286–98. |
[2] | Khristich AN , Mirkin SM . On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J Biol Chem. (2020) ;295: (13):4134–70. |
[3] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Prim. (2015) ;1: (1):15005. |
[4] | Orr HT , Zoghbi HY . Trinucleotide repeat disorders. Annu Rev Neurosci. (2007) ;30: (1):575–621. |
[5] | Coutelier M , Coarelli G , Monin ML , Konop J , Davoine CS , Tesson C , et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelo-pathies. Brain. (2017) ;140: (6):1579–94. |
[6] | Ciosi M , Maxwell A , Cumming SA , Hensman Moss DJ , Alshammari AM , Flower MD , et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. (2019) ;48: :568–80. |
[7] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900.e14. |
[8] | Wright GEB , Collins JA , Kay C , McDonald C , Dolzhenko E , Xia Q , et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet. (2019) ;104: (6):1116–26. |
[9] | Swinnen B , Robberecht W , Van Den Bosch L . RNA toxicity in non-coding repeat expansion disorders. EMBO J. (2020) ;39: (1):e101112. |
[10] | Bañez-Coronel M , Ayhan F , Tarabochia AD , Zu T , Perez BA , Tusi SK , et al. RAN translation in Huntington disease. Neuron. (2015) ;88: (4):667–77. |
[11] | Neueder A , Landles C , Ghosh R , Howland D , Myers RH , Faull RLM , et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. (2017) ;7: (1):1–10. |
[12] | Barnat M , Capizzi M , Aparicio E , Boluda S , Wennagel D , Kacher R , et al. Huntington’s disease alters human neurodevelopment. Science. (2020) ;369: (6505):787–93. |
[13] | Castaldo I , De Rosa M , Romano A , Zuchegna C , Squitieri F , Mechelli R , et al. DNA damage signatures in peripheral blood cells as biomarkers in prodromal Huntington disease. Ann Neurol. (2019) ;85: (2):296–301. |
[14] | Askeland G , Dosoudilova Z , Rodinova M , Klempir J , Liskova I , Kuśnierczyk A , et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci Rep. (2018) ;8: (1):1. |
[15] | Andrew SE , Goldberg YP , Kremer B , Telenius H , Theilmann J , Adam S , et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. (1993) ;4: (4):398–403. |
[16] | MacMillan JC , Snell RG , Tyler A , Houlihan GD , Fenton I , Cheadle JP , et al. Molecular analysis and clinical correlations of the Huntington’s disease mutation. Lancet. (1993) ;342: (8877):954–8. |
[17] | Duyao M , Ambrose C , Myers R , Novelletto A , Persichetti F , Frontali M , et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. (1993) ;4: (4):387–92. |
[18] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. |
[19] | Moss DJH , Tabrizi SJ , Mead S , Lo K , Pardiñas AF , Holmans P , et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. (2017) ;16: (9):1. |
[20] | Hong E , MacDonald M , Wheeler V , Jones L , Holmans P , Orth M . Huntington’s disease pathogenesis: Two sequential components. J Huntingtons Dis. 2020; doi: 10.3233/JHD-200427. |
[21] | Lee J-MM , Chao MJ , Harold D , Elneel KA , Gillis T , Holmans P , et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet. (2017) ;26: (19):3859–67. |
[22] | Goold R , Flower M , Moss DH , Medway C , Wood-Kaczmar A , Andre R , et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. (2019) ;28: (4):650–61. |
[23] | Kim KH , Hong EP , Shin JW , Chao MJ , Loupe J , Gillis T , et al. Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am J Hum Genet. (2020) ;107: (1):96–110. |
[24] | Shelbourne PF , Keller-McGandy C , Bi WL , Yoon S-R , Dubeau L , Veitch NJ , et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. (2007) ;16: (10):1133–42. |
[25] | Gonitel R , Moffitt H , Sathasivam K , Woodman B , Detloff PJ , Faull RLM , et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci U S A. (2008) ;105: (9):3467–72. |
[26] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) ;6: (4):409–14. |
[27] | De Rooij KE , De Koning Gans PAM , Roos RAC , Van Ommen GJB , Den Dunnen JT . Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum Genet. (1995) ;95: (3):270–4. |
[28] | Kennedy L , Evans E , Chen C-M , Craven L , Detloff PJ , Ennis M , et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. (2003) ;12: (24):3359–67. |
[29] | Swami M , Hendricks AE , Gillis T , Massood T , Mysore J , Myers RH , et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. (2009) ;18: (16):3039–47. |
[30] | Bourn RL , de Biase I , Pinto RM , Sandi C , Al-Mahdawi S , Pook MA , et al. Pms2 suppresses large expansions of the (GAA·TTC)n sequence in neuronal tissues. PLoS One. (2012) ;7: (10):e47085. |
[31] | Schmidt MHM , Pearson CE . Disease-associated repeat instability and mismatch repair. DNA Repair (Amst). (2016) ;38: (1):117–26. |
[32] | Wheeler V , Dion V . Modifiers of CAG repeat instability: insights from model systems. J Huntingtons Dis. 2020; doi: 10.3233/JHD-200426. |
[33] | Massey TH , Jones L . The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. (2018) ;11: (1):dmm031930. |
[34] | Kaplan S , Itzkovitz S , Shapiro E . A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput Biol. (2007) ;3: (11):e235. |
[35] | Langbehn DR , Stout JC , Gregory S , Mills JA , Durr A , Leavitt BR , et al. Association of CAG repeats with long-term progression in Huntington disease. JAMA Neurol. (2019) ;76: (11):1375–85. |
[36] | Paulsen JS , Hayden M , Stout JC , Langbehn DR , Aylward E , Ross CA , et al. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. (2006) ;63: (6):883–90. |
[37] | Paulsen JS , Long JD , Ross CA , Harrington DL , Erwin CJ , Williams JK , et al. Prediction of manifest Huntington’s disease with clinical and imaging measures: A prospective observational study. Lancet Neurol. (2014) ;13: (12):1193–201. |
[38] | Tabrizi SJ , Scahill RI , Owen G , Durr A , Leavitt BR , Roos RA , et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. (2013) ;12: (7):637–49. |
[39] | Scahill RI , Zeun P , Osborne-Crowley K , Johnson EB , Gregory S , Parker C , et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. (2020) ;19: (6):502–12. |
[40] | Klöppel S , Gregory S , Scheller E , Minkova L , Razi A , Durr A , et al. Compensation in preclinical Huntington’s disease: evidence from the Track-On HD Study. EBioMedicine. (2015) ;2: (10):1420–9. |
[41] | Gregory S , Long JD , Klöppel S , Razi A , Scheller E , Minkova L , et al. Testing a longitudinal compensation model in premanifest Huntington’s disease. Brain. (2018) ;141: (7):2156–66. |
[42] | Lloret A , Dragileva E , Teed A , Espinola J , Fossale E , Gillis T , et al. Genetic background modifies nuclear mutant huntingtin accumulation and HD CAG repeat instability in Huntington’s disease knock-in mice. Hum Mol Genet. (2006) ;15: (12):2015–24. |
