
Effects of Pridopidine on Functional Capacity in Early-Stage Participants from the PRIDE-HD Study
Abstract
Background:
No pharmacological treatment has been demonstrated to provide a functional benefit for persons with Huntington’s disease (HD). Pridopidine is a sigma-1-receptor agonist shown to have beneficial effects in preclinical models of HD.
Objective:
To further explore the effect of pridopidine on Total Functional Capacity (TFC) in the recent double-blind, placebo-controlled PRIDE-HD study.
Methods:
We performed post-hoc analyses to evaluate the effect of pridopidine on TFC at 26 and 52 weeks. Participants were stratified according to baseline TFC score and analyzed using repeated measures (MMRM) and multiple imputation assuming missing not-at-random (MNAR) and worst-case scenarios.
Results:
The pridopidine 45 mg bid dosage demonstrated a beneficial effect on TFC for the entire population at week 52 of 0.87 (nominal p = 0.0032). The effect was more pronounced for early HD participants (HD1/HD2, TFC = 7–13), with a change from placebo of 1.16 (nominal p = 0.0003). This effect remained nominally significant using multiple imputation with missing not at random assumption as a sensitivity analysis. Responder analyses showed pridopidine 45 mg bid reduced the probability of TFC decline in early HD patients at Week 52 (nominal p = 0.02).
Conclusion:
Pridopidine 45 mg bid results in a nominally significant reduction in TFC decline at 52 weeks compared to placebo, particularly in patients with early-stage HD.
INTRODUCTION
Huntington’s disease (HD) is a progressive, fatal neurodegenerative condition characterized by behavioral, cognitive, and movement dysfunction [1]. These features contribute to gradual clinical worsening and subsequent functional decline. Current treatment approaches address choreiform movements and behavioral symptoms with limited success, but none to date are capable of modifying the evolving functional deficits that are inexorably observed with disease progression. New treatments that maintain or delay functional disability are of paramount interest and represent the major unmet medical need in the treatment of HD.
An emerging pathway with robust preclinical data suggestive of potential to provide benefit in HD is the Sigma-1 Receptor (S1R), a transmembrane chaperone protein located in mitochondria-associated membrane domains of the endoplasmic reticulum (ER) [2]. S1R regulates protein folding and degradation, with critical roles in calcium signaling, mitochondrial function, neuronal survival, synaptic plasticity, and activation of trophic factors [3, 4]. Pridopidine, a small molecule in clinical development for HD, was originally postulated to influence only motor symptoms via low-affinity dopamine D2 receptor antagonism [5]. More recent in-vitro binding assays and in-vivo PET imaging studies in rats show that pridopidine acts primarily via the S1R, where it demonstrates 100- to 500-fold greater binding affinity compared to dopamine D2 receptors [6, 7]. Pridopidine activates neuroprotective pathways known to be disturbed in HD (PI3k/Akt, BDNF, calbindin) and demonstrates protective properties in several in-vivo and in-vitro HD models mediated by the S1R [2, 8, 9]. A robust and dose-dependent neuroprotective effect against mutant huntingtin (mHTT)-induced cell death is observed after pridopidine treatment in induced pluripotent stem cells (iPSCs) from humans with HD and murine HD neurons. These effects are completely abolished by pharmacological inhibition of the S1R or deletion of the S1R gene [10]. In medium spiny neurons from the YAC128 model, pridopidine increases spine density and prevents aberrant calcium signaling, both known features of HD. This effect is abolished in S1R- deleted neurons [8, 11]. Pridopidine also reduces striatal aggregate size and upregulates brain-derived neurotrophic factor (BDNF) in the R6/2 HD murine model [12]. Collectively, these data support a S1R-mediated beneficial effect for pridopidine in HD.
Pridopidine has been investigated as a treatment for HD in three randomized, double-blind, placebo-controlled clinical trials: HART, MermaiHD, and PRIDE-HD [13–15]. These studies were initially designed to focus on symptomatic motor effects based on the hypothesis of dopamine modulation as pridopidine’s chief mechanism of action. Indeed, based on suggestion of motor improvement in HART and MermaiHD, the PRIDE-HD study tested four doses of pridopidine for their effects on the Total Motor Score of the Unified Huntington’s Disease Rating Scale (UHDRS-TMS). The Total Functional Capacity (TFC) score was also a pre-specified outcome measure [15, 16]. The TFC is a broad assay of functional status, consisting of five domains reflecting major lifestyle elements (capacity for work, finances, domestic chores, activities of daily living, home and caregiver status). It has established interrater reliability and validity, and served as the primary outcome measure for numerous HD trials [17–21]. Total scores range from 0 to 13, with higher scores indicating a greater capacity for independent function. TFC scores allow staging into early (HD1, TFC 11–13), early-mid (HD2, TFC 7–10), and more advanced disease states (HD3, TFC 4–6 and HD4, TFC 0–3). The TFC has attracted particular attention for HD studies based on willingness of the FDA and European regulatory agencies to accept this measure as a primary endpoint for clinical trials.
Based on an evolving understanding of pridopidine’s potent agonism at the S1R and regulatory agreement that TFC can serve as a single primary endpoint in pivotal trials, PRIDE-HD was extended from its original length of 26 weeks to 52 weeks [15]. This report provides post-hoc analyses on data from the PRIDE-HD trial, with new insights that can inform participant selection and treatment duration in future studies.
