
DNA Repair in Huntington’s Disease and Spinocerebellar Ataxias: Somatic Instability and Alternative Hypotheses
Abstract
The use of genome wide association studies (GWAS) in Huntington’s disease (HD) research, driven by unbiased human data analysis, has transformed the focus of new targets that could affect age at onset. While there is a significant depth of information on DNA damage repair, with many drugs and drug targets, most of this development has taken place in the context of cancer therapy. DNA damage repair in neurons does not rely on DNA replication correction mechanisms. However, there is a strong connection between DNA repair and neuronal metabolism, mediated by nucleotide salvaging and the poly ADP-ribose (PAR) response, and this connection has been implicated in other age-onset neurodegenerative diseases. Validation of leads including the mismatch repair protein MSH3, and interstrand cross-link repair protein FAN1, suggest the mechanism is driven by somatic CAG instability, which is supported by the protective effect of CAA substitutions in the CAG tract. We currently do not understand: how somatic instability is triggered; the state of DNA damage within expanding alleles in the brain; whether this damage induces mismatch repair and interstrand cross-link pathways; whether instability mediates toxicity, and how this relates to human ageing. We discuss DNA damage pathways uncovered by HD GWAS, known roles of other polyglutamine disease proteins in DNA damage repair, and a panel of hypotheses for pathogenic mechanisms.
HD GWAS BRINGS UNBIASED HYPOTHESES BACK TO HUNTINGTON’S DISEASE
With the 2015 Huntington’s disease (HD) genome-wide association study (GWAS) for genetic modifiers of age at onset [1, 2], HD research once again returned to an unbiased investigation of human genetics to uncover new genes and pathways directly relevant to human disease. Perhaps surprising to the HD research community was that most of the modifiers of disease appeared to be involved in DNA damage repair, mitochondrial health, and redox pathways, and not a single pathway related to protein homeostasis was significant in modifying age at onset in HD. Analysis beyond HD to other polyglutamine diseases revealed a commonality of some modifier genes, suggesting that a common mechanism may exist among polyglutamine diseases at the level of DNA repair [3]. This data was complemented by two studies in which the most profound modifier of disease was the presence of an interrupting CAA codon in HTT at the 3’ end of the CAG expansion [4, 5].
DNA repair pathways identified as disease modifiers from GWAS, such as mismatch repair (MMR), can influence somatic instability of the CAG tract, and this is also possible for the interrupting CAA allele. Subsequent studies have correlated disease progression with somatic instability in HD patient blood [6], and implicated the interstrand crosslink repair protein FAN1 in the mechanism of CAG expansion [7]. However, general DNA damage in various tissue types has also been implicated in HD (reviewed in [8] and [9]). As early as the 1980s, cells from HD patients were observed to be hypersensitive to DNA damaging agents, and accrue more DNA damage [10–12]. Elevated DNA damage levels were later corroborated in fibroblasts [13] and peripheral blood mononuclear cells [14, 15] from HD patients, and have been observed in HD mouse models [16, 17]. The fact that DNA damage is observed in pre-manifest individuals [15], and precedes mitochondrial dysfunction in patient cells [14] and aggregate formation in a mouse model [17], suggests that DNA damage is not a downstream consequence, but may contribute to pathogenesis. It is therefore unclear if the DNA repair pathways implicated in HD cause generalized damage before or after increased somatic expansion, or to what extent each damaging effect contributes to disease progression [8, 9, 18]. If reducing somatic expansion ameliorates disease, this would suggest that somatic expansion has a key role in disease pathology. Naphthyridine-azaquinolone was found to specifically bind and stabilize secondary CAG DNA structures, and induced contractions of the expanded repeat through transcription-dependent repair, not replication [19]. The specificity and efficacy of this molecule was important to establish that repeat contraction is possible as a therapeutic mechanism, but it is yet unclear if the repeat-contracting effect is beneficial to disease progression. What also remains unknown is the pathogenic mechanism of somatic repeat expansion. In theory, this would cause the production of very large, highly toxic protein, but the difficulty associated with measuring protein size in the cells bearing repeat expansions has precluded the detection of such protein. In practice, increased polyglutamine length severely reduces huntingtin expression levels [20], which could point to haploinsufficiency or loss of function as the pathogenic driver.
