
DNA Mismatch Repair and its Role in Huntington’s Disease
Abstract
DNA mismatch repair (MMR) is a highly conserved genome stabilizing pathway that corrects DNA replication errors, limits chromosomal rearrangements, and mediates the cellular response to many types of DNA damage. Counterintuitively, MMR is also involved in the generation of mutations, as evidenced by its role in causing somatic triplet repeat expansion in Huntington’s disease (HD) and other neurodegenerative disorders. In this review, we discuss the current state of mechanistic knowledge of MMR and review the roles of key enzymes in this pathway. We also present the evidence for mutagenic function of MMR in CAG repeat expansion and consider mechanistic hypotheses that have been proposed. Understanding the role of MMR in CAG expansion may shed light on potential avenues for therapeutic intervention in HD.
INTRODUCTION
DNA repair processes are characterized by their ubiquity across all domains of life [1]. These mechanisms have evolved to maintain genomic stability, especially in light of the high levels of DNA damage generated by exogenous and endogenous agents such as ultraviolet light, ionizing radiation, and reactive chemical species. Errors made by DNA synthetic enzymes (either during the normal course of cell division or perhaps while carrying out DNA repair itself) further imperil the integrity of the genetic information [1, 2]. Replicative DNA polymerases make base insertion errors infrequently (at a rate of ∼10–4–10–5), and their fidelity is further enhanced by ∼100 fold by the exonucleolytic proofreading machinery associated with many such enzymes [2]. Errors that escape these mechanisms are rectified by the highly conserved DNA mismatch repair (MMR) pathway, which confers an additional ∼100–1000 fold enhancement in fidelity [3–7]. In addition to its role in error correction and replication fidelity, MMR also plays important roles in preventing chromosomal rearrangements and mediating the cellular response to several types of DNA damage [8–10].
The importance of MMR is exemplified by the profound consequences of its loss. MMR defects result in a 100–1000 fold elevation in mutation rate in most organisms [3–7], and modulation of MMR has been suggested to be a powerful evolutionary survival mechanism [11, 12]. Loss of MMR function in humans is the cause of Lynch syndrome, a hereditary cancer predisposition condition that is characterized by an increased risk of gastrointestinal, uterine, and ovarian tumors [13, 14]. More recently, biallelic inactivation of MMR has been linked to constitutive mismatch repair deficiency syndrome (CMMR-D), a rare childhood/young adult condition associated with a higher propensity for developing colorectal, brain, and blood cancers [15–17]. In addition, MMR defects underlie a significant subset of sporadic tumors across various tissues [18]. Current evidence suggests that the high mutation load of MMR-deficient tumors results in neoantigen production, rendering such cancers highly sensitive to immune checkpoint inhibitors such as pembrolizumab and nivolumab, resulting in dramatic improvements in patient survival rates [19].
Counterintuitively, this mutation prevention mechanism can in some instances be subverted to produce mutations. This phenomenon is exemplified by the requirement of MMR proteins for somatic hypermutation, a mutagenic process in B lymphocytes that generates immunoglobulin diversity [20–22]. This type of MMR-dependent mutagenesis has been attributed to a non-canonical MMR (ncMMR) mechanism which is activated in a variety of cell types, and is independent of DNA replication [23–25]. A number of MMR proteins have also been implicated in triplet repeat expansions that underlie several neurodegenerative diseases, including Huntington’s disease (HD) [26–28]. Herein, we review what is known about the molecular mechanisms of MMR, and consider its role in somatic CAG triplet repeat expansion in HD.
MECHANISM OF MISMATCH REPAIR
Mismatches, strand slippage, and extrahelical extrusions
The best understood function of MMR is its ability to efficiently correct mistakes made by DNA polymerases during DNA synthesis [3, 5–7, 29]. A number of factors involved in the error-correction reaction have been identified and are listed in Table 1. Replication errors may be base substitution errors that result in a base-base mismatch, or strand slippage errors that generate an extrahelical extrusion or loop. Inability to rectify these errors result in transitions, transversions, and small insertion/deletion mutations. Although the bulk of base insertion errors occur during DNA replication, they may also occur during repair DNA synthesis in non-replicating cells, and such mistakes if left unrectified could be a significant source of mispairs.
Table 1
MMR proteins and their functions
| Protein | Representation | Activity | Function |
| MutSα |  | ATPase, DNA binding | Mismatch recognition (base-base and small extrusions) |
| MutSβ |  | ATPase, DNA binding | Mismatch recognition (extrusions only) |
| MutLα |  | ATPase, Endonuclease | Introduction of strand breaks |
| MutLγ |  | ATPase, Endonuclease | DNA structure- and strand-specific nicking |
| MutLβ |  | ATPase | Unknown (suggested to be an accessory factor) |
| PCNA |  | Sliding clamp | Strand-directionality factor |
| RFC |  | ATPase | Loads PCNA onto the DNA |
| EXOI |  | 5’–3’ exonuclease | Exonucleolytic removal of error-containing strand |
| Polδ |  | DNA polymerase | Strand synthesis (also carries out strand displacement) |
| LIG1 | Not shown | DNA ligase | Ligation of strand-breaks post repair DNA synthesis |
Strand-slippage errors occur with high frequency within microsatellite sequences (e.g., mono-, di-, and trinucleotide repeats), the repetitive nature of which renders them particularly prone to the formation of extrahelical extrusions by strand misalignment [30–33]. In principle, two DNA strands can misalign not only during DNA synthesis but also whenever the duplex strands separate and reanneal during DNA metabolic processes such as transcription. Helix opening can also be driven by the energy of negative supercoiling. While superhelical tension in eukaryotic cells is normally kept restrained by nucleosomes, release of negative superhelicity upon nucleosome disassembly can cause transient helix destabilization [34]. Likewise, helix opening due to accumulation of negative superhelical tension within the underwound DNA in the wake of a translocating RNA polymerase has been documented [35, 36]. Thus, duplex melting within repetitive DNA tracts can occur under a variety of circumstances, resulting in the formation of extrahelically extruded slipped-strand structures. It is noteworthy in this regard that long CTG repeat tracts are among the strongest nucleosome binding sequences known due to their high intrinsic negative superhelical writhe [37, 38]. These observations may explain why CTG repeats may be particularly prone to supercoil-induced spontaneous helix opening and formation of slipped-strand structures. In fact, slipped-strand structures readily form both in vitro and in vivo, are structurally heterogeneous, and thermodynamically stable [33, 39, 40]. Thus, extrahelically extruded slipped strand structures could form within resting DNA repeat tracts in non-dividing cells such as neurons during DNA repair, transcription, or chromatin remodeling.
Mismatch recognition
To deal with the diversity of base-base mismat-ches and extrahelical loops that are generated during DNA metabolic processes [2], eukaryotic MMR has evolved a modular approach to mispair recognition, employing one of two heterodimeric MutS homologs, MutSα (MSH2-MSH6) and MutSβ (MSH2-MSH3) (Fig. 1). MutSα recognizes base-base mismatches and small DNA extrusions (1–4 extrahelical residues), and MutSβ exclusively recognizes DNA extrusions of 2 to about 10 extrahelical residues [3]. The structural basis for the mismatch recognition has been clarified by crystallographic studies of MutSα, MutSβ, and bacterial MutS homologs in complex with mispair- or lesion-containing DNA substrates [41–44]. These studies have shown that mismatch recognition is asymmetric: whereas MSH2 makes limited contact with the DNA in both MutSα and MutSβ, extensive interactions occur between residues in MSH3 and MSH6 with the heteroduplex [41, 42].
Because of their overlapping substrate recognition specificities, MutSα can largely compensate for the absence of MutSβ, and the overwhelming majority of mispairs are recognized and rectified by a MutSα-initiated pathway. Hence, MutSα is critical for mutation avoidance, and its inactivation (by mutations in MSH2 or MSH6) is a risk factor for carcinogenesis [13, 14]. By contrast, loss of MutSβ function (by inactivation of MSH3) has not been conclusively linked to an elevated cancer risk in humans [45]. Consistent with these observations, mice lacking MSH2 or MSH6 display increased susceptibility to spontaneous tumorigenesis and an attenuated lifespan, whereas MSH3 knockout mice are indistinguishable from wild-type mice both in terms of tumorigenicity and life expectancy [46–49]. These striking differences are particularly relevant in light of the observation (considered below) that CAG somatic expansion is attenuated in HD mice lacking Msh2 or Msh3 (but not Msh6), thus suggesting that pharmacological modulation of MSH3 may pose a lower risk than targeting MSH2 or MSH6.
In addition to their DNA binding functions, MutSα and MutSβ possess ATP hydrolytic activities and belong to the ABC (ATP Binding Cassette) ATPase superfamily [41, 42]. The MutSα and MutSβ ATPases are stimulated by heteroduplex DNA, an effect that is indicative of long-range conformational changes elicited by DNA cofactor binding at a site distal to the nucleotide pocket [50–55]. Although the functional significance of ATP binding/hydrolysis of MutS homologs has been the subject of intense debate for over three decades, there is general agreement that inactivation of the ATPase function is deleterious for overall mismatch repair [3, 4]. Consequently, such mutations result in elevation in mutation rate in bacteria [56] and a higher cancer predisposition risk in humans [42].
Formation of mismatch repair protein assemblies and mismatch removal
Mismatch recognition by MutSα/β is followed by the recruitment of the heterodimeric MutLα (MLH1-PMS2). MutL homologs belong to the GHKL ATPase superfamily that is characterized by the unique Bergerat ATP-binding fold [57]. Although detailed structure-function studies of the MutL ATPases are lacking, structures of both MLH1 and PMS2 ATPase domains are available [58–60], and biochemical and genetic studies have shown that the MutL ATPase function is required for MMR [61–63]. Interaction between MutSα/β and MutLα results in the formation of a dynamic ATP-dependent DNA-MutSα/β-MutLα ternary complex that is required for MMR [64–72]. These observations have led to the notion that ATP binding/hydrolysis drives conformational changes in MutS homologs (and presumably in MutL homologs as well), thereby facilitating ternary complex assembly.
The PMS2 subunit of MutLα harbors a latent zinc-dependent endonuclease that is activated by DNA-loaded replication sliding clamp PCNA (proliferating cell nuclear antigen) in the presence of MutSα/β, a mispair, and ATP-Mg+2 [73–75]; this activity depends on the integrity of a conserved metal-binding DQHA(X)2E(X)4E motif and requires a physical interaction between PMS2 and PCNA [74–76]. The MutLα-catalyzed strand breaks are restricted to the strand harboring a pre-existing break (and hence assumed to harbor the incorrect base) and bracket the mismatch on both 3′ and 5′ sides (Fig. 1); these breaks provide initiation sites for excision of the strand harboring the incorrect base as discussed below. The importance of MutLα in the MMR reaction is highlighted by the observation that loss of MLH1 or PMS2 not only results in elevation of mutation rate, but also increases the lifetime risk of tumorigenesis in both mice and humans [77]. Interestingly, both MLH1 and PMS2 have been identified as onset modifiers of HD, and loss of Mlh1 or Pms2 substantially attenuates somatic triplet repeat expansion in animal and cellular models of HD, myotonic dystrophy type 1 (DM1), and Fragile X-related disorders (FXDs) [78–80].
Fig. 1
Mechanisms of 5′ and 3′ human mismatch repair. Distinct molecular mechanisms mediate mismatch repair, depending on strand-break polarity. Left, DNA mismatches or extrahelical extrusions are recognized by MutSα or MutSβ. When the strand-break is located 5′ to the mismatch, MutSα/β activates the processive 5′–3′ exonuclease activity of ExoI in an ATP-dependent manner. The ensuing gap is protected by the single-stranded DNA binding protein complex RPA, followed by DNA resynthesis across the gap by DNA polymerase δ, aided by the replication sliding clamp PCNA and the clamp loader RFC. Right, if the strand-break is located 3′ to the mispair, error correction relies on oriented loading of PCNA by RFC at the strand break. Thus, MutSα/β recruits MutLα in an ATP-dependent manner, resulting in the activation of a latent endonuclease function in MutLα in the presence of DNA-loaded PCNA. The additional strand-breaks catalyzed by MutLα bracket the mismatch, and facilitate processive 5′–3′ hydrolysis of the nicked strand by MutSα-activated ExoI. Gap protection and filling occur as in the 5′ nick-directed reaction.