MATERIALS AND METHODS
The statistical analysis for PRIDE-HD is described in the primary manuscript [15]. In the present analysis, we examined the 45 mg BID dosage for all participants and in early HD participants with baseline TFC scores of 7–13 (HD1 and HD2). HD1 are defined as participants with baseline TFC 11–13, and HD2 are defined as participants with baseline TFC7–10. Dosages of pridopidine other than 45 mg BID showed progressive loss of benefit as dosage and drug exposure increased. This finding is consistent with the known bell-shaped effects of S1R agonists. Therefore, the 45 mg BID dosage was selected for more detailed analysis. A mixed model of repeated measures (MMRM) was used to evaluate data from the full analysis set at 26 and 52 weeks for these groups. Sensitivity analyses were performed using the multiple imputation method assuming Missing Not At Random (MNAR) and the ‘worst-case scenario’, where placebo values were imputed for missing data from all patients who discontinued therapy. TFC was a pre-specified endpoint in PRIDE. We performed post-hoc analysis on the early HD subgroup, not corrected for multiple observations; p-values are presented for descriptive purposes only.
In PRIDE-HD, a total of 323 patients (from all treatment arms combined) completed 26 weeks of treatment. Out of these 323 patients, 262 (81%) continued to the second treatment period up to 52 weeks. The 61 participants (out of 323) who did not continue were enrolled into either Open-HART or Open-PRIDE. These participants had completed 26 weeks of treatment before the IRB approval for the extension of the trial to 52 weeks was granted.
RESULTS
The PRIDE-HD study was initially designed to assess the effect of pridopidine on TMS at 26 weeks [15]. Due to recognition of the S1R as pridopidine’s main target after initiation of the study, suggesting a therapeutic potential beyond motor function, the ongoing trial was extended from 26 weeks to 52 weeks. Approximately 19% of participants reached the 26-week endpoint prior to their institution obtaining IRB approval for study extension (61 out of 323 participants). These participants went directly into the open-label extension study. Of the 323 participants who completed 26 weeks of treatment, 262 (81%) entered the second study period and continued treatment for 52 weeks.
Baseline demographic characteristics were similar between early HD participants (HD1 + HD2, TFC 7–13) who completed 26 weeks of treatment and those who completed 52 weeks of treatment (Table 1). There were no notable demographic differences between early HD participants who completed the full 52 weeks of treatment and those who dropped out (52-week non-completers) (Table 1). The dropout rates between the early HD (TFC 7–13) placebo and 45 mg BID groups were comparable (Table 2). 89% (55/62) of the placebo early HD participants completed the first 26 weeks of the study, vs. 81% (48/59) patients in the 45 mg BID group. 76% (42/55) of early HD placebo participants and 77% (37/48) of 45 mg BID participants who completed 26 weeks started the second treatment period, while 97% (41/42) of the early HD placebo group and 100% (37/37) of the 45 mg BD group who initiated the second study period completed 52 weeks (Table 2).
Table 1
Demographic characteristics of Early HD participants (baseline TFC 7-13) completing 26 weeks and 52 weeks and those who did not complete 52 weeks
| Parameter | Early HD 26-weeks Completers | Early HD 52-weeks Completers | Early HD 52-weeks Non-completers | |||
| Placebo | 45 mg bid | Placebo, | 45 mg BID, | Placebo, | 45 mg BID, | |
| n = 55 | n = 48 | n = 41 | n = 37 | n = 21 | n = 22 | |
| Baseline TFC mean (SD) | 9.0 (1.8) | 9.3 (1.8) | 8.9 (1.7) | 9.2 (1.9) | 8.9 (1.9) | 9.0 (1.7) |
| CAG mean (SD) | 44.7 (3.4) | 44.2 (4.7) | 45.0 (3.8) | 43.7 (4.4) | 43.8 (1.8) | 44.5 (4.2) |
| Age (Y) mean (SD) | 49.2 (11.8) | 50.2 (12.6) | 48.3 (12.7) | 51.4 (12.5) | 50.8 (8.0) | 50.6 (12.6) |
| Gender | M, 26 (47.3%) | M, 22 (45.8%) | F, 23(56%) | F, 19 (51.4%) | F, 8 (38%) | F, 11 (50%) |
| N (%) | F, 29 (52.7%) | F, 26 (54.2%) | M, 18 (44%) | M, 18 (48.6%) | M, 13 (62%) | M, 11 (50%) |
| Height (cm) mean (SD) | 170.4 (9.6) | 170.5 (10.2) | 169.5 (9.5) | 170.3 (8.0) | 174.5 (10.1) | 170.6 (13.8) |
| Weight (kg) mean (SD) | 73.0 (12.7) | 70.8 (15.0) | 73.2 (13.1) | 71.2 (15.2) | 72.8 (9.7) | 69.3 (13.4) |
| BMI mean (SD) | 25.2 (4.3) | 24.2 (4.0) | 25.5 (4.3) | 24.5 (4.4) | 24.1 (4.0) | 23.7 (2.0) |
| Neuroleptics | Yes, 21 (38.2%) | Yes, 15 (31.3%) | Yes, 17 (41.5)% | Yes, 11 (29.7%) | Yes, 6 (28.6%) | Yes, 8 (36.4%) |
| N (%) | No, 34 (62%) | No, 33 (68.7%) | No, 24 (58.5%) | No, 26 (70.3%) | No, 15 (71.4%) | No, 14 (63.6%) |
Table 2
Disposition of Early HD Participants in Placebo and 45 mg BID Pridopidine Groups
| Placebo | Pridopidine 45 mg BID | |
| N = 62 | N = 59 | |
| Completed 26 weeks | 55/62 (89%) | 48/59 (81%) |
| Started 2nd study period of 52 weeks | 42/55 (76%) | 37/48 (77%) |
| Completed 52 weeks | 41/42 (97%) | 37/37 (100%) |
Source: PRIDE-HD data.