Somatic CAG tract instability is an exciting and validated course of investigation, and has been reviewed elsewhere [21, 22]. The purpose of this review is to discuss alternate hypotheses, as well as the importance of neuronal metabolism in the context of defective DNA damage repair.
CAA INTERRUPTIONS IN THE CAG EXPANDED ALLELE
Two studies reported that the loss of an interrupting CAA codon at the end of the HTT CAG tract hastens disease progression, while individuals with duplicate interrupting alleles had much later disease onset than expected based on their CAG length [4, 5]. The 2019 GeM-HD study showed that mis-calling of the pure CAG length resulted in the strong chromosome 4 HTT signal, which was significantly reduced after accounting for the length of the pure CAG tract. Thus, pure CAG length is critical in determining onset. The CAA codon, like CAG, is translated to a glutamine residue, supporting the hypothesis that DNA content in the HTT exon1 expansion is more important than the actual glutamine tract length in the protein. While loss of the interrupting codon can influence somatic instability at the DNA level [4, 23], and this is consistent with a possible pathogenic mechanism [4–6], the role of the interruptions on instability remains unclear.
The protective effect of the CAA codon could also be a result of reduced RNA toxicity, or slower translation rates resulting in improved co-translational protein folding. RNA from expanded HTT exon 1 shows less cellular toxicity when CAA codons are used rather than CAG codons in human cell lines [24], and CAA interruptions may beneficially modify transcript folding [25]. At the protein level, altered products from repeat-associated non-ATG (RAN) translation [26] of interrupted alleles may be less toxic than their uninterrupted counterparts. Synonymous codons do not have the same ribosomal residency time during translation [27]. Optimal codons appear more frequently within a genome and are translated faster than non-optimal codons, which allows species to fine-tune translation rates to achieve accurate co-translational folding [28, 29]. In humans, CAG is the optimal codon for glutamine, suggesting CAA codons are translated more slowly [30]. Thus, despite the same number of glutamine residues in the protein, the presence of a CAA codon may lead to a slower translation rate, hence different folding, than protein translated from a pure CAG allele. This suggests two different protein structures may be generated despite the similar genetic code. The 2018 cryo-EM structure of the huntingtin/HAP40 complex [31] highlighted the role of co-translational folding of huntingtin, as independently purified HAP40 and huntingtin cannot form a complex when mixed, and need to be co-expressed and co-translated [20, 31]. Regardless of mechanism, the CAA allele data uncovered a historic gap in clinical information in HD that was already known to be important with CAT interruptions in spinocerebellar ataxia type 1 [32]; that DNA sequence information is important, not just DNA expansion length defined by PCR-based assays.
INTERSTRAND CROSSLINK REPAIR
The FAN1 gene was identified as one of the most significant modifiers of HD and spinocerebellar ataxia onset [1, 5]. Expression of the protein product, FAN1, is associated with delayed age at onset, and this has been attributed to the strong stabilizing effect of FAN1 on CAG tract somatic instability [6, 7].
In addition to its effect on somatic instability, FAN1 is known for its role in repairing cytotoxic interstrand crosslinks. In dividing cells, these lesions result in stalled replication forks, which are resolved by FAN1 in concert with proteins of the Fanconi anemia pathway [33]. In non-dividing cells, interstrand crosslinks must be recognized by either RNA polymerase collisions during transcription, or simply by their distortion of the DNA double helix [33, 34]. While Fanconi anemia proteins are dispensable for this replication-independent repair, mismatch repair and nucleotide excision repair proteins have been implicated [35]. A variety of in vitro and in vivo assays for interstrand crosslink repair have revealed the involvement of mismatch repair proteins such as MutSα (MSH2-MSH6), MutLα (MLH1-PMS2) and EXO1 [36], and their cooperation with nucleotide excision repair proteins [37, 38]. This is of note because both mismatch and nucleotide excision repair pathways were highlighted by GWAS pathway analysis. In much the same way that these functional associations are likely to play a role in somatic instability, they support a role for interstrand crosslink repair as a possible disease-modifying pathway in HD and spinocerebellar ataxias.