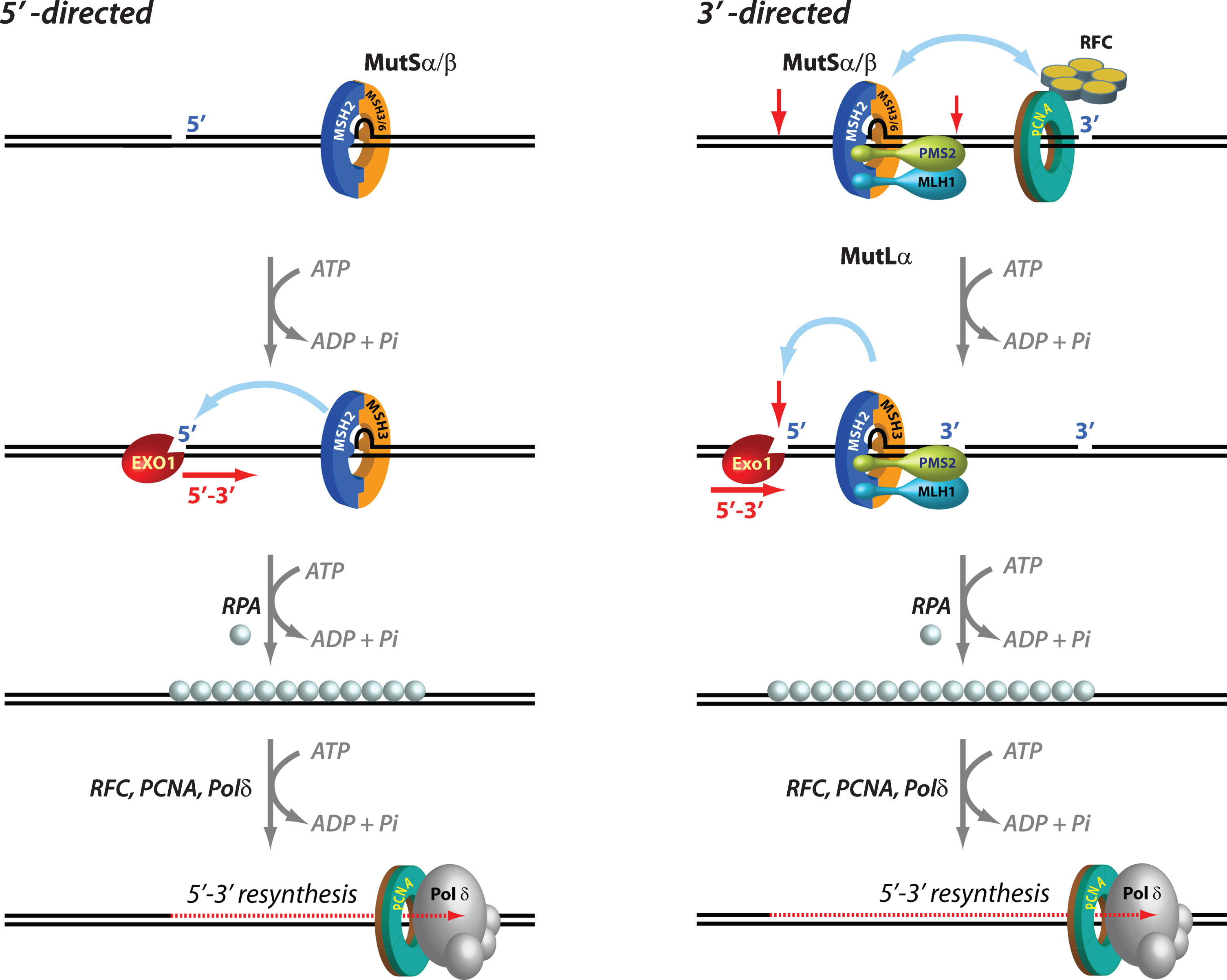
Mismatch excision is carried out by the 5′ to 3′ hydrolytic activity of EXO1, the only exonuclease that has been identified in eukaryotic MMR. EXO1, an otherwise distributive enzyme (i.e., it excises only a few nucleotides at a time from the DNA before it dissociates), is rendered highly processive (i.e., capable of removing hundreds of nucleotides without having to dissociate) when it associates with MutSα in the presence of a mispair and ATP. Loading of MutSα-activated EXO1 at 5′ DNA termini results in processive strand excision that proceeds until the mismatch is removed [81–83]. This MutSα-stimulated EXO1 reaction relies on pre-existing 5′ ends (but not 3′ ends) such as those that exist in Okazaki fragments on the lagging strand of DNA replication [3–7], and is notable in its lack of an absolute requirement for MutLα [84] (Fig. 1).
In the presence of MutLα however, the MMR system can utilize either pre-existing 5′ or 3′ DNA termini. In fact, the 3′ nick-directed reaction displays an absolute requirement for MutLα [74, 81, 85, 86] (Fig. 1). The additional strand-breaks introduced by the activated MutLα endonuclease serve as initiation sites for processive 5' to 3' excision by MutSα/β-activated Exo I [74, 81, 82, 87]. The ensuing EXO1-catalyzed gap is protected by the single-stranded DNA binding protein RPA, followed by resynthesis with high fidelity by DNA polymerase δ (Polδ), and ligation by DNA ligase I, thereby maintaining the integrity of the genetic information [83, 85, 86, 88]. Interestingly, EXO1-independent mismatch repair has also been documented wherein Pol δ conducts strand displacement synthesis initiated from 3′-OH DNA termini, thereby replacing the strand that contains the incorrect information [87].
Mammalian cells also express two other MutL complexes—MutLγ (MLH1-MLH3) and MutLβ (MLH1-PMS1). Both MLH3 and PMS1 are GHKL ATPases, but only MLH3 possesses the metal-binding endonuclease motif found in PMS2 [74]. However, although MutLγ endonuclease is activated by DNA-bound MutSβ, unlike MutLα this effect does not require DNA-loaded PCNA [89–91]. The primary function of MutLγ is in meiotic recombination wherein it plays an essential role wherein the MutLγ endonuclease is activated by MutSγ (MSH4-MSH5), and to a lesser extent by RFC, PCNA, and EXO1 [92–95]. MutLγ also plays a modest role in MMR since loss of MLH3 function results in a moderate increase in mutation rates, and in vitro complementation of MLH1-deficient cells with MutLγ-enriched fractions results in partial restoration of MMR function [96–100]. Consistent with a possible role for MLH3 in cancer avoidance [101], Mlh3 -/- mice display elevated mutation rates, increased tumor susceptibility, and reduced lifespans relative to wild type mice [99, 102]. Interestingly, whereas human GWA studies have not identified MLH3 as an HD onset modifier, Mlh3 is required for CAG and CGG somatic expansions in HD and FXD mouse models, respectively [80, 103, 104].
As noted above, PMS1 is the only MutL ortholog that does not contain the DHQA endonuclease motif. PMS1 interacts with MLH1 to form MutLβ, a heterodimer that associates with MutSα or MutSβ to form an ATP-dependent DNA-MutSα/β-MutLβ ternary complex [105–109]. However, MutLβ appears to have no role in canonical human MMR as judged by the inability of recombinant MutLβ to restore MMR activity to MLH1-deficient cell extracts [105]. Consistent with these findings, Pms1 knockout mice show no observable defects in either MMR or tumor susceptibility [77]. Nevertheless, studies with the yeast homolog have indicated a possible accessory role for MutLβ in the repair of a subset of heteroduplexes [92, 98, 109]. Thus, although PMS1 modifies HD onset age [103], given the limited understanding of the molecular function of PMS1 or MutLβ, the possible mechanisms underlying its role in HD remain to be investigated.
Strand directionality of mismatch repair
Upon encountering a mismatch, the MMR system must act with exquisite strand bias in order to specifically rectify the strand containing the incorrect information. This is achieved by using “strand signals” that mark one strand relative to the other, and thus enable the MMR system to distinguish between the template strand and the newly synthesized strand (Fig. 1). Whereas in E. coli, DNA hemi-methylation has been established as the mechanism that directs the system to the appropriate strand, the identity of eukaryotic strand signals has been intensely debated for over three decades. In in vitro biochemical experiments, strand-breaks serve effectively as strand signals, and mismatch repair of circular DNA substrates is restricted to the strand that contains a pre-existing break. When such strand-breaks are located 5′ to the mismatch, MutSα/β-activated EXO1 processively and exclusively excises the nicked strand with 5′-3′ polarity thereby conferring strand-directionality to the mismatch repair reaction. However, when the strand-break is located 3′ to the mismatch, strand directionality is conferred by the introduction of additional strand-breaks by MutLα in a reaction that also requires MutSα/β, a mispair, DNA-loaded PCNA, and ATP. These new strand-breaks are restricted to the nick-containing strand. The strand-directional activation of MutLα can also occur when the original strand-break is located 5′ to the mismatch. The strand bias of the MutLα endonuclease is determined by the unique spatial orientation (relative to 3′-OH termini) with which the donut-shaped PCNA (with its two non-equivalent faces; Fig. 2) is loaded at DNA strand discontinuities by the clamp loader replication factor C (RFC). This effect requires a physical association of oriented PCNA with the PMS2 subunit of MutLα [76]. Thus, it is relatively straightforward to envision PCNA loaded at DNA termini as strand signals for MMR in the context of active DNA replication in dividing cells (e.g., DNA ends of Okazaki fragments). In fact, recent studies have suggested that loaded PCNA continues to direct strand-specific MMR even after the removal of strand-discontinuities, and that a physical interaction between MutSα and PCNA enhances the temporal window for effective strand-directed MMR [110, 111]. Thus, persistence of DNA-loaded PCNA after completion of DNA synthesis can continue to provide strand-directionality “memory” to the MMR system [110].
Fig. 2
Models for involvement of mismatch repair in CAG/CTG repeat expansion. Strand slippage within long repetitive CAG/CTG tracts results in the formation of extrahelical extrusions that are not only recognized by MutSβ, but also serve as loading sites for PCNA even in the absence of strand-breaks. PCNA, a ring shaped homotrimeric protein with two distinct faces (inset, indicated in green and brown) preferentially associates with its partner proteins via residues on one face. Although the two faces of PCNA are functionally non-equivalent, due to the symmetry of the extrahelical extrusions, PCNA loading at such structures occurs in both possible spatial orientations. Since the strand directionality of the MutSβ-dependent activity of the MutLα endonuclease is determined by the orientation of DNA-loaded PCNA, the disoriented loading of PCNA misdirects MutLα catalyzed incisions to either DNA strand. Left, when incision occurs on the extrusion-containing strand (shown in blue), strand excision results in removal of the extrusion, and faithful repair by Polδ results in a contraction (not shown). However, error-prone gap resynthesis by Polη or Polβ as illustrated in the diagram may provide additional opportunities for strand slippage and formation of new extrahelical extrusions, which either result in a net increase in CAG repeat length (i.e., expansion) or trigger additional rounds of MutSβ-initiated incision/excision. Middle, when MutLα-mediated strand-breaks are formed on the complementary (red) strand, error-free resynthesis by Polδ results in inclusion of the extrusion, leading to a net increase in CAG repeat length (expansion). Gap resynthesis may also be driven by Polβ as on left, resulting in additional strand slippage (not shown). Right, extrusion-bound MutSβ can also activate MutLγ in a PCNA-independent manner. The incisions catalyzed by MutLγ are restricted to the complementary (red) strand opposite to the extrusion. DNA resynthesis results in inclusion of the extrusion, leading to a net increase in CAG repeat length (expansion).