Total Functional Capacity scores for all recipients on the 45 mg bid dosage (N = 75) at 26 and 52 weeks compared to placebo (N = 81) are displayed in Fig. 1. At 26 weeks, a trend towards improvement in change from baseline vs. placebo was seen in the pridopidine group (–0.49, SE 0.16 vs. –0.15, SE 0.17; difference between groups 0.34, nominal p = 0.15). At 52 weeks the difference between groups was nominally significant; placebo declined by –0.83 points (SE 0.20), while the treatment group was essentially unchanged from baseline (0.04, SE 0.22; difference between groups of 0.87, nominal p = 0.0032). Similar effects (data not shown) were seen with the analysis utilizing exposure instead of dosage. The TFC scale demonstrates ceiling and floor effects over the trajectory of HD, with steepest decline in earlier disease (HD1/HD2); therefore, we further analyzed the effect of 45 mg BID pridopidine on TFC after 26 and 52 weeks of treatment in participants with baseline early-stage HD [22]. Figure 1C-D shows TFC change from baseline to Week 26 and 52 in early HD. At 26 weeks, the difference between groups was 0.56 (nominal p = 0.036). At 52 weeks, a difference of 1.16 was seen (nominal p = 0.0003). Table 3 summarizes TFC change from baseline to Week 52 in early HD combined subgroups (HD1 + HD2, TFC 7–13), individual subgroups HD1 (TFC 11–13) and HD2 (TFC 7–10), and late-stage participants (HD3 + HD4, TFC 0–6). The observed beneficial effect in the combined HD1 and HD2 group is not driven by a single subset of patients, as both HD1 and HD2 contributed to the overall effect on TFC in the early HD population (HD1:1.89, nominal P = 0.0059; HD2, 0.94, nominal p = 0.009). No change was seen for later-stage participants (HD3 and HD4, treatment effect 0.07, nominal p = 0.91). Figure 1E demonstrate the TFC change over time for the placebo and 45 mg bid groups in early HD participants.
Fig. 1
TFC change from baseline vs. placebo for pridopidine 45 mg bid at Weeks 26 and 52 in All Participants (A,B) and Early HD patients (C,D). Mean±SEM; p-values are nominal and presented for descriptive purposes only. (E) Change in TFC plotted over time in early HD (TFC≥7); *p = 0.036; **p = 0.0003.
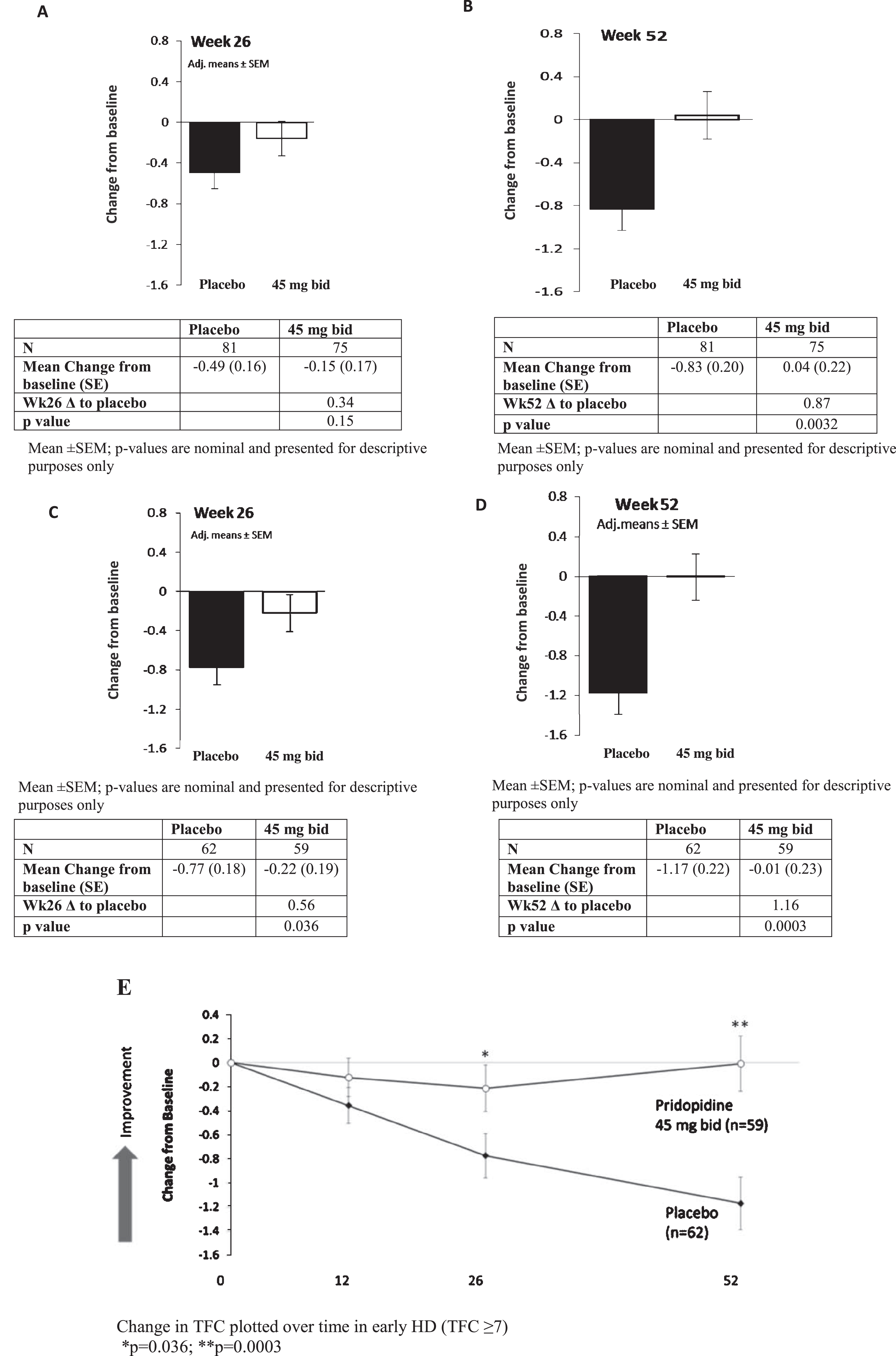
Table 3
TFC change from baseline compared to placebo in the 45 mg BID group at Week 52 by disease stage
| HD stage (TFC) | Placebo ΔTFC from baseline, mean (SE) | 45 mg BID ΔTFC from baseline, mean (SE) | 45 mg BID vs. placebo Mean change from baseline, | p |
| All | –0.83 (0.20) | +0.04 (0.22) | 0.87 | 0.0032 |
| Early HD1&HD2 (TFC 7–13) | –1.17 (0.22) | –0.01 (0.23) | 1.16 | 0.0003 |
| HD1 (TFC11–13) | –1.63 (0.51) | 0.26 (0.45) | 1.89 | 0.0059 |
| HD2 (TFC 7–10) | –0.95 (0.24) | –0.01 (0.27) | 0.94 | 0.009 |
| Late HD3&HD4 (TFC 0–6) | –0.14 (0.45) | –0.07 (0.51) | 0.07 | 0.91 |
p-values are nominal and presented for descriptive purposes only.