Another disease modifier identified by GWAS was the ribonucleotide reductase subunit RRM2B. Like huntingtin, RRM2B is transcriptionally activated by a regulator of DNA repair, the tumor suppressor TP53 [39]. Ribonucleotide reductase activity has been implicated in resolving interstrand crosslinks [40], which suggests that RRM2B could also work through this pathway. The possible role of interstrand crosslink repair may be linked to the DNA damage signaling cytokine, N6-furfuryladenine (N6FFA), which we previously identified as a therapeutic lead [9, 41]. N6FFA adducts, which occur naturally upon oxidative damage of DNA and must be repaired by excision [42], can generate crosslinks in the presence of reactive oxygen species (ROS) [43]. It is therefore possible that suboptimal excision of N6FFA increases the number of interstrand crosslinks, contributing to the mechanism of FAN1 and RRM2B disease modification in HD and spinocerebellar ataxias.
NUCLEOTIDE HOMEOSTASIS PATHWAYS IN NEURONS
Certain GWAS hits may also be considered through the lens of nucleotide homeostasis. The repair of damaged DNA is dependent on the availability of nucleotide building blocks and ATP, which must be synthesized de novo, or recycled from intermediates generated by DNA and RNA turnover in a process known as nucleotide salvaging [44]. The important nucleotide biosynthesis gene, DHFR, shares a promoter with MSH3, and was implicated by GWAS hits on chromosome 5. Notably, SNPs tagging one of the modifier haplotypes on chromosome 5 was associated with altered DHFR expression [5], suggesting that this gene may, in addition to MSH3 at this locus, modify HD onset.
While de novo synthesis is particularly important in dividing cells to support DNA and protein synthesis throughout the cell cycle [45], nucleotide salvage pathways are more energy efficient than de novo pathways and can help when energy demands are high [44], as is the case in neurons [46]. The brain has limited ability to generate sufficient purine and pyrimidine bases through de novo synthesis, and the nervous system becomes more reliant on salvaging into adulthood [47]. Nucleotide salvaging is therefore critical for maintaining nucleotide pools in non-dividing neuronal cells [48]. This process is particularly important in transient high-energy demand situations, such as upon DNA damage, which triggers a need for increased nucleotide triphosphate levels for DNA repair mechanisms. RRM2B catalyzes the conversion of ribonucleoside diphosphates into corresponding deoxynucleoside diphosphates necessary for DNA synthesis during cell division and DNA repair [49]. Thus, as a nucleotide salvaging protein that is induced specifically in response to DNA damage, RRM2B may mediate its disease-modifying effect through this pathway. Of note, mutations in RRM2B are the cause of mitochondrial DNA depletion syndrome, a severe condition characterized by brain dysfunction and muscle weakness (encephalomyopathy), in which defective maintenance and repair of mitochondrial DNA is thought to be associated with dysregulation of the mitochondrial nucleotide pool [50].
The link between DNA repair and nucleotide metabolism also extends to poly ADP-ribose (PAR) signalling. The production of poly ADP-ribose (PAR) from nicotinamide adenine dinucleotide (NAD+) by poly ADP-ribose polymerases (PARPs) is activated by binding of PARP to DNA lesions during DNA damage repair [51, 52]. PAR signalling primarily involves PARylation, a post-translational modification where PARP mediates the transfer of PAR onto proteins. PAR chains act as a scaffold for recruiting DNA repair factors, facilitating detection of DNA damage. Due to the high energy requirements for producing PAR by indirect depletion of ATP levels, rapid turnover and degradation by PAR glycohydrolase (PARG) is required to redistribute NAD+ metabolites. If PAR signalling persists, the depletion of NAD+ leads to necrosis [53] and PAR can act as a cell death signalling molecule for the caspase-free form of apoptosis, termed parthanatos [54, 55]. Although the PAR signalling pathway was not identified by GWAS, PAR dysregulation has been noted in several neurodegenerative diseases, including increased PARP1 activity in cerebellar ataxia [56], increased PAR immunostaining of frontal and temporal cortices in Alzheimer’s disease [57], in the motor neurons of spinal cord sections in amyotrophic lateral sclerosis [58], and elevated PAR levels in cerebrospinal fluid in Parkinson’s disease [59]. The PARP inhibitor INO-1001 was beneficial in the R6/2 mouse model [60], having a neuroprotective effect in striatal interneurons [61].