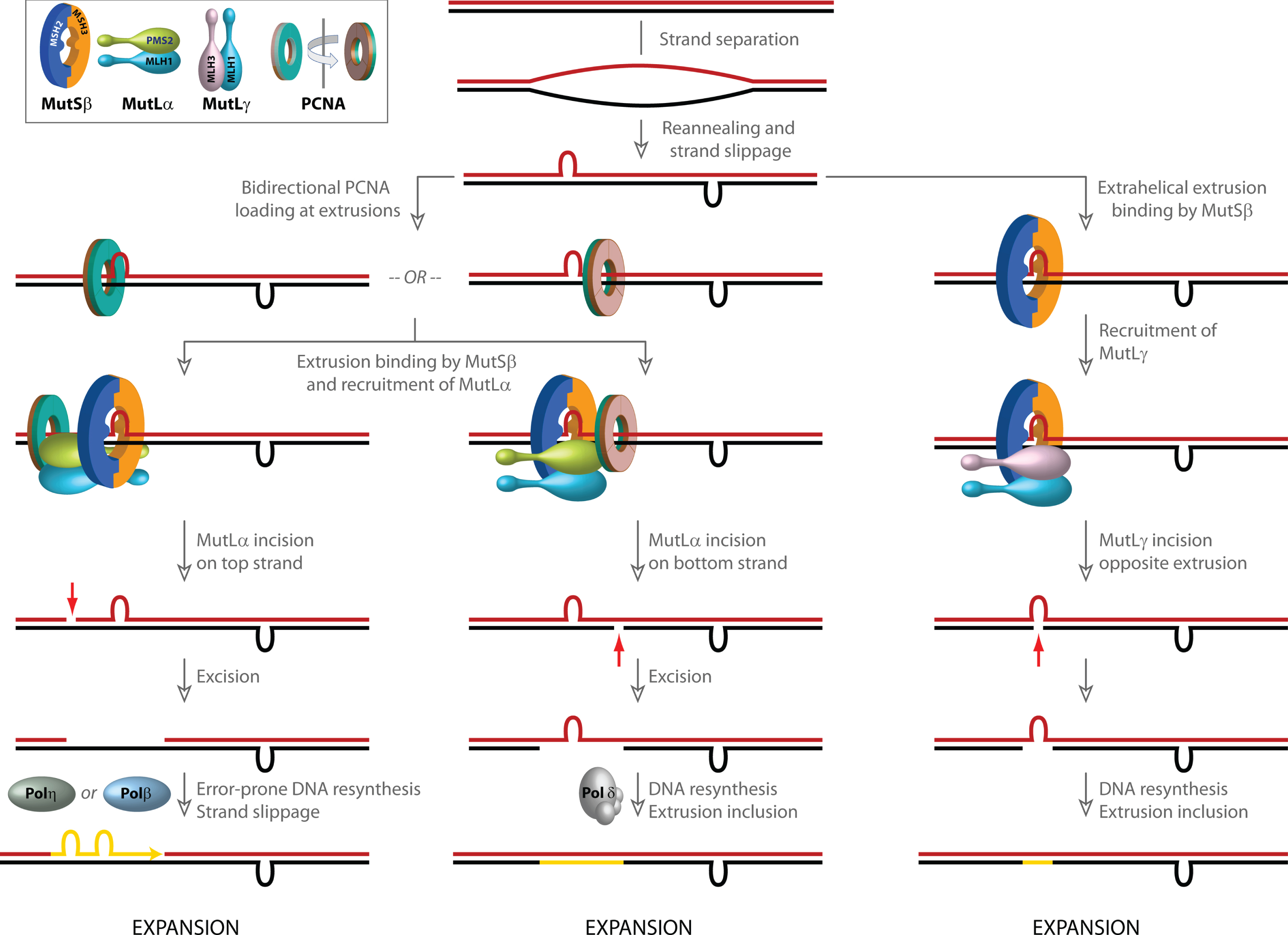
Sequences that adopt unusual DNA secondary structures are found throughout the human genome [112–115]. A substantial body of literature supports the idea that non B-DNA conformations have profound effects on DNA metabolic processes (reviewed in [26]). The DNA conformational transitions that govern the formation of these non B-DNA structures are energetically driven by negative superhelical tension, which in normal eukaryotic cells remains constrained within chromatin but can be unleashed during nucleosome disassembly. Interestingly, such structures may also affect the strand-directionality of MMR. DNA molecules that contain “open bubbles” (due to unpairing of a segment of the duplex) could provide sites for loading of PCNA even in the absence of strand-breaks. Such bubble structures contain single-strand/double-strand junctions with mirror symmetry and conformationally resemble extrahelical extrusions that form by strand slippage within repetitive DNA tracts (e.g., long CAG/CTG repeats). PCNA loading at such structures occurs in either spatial orientation (disoriented loading) and, consequently, MutSα/β-dependent activation of MutLα endonuclease on these DNAs also occurs without substantial strand bias [75, 116, 117]. The implications of these findings are three-fold. First, DNA-loaded, spatially oriented PCNA may direct the strand-specificity of MMR in post-replicative non-dividing cells. Second, PCNA loading at extrahelical extrusions such as those formed within long repetitive DNA tracts may misdirect the MMR system and cause it to act aberrantly on both DNA strands (see below). Third, strand-independent activation of ncMMR could involve PCNA ubiquitination and recruitment of error-prone DNA polymerases, resulting in repeat instability [23, 118].
Mismatch repair in the context of chromatin
The mechanistic dissection of the MMR reaction has relied largely on in vitro biochemical studies using a combination of purified proteins, cell extracts, and naked heteroduplex DNA substrates [6]. However, there has been growing interest in understanding how mismatch repair functions in the cellular context, especially within chromatin (for comprehensive review, see [119]. Studies have established that trimethylation of histone H3 at lysine 36 (H3K36me3) is required for recruitment of MutSα to replicating chromatin [120]. This effect is mediated by a physical interaction between the modified histone and the PWWP domain of the MSH6 subunit of MutSα. The importance of this histone modification for MMR is established by the observation that knockdown of SETD2 (a histone methyltransferase that trimethylates H3K36) causes a mutator phenotype characterized by high microsatellite instability and an elevated mutation rate [120]. Mutations in histone H3 (G34V/R/D) that block H3K36 trimethylation by SETD2 also inhibit the interaction between H3K36 and MutSα; this mutation not only causes a mutator phenotype in cells, but also has been identified as a driver of pediatric glioblastoma [121]. MMR modulation by histone modification helps maintain genomic stability of actively transcribed genes [122]. Not only are H3K36me3 and MutSα co-enriched in exons (relative to introns and non-transcribed regions), but also disruption of the H3K36me3-MutSα interaction elevates the spontaneous mutation rate in actively transcribed genes, with little influence on non-transcribed regions. These findings have implications for the role of MMR in maintaining genomic stability in non-dividing, but transcriptionally active, cells.
In addition to histone modification, interplay of the MMR proteins with nucleosome assembly and remodeling factors has also been documented [123–125]. MutSα and PCNA interact with the nucleosome assembly factor CAF-1, and these interactions inhibit the CAF-1 mediated nucleosome assembly on mispair-containing DNAs [123–125]. The formation of the MutSα-CAF-1 complex is mediated by residues within the MSH6 subunit of MutSα [123]. Investigations to date have focused on MutSα function in the context of chromatin dynamics. However, because MSH3 does not possess a PWWP domain [126], and since no studies have been carried out on MutSβ interactions with nucleosome assembly factors, the mechanisms that recruit MutSβ to chromatin are not known.
In the cellular environment, the MMR machinery is also regulated by post-translational modifications. Phosphorylation of several MMR proteins—including MutSα, MutLα, PCNA, RFC, ExoI, and Polδ—has been documented, and these modifications modulate protein-protein interactions, protein stability, as well as nuclear-cytoplasmic distribution [127]. A full mechanistic understanding of these diverse effects is still underway; nevertheless, based on available data it is expected that future investigations will unravel novel aspects of MMR regulation by post-translational modification.
Other functions of mismatch repair
In addition to its role in mismatch correction, the MMR machinery also mediates the cellular response to several types of DNA damage [8, 9, 128] and prevents illegitimate recombination [10]. However, these functions of MMR are not currently thought to play a role in triplet repeat instability [3, 26, 129], and will therefore not be considered further.
Mismatch repair in the central nervous system (CNS)
Our current understanding of canonical post-replicative DNA mismatch repair is based on fun-ctional studies in dividing cells complemented by mechanistic biochemical investigations. Because the primary phenotype of loss of mismatch repair activity is elevation in mutation rate, and since mutation rate measurements are (by definition) not feasible in non-replicating cells, the role of MMR in terminally differentiated cells such as neurons has not been studied extensively. Yet, the functional importance of MMR in the brain is underscored by the observation that ∼50% of CMMR-D patients who harbor biallelic germline mutations in one of four MMR genes (MSH2, MSH6, MLH1, and PMS2) develop brain tumors of various types including glioblastomas, astrocytomas, and oligodendrogliomas [16]. Cells derived from such tumors display low levels of MMR activity [130]. Immunohistochemistry and immunoblot analyses have established that MSH2, MSH6, MSH3, MLH1, and PMS2 are robustly expressed in normal human and rodent brains, suggesting that these proteins have functional significance [80, 131–136]. Insofar as MMR protein levels are reflective of the capacity of the brain to rectify mismatches, the lack of replicative errors in terminally differentiated brain cells suggest that mismatches must occur post-mitotically.
Because the brain consumes ∼20% of the total oxygen budget of the human body to support neuronal activities, it is also subjected to high levels of oxidative stress, a condition that has been correlated with oxidative DNA damage and accumulation of DNA strand breaks [137–140]. Therefore, base excision repair (BER), single-strand break repair (SSBR), and double-strand break repair (DSBR) mechanisms are highly active in the CNS, and in fact, defects in these repair pathways are the cause of several neurodegenerative diseases [139, 141, 142]. Physical and functional cross-talk between MMR and BER proteins has been documented [143], and therefore it is plausible that MMR factors may be recruited to sites of BER activity in neurons. It is also possible that even limited gap filling DNA synthesis during BER, DSBR, or SSBR may provide ample opportunities over several decades of human life for DNA polymerases to make errors that may then require the attention of the MMR system. Nevertheless, direct evidence for MMR activity in neurons is lacking, and although nuclear extracts prepared from rat brain retain mismatch binding activity [132], the capacity of whole brain or neuronal extracts to repair mismatches or extrahelical extrusions has not been studied, and brain- or neuron-specific MMR factors have not been identified. Therefore, biochemical and cellular studies of MMR function in brain-derived cells and tissues is likely to be a fruitful and instructive avenue of investigation.
ROLE OF MISMATCH REPAIR IN HUNTINGTON’S DISEASE
The diverse genome-stabilizing activities of MMR notwithstanding, this pathway has also been implicated in mutation production in the context of neurodegenerative disease. The most compelling corpus of data in this regard pertains to HD and will be considered here, although recent findings have also suggested a role for MMR in the repeat expansions underlying FXDs and Friedreich’s ataxia [78, 104, 144–147].