We performed multiple imputation analysis assuming Missing Not At Random (MNAR) and using the “worst case scenario” (Fig. 2). This method assumes that all missing data in the active treatment group follow the trajectory of the placebo group. Using MNAR for the entire population, the 45 mg bid pridopidine dose was superior to placebo at Week 52 (difference = 0.58; nominal p = 0.057). When this analysis was restricted to patients with early HD (HD1 and HD2), the MNAR analysis shows an effect of 0.79 (nominal p = 0.016). We also performed a post-hoc analysis to assess the effect of 45 mg bid on each of the five TFC sub-items in the early HD group (baseline TFC 7–13) (Table 4). Most TFC subscales contribute to the effect on total TFC score in early disease, with domestic chores, activity of daily living, care level, and finances each reaching nominal statistical significance. In an additional exploratory analysis, we defined “responders” as participants with a change from baseline in TFC≥0 at week 52 (i.e., no worsening), and “non-responders” as those with TFC decline of <0 points at Week 52 (i.e., worsening of any magnitude in TFC score) (Table 5). For the entire cohort, 47.3% of patients in the placebo group had worsening in TFC compared to 23.4% patients in the pridopidine group, with a nominally significant odds ratio (95% CI) of 0.32 (0.13–0.79, p = 0.01). In the early HD sub-group, 51.2% of patients in the placebo group showed worsening of TFC, compared to 18.9% of patients in the pridopidine group, with an odds ratio (95% CI) of 0.20 (0.07–0.56, nominal p = 0.002) (Table 5A). Among responders, there was also nominally significant improvement compared to placebo in the UHDRS Total Motor Score (UHDRS-TMS), UHDRS Functional Assessment (UHDRS-FA), Clinician Global Impression of Change (CGI-C), and the Clinician’s Interview-Based Impression of Change Plus (CIBIC+), demonstrating concordance between preservation of TFC and improvement in other clinical outcomes (Table 5B).
Fig. 2
Mean TFC change from baseline vs. placebo at Week 52 for all participants and early HD cohorts (TFC 7–13): comparison of MMRM to MNAR. Magnitude and p-value for the change in TFC, 45 mg BID vs. placebo, at 52 weeks.
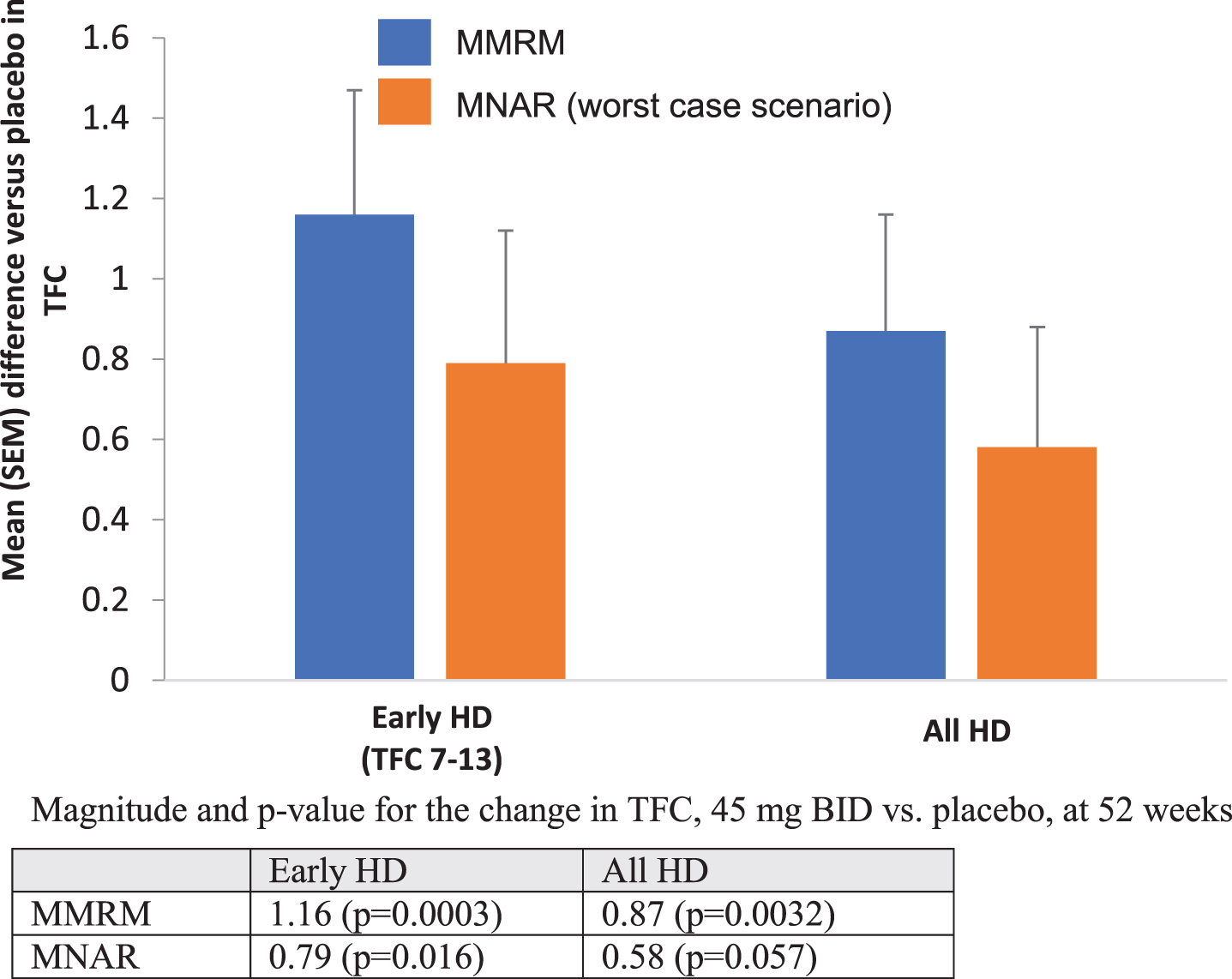
Table 4
Change from baseline to Week 52 in TFC domains for early HD subgroups (baseline TFC 7–13)
| Week 52 | ||
| Placebo | 45 mg BID | |
| Activity of Daily Living | ||
| LS Mean change from baseline (SE) | –0.32 (0.08) | 0.03 (0.08) |
| LS Mean difference | 0.35 | |
| p value | 0.002 | |
| Domestic Chores | ||
| LS Mean change from baseline (SE) | –0.23 (0.07) | 0.01 (0.07) |
| LS Mean difference | 0.24 | |
| p value | 0.02 | |
| Finance | ||
| LS Mean change from baseline (SE) | –0.37 (0.10) | –0.02 (0.11) |
| LS Mean difference | 0.35 | |
| p value | 0.017 | |
| Care level | ||
| LS Mean change from baseline (SE) | –0.09 (0.03) | 0.03 (0.03) |
| LS Mean difference | 0.12 | |
| p value | 0.004 | |
| Occupation | ||
| LS Mean change from baseline (SE) | –0.20 (0.09) | –0.07 (0.09) |
| LS Mean difference | 0.13 | |
| p value | 0.279 | |