Neuronal energy production is dependent on oxidative phosphorylation and efficient mitochondrial function. Increased oxidative stress and deficiencies in mitochondrial function have been implicated in HD and several other neurodegenerative disorders [62–66]. Hypoxanthine-guanine phosphoribosyl transferase (HPGRT), encoded by HPRT1, is a key component of the purine nucleotide salvage pathway [67]. Mutations in HPRT1 can lead to X-linked Lesch-Nyhan syndrome, a neurologic disease defined by low levels of GTP, defective dopaminergic pathways in the brain, and defective DNA damage repair [68]. Analysis of sex chromosomes, which was omitted in HD GWAS, may provide further insight into the importance of nucleotide salvagers such as HPGRT.
Fig. 1
Overview of the potential mechanisms presented in this review.
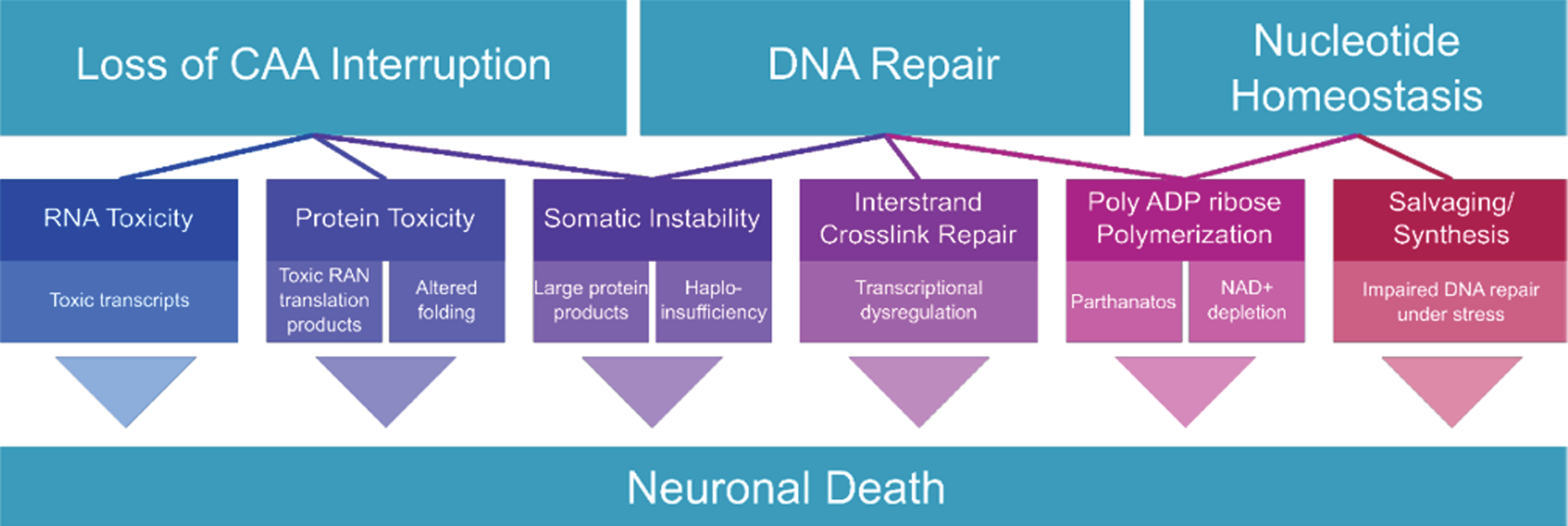
DNA DAMAGE REPAIR IN OTHER POLYGLUTAMINE DISEASES
DNA damage repair gene SNPs in HD have also been found to significantly impact age at symptom onset in a related group of CAG triplet repeat disorders, the spinocerebellar ataxias (SCAs) [3]. This suggests that CAG triplet repeat disorders could share a common pathogenic mechanism of disease rooted in DNA repair [3, 9].
Mirroring the situation with HD, somatic instability has also been observed in SCAs. Somatic expansion of alleles has been measured in both the central nervous system or periphery of SCA1, SCA2, SCA3, SCA7, and SCA17 patients [69–74]. The presence of CAA interruptions within CAG tracts of the ataxin-2 gene (ATXN2) and TATA box binding protein gene (TBP) appear to have a stabilizing effect, similar to that seen in HTT [6, 70, 75].