HD belongs to a family of slow-progressing neurodegenerative disorders caused by CAG triplet repeat expansions within the coding regions of distinct and unrelated genes [148]. A key feature of HD (and indeed other triplet repeat diseases as well) is the strong inverse correlation between the inherited length of the CAG repeat tract (located within exon1 of the Huntingtin HTT gene) and the age of disease onset [148, 149]. Moreover, HD gene expanded carriers (HDGEC) display high levels of CAG repeat expansion, not only in the germline, but also in somatic cells wherein such expansions occur in a tissue-specific manner: the greatest degree of expansion is observed in the terminally differentiated neurons of the striatum and cortex [131, 150–152] [153]. Recent studies have established that the inherited length of uninterrupted CAG repeats rather than the length of the encoded polyQ tract, (since both CAA and CAG can encode glutamine) modifies disease onset [103, 154–156]. Because uninterrupted repeats are thought to have a higher propensity to expand than repeat tracts harboring CAA interruptions, it has been suggested that somatic CAG instability is a key driver of CAG length dependent HD pathogenesis [103, 154, 155]. One implication of these observations is that factors that hasten or attenuate CAG-repeat instability may also be expected to affect disease onset. It is noteworthy that a recent study has suggested a role for huntingtin protein levels in promoting CAG repeat expansion in knock-in mouse models of HD and spinocerebellar ataxia type 2, as well as in HD patient iPS-derived medium spiny neurons, although the pathways underlying these effects are yet to be delineated [157].
Genetic evidence for the role of MMR in HD
Recent human genome-wide association studies (GWAS) have identified genetic modifiers of age at disease onset that map to a constellation of DNA repair genes that include the MMR genes MSH3, MLH1, PMS1, PMS2, as well as LIG1, and FAN1 (Table 2). In the case of MSH3, a genetic variant that is associated with reduced gene expression and/or function appears to not only delay age at onset, but also reduce somatic instability and disease severity in HD and DM1 patients [161]. Curiously, this MSH3 variant itself arises from instability of a 9-bp tandem repeat that codes for an alanine repeat element near the N-terminus of the MSH3 polypeptide [161]. Repeat instability measurements in the blood have revealed effects of polymorphisms in MSH3, MLH1, MLH3, and FAN1 on somatic CAG-repeat variation in HDGEC [155], and of MSH3 genetic variants on CTG repeat instability in DM1 patients [164]. Effects of MSH3 and FAN1 on HD progression have also been documented [148, 158, 161, 163]. These observations made in human HD patients suggest that the MMR pathway influences both disease onset and progression and, taken together with data from HD mouse models, imply that disease manifestation is modulated by somatic CAG expansion.
Table 2
Summary of genes implicated in HD. The ratios of observed versus expected genome variants for each of the genes was derived from the gnomAD database [211], and are reflective of how tolerant a gene is to genetic variation. A low o/e score is indicative of stronger selection for the gene and lower tolerance for loss of function (LoF). Phenotypic effects of variants, knockout, or knockdown of the listed genes in human GWA studies, HD mouse models, and cellular systems are listed, and pathological effects of LoF in other disease states are summarized. References are shown in parentheses
| Gene | LoF tolerance (o/e ratio) | Effects of variants in HD GWA studies | Effect of gene knockout or reduced expression | LoF pathology (other human diseases) | ||
| HD mouse models | Mouse models of other triplet repeat diseases | Cellular systems for triplet repeat expansion | ||||
| MSH2 | 0.192 | - | Attenuates CAG repeat expansion [167, 168, 170, 177, 212] | Attenuates CTG and CGG repeat expansions in DM1 and FXD mice, respectively [171, 172, 176, 213] | Knockdown attenuates CTG and GAA repeat expansions [145, 214–217] | Lynch syndrome [13, 18], CMMR-D [15-17] |
| MSH3 | 0.912 | Increased expression hastens onset; reduced expression delays onset/ progression [103, 161, 163] | Attenuates CAG repeat expansion [173, 175, 218] | Attenuates CTG and CGG repeat expansions in DM1 and FXD mice, respectively [144, 169, 174] | Knockdown attenuates CTG and GAA repeat expansions [145, 215, 216] | - |
| MSH6 | 0.336 | - | Limited effect on CAG repeat contraction [175] | No effect on CTG repeat expansion in DM1 mice [169] | Knockdown does not affect CTG or GAA repeat expansions [145, 215, 216] | Lynch syndrome [13, 18], CMMR-D [15-17] |
| MLH1 | 0.373 | Missense (LoF?) variant delays onset [103, 155, 159, 162] | Attenuates CAG repeat expansion [80, 205] | - | Knockdown attenuates GAA repeat expansions [146] | Lynch syndrome [13, 18], CMMR-D [15-17] |
| PMS2 | 0.976 | Delay of onset by variant with unknown effect; missense variant hastens onset [103, 160] | - | Attenuates CTG repeat expansion in DM1 mice [79] | Knockout attenuates CGG repeat expansion [78] | Lynch syndrome [13, 18], CMMR-D [15-17] |
| PMS1 | 0.755 | Reduced expression hastens onset [103] | - | - | Knockout attenuates CGG repeat expansion [78] | - |
| MLH3 | 0.411 | Variant associated with somatic CAG instability in blood [155] | Attenuates CAG repeat expansion [80] | Attenuates CGG repeat expansion in FXD mice [104] | Knockout attenuates CGG and GAA repeat expansions [78, 146] | - |
| LIG1 | 0.288 | Missense (LoF) variant delays onset; increased expression hastens onset [103] | - | No effect on somatic CTG expansion in DM1 mice, but promotes maternal intergenerational expansion [219] | - | Immune deficiency [220, 221] |
| FAN1 | 0.864 | Missense (LoF) variants hasten onset; increased expression delays onset and slows progression [103, 158–160] | Promotes CAG repeat expansion [205] | Promotes CGG expansion in FXD mice [206] | Overexpression of FAN1 attenuates CAG expansion; FAN1 knockdown increases CAG expansion [158] | Karyomegalic interstitial nephritis [222] |
A pathogenic role for a genome stabilizing DNA repair system in human disease may seem counterintuitive; nevertheless, multiple lines of evidence have converged in recent years to point to MMR as a likely culprit in CAG-repeat expansion. The earliest evidence that MMR genes may promote triplet repeat instability emerged from studies in E. coli, wherein inactivation of the MMR genes mutS, mutL, or mutH resulted in a dramatic reduction of CTG/CAG repeat instability [165, 166]. The mutagenic role of MMR in mammals was conclusively established by work in rodent models of HD and DM1 wherein inactivation of MMR strongly attenuated both somatic expansion of the CAG/CTG repeats in multiple tissues as well as intergenerational repeat expansion (although it is unclear whether somatic and intergenerational expansions are mediated by the same molecular mechanism(s)) [79, 80, 167–177]. Germline knockout of either of the MutS homologs Msh2 or Msh3 or the MutL homologs Mlh1 or Mlh3 in an Htt Q111 knock-in mouse model of HD blocks somatic CAG expansions in the striatum. Loss of Msh2, Msh3, or Mlh1 also reduces nuclear accumulation of mHTT protein, suggesting not only that CAG somatic expansions are associated with some aspects of mHTT pathology, but also that inactivation of MMR can mitigate such disease-associated signatures [80, 170, 175, 177]. Similarly, Msh2, Msh3, and Pms2 have been shown to drive CTG-repeat expansion in mouse models of DM1, suggesting that MutSβ and MutLα play a role in this process [79, 169, 171, 172, 176]. Interestingly, studies in a mouse model of FXD has identified Msh3 as a driver of somatic CGG repeat expansion [144].
Although the MMR system drives CAG repeat expansion, the overall increase in CAG repeat number may be a net consequence of individual expansion and contraction events. This type of expansion “biased” instability has been documented in the blood of DM1 patients [178, 179]. Bidirectional CAG somatic and intergenerational instability has also been observed in both DM1 and HD mouse models, with longer inherited CAG or CTG repeat tracts subject to higher rates of contraction versus expansion [170, 171, 175, 180]. Thus, it seems likely that factors that alter the balance between expansion and contraction may modulate disease onset. In fact, it has been suggested that genetic or pharmacological approaches to induce CAG repeat contractions might hold therapeutic promise [181, 182]. However, the mechanistic role of MMR proteins in controlling the balance between expansion and contraction remains poorly understood.
A striking feature of the tissue-specificity of somatic instability in HD is that the striatum and the cortex (the most severely affected parts of the CNS in HD patients) display high levels of CAG expansions; by contrast, CAG repeats are relatively stable in the cerebellum, which is pathologically unaffected [150, 151, 183]. In the periphery, CAG repeat expansions are observed in the liver, although this tissue does not display disease pathology. The simplest interpretation of these findings is that (a) the cellular milieu of striatal medium spiny neurons is more permissive to MMR-dependent CAG repeat expansion than the environment within cerebellar Purkinje or granule cells, and (b) cell vulnerability likely involves tissue- or cell-type specific factors that may render the striatum more susceptible to the consequence of CAG expansion than the liver. Since medium spiny neurons and Purkinje cells are both post-mitotic, differences in CAG instability between these cell types must arise due to factors other than cell division. It has been suggested that transcriptionally active genes are subject to higher levels of MMR than silent genes or non-transcribed regions of the genome [122, 184, 185]. In fact, transcription-mediated destabilization of CAG and CTG repeats has also been documented in in vitro systems [186–188], and a small-molecule that induces CAG repeat contractions in a transcription-dependent manner has been reported [182]. Thus, it is possible that locus-specific CAG repeat instability may be transcription-dependent. In summary, studies of MMR in different neuronal sub-types and its interface with transcription in these cells is likely to shed light on the possible mechanisms underlying CAG repeat instability in humans.
Mechanisms of MMR-mediated CAG repeat expansion
It has been known for over half a century that the two DNA strands within repetitive DNA elements have a high propensity to slip relative to one another, and that slippage-mediated mispairing of the DNA strands can give rise to changes in repeat tract length [30, 189, 190]. The probability of strand slippage is governed by the sequence composition as well as the number of repeating units within the repetitive element, meaning that a vital determinant of mutagenic propensity is encoded within the DNA sequence itself. An example of this phenomenon is the tendency of the CAG/CTG repeat in exon1 of the HTT gene to undergo length changes (thereby driving HD) by mechanisms presumed to involve strand slippage within the repeat tract. This results in the formation of extrahelical extrusions composed of CAG or CTG repeats, structures that are natural substrates for MutSβ. Studies from several laboratories have established that (CAG)1,2,3,4,7,or13 or (CTG)1,2,3,4,or13 extrahelical extrusions are recognized by MutSβ with high affinity in vitro, with reported dissociation constants ranging from 4–35 nM [51, 116, 173, 191]. Because MutSβ recognizes non-triplet repeat extrahelical extrusions with similar affinities, it is generally believed that the structure of the extrusion (rather than its sequence composition) governs the protein-DNA interaction [50, 51, 67, 173].
The general consensus is that extrahelical extrusions composed of 5 or more CTG or CAG units are rectified efficiently by a MMR-independent mechanism (as judged by robust repair of such extrusions by extracts of cells deficient in MSH2 or MLH1) [192–194]. These findings are consistent with previous observations that rectification of large loops (>12 nucleotides) in human cells is independent of canonical mismatch repair, although the molecular mechanisms governing these events have not been fully clarified [195–198]. By contrast, small extrahelical loops (1–4 triplet repeats) are not only high affinity substrates for MutSβ binding but are also subject to robust rectification by the MMR system in cell extracts, and repair activity is dependent on MutSβ [116, 199, 200]. Interestingly, repair of CAG and CTG extrusions also requires an MLH1-containing heterodimer since the reaction is not supported by extracts prepared from cells lacking MLH1 [116, 200]. Repair activity can be restored to MLH1-deficient extracts by supplementation with purified recombinant MutLα [116], indicating that this heterodimer suffices for this purpose. Furthermore, the inability of PMS2-deficient HEC-1A cell extracts to support repair of a (CTG)1 extrusion suggests that a PMS2-containing heterodimer is required for this process [200]. The inability of a PMS2 endonuclease-inactive MutLα derivative to restore (CAG)2 loop repair capacity to MLH1-deficient extract demonstrates that MutLα plays a catalytic role in extrusion repair, and that the strand incisions it catalyzes are critical intermediates in the MutSβ-dependent processing of small triplet repeat loops [116]. Thus, although CAG hairpins harboring 5 or more repeats are routinely invoked as hypothetical substrates that initiate repeat expansion, available evidence suggests that such hairpins are refractory to the MMR pathway. Because shorter CAG or CTG extrusions (1–4 repeats) not only trigger recognition by the MMR pathway, but also display preferential processing by a MutSβ- (rather than a MutSα-) initiated repair process, such extrahelical extrusions are the presumptive substrates that drive somatic CAG expansions in human cells.