[43] | Møllersen L , Rowe AD , Larsen E , Rognes T , Klungland A . Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. (2010) ;6: (12):1–11. |
[44] | Jonson I , Ougland R , Klungland A , Larsen E . Oxidative stress causes DNA triplet expansion in Huntington’s disease mouse embryonic stem cells. Stem Cell Res. (2013) ;11: (3):1264–71. |
[45] | Ross CA , Aylward EH , Wild EJ , Langbehn DR , Long JD , Warner JH , et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. (2014) ;10: :204–16. |
[46] | Crook ZR , Housman D . Huntington’s disease: can mice lead the way to treatment? Neuron. (2011) ;69: (3):423–35. |
[47] | Bates GP . HD mice: genetic and knock-in models. In: Precious SV , Rosser AE , Dunnett SB , eds. Methods in Molecular Biology: Huntington’s Disease. Humana Press, Totowa NJ, USA; (2018) . |
[48] | Du Montcel ST , Durr A , Bauer P , Figueroa KP , Ichikawa Y , Brussino A , et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain. (2014) ;137: (9):2444–55. |
[49] | Jacobi H , du Montcel ST , Bauer P , Giunti P , Cook A , Labrum R , et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and a longitudinal cohort study. Lancet Neurol. (2015) ;14: (11):1101–8. |
[50] | du Montcel ST , Durr A , Rakowicz M , Nanetti L , Charles P , Sulek A , et al. Prediction of the age at onset in spinocerebellar ataxia type 1, 2, 3 and 6. J Med Genet. (2014) ;51: (7):479–86. |
[51] | Menalled LB , Chesselet MF . Mouse models of Huntington’s disease. Trends Pharmacol Sci. (2002) ;23: (1):32–9. |
[52] | Mangiarini L , Sathasivam K , Seller M , Cozens B , Harper A , Hetherington C , et al. Exon I of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) ;87: (3):493–506. |
[53] | Mangiarini L , Sathasivam K , Mahal A , Mott R , Seller M , Bates GP . Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. (1997) ;15: (2):197–200. |
[54] | Hodgson JG , Agopyan N , Gutekunst CA , Leavitt BR , Lepiane F , Singaraja R , et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. (1999) ;23: (1):181–92. |
[55] | Zeron MM , Hansson O , Chen N , Wellington CL , Leavitt BR , Brundin P , et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. (2002) ;33: (6):849–60. |
[56] | Zeron MM , Fernandes HB , Krebs C , Shehadeh J , Wellington CL , Leavitt BR , et al. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol Cell Neurosci. (2004) ;25: (3):469–79. |
[57] | Laforet GA , Sapp E , Chase K , McIntyre C , Boyce FM , Campbell M , et al. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington’s disease. J Neurosci. (2001) ;21: (23):9112–23. |
[58] | Reddy PH , Williams M , Charles V , Garrett L , Pike-Buchanan L , Whetsell WO , et al. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. (1998) ;20: (2):198–202. |
[59] | White JK , Auerbach W , Duyao MP , Vonsattel JP , Gusella JF , Joyner AL , et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. (1997) ;17: (4):404–10. |
[60] | Wheeler VC , Auerbach W , White JK , Srinidhi J , Auerbach A , Ryan A , et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet. (1999) ;8: (1):115–22. |
[61] | Ciamei A , Detloff PJ , Morton AJ . Progression of behavioural despair in R6/2 and Hdh knock-in mouse models recapitulates depression in Huntington’s disease. Behav Brain Res. (2015) ;291: :140–6. |
[62] | Kumar A , Zhang J , Tallaksen-Greene S , Crowley MR , Crossman DK , Morton AJ , et al. Allelic series of Huntington’s disease knock-in mice reveals expression discorrelates. Hum Mol Genet. (2016) ;25: (8):1619–36. |
[63] | von Hörsten S , Schmitt I , Nguyen HP , Holzmann C , Schmidt T , Walther T , et al. Transgenic rat model of Huntington’s disease. Hum Mol Genet. (2003) ;12: (6):617–24. |
[64] | Levine MS , Klapstein GJ , Koppel A , Gruen E , Cepeda C , Vargas ME , et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res. (1999) ;58: (4):515–32. |
[65] | Menalled LB , Sison JD , Wu Y , Olivieri M , Li XJ , Li H , et al. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. J Neurosci. (2002) ;22: (18):8266–76. |
[66] | Li L , Murphy TH , Hayden MR , Raymond LA . Enhanced striatal NR2B-containing N-methyl-D-aspartate receptor-mediated synaptic currents in a mouse model of Huntington disease. J Neurophysiol. (2004) ;92: (5):2738–46. |
[67] | Shelbourne PF , Killeen N , Hevner RF , Johnston HM , Tecott L , Lewandoski M , et al. A Huntington’s disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. (1999) ;8: (5):763–74. |
[68] | Ishiguro H , Yamada K , Sawada H , Nishii K , Ichino N , Sawada M , et al. Age-dependent and tissue-specific CAG repeat instability occurs in mouse knock-in for a mutant Huntington’s disease gene. J Neurosci Res. (2001) ;65: (4):289–97. |
[69] | Sawada H , Ishiguro H , Nishii K , Yamada K , Tsuchida K , Takahashi H , et al. Characterization of neuron-specific huntingtin aggregates in human huntingtin knock-in mice. Neurosci Res. (2007) ;57: (4):559–73. |
[70] | Li H , Li SH , Yu ZX , Shelbourne P , Li XJ . Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci. (2001) ;21: (21):8473–81. |
[71] | Li H , Li SH , Johnston H , Shelbourne PF , Li XJ . Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. (2000) ;25: (4):385–9. |
[72] | Kennedy L , Shelbourne PF . Dramatic mutation instability in HD mouse striatum: Does polyglutamine load contribute to cell-specific vulnerability in Huntington’s disease? Hum Mol Genet. (2000) ;9: (17):2539–44. |
[73] | Schilling G , Becher MW , Sharp AH , Jinnah HA , Duan K , Kotzuk JA , et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. (1999) ;8: (3):397–407. |
[74] | Waldron-Roby E , Ratovitski T , Wang X , Jiang M , Watkin E , Arbez N , et al. Transgenic mouse model expressing the caspase 6 fragment of mutant huntingtin. J Neurosci. (2012) ;32: (1):183–93. |
[75] | Vatsavayai SC , Dallérac GM , Milnerwood AJ , Cummings DM , Rezaie P , Murphy KPSJ , et al. Progressive CAG expansion in the brain of a novel R6/1-89Q mouse model of Huntington’s disease with delayed phenotypic onset. Brain Res Bull. (2007) ;72: (2-3 SPEC. ISS.):98–102. |
[76] | Southwell AL , Warby SC , Carroll JB , Doty CN , Skotte NH , Zhang W , et al. A fully humanized transgenic mouse model of Huntington disease. Hum Mol Genet. (2013) ;22: (1):18–34. |
[77] | Kolodziejczyk K , Parsons MP , Southwell AL , Hayden MR , Raymond LA . Striatal synaptic dysfunction and hippocampal plasticity deficits in the Hu97/18 mouse model of huntington disease. PLoS One. (2014) ;9: (4):e94562. |
[78] | Gray M , Shirasaki DI , Cepeda C , André VM , Wilburn B , Lu XH , et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. (2008) ;28: (24):6182–95. |