Both HD and SCA patients have similar somatic instability mosaicism within brain tissues, with large allele expansions within the basal ganglia and striatum, and shorter expansions within the cerebellum [76–78]. Although this tissue-specific somatic expansion correlates with areas of neurodegeneration in HD, it does not with SCAs. Samples from SCA1, SCA2, SCA3 patient cerebellar cortices have contraction of CAG-containing alleles [72, 79, 80], with similar trends found in SCA7 transgenic mouse cerebellum samples [81]. It is unclear why this trend is observed across multiple spinocerebellar ataxias. One potential explanation is that somatic instability in Purkinje cells, the vulnerable population in SCAs, may escape detection. Since Purkinje cells are highly sensitive to any form of stress, and may be unable to compensate for the additional stress caused by somatic expansion, they may be lost prior to assessment of instability. The scarcity of this cell type also makes it difficult to detect when measuring instability in the cerebellum as a whole. Although somatic instability is a shared phenomenon across CAG triplet repeat diseases, the disconnect between areas of somatic expansion and neurodegeneration suggests that it is not likely the sole mechanism driving pathology across polyglutamine diseases.
Aside from repeat instability, additional aspects of DNA integrity pathways have been implicated in the polyglutamine diseases. Evidence has emerged tying DNA damage repair and oxidative stress to forms of SCA. Overexpression of DNA repair proteins HMGB1 and RPA1 improves the motor phenotype of SCA1 drosophila and mouse models [82, 83]. Knockdown of wild-type ataxin-2, the affected protein in SCA2, results in increased double-stranded break damage and RNA-DNA R-loop formation [84]. The SCA3 protein, ataxin-3, participates in transcription-coupled DNA repair, and is also implicated in double-stranded break repair and nonhomologous end-joining [85–87]. Oxidative damage to proteins and lipids correlates with disease severity and progression in SCA7 [88] and ataxin-7 is a member of the STAGA transcription complex which can interact with DNA repair factors [89]. The TATA box-binding protein, TBP, which has an expanded CAG tract in SCA17, preferentially binds cisplatin- and UV-damaged DNA sequences over TATA box sequences [90, 91]. The androgen receptor, expanded in spinal and bulbar muscle atrophy disease, is known to signal DNA damage repair factors for double-strand break repair [92, 93]. Thus, out of the nine polyglutamine disease proteins, seven have some known functional role in DNA damage repair. We anticipate additional findings to emerge as the SCA research field further investigates the connection between DNA repair genes and age at onset of ataxia symptoms.
DNA REPAIR AND CAG EXPANSION TARGETS AND TIMING ADMINISTRATION OF FUTURE THERAPIES
GWAS has revealed pathways and hit proteins that are being investigated and validated for therapeutic potential. While directly targeting these hits may not be practical (for example, increasing expression of FAN1), critical signaling nodes that affect age at onset will be important to pursue. Understanding how genes, proteins, and pathways identified by GWAS are regulated will reveal additional targets that may be more amenable to drug intervention, which usually involves the inhibition of enzymes. As perturbation of DNA repair pathways is the cause of many forms of cancer, strategies will need to be derived with utmost caution.
Given the intimate links between DNA repair, nucleotide metabolism, and energy homeostasis, correcting energy deficits remains a therapeutic avenue worth investigating [94]. Mitokinin Inc. (San Francisco) is currently developing N6FFA derivatives with improved potency and pharmacokinetics that could provide benefit by acting as neo-substrate ATP analogs to promote DNA repair [41] and mitochondrial health [95]. Research on disrupting PAR signalling is mostly focused on establishing cancer therapies; however, there are increasing therapeutic implications for neurodegeneration. Drugs that inhibit PARPs or PARGs can have similar benefits against cancer [96], suggesting there must be proper PAR response homeostasis, as either too little or too much PARylation may lead to disease. While cancer therapies focus on causing PAR dysregulation to promote cell death, neurodegenerative therapies will need to focus on correcting PAR dysregulation and energy deficits to restore neuronal health and survival.
Timing administration of future drugs to contract the CAG expansion, help correct ROS-damaged DNA, or restore energy homeostasis will be critical, as reverting any accumulated cellular damage may be more difficult than its prevention. A study using resting-state functional MRIs showed that children with CAG-expanded HTT have altered brain circuitry as early as age 6 [97]. Study of DNA and RNA status and genotoxic stresses in HD could provide us with very early biomarkers of disease and hence the ability to develop a prodromal HD therapeutic window. DNA repair in humans is carried out by inherently higher order complexes composed of both protein scaffolds and enzymes, where some proteins have both functions. Defining whether to target enzymatic function or protein levels will be essential for therapeutic development.