LS Mean, least square mean; Included all randomized patients with baseline TFC > = 7, who received at least one dose of study drug and had at least one post-baseline efficacy assesment; p-values are nominal and presented for descriptive purposes only.
Table 5A
Responder analyses for participants in placebo and 45 mg bid pridopidine groups. N(%) of participants with ΔTFC < 0 (worsening/non-responders) at 52 weeks
| Placebo | 45 mg bid | Odds Ratio (95% CI) (GLIMMIX model) | p | |
| ALL HD | 26 (47.3%) | 11 (23.4%) | 0.32 (0.13 –0.79) | 0.01 |
| Placebo, n = 55 | ||||
| 45 mg bid, n = 47 | ||||
| Early HD (TFC ≥ 7) | 21 (51.2%) | 7 (18.9%) | 0.20 (0.07 –0.56) | 0.002 |
| Placebo, n = 41 | ||||
| 45 mg bid, n = 37 |
p-values are nominal and presented for descriptive purposes only.
Table 5B
Change in TMS and Global Functional assessments in Early HD Responders (ΔTFC≥0) vs. Non-responders (ΔTFC < 0) at 52 weeks
| Assessment, Statistic | Respondersa | Non-Responders |
| (N = 99) | (N = 77) | |
| UHDRS-TMSb | ||
| n | 99 | 77 |
| Mean (SD) | –4.6 (8.04) | 2.5 (10.54) |
| p-value | <0.001 | |
| UHDRS-FAb | ||
| n | 98 | 77 |
| Mean (SD) | –0.0 (1.78) | –1.9 (3.03) |
| p-value | <0.001 | |
| CGI-C Ratingsc, n (%) | ||
| n | 94 (100) | 75 (100) |
| No change or improvementd | 68 (72) | 34 (45) |
| p-value | <0.001 | |
| CIBIC Ratings n (%) | ||
| n | 92 (100) | 77 (100) |
| No change or improvementd | 60 (65) | 35 (45) |
| p-value | 0.008 |
Source: PRIDE-HD CSR Post Hoc Summaries. Early HD from all treatment groups combined. aA responder is defined as a patient with a change in TFC from baseline > =0. bThe statistical test is an analysis of variance (ANOVA) with treatment group as a fixed effect. cThe statistical test is a Cochran-Mantel-Haenszel (CMH) test. dThis includes the following ratings: Very much improved; much improved; minimally improved, and no change. TFC, Total Functional Capacity; UHDRS, Unified Huntington’s Disease Rating Scale; TMS, Total Motor Score; FA, Functional Assessment; CGI-C, Clinical Global Impression of Change; CIBIC, Clinician’s Interview-based Impression of Change. p-values are nominal and presented for descriptive purposes only.
The composite UHDRS (cUHDRS) is a recent measure of interest, as its scoring system combines existing measurement scales to measure patient performance and function: UHDRS-TMS, UHDRS-TFC, SDMT (Symbol Digit Modality Test) and SWR (Stroop Word Reading Test). cUHDRS shows increased sensitivity over individual measures that is most obvious with increased duration [22]. PRIDE-HD did not measure SWR, but using available data the cUHDRS was calculated based on UHDRS-TMS, UHDRS-TFC and SDMT. Pridopidine shows a benefit in cUHDRS in early HD patients at 52 weeks (treatment effect of 0.6 points, nominal p = 0.04; Table 6).
Table 6
cUHDRS at Week 52 in Early Stage HD Patients from PRIDE-HD
| Early HD (TFC 7–13) | Placebo, | 45 mg bid, |
| n = 62 | n = 59 | |
| Change from baseline Mean (SEM) | –0.62 (0.20) | –0.07 (0.21) |
| Difference vs. placebo, Mean (SEM) | 0.6 (0.29) | |
| 95% CI | 0.03, 1.17 | |
| p-value | 0.04 |
CI, confidence interval; Note: In PRIDE-HD cUHDRS score is derived from UHDRS-TFC, UHDRS-TMS and SDMT (without SWR).