As noted earlier, activation of the MutLα endonuclease on bubble structures occurs without strand bias, presumably due to the disoriented loading of PCNA onto the DNA. Interestingly, triplet repeat extrahelical extrusions behave in much the same way as bubble structures. PCNA is loaded by RFC efficiently on DNAs harboring a (CTG)1–3 or (CAG)1–3 extrahelical extrusion, and in the presence of MutSβ, MutLα, and ATP, strand incision activity is directed to both DNA strands [116]. Thus, these extrahelical extrusions (although restricted to one strand, and lacking a juxtaposed complementary strand extrusion) not only provoke their recognition by MutSβ, but also dysregulate the strand-directionality of the MutLα endonuclease. Such MutLα catalyzed non-strand-specific breaks stimulates DNA synthetic activity on both DNA strands in cell extracts [116], opening up the possibility for further strand-slippage and expansion. Because these events are not coupled to DNA replication, the “dysregulated strand directionality” model (Fig. 2) has been suggested to play a role in CAG-repeat expansion in post-mitotic neurons in the striatum [26, 116]. In this model, expansions/contractions occur either due to the repair of double-strand breaks caused by MutLα-catalyzed incisions in close proximity to one another on the two strands (not shown), or during DNA re-synthesis of the gaps generated upon excision of the nicked strand. Whereas faithful resynthesis by Polδ results in a contraction (not shown), strand-slippage during error-prone resynthesis by Polβ or Polη results in an expansion. Alternatively, expansion can occur by inclusion of an extrahelical extrusion into the primary structure of the DNA. Overall expansion may be a net result of individual expansion and contraction events, as has been suggested to occur in both HD and DM1 [178, 179]. In fact, there is evidence that recruitment of DNA polymerase β (Polβ) by MutSβ to CAG or CTG extrusions triggers error-prone DNA resynthesis in vitro, resulting in expansions [201]. It has also been suggested that the MutSβ-Polβ complex may promote CAG-repeat expansion during base excision repair (BER) [202, 203], although the interplay between the MMR and BER pathways in CAG-repeat expansion remains to be fully dissected [26]. Also, given the limited processivity of Polβ (1–6 nucleotides) [204], it is unclear as to how it might carry out error-prone synthesis of disease-relevant CAG repeat tracts. Error-prone DNA synthesis has also been invoked to explain non-canonical MMR-mediated mutagenesis [23]. This process has been suggested to involve processive reiterative DNA synthesis of long triplet repeat tracts by Polη. However, the involvement of Polη in CAG strand slippage is yet to be experimentally validated.
An alternate model (Fig. 2) for CAG expansion has invoked PCNA-independent activation of the MutLγ endonuclease by MutSβ on CAG extrahelical extrusions. Whereas PCNA-directed MutLα endonuclease activity on such molecules produces nicks on both DNA strands [116], the MutLγ-catalyzed incisions are restricted to the strand complementary to the extrusion. Based on these observations, it has been proposed that DNA re-synthesis induced by the strand-break result in retention of the extrusion, leading to a net expansion of 1 CAG unit [91].
These models provide a mechanistic framework to understand the role of MutLα and MutLγ in CAG-repeat expansion; nevertheless, the GWAS have also suggested a role for PMS1 and FAN1 in modulating the process. Repeat instability measurements in cell lines have established that overexpression of wild type (but not catalytically inactive) FAN1 attenuates CAG-repeat expansion [158] (considered in detail elsewhere in this issue). Recent studies have shown that CAG somatic expansions are enhanced in an Htt Q111 knock-in mouse model of HD that also lacks Fan1 [205]. These instabilities are abolished by the additional knockout of Mlh1, suggesting that CAG expansions in the absence of Fan1 require functional Mlh1 [205]. Interestingly, knockout of Fan1 also exacerbates CGG repeat expansion in a mouse model of FXD [206]. These findings suggest a generalized protective role for FAN1 in repeat expansion, although the mechanistic bases remain unclear. Nevertheless, a physical interaction between FAN1 and MLH1 has been documented, and this interaction has been suggested to play a role in the DNA damage response [108, 207–209]. Also, it has been suggested that FAN1 binding to CAG extrusions may block access of MutSβ such structures, thus preventing CAG expansion [158, 210]. However, the functional relevance of these interactions either in the context of the error correction function of MMR or its mutagenic role in CAG expansion remains to be elucidated. Likewise, there is currently no data available on the role of PMS1 in modulating CAG repeat expansion. Future mechanistic studies evaluating the role of both FAN1 and PMS1 will be required before a mechanistic understanding accounting for all the implicated activities can be constructed.
CONCLUSION
Implications for therapeutic interventions in HD and/or other triplet repeat diseases
The convergence of evidence from human genetics, mouse models, and biochemical studies has highlighted the MMR pathway as a potential target for therapeutic intervention in HD. Because MutSβ-initiated processing of CAG and/or CTG extrusions by MutL homologs is the presumptive proximal step of the somatic expansion process, and since human genetics have provided evidence supporting the idea of somatic CAG expansion as a disease onset driver, inactivation of MutSβ and/or one or more of the MutL homologs is likely to impact disease. Given the wealth of the available structural and biochemical information on the MMR proteins, small molecule approaches for attenuation of somatic CAG expansion may be particularly tractable. Alternatively, recent advances in RNA-based gene inactivation approaches also provide hope that this avenue of therapeutically targeting MMR genes may meet with success. Insofar as diseases with such diverse pathophysiologies as HD, DM1, FXD, and Friedreich’s ataxia share related underlying mutational processes involving MMR, targeting this pathway may result in treatments that benefit a wide patient population.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest.
ACKNOWLEDGMENTS
We thank Vickers Burdett, Seung Kwak, Paul Modrich, Ignacio Munoz-Sanjuan, Simon Noble, Thomas Vogt, and Hilary Wilkinson for comments. AP’s research is supported by grants from the NIH (R03 NS114976), Hereditary Disease Foundation, and the Gies Foundation.
REFERENCES
[1] | Friedberg EC , Walker GC , Siede W , Wood RD , Schultz RA , Ellenberger T . DNA Repair and Mutagenesis. 2nd ed. Washington, D.C.: ASM Press; (2006) . |
[2] | Kunkel TA . Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. (2009) ;74: :91–101. |
[3] | Iyer RR , Pluciennik A , Burdett V , Modrich PL . DNA mismatch repair: functions and mechanisms. Chem Rev. (2006) ;106: (2):302–23. |
[4] | Hsieh P , Yamane K . DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. (2008) ;129: (7-8):391–407. |
[5] | Jiricny J . Postreplicative mismatch repair. Cold Spring Harb Perspect Biol. (2013) ;5: (4):a012633. |
[6] | Modrich P . Mechanisms in E. coli and Human Mismatch Repair (Nobel Lecture). Angew Chem Int Ed Engl. (2016) ;55: (30):8490–501. |
[7] | Kunkel TA , Erie DA . Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet.. (2015) ;49: :291–313. |
[8] | Gupta D , Heinen CD . The mismatch repair-dependent DNA damage response: mechanisms and implications. DNA Repair (Amst). (2019) ;78: :60–9. |
[9] | Li Z , Pearlman AH , Hsieh P . DNA mismatch repair and the DNA damage response. DNA Repair (Amst). (2016) ;38: :94–101. |
[10] | Spies M , Fishel R . Mismatch repair during homologous and homeologous recombination. Cold Spring Harb Perspect Biol. (2015) ;7: (3):a022657. |
[11] | Giraud A , Matic I , Tenaillon O , Clara A , Radman M , Fons M , et al. Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science. (2001) ;291: (5513):2606–8. |
[12] | Denamur E , Matic I . Evolution of mutation rates in bacteria. Mol Microbiol. (2006) ;60: (4):820–7. |
[13] | Peltomaki P . Update on Lynch syndrome genomics. Fam Cancer. (2016) ;15: (3):385–93. |
[14] | Cerretelli G , Ager A , Arends MJ , Frayling IM . Molecular pathology of Lynch syndrome. J Pathol. (2020) ;250: (5):518–31. |
[15] | Wimmer K , Etzler J . Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet ((2008) ;124: (2):105–22. |
[16] | Sijmons RH , Hofstra RMW . Review: Clinical aspects of hereditary DNA mismatch repair gene mutations. DNA Repair (Amst). (2016) ;38: :155–62. |
[17] | Abedalthagafi M . Constitutional mismatch repair-deficiency: current problems and emerging therapeutic strategies. Oncotarget. (2018) ;9: (83):35458–69. |
[18] | Peltomaki P . Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. (2003) ;21: (6):1174–9. |
[19] | Bever KM , Le DT . DNA repair defects and implications for immunotherapy. J Clin Invest. (2018) ;128: (10):4236–42. |
[20] | Zanotti KJ , Gearhart PJ . Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair (Amst). (2016) ;38: :110–6. |
[21] | Peled JU , Kuang FL , Iglesias-Ussel MD , Roa S , Kalis SL , Goodman MF , et al. The biochemistry of somatic hypermutation. Annu Rev Immunol. (2008) ;26: :481–511. |
[22] | Di Noia JM , Neuberger MS . Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. (2007) ;76: :1–22. |
[23] | Pena-Diaz J , Bregenhorn S , Ghodgaonkar M , Follonier C , Artola-Boran M , Castor D , et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. (2012) ;47: (5):669–80. |
[24] | Rodriguez GP , Romanova NV , Bao G , Rouf NC , Kow YW , Crouse GF . Mismatch repair-dependent mutagenesis in nondividing cells. Proc Natl Acad Sci U S A. (2012) ;109: (16):6153–8. |
[25] | Crouse GF . Non-canonical actions of mismatch repair. DNA Repair (Amst). (2016) ;38: :102–9. |
[26] | Iyer RR , Pluciennik A , Napierala M , Wells RD . DNA triplet repeat expansion and mismatch repair. Annu Rev Biochem. (2015) ;84: :199–226. |
[27] | Jones L , Houlden H , Tabrizi SJ . DNA repair in the trinucleotide repeat disorders. Lancet Neurol.. (2017) ;16: (1):88–96. |
[28] | Massey TH , Jones L . The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. (2018) ;11: (1):dmm031930. |
[29] | Fishel R . Mismatch repair. J Biol Chem. (2015) ;290: (44):26395–403. |
[30] | Streisinger G , Okada Y , Emrich J , Newton J , Tsugita A , Terzaghi E , et al. Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb Symp Quant Biol. (1966) ;31: :77–84. |
[31] | Sinden RR , Wells RD . DNA structure, mutations, and human genetic disease. Curr Opin Biotechnol. (1992) ;3: (6):612–22. |
[32] | Levinson G , Gutman GA . Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. (1987) ;4: (3):203–21. |
[33] | Sinden RR , Pytlos-Sinden MJ , Potaman VN . Slipped strand DNA structures. Front Biosci. (2007) ;12: :4788–99. |