CONFLICT OF INTEREST
RT is member of SAB and shareholder, Mitokinin Inc. No other authors have a conflict of interest to report.
ACKNOWLEDGMENTS
TM is a HDSA Berman Topper Fellow. The RT laboratory is supported by the Krembil Foundation, Canadian Institutes of Health Research (CIHR), National Science and Engineering Research Council (NSERC), Canadian Foundation for Innovation (CFI), Ontario Innovation Trust (OIT), Hereditary Disease Foundation, and The Huntington Society of Canada.
REFERENCES
[1] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: :516–26. |
[2] | Correia K , Harold D , Kim K-H , Holmans P , Jones L , Orth M , et al. The Genetic Modifiers of Motor OnsetAge (GeM MOA) website: Genome-wide association analysis for genetic modifiers of Huntington’s Disease. J Huntingtons Dis. (2015) ;4: :279–84. |
[3] | Bettencourt C , Hensman-Moss D , Flower M , Wiethoff S , Brice A , Goizet C , et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. (2016) ;79: :983–90. |
[4] | Wright GEB , Collins JA , Kay C , McDonald C , Dolzhenko E , Xia Q , et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet. (2019) ;104: :1116–26. |
[5] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: :887–900.e14. |
[6] | Ciosi M , Maxwell A , Cumming SA , Hensman Moss DJ , Alshammari AM , Flower MD , et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. (2019) ;48: :568–80. |
[7] | Goold R , Flower M , Moss DH , Medway C , Wood-Kaczmar A , Andre R , et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. (2019) ;28: :650–61. |
[8] | Massey TH , Jones L . The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. (2018) ;11: :dmm031930. |
[9] | Maiuri T , Suart CE , Hung CLK , Graham KJ , Barba Bazan CA , Truant R . DNA damage repair in Huntington’s disease and other neurodegenerative diseases. Neurotherapeutics. (2019) ;16: :948–56. |
[10] | Scudiero DA , Meyer SA , Clatterbuck BE , Tarone RE , Robbins JH . Hypersensitivity to N-methyl-N’-nitro-N-nitrosoguanidine in fibroblasts from patients with Huntington disease, familial dysautonomia, and other primary neuronal degenerations. Proc Natl Acad Sci U S A. (1981) ;78: :6451–5. |
[11] | Moshell AN , Tarone RE , Barrett SF , Robbins JH . Radiosensitivity in Huntington’s disease: Implications for pathogenesis and presymptomatic diagnosis. Lancet. (1980) ;1: :9–11. |
[12] | Robison SH , Bradley WG . DNA damage and chronic neuronal degenerations. J Neurol Sci. (1984) ;64: :11–20. |
[13] | Maiuri T , Mocle AJ , Hung CL , Xia J , van Roon-Mom WMC , Truant R . Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. (2017) ;26: :395–406. |
[14] | Askeland G , Dosoudilova Z , Rodinova M , Klempir J , Liskova I , Kuśnierczyk A , et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci Rep. (2018) ;8: :9817. |
[15] | Castaldo I , De Rosa M , Romano A , Zuchegna C , Squitieri F , Mechelli R , et al. DNA damage signatures in peripheral blood cells as biomarkers in prodromal huntington disease. Ann Neurol. (2019) ;85: :296–301. |
[16] | Lu X-H , Mattis VB , Wang N , Al-Ramahi I , van den Berg N , Fratantoni SA , et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med. (2014) ;6: :268ra178. |
[17] | Illuzzi J , Yerkes S , Parekh-Olmedo H , Kmiec EB . DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res. (2009) ;87: :733–47. |
[18] | PerezGrovas-Saltijeral A , Ochoa-Morales A , Miranda-Duarte A , Martínez-Ruano L , Jara-Prado A , Camacho-Molina A , et al. Telomere length analysis on leukocytes derived from patients with Huntington disease. Mech Ageing Dev. (2020) ;185: :111189. |
[19] | Nakamori M , Panigrahi GB , Lanni S , Gall-Duncan T , Hayakawa H , Tanaka H , et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo . Nat Genet. (2020) ;52: :146–59. |