DISCUSSION
This work is further analysis of TFC performance in the PRIDE-HD study, a randomized, placebo-controlled clinical trial of pridopidine in HD. PRIDE-HD was initially designed to assess the safety and efficacy of pridopidine on motor function at 26 weeks. After the trial started, emerging preclinical data indicated the primary target of pridopidine is the S1R, suggesting therapeutic potential beyond motor function. The ongoing trial was then extended from 26 weeks to 52 weeks to allow for more comprehensive assessment of outcomes that may require longer periods of time to show detectable therapeutic effects, including TFC.
Our analysis focuses on early-stage participants (HD1/HD2, TFC baseline 7–13) and the 45 mg bid dosage. Participants treated with 45 mg pridopidine BID had less TFC decline than placebo at week 52, demonstrating an almost 1-point difference (0.87, nominal p = 0.0032). A trend towards improvement was also noted at 26 weeks (difference of 0.34; nominal p = 0.15). Beneficial effects at 26 and 52 weeks were more pronounced in early-stage participants, with differences from baseline between active and placebo groups of 0.56 (nominal p = 0.036) and 1.16 points (nominal p = 0.0003), respectively. These beneficial effects on TFC were not derived from a single sub-population, as pridopidine showed a beneficial effect in both HD1 and HD2 groups separately. Most TFC sub-scales contributed to the overall effect on total TFC. We also observed higher numbers of participants in the 45 mg BID dosage group compared to placebo who did not deteriorate from baseline (change in UHDRS-TFC from baseline ≥0). This contrasts with the natural history of TFC scoring, which is known to progressively decline on an annual basis [23]. Responders in the 45 mg BID group with UHDRS-TFC change ≥0 at 52 weeks, there was nominally significant improvement vs. placebo in the UHDRS-TMS, UHDRS-FA, CGI-C and CIBIC+. The identified convergence between TFC and other clinical outcomes in responders strengthens the plausibility of a beneficial effect for this dosage. This broader effect is also seen in exploratory observations for the cUHDRS, a scoring system combining functional, motor and cognitive measures, where post-hoc analysis demonstrated a beneficial effect in early HD (treatment effect 0.6, nominal p = 0.04).
Due to the timing of independent review board (IRB) approval for extension of the study to 52 weeks, ∼19% of participants who completed 26 weeks of treatment did not continue treatment for 52 weeks (61 out of 323 participants). Demographic characteristics were similar between the placebo and 45 mg BID groups among early HD participants who completed 26 weeks, 52 weeks, or participants who discontinued over the duration of the study. Dropout rates between placebo and 45 BID were also comparable, suggesting these data are well-matched and suitable for TFC comparison at 52 weeks. To further validate the observed effect of pridopidine on functional decline at Week 52 with more conservative methods to address these missing data, multiple imputation was performed assuming MNAR and worst-case scenario, for which all missing data in the treatment group is assumed to follow the trajectory of the placebo group. Using this sensitivity analysis, pridopidine 45 mg BID showed an effect of 0.58 at week 52 (nominal p = 0.057) for all HD participants. Even with such a large proportion of missing data (∼19%) and using a highly conservative approach, a beneficial effect of pridopidine on TFC at week 52 was observed. When restricted to early HD (HD1 and HD2), MNAR showed a stronger effect (0.79, nominal p value = 0.016). These observations provide reinforcement of the MMRM analysis and suggest that pridopidine 45 mg BID may be associated with maintenance of functional capacity in HD.
It is noteworthy that participants receiving pridopidine 45 mg bid displayed virtually no decline in mean TFC over the course of 1 year, an effect particularly visible for patients with milder disease (TFC 7–13). This is very different than observations from natural history studies and placebo groups in previous clinical trials, where the rate of TFC decline for active treatments are also consistently similar to placebo. Early HD patients (TFC 7–13) naturally decline at a mean rate of 0.97 points/year, while TFC 3–6 (HD3) and HD4: 0–2 decline at 0.38 and 0.06 points/year, respectively, likely reflecting a floor effect in more advanced disease [23]. The greater magnitude of benefit noted in early HD patients treated with pridopidine 45 mg BID likely reflects sufficient numbers of residual neurons and functional reserve to respond to an intervention, compared to late stage participants (HD3 and HD4) for whom advanced disease processes may lessen the possibility of protection or functional rescue.
The observed treatment effects on TFC change from baseline appear to be dosage-specific. Differences from baseline vs. placebo are substantially greater in the 45 mg bid dose group compared to higher dosages [15]. This dose provides essentially complete binding of S1R throughout the human brain on imaging studies [24]. S1R agonists, including pridopidine, are known to modulate numerous important survival pathways (calcium homeostasis, attenuation of oxidative stress, mitochondrial function, lessening of reactive astrogliosis and microglial-induced injury) and are characterized by a bell-shaped dose-response curve in multiple preclinical models [25–37]. Treatment with pridopidine both increases BDNF secretion in B104 neuroblastoma cells (unpublished data) and restores impaired synaptic plasticity in HD cortical neurons with a bell-shaped dose-response curve [38]. In the 6-OHDA Parkinson’s disease mouse model, low-dose but not high-dose pridopidine increases neuroprotection of dopaminergic neurons and restores behavioral abnormalities [39]. Evidence of a bell-shaped curve for S1R agonists is also observed in clinical trials. In a 14-day open-label trial in 30 patients with major depression assessing two doses of igmesine were evaluated, a S1R agonist. The lower dose (25 mg) showed the most efficacious response (83%) compared with the higher 50 mg dose (50%) [40]. Further confirmation for these data was obtained in a 6-week, large scale, double-blind, placebo-controlled, Phase 2 trial of 350 patients, where the strongest anti-depressive effect was seen with 25 mg/day (p = 0.003) compared with both placebo and 100 mg/day (p > 0.05) [41]. The present data from the PRIDE study, demonstrating that 45 mg bid pridopidine is more effective for mitigating TFC decline than higher doses, is consistent with this phenomenon. It is biologically plausible to consider that the 45 mg bid dosage is optimal for S1R agonism because of bell-shaped pharmacokinetics, an effect that appears most robust in early stages of HD.