[79] | Menalled L , El-Khodor BF , Patry M , Suárez-Fariñas M , Orenstein SJ , Zahasky B , et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol Dis. (2009) ;35: (3):319–36. |
[80] | Pouladi MA , Stanek LM , Xie Y , Franciosi S , Southwell AL , Deng Y , et al. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum Mol Genet. (2012) ;21: (10):2219–32. |
[81] | Abada YSK , Schreiber R , Ellenbroek B . Motor, emotional and cognitive deficits in adult BACHD mice: A model for Huntington’s disease. Behav Brain Res. (2013) ;238: (1):243–51. |
[82] | Abada Y , Nguyen HP , Schreiber R , Ellenbroek B . Assessment of motor function, sensory motor gating and recognition memory in a novel BACHD transgenic rat model for Huntington disease. PLoS One. (2013) ;8: (7):e68584. |
[83] | Abada YSK , Nguyen HP , Ellenbroek B , Schreiber R . Reversal learning and associative memory impairments in a BACHD rat model for huntington disease. PLoS One. (2013) ;8: (11):e71633. |
[84] | Lee J-M , Ramos EM , Lee J-H , Gillis T , Mysore JS , Hayden MR , et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurol. (2012) ;78: (10):690–5. |
[85] | Nakamori M , Pearson CE , Thornton CA . Bidirectional transcription stimulates expansion and contraction of expanded (CTG)•(CAG) repeats. Hum Mol Genet. (2011) ;20: (3):580–8. |
[86] | Nakamori M , Panigrahi GB , Lanni S , Gall-Duncan T , Hayakawa H , Tanaka H , et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet. (2020) ;52: (2):146–59. |
[87] | Lin Y , Hubert L , Wilson JH . Transcription destabilizes triplet repeats. Mol Carcinog. (2009) ;48: (4):350–61. |
[88] | Larson E , Fyfe I , Morton AJ , Monckton DG . Age-, tissue- and length-dependent bidirectional somatic CAG•CTG repeat instability in an allelic series of R6/2 Huntington disease mice. Neurobiol Dis. (2015) ;76: :98–111. |
[89] | Dragatsis I , Goldowitz D , Del Mar N , Deng YP , Meade CA , Liu L , et al. CAG repeat lengths ≥ 335 attenuate the phenotype in the R6/2 Huntington’s disease transgenic mouse. Neurobiol Dis. (2009) ;33: (3):315–30. |
[90] | Morton AJ , Glynn D , Leavens W , Zheng Z , Faull RLM , Skepper JN , et al. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiol Dis. (2009) ;33: (3):331–41. |
[91] | Cummings DM , Alaghband Y , Hickey MA , Joshi PR , Hong SC , Zhu C , et al. A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington’s disease. J Neurophysiol. (2012) ;107: (2):677–91. |
[92] | Aviolat H , Pinto RM , Godschall E , Murtha R , Richey HE , Sapp E , et al. Assessing average somatic CAG repeat instability at the protein level. Sci Rep. (2019) ;9: (1):1–14. |
[93] | Neueder A , Dumas AA , Benjamin AC , Bates GP . Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat Commun. (2018) ;9: (1):3955. |
[94] | Cervera R , Shoenfeld Y . Pathogenic mechanisms. In: Bates GP , Tabrizi SJ , Jones L , eds. Autoantibodies. 4th ed. New York: Oxford University Press; (1996) . p. 607–17. |
[95] | Benn CL , Landles C , Li H , Strand AD , Woodman B , Sathasivam K , et al. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington’s disease. Hum Mol Genet. (2005) ;14: (20):3065–78. |
[96] | Landles C , Milton RE , Ali N , Flomen R , Flower M , Schindler F , et al. Subcellular localization and formation of Huntingtin aggregates correlates with symptom onset and progression in a Huntington’s disease model. Brain Commun. (2020) ;2: (2):fcaa066. |
[97] | Alexaki A , Kames J , Holcomb DD , Athey J , Santana-Quintero L V , Lam PVN et al. Codon and Codon-Pair Usage Tables (CoCoPUTs): facilitating genetic variation analyses and recombinant gene design. J Mol Biol. (2019) ;431: (13):2434–41. |
[98] | Brooks SP , Janghra N , Higgs GV , Bayram-Weston Z , Heuer A , Jones L , et al. Selective cognitive impairment in the YAC128 Huntington’s disease mouse. Brain Res Bull. (2012) ;88: (2-3):121–9. |
[99] | Trueman RC , Brooks SP , Jones L , Dunnett SB . Time course of choice reaction time deficits in the HdhQ92 knock-in mouse model of Huntington’s disease in the operant Serial Implicit Learning Task (SILT). Behav Brain Res. (2008) ;189: (2):317–24. |
[100] | Fielding SA , Brooks SP , Klein A , Bayram-Weston Z , Jones L , Dunnett SB . Profiles of motor and cognitive impairment in the transgenic rat model of Huntington’s disease. Brain Res Bull. (2012) ;88: (2-3):223–36. |
[101] | Fisher EMC , Bannerman DM . Mouse models of neurodegeneration: Know your question, know your mouse. Sci Transl Med. (2019) ;11: (493):1. |
[102] | Lin CH , Tallaksen-Greene S , Chien WM , Cearley JA , Jackson WS , Crouse AB , et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. (2001) ;10: (2):137–44. |
[103] | Vijg J , Suh Y . Genome instability and aging. Annu Rev Physiol. (2013) ;75: (1):645–68. |
[104] | Brooks SP , Jones L , Dunnett SB . Comparative analysis of pathology and behavioural phenotypes in mouse models of Huntington’s disease. Brain Res Bull. (2012) ;88: (2-3):81–93. |
[105] | Sun Z , Ghosh S , Li Y , Cheng Y , Mohan A , Sampaio C , et al. A probabilistic disease progression modeling approach and its application to integrated Huntington’s disease observational data. JAMIA Open. (2019) ;2: (1):123–30. |
[106] | Ellis N , Tee A , McAllister B , Massey T , McLauchlan D , Stone T , et al. Genetic risk underlying psychiatric and cognitive symptoms in Huntington’s disease. Biol Psychiatry. (2020) ;87: (9):857–65. |
[107] | Gymrek M , Willems T , Guilmatre A , Zeng H , Markus B , Georgiev S , et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet. (2015) ;48: (1):22–9. |
[108] | Trost B , Engchuan W , Nguyen CM , Thiruvahindrapuram B , Dolzhenko E , Backstrom I , et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature. (2020) ;586: (7827):80–6. |
[109] | Jones L , Houlden H , Tabrizi SJ . DNA repair in the trinucleotide repeat disorders. Lancet Neurol. (2017) ;16: (1):88. |
[110] | Tsuji S . Dentatorubral-pallidoluysian atrophy. In: Handbook of Clinical Neurology. 2012. p. 587-94. |
[111] | Todd PK , Paulson HL . RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. (2010) ;67: (3):291–300. |
[112] | Day FR , Ruth KS , Thompson DJ , Lunetta KL , Pervjakova N , Chasman DI , et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat Genet. (2015) ;47: (11):1294–303. |
[113] | Bettencourt C , Hensman-Moss D , Flower M , Wiethoff S , Brice A , Goizet C , et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. (2016) ;79: (6):983–90. |
[114] | Maiuri T , Suart CE , Hung CLK , Graham KJ , Barba Bazan CA , Truant R . DNA damage repair in Huntington’s disease and other neurodegenerative diseases. Neurotherapeutics. (2019) ;16: (4):948–56. |