[20] | Harding RJ , Loppnau P , Ackloo S , Lemak A , Hutchinson A , Hunt B , et al. Design and characterization of mutant and wildtype huntingtin proteins produced from a toolkit of scalable eukaryotic expression systems. J Biol Chem. (2019) ;294: :6986–7001. |
[21] | Massey T , McAllister B , Jones L . Methods for assessing DNA repair and repeat expansion in Huntington’s disease. Methods Mol Biol. (2018) ;1780: :483–95. |
[22] | Schmidt MHM , Pearson CE . Disease-associated repeat instability and mismatch repair. DNA Repair. (2016) ;38: :117–26. |
[23] | Xu P , Pan F , Roland C , Sagui C , Weninger K . Dynamics of strand slippage in DNA hairpins formed by CAG repeats: Roles of sequence parity and trinucleotide interrupts. Nucleic Acids Res. (2020) ;48: :2232–45. |
[24] | Bañez-Coronel M , Porta S , Kagerbauer B , Mateu-Huertas E , Pantano L , Ferrer I , et al. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. (2012) ;8: :e1002481. |
[25] | Busan S , Weeks KM . Role of context in RNA structure: Flanking sequences reconfigure CAG motif folding in huntingtin exon 1 transcripts. Biochemistry. (2013) ;52: :8219–25. |
[26] | Bañez-Coronel M , Ayhan F , Tarabochia AD , Zu T , Perez BA , Tusi SK , et al. RAN translation in Huntington disease. Neuron. (2015) ;88: :667–77. |
[27] | Chaney JL , Clark PL . Roles for synonymous codon usage in protein biogenesis. Annu Rev Biophys. (2015) ;44: :143–66. |
[28] | O’Brien EP , Ciryam P , Vendruscolo M , Dobson CM . Understanding the influence of codon translation rates on cotranslational protein folding. Acc Chem Res. (2014) ;47: :1536–44. |
[29] | Riba A , Di Nanni N , Mittal N , Arhné E , Schmidt A , Zavolan M . Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc Natl Acad Sci U S A. (2019) ;116: :15023–32. |
[30] | Athey J , Alexaki A , Osipova E , Rostovtsev A , Santana-Quintero LV , Katneni U , et al. A new and updated resource for codon usage tables. BMC Bioinformatics. (2017) ;18: :391. |
[31] | Guo Q , Bin Huang , Cheng J , Seefelder M , Engler T , Pfeifer G , et al. The cryo-electron microscopy structure of huntingtin. Nature. (2018) ;555: :117–20. |
[32] | Quan F , Janas J , Popovich BW . A novel CAG repeat configuration in the SCA1 gene: Implications for the molecular diagnostics of spinocerebellar ataxia type 1. Hum Mol Genet. (1995) ;4: :2411–3. |
[33] | Jin H , Cho Y . Structural and functional relationships of FAN1. DNA Repair. (2017) ;56: :135–43. |
[34] | Williams HL , Gottesman ME , Gautier J . The differences between ICL repair during and outside of S phase. Trends Biochem Sci. (2013) ;38: :386–93. |
[35] | Datta A , Brosh RM Jr. . Holding all the cards-how Fanconi anemia proteins deal with replication stress and preserve genomic stability. Genes. (2019) ;10: :170. |
[36] | Kato N , Kawasoe Y , Williams H , Coates E , Roy U , Shi Y , et al. Sensing and processing of DNA interstrand crosslinks by the mismatch repair pathway. Cell Rep. (2017) ;21: :1375–85. |
[37] | Zhang N , Lu X , Zhang X , Peterson CA , Legerski RJ . hMutSbeta is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol. (2002) ;22: :2388–97. |
[38] | Zhao J , Jain A , Iyer RR , Modrich PL , Vasquez KM . Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic Acids Res. (2009) ;37: :4420–9. |
[39] | Feng Z , Jin S , Zupnick A , Hoh J , de Stanchina E , Lowe S , et al. p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene. (2006) ;25: :1–7. |
[40] | Fujii N , Evison BJ , Actis ML , Inoue A . A novel assay revealed that ribonucleotide reductase is functionally important for interstrand DNA crosslink repair. Bioorg Med Chem. (2015) ;23: :6912–21. |
[41] | Bowie LE , Maiuri T , Alpaugh M , Gabriel M , Arbez N , Galleguillos D , et al. N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc Natl Acad Sci U S A. (2018) ;115: :E7081–90. |