Improvement in functional capacity—a measure which synthesizes motor, cognitive, and behavioral ability into relevant daily activities—is perhaps the most pressing unmet therapeutic need in HD. On September 22, 2015, the FDA held a public meeting to hear perspectives from people living with HD about disease symptoms, the impact of HD on their daily life, and their experiences with currently available therapies [42]. Participants strongly emphasized that disease burden left them or their loved ones unable to perform many, if not all, meaningful daily activities (working, driving, self-care, upkeep of household, etc.). The UHDRS-TFC captures these concerns, as it reflects elements of function with meaningful impact on patients’ lives. Thus, a therapy with the ability to beneficially modify TFC decline would be of significant therapeutic value. It may be the case that multiple different mechanisms will be required to optimize slowing of functional decline (e.g., huntingtin-lowering, growth-factor enhancing, anti-inflammatory, antioxidant, etc.). Given this uncertainty, it is critical that compounds with potential to lessen functional decline continue to be sought and tested in appropriately designed clinical trials. Longer pridopidine studies than those already conducted (12–26 weeks for HART, MermaiHD, and PRIDE before extension) may be required to see cumulative beneficial effects on functional outcomes that change slowly, like the TFC. The analyses described in this report support further investigation with a prospective, long term, placebo-controlled trial testing pridopidine 45 mg BID in early-stage HD using UHDRS-TFC as the primary outcome measure.
CONFLICT OF INTEREST
AM and KK have previously received grant support from Teva. MG and MH are previous employees of Teva. AM, KK, CWO are consultants for Prilenia therapeutics.
ACKNOWLEDGMENTS
The authors are grateful for the commitment of the PRIDE-HD study participants and their caregivers, without whom this work would not be possible. The original PRIDE-HD study was supported by Teva Pharmaceuticals.
REFERENCES
[1] | McGarry A , Biglan K , Marshall F . Huntington’s disease. Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease. London; Elsevier, (2015) . |
[2] | Nguyen L , Lucke-Wold BP , Mookerjee SA , Cavendish JZ , Robson MJ , Scandinaro AL , et al. Role of sigma-1 receptors in neurodegenerative diseases. J Pharmacol Sci. (2015) ;127: :17–29. |
[3] | Hayashi T , Su TP . Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca (2+) signaling and cell survival. Cell. (2007) ;131: (3):596–610. |
[4] | Kourrich S , Su TP , Fujimoto M , Bonci A . The sigma-1 receptor: Roles in neuronal plasticity and disease. Trends Neurosci. (2012) ;35: (12):762–71. |
[5] | Ponten H , Kullingsjö J , Lagerkvist S , Martin P , Pettersson F , Sonesson C , et al. In vivo pharmacology of the dopaminergic stabilizer pridopidine. Eur J Pharmacol. (2010) ;644: :88–95. |
[6] | Sahlholm K , Århem P , Fuxe K , Marcellino D . The dopamine stabilizers ACR16 and (–) OSU6162 display nanomolar affinities at the sigma-1 receptor. Mol Psychiatry. (2013) ;18: :12–14. |
[7] | Sahlholm K , Sijbesma JW , Maas B , Kwizera C , Marcellino D , Ramakrishnan NK , et al. Pridopidine selectively occupies sigma-1 rather than dopamine D2 receptors at behaviorally active doses. Psychopharmacology (Berl). (2015) ;232: (18):3443–53. |
[8] | Ryskamp D , Wu J , Geva M , Kusko R , Grossman I , Hayden M , et al. The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol Dis. (2016) ;97: (Pt A):46–59. |
[9] | Geva M , Kusko R , Soares H , Fowler KD , Birnberg T , Barash S , et al. Pridopidine activates neuroprotective pathways impaired in Huntington disease. Hum Med Gen. (2016) ;25: (18):3975–87. |
[10] | Eddings CR , Arbez N , Akimov S . Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor. Neurobiol Dis. (2019) ;129: :118–29. |
[11] | Wu J , Ryskamp DA , Liang X , Egorova P , Zakharova O , Hung G , et al. Enhanced store-operated calcium entry leads to striatal synaptic loss in a Huntington’s disease mouse model. J Neurosci. (2016) ;36: :125–41. |
[12] | Squitieri F , Di Pardo A , Favellato M , Amico E , Maglione V , Frati L . Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J Cell Mol Med. (2015) ;19: (11):2540–8. |
[13] | Huntington Study Group HART Investigators. A randomized, double-blind, placebo controlled trial of pridopidine in Huntington’s disease. Mov Disord. (2013) ;28: (10):1407–15. |
[14] | de Yebenes JG , Landwehrmeyer B , Squitieri F , Reilmann R , Rosser A , Barker RA , et al. Pridopidine for the treatment of motor function in patients with Huntington’s Disease (MermaiHD): A phase 3, randomized, double-blind, placebo-controlled trial. Lancet Neurol. (2011) ;10: (12):1049–57. |
[15] | Reilmann R , McGarry A , Grachev ID , Savola JM , Borowsky B , Eyal E , et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE HD): A phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. (2019) ;18: (2):165–76. |
[16] | Huntington Study Group. The Unified Huntington’s Disease Rating Scale: Reliability and consistency. Mov Disord. (1996) ;11: :136–42. |