[115] | Ishikawa K , Watanabe M , Yoshizawa K , Fujita T , Iwamoto H , Yoshizawa T , et al. Clinical, neuropathological, and molecular study in two families with spinocerebellar ataxia type 6 (SCA6). J Neurol Neurosurg Psychiatry. (1999) ;67: (1):86–9. |
[116] | Sun YM , Lu C , Wu ZY . Spinocerebellar ataxia: relationship between phenotype and genotype - a review. Clin Genet. (2016) ;90: (4):305–14. |
[117] | Wiethoff S , O’Connor E , Haridy NA , Nethisinghe S , Wood N , Giunti P , et al. Sequencing analysis of the SCA6 CAG expansion excludes an influence of repeat interruptions on disease onset. J Neurol Neurosurg Psychiatry. (2018) ;89: (11):1226. |
[118] | Kordasiewicz HB , Thompson RM , Clark HB , Gomez CM . C-termini of P/Q-type Ca2+ channel α1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum Mol Genet. (2006) ;15: (10):1587–99. |
[119] | Ishiguro T , Ishikawa K , Takahashi M , Obayashi M , Amino T , Sato N , et al. The carboxy-terminal fragment of α1A calcium channel preferentially aggregates in the cytoplasm of human spinocerebellar ataxia type 6 Purkinje cells. Acta Neuropathol. (2010) ;119: (4):447–64. |
[120] | Giunti P , Mantuano E , Frontali M , Veneziano L . Molecular mechanism of Spinocerebellar Ataxia type Glutamine repeat disorder, channelopathy and transcriptional dysregulation. The multifaceted aspects of a single mutation. Front Cell Neurosci. (2015) ;9: (FEB):5. |
[121] | Stoyas CA , La Spada AR . The CAG-polyglutamine repeat diseases: a clinical, molecular, genetic, and pathophysiologic nosology. In: Handbook of Clinical Neurology. 2018. p. 143-70. |
[122] | Ansved T , Lundin A , Anvret M . Larger CAG expansions in skeletal muscle compared with lymphocytes in Kennedy disease but not in Huntington disease. Neurology. (1998) ;51: (5):1442–4. |
[123] | Orr HT , Chung MY , Banfi S , Kwiatkowski TJ , Servadio A , Beaudet AL , et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. (1993) ;4: (3):221–6. |
[124] | Zühlke C , Dalski A , Hellenbroich Y , Bubel S , Schwinger E , Bürk K . Spinocerebellar ataxia type 1 (SCA1): Phenotype-genotype correlation studies in intermediate alleles. Eur J Hum Genet. (2002) ;10: (3):204–9. |
[125] | Sobczak K , Krzyzosiak WJ . Patterns of CAG repeat interruptions in SCA1 and SCA2 genes in relation to repeat instability. Hum Mutat. (2004) ;24: (3):236–47. |
[126] | Menon RP , Nethisinghe S , Faggiano S , Vannocci T , Rezaei H , Pemble S , et al. The role of interruptions in polyQ in the pathology of SCA1. PLoS Genet. (2013) ;9: (7):e1003648. |
[127] | Nethisinghe S , Pigazzini ML , Pemble S , Sweeney MG , Labrum R , Manso K , et al. PolyQ tract toxicity in SCA1 is length dependent in the absence of CAG repeat interruption. Front Cell Neurosci. (2018) ;12: :200. |
[128] | Quan F , Janas J , Popovich BW . A novel CAG repeat configuration in the SCA1 gene: Implications for the molecular diagnostics of spinocerebellar ataxia type 1. Hum Mol Genet. (1995) ;4: (12):2411–3. |
[129] | Chong SS , McCall AE , Cota J , Subramony SH , Orr HT , Hughes MR , et al. Gametic and somatic tissue– specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. (1995) ;10: (3):344–50. |
[130] | Goldfarb LG , Vasconcelos O , Platonov FA , Lunkes A , Kipnis V , Kononova S , et al. Unstable triplet repeat and phenotypic variability of spinocerebellar ataxia type 1. Ann Neurol. (1996) ;39: (4):500–6. |
[131] | Lopes-Cendes I , Maciel P , Kish S , Gaspar C , Robitaille Y , Brent Clark H , et al. Somatic mosaicism in the central nervous system in spinocerebellar ataxia type 1 and Machado-Joseph disease. Ann Neurol. (1996) ;40: (2):199–206. |
[132] | Maciel P , Lopes-Cendes I , Kish S , Sequeiros J , Rouleau GA . Mosaicism of the CAG repeat in CNS tissue in relation to age at death in spinocerebellar ataxia type 1 and Machado-Joseph disease patients. Am J Hum Genet. (1997) ;60: (4):993–6. |
[133] | Hashida H , Goto J , Kurisaki H , Mizusawa H , Kanazawa I . Brain regional differences in the expansion of a CAG repeat in the spinocerebellar ataxias: Dentatorubral-pallidoluysian atrophy, Machado-Joseph disease, and spinocerebellar ataxia type 1. Ann Neurol. (1997) ;41: (4):505–11. |
[134] | Cancel G , Gourfinkel-An I , Stevanin G , Didierjean O , Abbas N , Hirsch E , et al. Somatic mosaicism of the CAG repeat expansion in spinocerebellar ataxia type 3/Machado-Joseph disease. Hum Mutat. (1998) ;11: (1):23–7. |
[135] | Matsuyama Z , Izumi Y , Kameyama M , Kawakami H , Nakamura S . The effect of CAT trinucleotide interruptions on the age at onset of spinocerebellar ataxia type 1 (SCA1). J Med Genet. (1999) ;36: (7):546–8. |
[136] | Zhou YX , Qiao WH , Gu WH , Xie H , Tang BS , Zhou LS , et al. Spinocerebellar ataxia type 1 in China molecular analysis and Genotype-Phenotype correlation in 5 families. Arch Neurol. (2001) ;58: (5):789–94. |
[137] | Ramos EM , Martins S , Alonso I , Emmel VE , Saraiva-Pereira ML , Jardim LB , et al. Common origin of pure and interrupted repeat expansions in spinocerebellar ataxia type 2 (SCA2). Am J Med Genet Part B Neuropsychiatr Genet. (2010) ;153B: (2):524–31. |
[138] | Corrado L , Mazzini L , Oggioni GD , Luciano B , Godi M , Brusco A , et al. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet. (2011) ;130: (4):575–80. |
[139] | Wang C , Xu Y , Feng X , Ma J , Xie S , Zhang Y , et al. Linkage analysis and whole-exome sequencing exclude extra mutations responsible for the parkinsonian phenotype of spinocerebellar ataxia-2. Neurobiol Aging. (2015) ;36: (1):545.e1–545.e7. |
[140] | Matsuura T , Sasaki H , Yabe I , Hamada K , Hamada T , Shitara M , et al. Mosaicism of unstable CAG repeats in the brain of spinocerebellar ataxia type 2. J Neurol. (1999) ;246: (9):835–9. |
[141] | Yu Z , Zhu Y , Chen-Plotkin AS , Clay-Falcone D , McCluskey L , Elman L , et al. PolyQ repeat expansions in ATXN2 associated with ALS are CAA interrupted repeats. PLoS One. (2011) ;6: (3):e17951. |
[142] | Pulst SM , Nechiporuk A , Nechiporuk T , Gispert S , Chen XN , Lopes-Cendes I , et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinooerebellar ataxia type 2. Nat Genet. (1996) ;14: (3):269–76. |
[143] | Sanpei K , Takano H , Igarashi S , Sato T , Oyake M , Sasaki H , et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. (1996) ;14: (3):277–84. |
[144] | Imbert G , Saudou F , Yvert G , Devys D , Trottier Y , Garnier JM , et al. Cloning of the gene for spinooerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. (1996) ;14: (3):285–91. |