[34] | Corless S , Gilbert N . Effects of DNA supercoiling on chromatin architecture. Biophys Rev. (2016) ;8: (3):245–58. |
[35] | Skarstad K , Baker TA , Kornberg A . Strand separation required for initiation of replication at the chromosomal origin of E. coli is facilitated by a distant RNA–DNA hybrid. EMBO J. (1990) ;9: (7):2341–8. |
[36] | Baker TA , Kornberg A . Transcriptional activation of initiation of replication from the E. coli chromosomal origin: an RNA-DNA hybrid near oriC. Cell. (1988) ;55: (1):113–23. |
[37] | Bacolla A , Gellibolian R , Shimizu M , Amirhaeri S , Kang S , Ohshima K , et al. Flexible DNA: genetically unstable CTG. CAG and CGG.CCG from human hereditary neuromuscular disease genes. J Biol Chem. (1997) ;272: (27):16783–92. |
[38] | Wang YH , Amirhaeri S , Kang S , Wells RD , Griffith JD . Preferential nucleosome assembly at DNA triplet repeats from the myotonic dystrophy gene. Science. (1994) ;265: (5172):669–71. |
[39] | Pearson CE , Sinden RR . Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry. (1996) ;35: (15):5041–53. |
[40] | Axford MM , Wang YH , Nakamori M , Zannis-Hadjopoulos M , Thornton CA , Pearson CE . Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLoS Genet. (2013) ;9: (12):e1003866. |
[41] | Gupta S , Gellert M , Yang W . Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops. Nat Struct Mol Biol. (2012) ;19: (1):72–8. |
[42] | Warren JJ , Pohlhaus TJ , Changela A , Iyer RR , Modrich PL , Beese LS . Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. (2007) ;26: (4):579–92. |
[43] | Lamers MH , Perrakis A , Enzlin JH , Winterwerp HH , de Wind N , Sixma TK . The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature. (2000) ;407: (6805):711–7. |
[44] | Obmolova G , Ban C , Hsieh P , Yang W . Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. (2000) ;407: (6805):703–10. |
[45] | Valle L , de Voer RM , Goldberg Y , Sjursen W , Forsti A , Ruiz-Ponte C , et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol Aspects Med. (2019) ;69: :10–26. |
[46] | Reitmair AH , Schmits R , Ewel A , Bapat B , Redston M , Mitri A , et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. (1995) ;11: (1):64–70. |
[47] | de Wind N , Dekker M , Berns A , Radman M , te Riele H . Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. (1995) ;82: (2):321–30. |
[48] | Edelmann W , Yang K , Umar A , Heyer J , Lau K , Fan K , et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. (1997) ;91: (4):467–77. |
[49] | de Wind N , Dekker M , Claij N , Jansen L , van Klink Y , Radman M , et al. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet. (1999) ;23: (3):359–62. |
[50] | Wilson T , Guerrette S , Fishel R . Dissociation of mismatch recognition and ATPase activity by hMSH2-hMSH3. J Biol Chem. (1999) ;274: (31):21659–64. |
[51] | Tian L , Gu L , Li GM . Distinct nucleotide binding/hydrolysis properties and molar ratio of MutSalpha and MutSbeta determine their differential mismatch binding activities. J Biol Chem. (2009) ;284: (17):11557–62. |
[52] | Tian L , Hou C , Tian K , Holcomb NC , Gu L , Li GM . Mismatch recognition protein MutSbeta does not hijack (CAG)n hairpin repair in vitro. J Biol Chem. . (2009) ;284: (31):20452–6. |
[53] | Gradia S , Acharya S , Fishel R . The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. (1997) ;91: (7):995–1005. |
[54] | Blackwell LJ , Bjornson KP , Modrich P . DNA-dependent activation of the hMutSalpha ATPase. J Biol Chem. (1998) ;273: (48):32049–54. |
[55] | Hess MT , Gupta RD , Kolodner RD . Dominant Saccharomyces cerevisiae msh6 mutations cause increased mispair binding and decreased dissociation from mispairs by Msh2-Msh6 in the presence of ATP. J Biol Chem. (2002) ;277: (28):25545–53. |
[56] | Wu TH , Marinus MG . Dominant negative mutator mutations in the mutS gene of Escherichia coli. J Bacteriol. (1994) ;176: (17):5393–400. |
[57] | Dutta R , Inouye M . GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. (2000) ;25: (1):24–8. |
[58] | Guarne A , Junop MS , Yang W . Structure and function of the N-terminal 40 kDa fragment of human PMS a monomeric GHL ATPase. EMBO J. (2001) ;20: (19):5521–31. |
[59] | Arana ME , Holmes SF , Fortune JM , Moon AF , Pedersen LC , Kunkel TA . Functional residues on the surface of the N-terminal domain of yeast Pms1. DNA Repair (Amst). (2010) ;9: (4):448–57. |
[60] | Wu H , Zeng H , Lam R , Tempel W , Kerr ID , Min J . Structure of the human MLH1 N-terminus: implications for predisposition to Lynch syndrome. Acta Crystallogr F Struct Biol Commun. (2015) ;71: (Pt 8):981–5. |
[61] | Spampinato C , Modrich P . The MutL ATPase is required for mismatch repair. J Biol Chem. (2000) ;275: (13):9863–9. |
[62] | Avdievich E , Reiss C , Scherer SJ , Zhang Y , Maier SM , Jin B , et al. Distinct effects of the recurrent Mlh1G67R mutation on MMR functions, cancer, and meiosis. Proc Natl Acad Sci U S A. (2008) ;105: (11):4247–52. |
[63] | Johnson JR , Erdeniz N , Nguyen M , Dudley S , Liskay RM . Conservation of functional asymmetry in the mammalian MutLalpha ATPase. DNA Repair (Amst). (2010) ;9: (11):1209–13. |
[64] | Habraken Y , Sung P , Prakash L , Prakash S . ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2-MSH6 and MLH1-PMS1 protein complexes. J Biol Chem. (1998) ;273: (16):9837–41. |
[65] | Blackwell LJ , Wang S , Modrich P . DNA chain length dependence of formation and dynamics of hMutSalpha. hMutLalpha.heteroduplex complexes. J Biol Chem. (2001) ;276: (35):33233–40. |
[66] | Habraken Y , Sung P , Prakash L , Prakash S . Enhancement of MSH2-MSH3-mediated mismatch recognition by the yeast MLH1-PMS1 complex. Curr Biol. (1997) ;7: (10):790–3. |
[67] | Iyer RR , Pluciennik A , Genschel J , Tsai MS , Beese LS , Modrich P . MutLalpha and proliferating cell nuclear antigen share binding sites on MutSbeta. J Biol Chem. (2010) ;285: (15):11730–9. |
[68] | Mendillo ML , Mazur DJ , Kolodner RD . Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem. (2005) ;280: (23):22245–57. |
[69] | Mendillo ML , Hargreaves VV , Jamison JW , Mo AO , Li S , Putnam CD , et al. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc Natl Acad Sci U S A. (2009) ;106: (52):22223–8. |
[70] | Srivatsan A , Bowen N , Kolodner RD . Mispair-specific recruitment of the Mlh1-Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2-Msh3 complex. J Biol Chem. (2014) ;289: (13):9352–64. |
[71] | Baitinger C , Burdett V , Modrich P . Hydrolytically deficient MutS E694A is defective in the MutL-dependent activation of MutH and in the mismatch-dependent assembly of the MutS. MutL.heteroduplex complex. J Biol Chem. (2003) ;278: (49):49505–11. |
[72] | Graham WJt , Putnam CD , Kolodner RD . The properties of Msh2-Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair. J Biol Chem. (2018) ;293: (47):18055–70. |
[73] | Sherrer SM , Penland E , Modrich P . The mutagen and carcinogen cadmium is a high-affinity inhibitor of the zinc-dependent MutLalpha endonuclease. Proc Natl Acad Sci U S A. (2018) ;115: (28):7314–9. |
[74] | Kadyrov FA , Dzantiev L , Constantin N , Modrich P . Endonucleolytic function of MutLalpha in human mismatch repair. Cell. (2006) ;126: (2):297–308. |
[75] | Pluciennik A , Dzantiev L , Iyer RR , Constantin N , Kadyrov FA , Modrich P . PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci U S A. (2010) ;107: (37):16066–71. |
[76] | Genschel J , Kadyrova LY , Iyer RR , Dahal BK , Kadyrov FA , Modrich P . Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLalpha activation and function in mismatch repair. Proc Natl Acad Sci U S A. (2017) ;114: (19):4930–5. |
[77] | Prolla TA , Baker SM , Harris AC , Tsao JL , Yao X , Bronner CE , et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. (1998) ;18: (3):276–9. |
[78] | Miller CJ , Kim GY , Zhao X , Usdin K . All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet. (2020) ;16: (6):e1008902. |
[79] | Gomes-Pereira M , Fortune MT , Ingram L , McAbney JP , Monckton DG . Pms2 is a genetic enhancer of trinucleotide CAG•CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet. (2004) ;13: (16):1815–25. |
[80] | Pinto RM , Dragileva E , Kirby A , Lloret A , Lopez E , St Claire J , et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. (2013) ;9: (10):e1003930. |
[81] | Genschel J , Bazemore LR , Modrich P . Human exonuclease I is required for 5’ and 3’ mismatch repair. J Biol Chem. (2002) ;277: (15):13302–11. |
[82] | Genschel J , Modrich P . Mechanism of 5’-directed excision in human mismatch repair. Mol Cell. (2003) ;12: (5):1077–86. |
[83] | Zhang Y , Yuan F , Presnell SR , Tian K , Gao Y , Tomkinson AE , et al. Reconstitution of 5’-directed human mismatch repair in a purified system. Cell. (2005) ;122: (5):693–705. |
[84] | Genschel J , Modrich P . Functions of MutLalpha, replication protein A (RPA), and HMGB1 in 5’-directed mismatch repair. J Biol Chem. (2009) ;284: (32):21536–44. |
[85] | Dzantiev L , Constantin N , Genschel J , Iyer RR , Burgers PM , Modrich P . A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell. (2004) ;15: (1):31–41. |
[86] | Constantin N , Dzantiev L , Kadyrov FA , Modrich P . Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J Biol Chem. (2005) ;280: (48):39752–61. |
[87] | Kadyrov FA , Genschel J , Fang Y , Penland E , Edelmann W , Modrich P . A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc Natl Acad Sci U S A. (2009) ;106: (21):8495–500. |
[88] | Longley MJ , Pierce AJ , Modrich P . DNA polymerase delta is required for human mismatch repair in vitro. J Biol Chem. (1997) ;272: (16):10917–21. |
[89] | Rogacheva MV , Manhart CM , Chen C , Guarne A , Surtees J , Alani E . Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J Biol Chem. (2014) ;289: (9):5664–73. |
[90] | Ranjha L , Anand R , Cejka P . The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem. (2014) ;289: (9):5674–86. |
[91] | Kadyrova LY , Gujar V , Burdett V , Modrich PL , Kadyrov FA . Human MutLgamma, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc Natl Acad Sci U S A. (2020) ;117: (7):3535–42. |
[92] | Wang TF , Kleckner N , Hunter N . Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci U S A. (1999) ;96: (24):13914–9. |
[93] | Lipkin SM , Moens PB , Wang V , Lenzi M , Shanmugarajah D , Gilgeous A , et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. (2002) ;31: (4):385–90. |
[94] | Santucci-Darmanin S , Neyton S , Lespinasse F , Saunieres A , Gaudray P , Paquis-Flucklinger V . The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum Mol Genet. (2002) ;11: (15):1697–706. |
[95] | Cannavo E , Sanchez A , Anand R , Ranjha L , Hugener J , Adam C , et al. Regulation of the MLH1-MLH3 endonuclease in meiosis. Nature. (2020) ;586: :618–22. |
[96] | Harrington JM , Kolodner RD . Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol Cell Biol. (2007) ;27: (18):6546–54. |
[97] | Flores-Rozas H , Kolodner RD . The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc Natl Acad Sci U S A. (1998) ;95: (21):12404–9. |