[17] | Shoulson I , Kurlan R , Rubin AJ Assessment of functional capacity in neurodegenerative movement disorders: Huntington’s disease as a prototype. In: Munsat TL, editors. Quantification of neurological deficit. Boston: Butterworths; 1989. pp. 271-283. |
[18] | The Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s Disease. Neurology. (2001) ;57: :397–404. |
[19] | Landwehrmeyer GB , Dubois B , de Yébenes JG , Kremer B , Gaus W , Kraus PH , et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann Neurol. (2007) ;62: (3):262–72. |
[20] | McGarry A , McDermott M , Kieburtz K , de Blieck EA , Beal F , Marder K , et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology. (2017) ;88: (2):152–9. |
[21] | Hersch SM , Schifitto G , Oakes D , Bredlau AL , Meyers CM , Nahin R , et al. The CREST-E study of creatine for Huntington disease: A randomized controlled trial. Neurology. (2017) ;89: (6):594–601. |
[22] | Schobel SA , Palermo G , Auinger P , Long JD , Ma S , Khwaja OS , et al. Motor, cognitive, and functional declines contribute to a single progressive factor in early HD. Neurology. (2017) ;89: (24):2495–502. |
[23] | Marder K , Zhao H , Myers RH , Cudkowicz M , Kayson E , Kieburtz K , et al. Rate of functional decline in Huntington’s disease. Neurology. (2000) ;54: (2):452–8. |
[24] | Reilmann R , Olanow CW , McGarry A , Geva M , Leinonen M , Hayden M , et al. Novel PET data and analysis of early HD from PRIDE-HD. International Parkinson and Movement Disorder Society Congress, Nice, France, September 2-26, 2019. |
[25] | Lucas G , Rymar VV , Sadikot AF , Debonnel G . Further evidence for an antidepressant potential of the selective s1 agonist SAElectrophysiological, morphological and behavioural studies. Int J Neuropsychopharmacol. (2008) ;11: :485–95. |
[26] | Maurice T , Hiramatsu M , Itoh J , Kameyama T , Hasegawa T , Nabeshima T . Behavioral evidence for a modulating role of sigma ligands in memory processes. I: Attenuation of dizocilpine (MK-801)-induced amnesia. Brain Res. (1994) ;647: :44–56. |
[27] | Maurice T , Lockhart BP . Neuroprotective and anti-amnesic potentials of sigma (σ) receptor ligands. Prog Neuropsychopharmacol Biol Psychiatry. (1997) ;21: (1):69–102. |
[28] | Maurice T , Urani A , Phan VL , Romieu P . The interaction between neuroactive steroids and the σ1 receptor function: Behavioral consequences and therapeutic opportunities. Brain Res Rev. (2001) ;37: (1-3):116–32. |
[29] | Monnet FP , Maurice T . The sigma 1 protein as a target for the non-genomic effects of neuro (active) steroids: Molecular, physiological, and behavioral aspects. J Pharmacol Sci. (2006) ;100: (2):93–118. |
[30] | Matsuno K , Senda T , Matsunaga K , Mita S , Kaneto H . Similar ameliorating effects of benzomorphans and 5-HT 2 antagonists on drug-induced impairment of passive avoidance response in mice: Comparison with acetylcholinesterase inhibitors. Psychopharmacology (Berl). (1993) ;112: (1):134–41. |
[31] | Matsuno K , Senda T , Matsunaga K , Mita S . Ameliorating effects of σ receptor ligands on the impairment of passive avoidance tasks in mice: Involvement in the central acetylcholinergic system. Eur J Pharmacol. (1994) ;261: (1-2):43–51. |
[32] | Monnet FP , Debonnel G , Junien JL , De Montigny C . N-methyl-D- aspartate-induced neuronal activation is selectively modulated by sigma receptors. Eur J Pharmacol. (1990) ;179: :441–5. |
[33] | Monnet FP , Blier P , Debonnel G , de Montigny C . Modulation by sigma ligands of N-methyl-D-aspartate-induced [3 H] noradrenaline release in the rat hippocampus: G-protein dependency. Naunyn Schmiedebergs Arch Pharmaco. (1992) ;346: (1):32–9. |
[34] | Bermack JE , Debonnel G . The role of sigma receptors in depression. J Pharmacol Sci. (2005) ;97: (3):317–36. |
[35] | Hayashi T , Maurice T , Su TP . Ca2+ signaling via ς1-receptors: Novel regulatory mechanism affecting intracellular Ca2+ concentration. J Pharmacol Exp Ther.. (2000) ;293: (3):788–98. |
[36] | Hong W , Nuwayhid SJ , Werling LL . Modulation of bradykinin-induced calcium changes in SH-SY5Y cells by neurosteroids and sigma receptor ligands via a shared mechanism. Synapse. (2004) ;54: (2):102–10. |
[37] | Urani A , Romieu P , Roman FJ , Yamada K , Noda Y , Kamei H , et al. Enhanced antidepressant efficacy of σ1 receptor agonists in rats after chronic intracerebroventricular infusion of β-amyloid-(1– 40) protein. Eur J Pharmacol. (2004) ;486: (2):151–61. |
[38] | Smith-Dijak AI , Sepers MD , Raymond LA . Alterations in synaptic function and plasticity in Huntington disease. J Neurochem. (2019) ;150: (4):346–65. |
[39] | Francardo V , Geva M , Bez F , Denis Q , Steiner L , Hayden MR , et al. Pridopidine induces functional neurorestoration via the sigma-1 receptor in a mouse model of Parkinson’s disease. Neurotherapeutics. (2019) ;16: (2):465–79. |
[40] | Pande AC , Genève J , Scherrer B , Smith F , Leadbetter RA , de Meynard C . A placebo-controlled trial of igmesine in the treatment of major depression. Eur Neuropsychopharmacol. (1999) ;9: (Suppl 5):S138. |
[41] | Volz HP , Stoll KD . Clinical trials with sigma ligands. Pharmacopsychiatry. (2004) ;37: (S3):214–20. |
[42] | The Voice of the Patient: Huntington’s Disease. https://www.fda.gov/media/96196/download |