[145] | Cancel G , Dürr A , Didierjean O , Imbert G , Bürk K , Lezin A , et al. Molecular and clinical correlations in spinocerebellar ataxia A study of 32 families. Hum Mol Genet. (1997) ;6: (5):709–15. |
[146] | Mizushima K , Watanabe M , Kondo I , Okamoto K , Shizuka M , Abe K , et al. Analysis of spinocerebellar ataxia type 2 gene and haplotype analysis: (CCG)1-2 polymorphism and contribution to founder effect. J Med Genet. (1999) ;36: (2):112–4. |
[147] | Choudhry S , Mukerji M , Srivastava AK , Jain S , Brahmachari SK . CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum Mol Genet. (2001) ;10: (21):2437–46. |
[148] | Furtado S , Payami H , Lockhart PJ , Hanson M , Nutt JG , Singleton AA , et al. Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2). Mov Disord. (2004) ;19: (6):622–9. |
[149] | Charles P , Camuzat A , Benammar N , Sellal F , Destée A , Bonnet AM , et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology. (2007) ;69: (21):1970–5. |
[150] | Kim JM , Hong S , Gyoung PK , Yoon JC , Yu KK , Sung SP , et al. Importance of low-range CAG expansion and CAA interruption in SCA2 parkinsonism. Arch Neurol. (2007) ;64: (10):1510–8. |
[151] | Kawaguchi Y , Okamoto T , Taniwaki M , Aizawa M , Inoue M , Katayama S , et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. (1994) ;8: (3):221–8. |
[152] | Cancel G , Abbas N , Stevanin G , Dürr A , Chneiweiss H , Néri C , et al. Marked phenotypic heterogeneity associated with expansion of a CAG repeat sequence at the spinocerebellar ataxia 3/Machado-Joseph disease locus. Am J Hum Genet. (1995) ;57: (4):809–16. |
[153] | Tanaka F , Sobue G , Doyu M , Ito Y , Yamamoto M , Shimada N , et al. Differential pattern in tissue-specific somatic mosaicism of expanded CAG trinucleotide repeat in dentatorubral-pallidoluysian atrophy, Machado-Joseph disease, and X-linked recessive spinal and bulbar muscular atrophy. J Neurol Sci. (1996) ;135: (1):43–50. |
[154] | Ito Y , Tanaka F , Yamamoto M , Doyu M , Nagamatsu M , Riku S , et al. Somatic mosaicism of the expanded CAG trinucleotide repeat in mRNAs for the responsible gene of Machado-Joseph disease (MJD), dentatorubral-pallidoluysian atrophy (DRPLA), and spinal and bulbar muscular atrophy (SBMA). Neurochem Res. (1998) ;23: (1):25–32. |
[155] | Zhang S , Wang J , Xu Q , Li X , Lei L , Jiang H , et al. Detection of the CAG trinucleotide repeats of MJD1 gene by recombinant DNA technology. Chinese J Med Genet. (2009) ;26: (4):406–9. |
[156] | David G , Abbas N , Stevanin G , Dürr A , Yvert G , Cancel G , et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. (1997) ;17: (1):65–70. |
[157] | David G , Dürr A , Stevanin G , Cancel G , Abbas N , Benomar A , et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet. (1998) ;7: (2):165–70. |
[158] | Gouw LG , Castañeda MA , McKenna CK , Digre KB , Pulst SM , Perlman S , et al. Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum Mol Genet. (1998) ;7: (3):525–32. |
[159] | Gu W , Wang Y , Liu X , Zhou B , Zhou Y , Wang G . Molecular and clinical study of spinocerebellar ataxia type 7 in Chinese kindreds. Arch Neurol. (2000) ;57: (10):1513–8. |
[160] | García-Velázquez LE , Canizales-Quinteros S , Romero-Hidalgo S , Ochoa-Morales A , Martínez-Ruano L , Márquez-Luna C , et al. Founder effect and ancestral origin of the spinocerebellar ataxia type 7 (SCA7) mutation in Mexican families. Neurogenetics. (2014) ;15: (1):13–7. |
[161] | Katagiri S , Hayashi T , Takeuchi T , Yamada H , Gekka T , Kawabe K , et al. Somatic instability of expanded CAG repeats of ATXN7 in Japanese patients with spinocerebellar ataxia type 7. Doc Ophthalmol. (2015) ;130: (3):189–95. |
[162] | Trang H , Stanley SY , Thorner P , Faghfoury H , Schulze A , Hawkins C , et al. Massive CAG repeat expansion and somatic instability in maternally transmitted infantile spinocerebellar ataxia type 7. JAMA Neurol. (2015) ;72: (2):219–23. |
[163] | Holmes SE , O’Hearn EE , McInnis MG , Gorelick-Feldman DA , Kleiderlein JJ , Callahan C , et al. Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat Genet. (1999) ;23: (4):391–2. |
[164] | Fujigasaki H , Verma IC , Camuzat A , Margolis RL , Zander C , Lebre AS , et al. SCA12 is a rare locus for autosomal dominant cerebellar ataxia: A study of an Indian family. Ann Neurol. (2001) ;49: (1):117–21. |
[165] | Srivastava AK , Takkar A , Garg A , Faruq M . Clinical behaviour of spinocerebellar ataxia type 12 and intermediate length abnormal CAG repeats in PPP2R2B. Brain. (2016) ;140: (1):27–36. |
[166] | Gao R , Matsuura T , Coolbaugh M , Zühlke C , Nakamura K , Rasmussen A , et al. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet. (2008) ;16: (2):215–22. |
[167] | Nielsen TT , Mardosiene S , Løkkegaard A , Stokholm J , Ehrenfels S , Bech S , et al. Severe and rapidly progressing cognitive phenotype in a SCA17-family with only marginally expanded CAG/CAA repeats in the TATA-box binding protein gene: A case report. BMC Neurol. (2012) ;12: :73. |
[168] | Koide R , Ikeuchi T , Onodera O , Tanaka H , Igarashi S , Endo K , et al. Unstable expansion of CAG repeat in hereditary dentatorubral– pallidoluysian atrophy (DRPLA). Nat Genet. (1994) ;6: (1):9–13. |
[169] | Ueno S , Kondoh K , Komure Y , Komure O , Kuno S , Kawai J , et al. Somatic mosaicism of CAG repeat in dentatorubral-pallidoluysian atrophy (DRPLA). Hum Mol Genet. (1995) ;4: (4):663–6. |
[170] | Takano H , Onodera O , Takahashi H , Igarashi S , Yamada M , Oyake M , et al. Somatic mosaicism of expanded CAG repeats in brains of patients with dentatorubral-pallidoluysian atrophy: Cellular population-dependent dynamics of mitotic instability. Am J Hum Genet. (1996) ;58: (6):1212–22. |
[171] | Aoki M , Abe K , Tobita M , Kameya T , Watanabe M , Itoyama Y . Reduction of CAG expansions in cerebellar cortex and spinal cord of DRPLA. Clin Genet. (1996) ;50: (4):199–201. |
[172] | Watanabe H , Tanaka F , Doyu M , Riku S , Yoshida M , Hashizume Y , et al. Differential somatic CAG repeat instability in variable brain cell lineage in dentatoru-bral pallidoluysian atrophy (DRPLA): A laser-captured microdissection (LCM)-based analysis. Hum Genet. (2000) ;107: (5):452–7. |
[173] | Hashida H , Goto J , Suzuki T , Jeong SY , Masuda N , Ooie T , et al. Single cell analysis of CAG repeat in brains of dentatorubral-pallidoluysian atrophy (DRPLA). J Neurol Sci. (2001) ;190: (1-2):87–93. |
[174] | La Spada AR , Wilson EM , Lubahn DB , Harding AE , Fischbeck KH . Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. (1991) ;352: (6330):77–9. |
[175] | Watanabe M , Abe K , Aoki M , Yasuo K , Itoyama Y , Shoji M , et al. Mitotic and meiotic stability of the CAG repeat in the X-linked spinal and bulbar muscular atrophy gene. Clin Genet. (1996) ;50: (3):133–7. |