[98] | Harfe BD , Minesinger BK , Jinks-Robertson S . Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr Biol. (2000) ;10: (3):145–8. |
[99] | Chen PC , Dudley S , Hagen W , Dizon D , Paxton L , Reichow D , et al. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. (2005) ;65: (19):8662–70. |
[100] | Cannavo E , Marra G , Sabates-Bellver J , Menigatti M , Lipkin SM , Fischer F , et al. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. (2005) ;65: (23):10759–66. |
[101] | Olkinuora A , Nieminen TT , Martensson E , Rohlin A , Ristimaki A , Koskenvuo L , et al. Biallelic germline nonsense variant of MLH3 underlies polyposis predisposition. Genet Med. (2019) ;21: (8):1868–73. |
[102] | Lipkin SM , Wang V , Jacoby R , Banerjee-Basu S , Baxevanis AD , Lynch HT , et al. MLH a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. (2000) ;24: (1):27–35. |
[103] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900 e14. |
[104] | Zhao X , Zhang Y , Wilkins K , Edelmann W , Usdin K . MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. (2018) ;14: (10):e1007719. |
[105] | Raschle M , Marra G , Nystrom-Lahti M , Schar P , Jiricny J . Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J Biol Chem. (1999) ;274: (45):32368–75. |
[106] | Leung WK , Kim JJ , Wu L , Sepulveda JL , Sepulveda AR . Identification of a second MutL DNA mismatch repair complex (hPMS1 and hMLH1) in human epithelial cells. J Biol Chem. (2000) ;275: (21):15728–32. |
[107] | Plotz G , Raedle J , Brieger A , Trojan J , Zeuzem S . hMutSalpha forms an ATP-dependent complex with hMutLalpha and hMutLbeta on DNA. Nucleic Acids Res. (2002) ;30: (3):711–8. |
[108] | Cannavo E , Gerrits B , Marra G , Schlapbach R , Jiricny J . Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J Biol Chem. (2007) ;282: (5):2976–86. |
[109] | Campbell CS , Hombauer H , Srivatsan A , Bowen N , Gries K , Desai A , et al. Mlh2 is an accessory factor for DNA mismatch repair in Saccharomyces cerevisiae. PLoS Genet. (2014) ;10: (5):e1004327. |
[110] | Kawasoe Y , Tsurimoto T , Nakagawa T , Masukata H , Takahashi TS . MutSalpha maintains the mismatch repair capability by inhibiting PCNA unloading. Elife. (2016) ;5: :e15155. |
[111] | Paul Solomon Devakumar LJ , Gaubitz C , Lundblad V , Kelch BA , Kubota T . Effective mismatch repair depends on timely control of PCNA retention on DNA by the Elg1 complex. Nucleic Acids Res. (2019) ;47: (13):6826–41. |
[112] | Wells RD . Non-B DNA conformations, mutagenesis and disease. Trends Biochem Sci. (2007) ;32: (6):271–8. |
[113] | Bacolla A , Wells RD . Non-B DNA conformations as determinants of mutagenesis and human disease. Mol Carcinog. (2009) ;48: (4):273–85. |
[114] | Choi J , Majima T . Conformational changes of non-B DNA. Chem Soc Rev. (2011) ;40: (12):5893–909. |
[115] | Gemayel R , Vinces MD , Legendre M , Verstrepen KJ . Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. (2010) ;44: :445–77. |
[116] | Pluciennik A , Burdett V , Baitinger C , Iyer RR , Shi K , Modrich P . Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLalpha endonuclease activation. Proc Natl Acad Sci U S A. (2013) ;110: (30):12277–82. |
[117] | Bowman GD , O’Donnell M , Kuriyan J . Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. (2004) ;429: (6993):724–30. |
[118] | Pena-Diaz J , Jiricny J . Mammalian mismatch repair: error-free or error-prone? Trends Biochem Sci. (2012) ;37: (5):206–14. |
[119] | Huang Y , Li GM . DNA mismatch repair in the context of chromatin. Cell Biosci. (2020) ;10: :10. |
[120] | Li F , Mao G , Tong D , Huang J , Gu L , Yang W , et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. (2013) ;153: (3):590–600. |
[121] | Fang J , Huang Y , Mao G , Yang S , Rennert G , Gu L , et al. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proc Natl Acad Sci U S A. (2018) ;115: (38):9598–603. |
[122] | Huang Y , Gu L , Li GM . H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J Biol Chem. (2018) ;293: (20):7811–23. |
[123] | Schopf B , Bregenhorn S , Quivy JP , Kadyrov FA , Almouzni G , Jiricny J . Interplay between mismatch repair and chromatin assembly. Proc Natl Acad Sci U S A. (2012) ;109: (6):1895–900. |
[124] | Kadyrova LY , Blanko ER , Kadyrov FA . CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. Proc Natl Acad Sci U S A. (2011) ;108: (7):2753–8. |
[125] | Rodriges Blanko E , Kadyrova LY , Kadyrov FA . DNA mismatch repair interacts with CAF-1- and ASF1A-H3-H4-dependent histone (H3-H4)2 tetramer deposition. J Biol Chem. (2016) ;291: (17):9203–17. |
[126] | Laguri C , Duband-Goulet I , Friedrich N , Axt M , Belin P , Callebaut I , et al. Human mismatch repair protein MSH6 contains a PWWP domain that targets double stranded DNA. Biochemistry. (2008) ;47: (23):6199–207. |
[127] | Wessbecher IM , Brieger A . Phosphorylation meets DNA mismatch repair. DNA Repair (Amst). (2018) ;72: :107–14. |
[128] | Head RJ , Fay MF , Cosgrove L , K YCF , Rundle-Thiele D , Martin JH . Persistence of DNA adducts, hypermutation and acquisition of cellular resistance to alkylating agents in glioblastoma. Cancer Biol Ther. (2017) ;18: (12):917–26. |
[129] | Schmidt MHM , Pearson CE . Disease-associated repeat instability and mismatch repair. DNA Repair (Amst). (2016) ;38: :117–26. |
[130] | Shuen AY , Lanni S , Panigrahi GB , Edwards M , Yu L , Campbell BB , et al. Functional repair assay for the diagnosis of constitutional mismatch repair deficiency from non-neoplastic tissue. J Clin Oncol. (2019) ;37: (6):461–70. |
[131] | Gonitel R , Moffitt H , Sathasivam K , Woodman B , Detloff PJ , Faull RL , et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci U S A. (2008) ;105: (9):3467–72. |
[132] | Marietta C , Palombo F , Gallinari P , Jiricny J , Brooks PJ . Expression of long-patch and short-patch DNA mismatch repair proteins in the embryonic and adult mammalian brain. Brain Res Mol Brain Res. (1998) ;53: (1-2):317–20. |
[133] | Cardozo-Pelaez F , Brooks PJ , Stedeford T , Song S , Sanchez-Ramos J . DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free Radic Biol Med. (2000) ;28: (5):779–85. |
[134] | Tome S , Simard JP , Slean MM , Holt I , Morris GE , Wojciechowicz K , et al. Tissue-specific mismatch repair protein expression: MSH3 is higher than MSH6 in multiple mouse tissues. DNA Repair (Amst). (2013) ;12: (1):46–52. |
[135] | Uhlen M , Fagerberg L , Hallstrom BM , Lindskog C , Oksvold P , Mardinoglu A , et al. Tissue-based map of the human proteome. Science. (2015) ;347: (6220):1260419. |
[136] | Belloni M , Uberti D , Rizzini C , Ferrari-Toninelli G , Rizzonelli P , Jiricny J , et al. Distribution and kainate-mediated induction of the DNA mismatch repair protein MSH2 in rat brain. Neuroscience. (1999) ;94: (4):1323–31. |
[137] | Iyama T , Wilson DM 3rd . DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). (2013) ;12: (8):620–36. |
[138] | Cobley JN , Fiorello ML , Bailey DM . 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. (2018) ;15: :490–503. |
[139] | Madabhushi R , Pan L , Tsai LH . DNA damage and its links to neurodegeneration. Neuron. (2014) ;83: (2):266–82. |
[140] | Rink C , Khanna S . Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid Redox Signal. (2011) ;14: (10):1889–903. |
[141] | Hegde ML , Mantha AK , Hazra TK , Bhakat KK , Mitra S , Szczesny B . Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev. (2012) ;133: (4):157–68. |
[142] | Proukakis C . Somatic mutations in neurodegeneration: An update. Neurobiol Dis. (2020) ;144: :105021. |
[143] | Gu Y , Parker A , Wilson TM , Bai H , Chang DY , Lu AL . Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J Biol Chem. (2002) ;277: (13):11135–42. |
[144] | Zhao XN , Kumari D , Gupta S , Wu D , Evanitsky M , Yang W , et al. Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum Mol Genet. (2015) ;24: (24):7087–96. |
[145] | Halabi A , Ditch S , Wang J , Grabczyk E . DNA mismatch repair complex MutSbeta promotes GAA. TTC repeat expansion in human cells. J Biol Chem. (2012) ;287: (35):29958–67. |
[146] | Halabi A , Fuselier KTB , Grabczyk E . GAA*TTC repeat expansion in human cells is mediated by mismatch repair complex MutLgamma and depends upon the endonuclease domain in MLH3 isoform one. Nucleic Acids Res. (2018) ;46: (8):4022–32. |
[147] | Ezzatizadeh V , Pinto RM , Sandi C , Sandi M , Al-Mahdawi S , Te Riele H , et al. The mismatch repair system protects against intergenerational GAA repeat instability in a Friedreich ataxia mouse model. Neurobiol Dis. (2012) ;46: (1):165–71. |
[148] | Tabrizi SJ , Flower MD , Ross CA , Wild EJ . Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol. (2020) ;16: (10):529–46. |
[149] | Keum JW , Shin A , Gillis T , Mysore JS , Abu Elneel K , Lucente D , et al. The HTT CAG-expansion mutation determines age at death but not disease duration in Huntington disease. Am J Hum Genet. (2016) ;98: (2):287–98. |
[150] | Kennedy L , Evans E , Chen CM , Craven L , Detloff PJ , Ennis M , et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. (2003) ;12: (24):3359–67. |
[151] | Shelbourne PF , Keller-McGandy C , Bi WL , Yoon SR , Dubeau L , Veitch NJ , et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. (2007) ;16: (10):1133–42. |
[152] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) ;6: (4):409–14. |
[153] | Swami M , Hendricks AE , Gillis T , Massood T , Mysore J , Myers RH , et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. (2009) ;18: (16):3039–47. |
[154] | Wright GEB , Collins JA , Kay C , McDonald C , Dolzhenko E , Xia Q , et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet. (2019) ;104: (6):1116–26. |
[155] | Ciosi M , Maxwell A , Cumming SA , Hensman Moss DJ , Alshammari AM , Flower MD , et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. (2019) ;48: :568–80. |
[156] | Findlay Black H , Wright GEB , Collins JA , Caron N , Kay C , Xia Q , et al. Frequency of the loss of CAA interruption in the HTT CAG tract and implications for Huntington disease in the reduced penetrance range. Genet Med. 2020. DOI: 10.1038/s41436-020-0917-z. |
[157] | Coffey SR , Andrew M , Ging H , Hamilton J , Flower M , Kovalenko M , et al. Huntingtin lowering reduces somatic instability at CAG-expanded loci. bioRxiv. 2020. DOI: 10.1101/2020.07.23.218347. |
[158] | Goold R , Flower M , Moss DH , Medway C , Wood-Kaczmar A , Andre R , et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. (2019) ;28: (4):650–61. |
[159] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. |
[160] | Bettencourt C , Hensman-Moss D , Flower M , Wiethoff S , Brice A , Goizet C , et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. (2016) ;79: (6):983–90. |
[161] | Flower M , Lomeikaite V , Ciosi M , Cumming S , Morales F , Lo K , et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain. (2019) ;142: (7):1876–86. |
[162] | Lee JM , Chao MJ , Harold D , Abu Elneel K , Gillis T , Holmans P , et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet. (2017) ;26: (19):3859–67. |
[163] | Hensman Moss DJ , Pardinas AF , Langbehn D , Lo K , Leavitt BR , Roos R , et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. (2017) ;16: (9):701–11. |
[164] | Morales F , Vasquez M , Santamaria C , Cuenca P , Corrales E , Monckton DG . A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst). (2016) ;40: :57–66. |
[165] | Jaworski A , Rosche WA , Gellibolian R , Kang S , Shimizu M , Bowater RP , et al. Mismatch repair in Escherichia coli enhances instability of (CTG)n triplet repeats from human hereditary diseases. Proc Natl Acad Sci U S A. (1995) ;92: (24):11019–23. |
[166] | Wells RD , Parniewski P , Pluciennik A , Bacolla A , Gellibolian R , Jaworski A . Small slipped register genetic instabilities in Escherichia coli in triplet repeat sequences associated with hereditary neurological diseases. J Biol Chem. (1998) ;273: (31):19532–41. |
[167] | Manley K , Shirley TL , Flaherty L , Messer A . Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. (1999) ;23: (4):471–3. |
[168] | Kovtun IV , McMurray CT . Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet. (2001) ;27: (4):407–11. |
[169] | van den Broek WJ , Nelen MR , Wansink DG , Coerwinkel MM , te Riele H , Groenen PJ , et al. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. (2002) ;11: (2):191–8. |
[170] | Wheeler VC , Lebel LA , Vrbanac V , Teed A , te Riele H , MacDonald ME . Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. (2003) ;12: (3):273–81. |
[171] | Savouret C , Brisson E , Essers J , Kanaar R , Pastink A , te Riele H , et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. (2003) ;22: (9):2264–73. |
[172] | Savouret C , Garcia-Cordier C , Megret J , te Riele H , Junien C , Gourdon G . MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol Cell Biol. (2004) ;24: (2):629–37. |
[173] | Owen BA , Yang Z , Lai M , Gajec M , Badger JD 2nd , Hayes JJ , et al. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. (2005) ;12: (8):663–70. |
[174] | Foiry L , Dong L , Savouret C , Hubert L , te Riele H , Junien C , et al. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet. (2006) ;119: (5):520–6. |
[175] | Dragileva E , Hendricks A , Teed A , Gillis T , Lopez ET , Friedberg EC , et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. (2009) ;33: (1):37–47. |
[176] | Tome S , Holt I , Edelmann W , Morris GE , Munnich A , Pearson CE , et al. MSH2 ATPase domain mutation affects CTG•CAG repeat instability in transgenic mice. PLoS Genet. (2009) ;5: (5):e1000482. |
[177] | Kovalenko M , Milnerwood A , Giordano J , St Claire J , Guide JR , Stromberg M , et al. HttQ111/+Huntington’s disease knock-in mice exhibit brain region-specific morphological changes and synaptic dysfunction. J Huntingtons Dis. (2018) ;7: (1):17–33. |
[178] | Higham CF , Monckton DG . Modelling and inference reveal nonlinear length-dependent suppression of somatic instability for small disease associated alleles in myotonic dystrophy type 1 and Huntington disease. J R Soc Interface. (2013) ;10: (88):20130605. |
[179] | Morales F , Vasquez M , Corrales E , Vindas-Smith R , Santamaria-Ulloa C , Zhang B , et al. Longitudinal increases in somatic mosaicism of the expanded CTG repeat in myotonic dystrophy type 1 are associated with variation in age-at-onset. Hum Mol Genet. 2020. |
[180] | Larson E , Fyfe I , Morton AJ , Monckton DG . Age-, tissue- and length-dependent bidirectional somatic CAG•CTG repeat instability in an allelic series of R6/2 Huntington disease mice. Neurobiol Dis. (2015) ;76: :98–111. |
[181] | Cinesi C , Aeschbach L , Yang B , Dion V . Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat Commun. (2016) ;7: :13272. |
[182] | Nakamori M , Panigrahi GB , Lanni S , Gall-Duncan T , Hayakawa H , Tanaka H , et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet. (2020) ;52: (2):146–59. |
[183] | Mouro Pinto R , Arning L , Giordano JV , Razghandi P , Andrew MA , Gillis T , et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet. (2020) ;29: (15):2551–67. |
[184] | Huang Y , Li GM . DNA mismatch repair preferentially safeguards actively transcribed genes. DNA Repair (Amst). (2018) ;71: :82–6. |
[185] | Georgakopoulos-Soares I , Koh G , Momen SE , Jiricny J , Hemberg M , Nik-Zainal S . Transcription-coupled repair and mismatch repair contribute towards preserving genome integrity at mononucleotide repeat tracts. Nat Commun. (2020) ;11: (1):1980. |
[186] | Lin Y , Hubert L Jr , Wilson JH . Transcription destabilizes triplet repeats. Mol Carcinog. (2009) ;48: (4):350–61. |
[187] | Lin Y , Dent SY , Wilson JH , Wells RD , Napierala M . R loops stimulate genetic instability of CTG. CAG repeats. Proc Natl Acad Sci U S A. (2010) ;107: (2):692–7. |
[188] | Lin Y , Wilson JH . Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble. Cell Cycle. (2011) ;10: (4):611–8. |
[189] | Kornberg A , Bertsch LL , Jackson JF , Khorana HG . Enzymatic synthesis of deoxyribonucleic acid, Xvi. Oligonucleotides as templates and the mechanism of their replication. Proc Natl Acad Sci U S A. (1964) ;51: :315–23. |
[190] | Wells RD , Jacob TM , Narang SA , Khorana HG . Studies on polynucleotides. LXIX. Synthetic deoxyribopolynucleotides as templates for the DNA polymerase of Escherichia coli: DNA-like polymers containing repeating trinucleotide sequences. J Mol Biol. (1967) ;27: (2):237–63. |
[191] | Lang WH , Coats JE , Majka J , Hura GL , Lin Y , Rasnik I , et al. Conformational trapping of mismatch recognition complex MSH2/MSH3 on repair-resistant DNA loops. Proc Natl Acad Sci U S A. (2011) ;108: (42):E837–44. |
[192] | Panigrahi GB , Lau R , Montgomery SE , Leonard MR , Pearson CE . Slipped (CTG)•(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat Struct Mol Biol. (2005) ;12: (8):654–62. |
[193] | Hou C , Chan NL , Gu L , Li GM . Incision-dependent and error-free repair of (CAG)(n)/(CTG)(n) hairpins in human cell extracts. Nat Struct Mol Biol. (2009) ;16: (8):869–75. |
[194] | Zhang T , Huang J , Gu L , Li GM . In vitro repair of DNA hairpins containing various numbers of CAG/CTG trinucleotide repeats. DNA Repair (Amst). (2012) ;11: (2):201–9. |
[195] | Genschel J , Littman SJ , Drummond JT , Modrich P . Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. (1998) ;273: (31):19895–901. |
[196] | Littman SJ , Fang WH , Modrich P . Repair of large insertion/deletion heterologies in human nuclear extracts is directed by a 5’ single-strand break and is independent of the mismatch repair system. J Biol Chem. (1999) ;274: (11):7474–81. |
[197] | Umar A , Boyer JC , Kunkel TA . DNA loop repair by human cell extracts. Science. (1994) ;266: (5186):814–6. |
[198] | McCulloch SD , Gu L , Li GM . Nick-dependent and -independent processing of large DNA loops in human cells. J Biol Chem. (2003) ;278: (50):50803–9. |
[199] | Panigrahi GB , Slean MM , Simard JP , Gileadi O , Pearson CE . Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSbeta, but clustered slip-outs are poorly repaired. Proc Natl Acad Sci U S A. (2010) ;107: (28):12593–8. |
[200] | Panigrahi GB , Slean MM , Simard JP , Pearson CE . Human mismatch repair protein hMutLalpha is required to repair short slipped-DNAs of trinucleotide repeats. J Biol Chem. (2012) ;287: (50):41844–50. |
[201] | Guo J , Gu L , Leffak M , Li GM . MutSbeta promotes trinucleotide repeat expansion by recruiting DNA polymerase beta to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res. (2016) ;26: (7):775–86. |
[202] | Lai Y , Budworth H , Beaver JM , Chan NL , Zhang Z , McMurray CT , et al. Crosstalk between MSH2-MSH3 and polbeta promotes trinucleotide repeat expansion during base excision repair. Nat Commun. (2016) ;7: :12465. |
[203] | Kovtun IV , Liu Y , Bjoras M , Klungland A , Wilson SH , McMurray CT . OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. (2007) ;447: (7143):447–52. |
[204] | Beard WA , Wilson SH . DNA polymerase beta and other gap-filling enzymes in mammalian base excision repair. Enzymes. (2019) ;45: :1–26. |
[205] | Loupe JM , Pinto RM , Kim KH , Gillis T , Mysore JS , Andrew MA , et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet. 2020. |
[206] | Zhao XN , Usdin K . FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst). (2018) ;69: :1–5. |
[207] | Smogorzewska A , Desetty R , Saito TT , Schlabach M , Lach FP , Sowa ME , et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell. (2010) ;39: (1):36–47. |
[208] | MacKay C , Declais AC , Lundin C , Agostinho A , Deans AJ , MacArtney TJ , et al. Identification of KIAA/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. (2010) ;142: (1):65–76. |
[209] | Rikitake M , Fujikane R , Obayashi Y , Oka K , Ozaki M , Hidaka M . MLH1-mediated recruitment of FAN1 to chromatin for the induction of apoptosis triggered by O(6)-methylguanine. Genes Cells. (2020) ;25: (3):175–86. |
[210] | Kim KH , Hong EP , Shin JW , Chao MJ , Loupe J , Gillis T , et al. Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am J Hum Genet. (2020) ;107: (1):96–110. |
[211] | Karczewski KJ , Francioli LC , Tiao G , Cummings BB , Alföldi J , Wang Q , et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) ;581: (7809):434–43. |
[212] | Kovtun IV , Thornhill AR , McMurray CT . Somatic deletion events occur during early embryonic development and modify the extent of CAG expansion in subsequent generations. Hum Mol Genet. (2004) ;13: (24):3057–68. |
[213] | Lokanga RA , Zhao XN , Usdin K . The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum Mutat. (2014) ;35: (1):129–36. |
[214] | Du J , Campau E , Soragni E , Jespersen C , Gottesfeld JM . Length-dependent CTG. CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum Mol Genet. (2013) ;22: (25):5276–87. |
[215] | Nakatani R , Nakamori M , Fujimura H , Mochizuki H , Takahashi MP . Large expansion of CTG*CAG repeats is exacerbated by MutSbeta in human cells. Sci Rep. (2015) ;5: :11020. |
[216] | Gannon AM , Frizzell A , Healy E , Lahue RS . MutSbeta and histone deacetylase complexes promote expansions of trinucleotide repeats in human cells. Nucleic Acids Res. (2012) ;40: (20):10324–33. |
[217] | Du J , Campau E , Soragni E , Ku S , Puckett JW , Dervan PB , et al. Role of mismatch repair enzymes in GAA•TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem. (2012) ;287: (35):29861–72. |
[218] | Tome S , Manley K , Simard JP , Clark GW , Slean MM , Swami M , et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. (2013) ;9: (2):e1003280. |
[219] | Tome S , Panigrahi GB , Lopez Castel A , Foiry L , Melton DW , Gourdon G , et al. Maternal germline-specific effect of DNA ligase I on CTG/CAG instability. Hum Mol Genet. (2011) ;20: (11):2131–43. |
[220] | Howes TR , Tomkinson AE . DNA ligase I, the replicative DNA ligase. Subcell Biochem. (2012) ;62: :327–41. |
[221] | Maffucci P , Chavez J , Jurkiw TJ , O’Brien PJ , Abbott JK , Reynolds PR , et al. Biallelic mutations in DNA ligase 1 underlie a spectrum of immune deficiencies. J Clin Invest. (2018) ;128: (12):5489–504. |
[222] | Zhou W , Otto EA , Cluckey A , Airik R , Hurd TW , Chaki M , et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. (2012) ;44: (8):910–5. |