[176] | Tanaka F , Reeves MF , Ito Y , Matsumoto M , Li M , Miwa S , et al. Tissue-specific somatic mosaicism in spinal and bulbar muscular atrophy is dependent on CAG-repeat length and androgen receptor-gene expression level. Am J Hum Genet. (1999) ;65: (4):966–73. |
[177] | Fratta P , Collins T , Pemble S , Nethisinghe S , Devoy A , Giunti P , et al. Sequencing analysis of the spinal bulbar muscular atrophy CAG expansion reveals absence of repeat interruptions. Neurobiol Aging. (2014) ;35: (2):443.e1–443.e3. |
[178] | Fratta P , Nirmalananthan N , Masset L , Skorupinska I , Collins T , Cortese A , et al. Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology. (2014) ;82: (23):2077–84. |
[179] | Mouro Pinto R , Arning L , Giordano J V , Razghandi P , Andrew MA , Gillis T , et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet. (2020) ;29: (15):2551–67. |
[180] | Rüb U , Schöls L , Paulson H , Auburger G , Kermer P , Jen JC , et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol. (2013) ;104: (1):38–66. |
[181] | Walløe S , Pakkenberg B , Fabricius K . Stereological estimation of total cell numbers in the human cerebral and cerebellar cortex. Front Hum Neurosci. (2014) ;8: (JULY):508. |
[182] | Nalls M , Blauwendraat C , Vallerga C , Heilbron K , Bandres-Ciga S , Chang D , et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. bioRxiv. 2018;388165. |
[183] | Rousseaux MWC , Tschumperlin T , Lu HC , Lackey EP , Bondar V V , Wan YW et al. ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron. (2018) ;97: (6):1235–1243.e5. |
[184] | Wang L , Aasly JO , Annesi G , Bardien S , Bozi M , Brice A , et al. Large-scale assessment of polyglutamine repeat expansions in Parkinson disease. Neurology. (2015) ;85: (15):1283–92. |
[185] | Cui Y , Yang S , Li XJ , Li S . Genetically modified rodent models of SCA17. J Neurosci Res. (2017) ;95: (8):1540–7. |
[186] | Stevanin G , Brice A . Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4). Cerebellum. (2008) ;7: (2):170–8. |
[187] | Fortune MT , Vassilopoulos C , Coolbaugh MI , Siciliano MJ , Monckton DG . Dramatic, expansion-biased, age-dependent, tissue-specific somatic mosaicism in a transgenic mouse model of triplet repeat instability. Hum Mol Genet. (2000) ;9: (3):439–45. |
[188] | Ross CA . When more is less: Pathogenesis of glutamine repeat neurodegenerative diseases. Neuron. (1995) ;15: (3):493–6. |
[189] | Lin Y , Dent SYR , Wilson JH , Wells RD , Napierala M . R loops stimulate genetic instability of CTG·CAG repeats. Proc Natl Acad Sci U S A. (2010) ;107: (2):692–7. |
[190] | Neil AJ , Liang MU , Khristich AN , Shah KA , Mirkin SM . RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res. (2018) ;46: (7):3487–97. |
[191] | Shah KA , McGinty RJ , Egorova VI , Mirkin SM . Coupling transcriptional state to large-scale repeat expansions in yeast. Cell Rep. (2014) ;9: (5):1594–602. |
[192] | Goula AV , Stys A , Chan JPK , Trottier Y , Festenstein R , Merienne K . Transcription elongation and tissue-specific somatic CAG instability. PLoS Genet. (2012) ;8: (11):e1003051. |
[193] | Cemal CK , Carroll CJ , Lawrence L , Lowrie MB , Ruddle P , Al-Mahdawi S , et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet. (2002) ;11: (9):1075–94. |
[194] | Luthi-Carter R , Strand AD , Hanson SA , Kooperberg C , Schilling G , La Spada AR , et al. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum Mol Genet. (2002) ;11: (17):1927–37. |
[195] | Schilling G , Wood JD , Duan K , Slunt HH , Gonzales V , Yamada M , et al. Nuclear accumulation of truncated atrophin-1 fragments in a transgenic mouse model of DRPLA. Neuron. (1999) ;24: (1):275–86. |
[196] | Sato T , Oyake M , Nakamura K , Nakao K , Fukusima Y , Onodera O , et al. Transgenic mice harboring a full-length human mutant DRPLA gene exhibit age-dependent intergenerational and somatic instabilities of CAG repeats comparable with those in DRPLA patients. Hum Mol Genet. (1999) ;8: (1):99–106. |
[197] | Suzuki K , Zhou J , Sato T , Takao K , Miyagawa T , Oyake M , et al. DRPLA transgenic mouse substrains carrying single copy of full-length mutant human DRPLA gene with variable sizes of expanded CAG repeats exhibit CAG repeat length- and age-dependent changes in behavioral abnormalities and gene expression profiles. Neurobiol Dis. (2012) ;46: (2):336–50. |
[198] | Sato T , Miura M , Yamada M , Yoshida T , Wood JD , Yazawa I , et al. Severe neurological phenotypes of Q129 DRPLA transgenic mice serendipitously created by en masse expansion of CAG repeats in Q76 DRPLA mice. Hum Mol Genet. (2009) ;18: (4):723–36. |
[199] | Ying M , Xu R , Wu X , Zhu H , Zhuang Y , Han M , et al. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem. (2006) ;281: (18):12580–6. |
[200] | Sakai K , Yamada M , Sato T , Yamada M , Tsuji S , Takahashi H . Neuronal atrophy and synaptic alteration in a mouse model of dentatorubral-pallidoluysian atrophy. Brain. (2006) ;129: (9):2353–62. |
[201] | Lorenzetti D , Watase K , Xu B , Matzuk MM , Orr HT , Zoghbi HY . Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus. Hum Mol Genet. (2000) ;9: (5):779–85. |
[202] | Burright EN , Brent Clark H , Servadio A , Matilla T , Feddersen RM , Yunis WS , et al. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. (1995) ;82: (6):937–48. |
[203] | Clark HB , Burright EN , Yunis WS , Larson S , Wilcox C , Hartman B , et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. (1997) ;17: (19):7385–95. |
[204] | Lin X , Antalffy B , Kang D , Orr HT , Zoghbi HY . Poly-glutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. (2000) ;3: (2):157–63. |
[205] | Giovannoni R , Maggio N , Rosaria Bianco M , Cavaliere C , Cirillo G , Lavitrano M , et al. Reactive astrocytosis and glial glutamate transporter clustering are early changes in a spinocerebellar ataxia type 1 transgenic mouse model. Neuron Glia Biol. (2007) ;3: (4):335–51. |
[206] | Watase K , Weeber EJ , Xu B , Antalffy B , Yuva-Paylor L , Hashimoto K , et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. (2002) ;34: (6):905–19. |
[207] | Watase K , Venken KJT , Sun Y , Orr HT , Zoghbi HY . Regional differences of somatic CAG repeat instability do not account for selective neuronal vulnerability in a knock-in mouse model of SCA1. Hum Mol Genet. (2003) ;12: (21):2789–95. |
[208] | Damrath E , Heck M V , Gispert S , Azizov M , Nowock J , Seifried C , et al. ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. (2012) ;8: (8):e1002920. |
[209] | Huynh DP , Figueroa K , Hoang N , Pulst SM . Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet. (2000) ;26: (1):44–50. |
[210] | Liu J , Tang TS , Tu H , Nelson O , Herndon E , Huynh DP , et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. (2009) ;29: (29):9148–62. |
[211] | Dansithong W , Paul S , Figueroa KP , Rinehart MD , Wiest S , Pflieger LT , et al. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. (2015) ;11: (4):e1005182. |
[212] | Aguiar J , Fernández J , Aguilar A , Mendoza Y , Vázquez M , Suárez J , et al. Ubiquitous expression of human SCA2 gene under the regulation of the SCA2 self promoter cause specific Purkinje cell degeneration in transgenic mice. Neurosci Lett. (2006) ;392: (3):202–6. |
[213] | Sen NE , Canet-Pons J , Halbach M V , Arsovic A , Pilatus U , Chae WH , et al. Generation of an Atxn2-CAG100 knock-in mouse reveals N-acetylaspartate production deficit due to early Nat8l dysregulation. Neurobiol Dis. (2019) ;132: (1):104559. |
[214] | Hansen ST , Meera P , Otis TS , Pulst SM . Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. (2013) ;22: (2):271–83. |
[215] | Torashima T , Koyama C , Iizuka A , Mitsumura K , Takayama K , Yanagi S , et al. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. (2008) ;9: (4):393–9. |
[216] | Menzies FM , Huebener J , Renna M , Bonin M , Riess O , Rubinsztein DC . Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. (2010) ;133: (1):93–104. |
[217] | Bichelmeier U , Schmidt T , Hübener J , Boy J , Rüttiger L , Häbig K , et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA in vivo evidence. J Neurosci. (2007) ;27: (28):7418–28. |
[218] | Goti D , Katzen SM , Mez J , Kurtis N , Kiluk J , Ben-Haïem L , et al. A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J Neurosci. (2004) ;24: (45):10266–79. |
[219] | Boy J , Schmidt T , Wolburg H , Mack A , Nuber S , Böttcher M , et al. Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum Mol Genet. (2009) ;18: (22):4282–95. |
[220] | Ikeda H , Yamaguchi M , Sugai S , Aze Y , Narumiya S , Kakizuka A . Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet. (1996) ;13: (2):196–202. |
[221] | Chou AH , Yeh TH , Ouyang P , Chen YL , Chen SY , Wang HL . Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol Dis. (2008) ;31: (1):89–101. |
[222] | Ramani B , Harris GM , Huang R , Seki T , Murphy GG , Costa M do C , et al. A knockin mouse model of spino-cerebellar ataxia type 3 exhibits prominent aggregate pathology and aberrant splicing of the disease gene transcript. Hum Mol Genet. (2015) ;24: (5):1211–24. |
[223] | Chen X , Tang TS , Tu H , Nelson O , Pook M , Hammer R , et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. (2008) ;28: (48):12713–24. |
[224] | Switonski PM , Szlachcic WJ , Krzyzosiak WJ , Figiel M . A new humanized ataxin-3 knock-in mouse model combines the genetic features, pathogenesis of neurons and glia and late disease onset of SCA3/MJD. Neurobiol Dis. (2015) ;73: :174–88. |
[225] | Silva-Fernandes A , Costa M do C , Duarte-Silva S , Oliveira P , Botelho CM , Martins L , et al. Motor uncoordination and neuropathology in a transgenic mouse model of Machado-Joseph disease lacking intranuclear inclusions and ataxin-3 cleavage products. Neurobiol Dis. (2010) ;40: (1):163–76. |
[226] | Silva-Fernandes A , Duarte-Silva S , Neves-Carvalho A , Amorim M , Soares-Cunha C , Oliveira P , et al. Chronic treatment with 17-DMAG improves balance and coordination in a new mouse model of Machado-Joseph disease. Neurotherapeutics. (2014) ;11: (2):433–49. |
[227] | Boy J , Schmidt T , Schumann U , Grasshoff U , Unser S , Holzmann C , et al. A transgenic mouse model of spinocerebellar ataxia type 3 resembling late disease onset and gender-specific instability of CAG repeats. Neurobiol Dis. (2010) ;37: (2):284–93. |
[228] | Haas E , Incebacak R , Hentrich T , Maringer Y , Schmidt T , Zimmermann F , et al. A novel ataxin-3 knock-in mouse model mimics the human SCA3 disease phenotype including neuropathological, behavioral, and transcriptional abnormalities. bioRxiv. 2020;2020.02.28.968024. |
[229] | Mark MD , Krause M , Boele HJ , Kruse W , Pollok S , Kuner T , et al. Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J Neurosci. (2015) ;35: (23):8882–95. |
[230] | Saegusa H , Wakamori M , Matsuda Y , Wang J , Mori Y , Zong S , et al. Properties of human Cav2. 1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol Cell Neurosci. (2007) ;34: (2):261–70. |
[231] | Watase K , Barrett CF , Miyazaki T , Ishiguro T , Ishikawa K , Hu Y , et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2. 1 channels. Proc Natl Acad Sci U S A. (2008) ;105: (33):11987–92. |
[232] | Jayabal S , Ljungberg L , Erwes T , Cormier A , Quilez S , El Jaouhari S , et al. Rapid onset of motor deficits in a mouse model of spinocerebellar ataxia type 6 precedes late cerebellar degeneration. eNeuro. (2015) ;2: (6):ENEURO. 0094-15.2015. |
[233] | Unno T , Wakamori M , Koike M , Uchiyama Y , Ishikawa K , Kubota H , et al. Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc Natl Acad Sci U S A. (2012) ;109: (43):17693–8. |
[234] | Chou A-H , Chen C-Y , Chen S-Y , Chen W-J , Chen Y-L , Weng Y-S , et al. Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem Int. (2010) ;56: (2):329–39. |
[235] | Wang H-L , Chou A-H , Lin A-C , Chen S-Y , Weng Y-H , Yeh T-H . Polyglutamine-expanded ataxin-7 upregulates Bax expression by activating p53 in cerebellar and inferior olivary neurons. Exp Neurol. (2010) ;224: (2):486–94. |
[236] | Yvert G , Lindenberg KS , Picaud S , Landwehrmeyer GB , Sahel JA , Mandel JL . Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet. (2000) ;9: (17):2491–506. |
[237] | Helmlinger D , Abou-Sleymane G , Yvert G , Rousseau S , Weber C , Trottier Y , et al. Disease progression despite early loss of polyglutamine protein expression in SCA7 mouse model. J Neurosci. (2004) ;24: (8):1881–7. |
[238] | La Spada AR , Fu YH , Sopher BL , Libby RT , Wang X , Li LY , et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. (2001) ;31: (6):913–27. |
[239] | Garden GA , Libby RT , Fu YH , Kinoshita Y , Huang J , Possin DE , et al. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous Purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. (2002) ;22: (12):4897–905. |
[240] | Libby RT , Monckton DG , Fu YH , Martinez RA , McAbney JP , Lau R , et al. Genomic context drives SCA7 CAG repeat instability, while expressed SCA7 cDNAs are intergenerationally and somatically stable in transgenic mice. Hum Mol Genet. (2003) ;12: (1):41–50. |
[241] | Custer SK , Garden GA , Gill N , Rueb U , Libby RT , Schultz C , et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. (2006) ;9: (10):1302–11. |
[242] | Furrer SA , Mohanachandran MS , Waldherr SM , Chang C , Damian VA , Sopher BL , et al. Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non-cell-autonomous Bergmann glia degeneration. J Neurosci. (2011) ;31: (45):16269–78. |
[243] | Chen YC , Gatchel JR , Lewis RW , Mao C-A , Grant PA , Zoghbi HY , et al. Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet. (2012) ;21: (2):394–405. |
[244] | Yvert G , Lindenberg KS , Devys D , Helmlinger D , Landwehrmeyer GB , Mandel J-L . SCA7 mouse models show selective stabilization of mutant ataxin-7 and similar cellular responses in different neuronal cell types. Hum Mol Genet. (2001) ;10: (16):1679–92. |
[245] | Yoo SY , Pennesi ME , Weeber EJ , Xu B , Atkinson R , Chen S , et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. (2003) ;37: (3):383–401. |
[246] | Kelp A , Koeppen AH , Petrasch-Parwez E , Calaminus C , Bauer C , Portal E , et al. A novel transgenic rat model for spinocerebellar ataxia type 17 recapitulates neuropathological changes and supplies in vivo imaging biomarkers. J Neurosci. (2013) ;33: (21):9068–81. |
[247] | Friedman MJ , Shah AG , Fang ZH , Ward EG , Warren ST , Li S , et al. Polyglutamine domain modulates the TBP-TFIIB interaction: Implications for its normal function and neurodegeneration. Nat Neurosci. (2007) ;10: (12):1519–28. |
[248] | Chang YC , Lin CY , Hsu CM , Lin HC , Chen YH , Lee-Chen GJ , et al. Neuroprotective effects of granulocyte-colony stimulating factor in a novel transgenic mouse model of SCA17. J Neurochem. (2011) ;118: (2):288–303. |
[249] | Bingham PM , Scott MO , Wang S , McPhaul MJ , Wilson EM , Garbern JY , et al. Stability of an expanded trinucleotide repeat in the androgen receptor gene in transgenic mice. Nat Genet. (1995) ;9: (2):191–6. |
[250] | La Spada AR , Peterson KR , Meadows SA , McClain ME , Jeng G , Chmelar RS , et al. Androgen receptor YAC transgenic mice carrying CAG 45 alleles show trinucleotide repeat instability. Hum Mol Genet. (1998) ;7: (6):959–67. |
[251] | Albertelli MA , Scheller A , Brogley M , Robins DM . Replacing the mouse androgen receptor with human alleles demonstrates glutamine tract length-dependent effects on physiology and tumorigenesis in mice. Mol Endocrinol. (2006) ;20: (6):1248–60. |
[252] | McManamny P , Chy HS , Finkelstein DI , Craythorn RG , Crack PJ , Kola I , et al. A mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. (2002) ;11: (18):2103–11. |
[253] | Katsuno M , Adachi H , Kume A , Li M , Nakagomi Y , Niwa H , et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. (2002) ;35: (5):843–54. |
[254] | Sopher BL , Thomas PS , Lafevre-Bernt MA , Holm IE , Wilke SA , Ware CB , et al. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. (2004) ;41: (5):687–99. |
[255] | Chevalier-Larsen ES , O’Brien CJ , Wang H , Jenkins SC , Holder L , Lieberman AP , et al. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. (2004) ;24: (20):4778–86. |
[256] | Yu Z , Dadgar N , Albertelli M , Gruis K , Jordan C , Robins DM , et al. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. (2006) ;116: (10):2663–72. |
[257] | Yu Z , Dadgar N , Albertelli M , Scheller A , Albin RL , Robins DM , et al. Abnormalities of germ cell maturation and Sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am J Pathol. (2006) ;168: (1):195–204. |
[258] | Adachi H , Kume A , Li M , Nakagomi Y , Niwa H , Do J , et al. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. (2001) ;10: (10):1039–48. |
[259] | Jafar-Nejad P , Ward CS , Richman R , Orr HT , Zoghbi HY . Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3ɛ haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci U S A. (2011) ;108: (5):2142–7. |
[260] | Vonsattel JPG , DiFiglia M . Huntington disease. J Neuropathol Exp Neurol. (1998) ;57: (5):369–84. |
[261] | Waldvogel HJ , Kim EH , Tippett LJ , Vonsattel J-PG , Faull RLM , Bates G , et al. Neuropathology in the human brain. In: Huntington’s Disease. 4th ed. New York: Oxford University Press; (2014) . p. 185–217. |
[262] | Waldvogel HJ , Kim EH , Thu DCV , Tippett LJ , Faull RLM . New perspectives on the neuropathology in Huntington’s disease in the human brain and its relation to symptom variation. J Huntingtons Dis. (2012) ;1: (2):143–53. |
[263] | Wijeratne PA , Johnson EB , Eshaghi A , Aksman L , Gregory S , Johnson HJ ,et al.. Robust markers and sample sizes for multicenter trials of Huntington disease. Ann Neurol. (2020) ;87: (5):751–62. |
[264] | Höijer I , Tsai YC , Clark TA , Kotturi P , Dahl N , Stattin EL ,et al.. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum Mutat. (2018) ;39: (9):1262–72. |
[265] | Massey T , McAllister B , Jones L . Methods for assessing DNA repair and repeat expansion in Huntington’s disease. Methods Mol Biol. (2018) ;1780: (1):483–95. |
[266] | Miyazaki H , Yamanaka T , Oyama F , Kino Y , Kurosawa M , Yamada-Kurosawa M ,et al.. FACS-array-based cell purification yields a specific transcriptome of striatal medium spiny neurons in a murine Huntington disease model. J Biol Chem. (2020) ;295: (29):9768–85. |
[267] | Macaulay IC , Haerty W , Kumar P , Li YI , Hu TX , Teng MJ ,et al.. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. (2015) ;12: (6):519–22. |
[268] | Nam AS , Kim K-T , Chaligne R , Izzo F , Ang C , Taylor J ,et al.. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature. (2019) ;571: (7765):355–60. |
[269] | Frost B , Diamond MI . Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. (2010) ;11: (3):155–9. |
[270] | Levine MS , Wang EA , Chen JY , Cepeda C , Andre VM . Altered neuronal circuitry. In: Huntington’s disease. 2014. p. 218-40. |
[271] | Wang N , Gray M , Lu XH , Cantle JP , Holley SM , Greiner E ,et al.. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. (2014) ;20: (5):536–41. |
[272] | Purcell SM , Wray NR , Stone JL , Visscher PM , O’Donovan MC , Sullivan PF ,et al.. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. (2009) ;460: (7256):748–52. |
[273] | Long JD , Mills JA . Joint modeling of multivariate longitudinal data and survival data in several observational studies of Huntington’s disease. BMC Med Res Methodol. (2018) ;18: (1):138. |
[274] | Ciosi M , Cumming S , Chatzi A , Larson E , Tottey W , Lomeikaite V . Approaches to sequence the HTT CAG repeat expansion and quantify repeat length variation. J Huntingtons Dis. 2020; doi: 10.3233/JHD-200433. |
[275] | Benn C , Gibson K , Reynolds D . Drugging DNA damage repair pathways for trinucleotide repeat expansion diseases. J Huntington’s Dis. 2020; doi: 10.3233/JHD-200421. |