[42] | Barciszewski J , Siboska GE , Pedersen BO , Clark BF , Rattan SI . A mechanism for the in vivo formation of N6-furfuryladenine, kinetin, as a secondary oxidative damage product of DNA. FEBS Lett. (1997) ;414: :457–60. |
[43] | Carrette LLG , Gyssels E , De Laet N , Madder A . Furan oxidation based cross-linking: A new approach for the study and targeting of nucleic acid and protein interactions. Chem Commun. (2016) ;52: :1539–54. |
[44] | Nucleotide Synthesis via Salvage Pathway. eLS. Chichester, UK: John Wiley & Sons, Ltd; 2001. pp. 51. |
[45] | Sigoillot FD , Berkowski JA , Sigoillot SM , Kotsis DH , Guy HI . Cell cycle-dependent regulation of pyrimidine biosynthesis. J Biol Chem. (2003) ;278: :3403–9. |
[46] | Hyder F , Rothman DL , Bennett MR . Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci U S A. (2013) ;110: :3549–54. |
[47] | Federico A , Cardaioli E , Da Pozzo P , Formichi P , Gallus GN , Radi E . Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. (2012) ;322: :254–62. |
[48] | Fasullo M , Endres L . Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int J Mol Sci. (2015) ;16: :9431–49. |
[49] | Herrick J , Sclavi B . Ribonucleotide reductase and the regulation of DNA replication: An old story and an ancient heritage. Mol Microbiol. (2007) ;63: :22–34. |
[50] | Pontarin G , Ferraro P , Bee L , Reichard P , Bianchi V . Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc Natl Acad Sci U S A. (2012) ;109: :13302–7. |
[51] | Benjamin RC , Gill DM . ADP-ribosylation in mammalian cell ghosts. Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J Biol Chem. (1980) ;255: :10493–501. |
[52] | Benjamin RC , Gill DM . Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA molecules containing different types of strand breaks. J Biol Chem. (1980) ;255: :10502–8. |
[53] | Ha HC , Snyder SH . Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. (1999) ;96: :13978–82. |
[54] | Andrabi SA , Kim NS , Yu S-W , Wang H , Koh DW , Sasaki M , et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. (2006) ;103: :18308–13. |
[55] | Yu S-W , Andrabi SA , Wang H , Kim NS , Poirier GG , Dawson TM , et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. (2006) ;103: :18314–9. |
[56] | Hoch NC , Hanzlikova H , Rulten SL , Tétreault M , Komulainen E , Ju L , et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. (2017) ;541: :87–91. |
[57] | Love S , Barber R , Wilcock GK . Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain. (1999) ;122: (Pt 2):247–53. |
[58] | McGurk L , Mojsilovic-Petrovic J , Van Deerlin VM , Shorter J , Kalb RG , Lee VM , et al. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol Commun. (2018) ;6: :84. |
[59] | Kam T-I , Mao X , Park H , Chou S-C , Karuppagounder SS , Umanah GE , et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science. (2018) ;362: :eaat8407. |
[60] | Cardinale A , Paldino E , Giampà C , Bernardi G , Fusco FR . PARP-1 inhibition is neuroprotective in the R6/2 mouse model of Huntington’s disease. PLoS One. (2015) ;10: :e0134482. |
[61] | Paldino E , Cardinale A , D’Angelo V , Sauve I , Giampà C , Fusco FR . Selective sparing of striatal interneurons after poly (ADP-Ribose) polymerase 1 inhibition in the R6/2 mouse model of Huntington’s disease. Front Neuroanat. (2017) ;11: :61. |
[62] | Tabrizi SJ , Workman J , Hart PE , Mangiarini L , Mahal A , Bates G , et al. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. (2000) ;47: :80–6. |
[63] | Zhou C , Huang Y , Przedborski S . Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci. (2008) ;1147: :93–104. |
[64] | Rodinova M , Krizova J , Stufkova H , Bohuslavova B , Askeland G , Dosoudilova Z , et al. Deterioration of mitochondrial bioenergetics and ultrastructure impairment in skeletal muscle of a transgenic minipig model in the early stages of Huntington’s disease. Dis Model Mech. (2019) ;12: :dmm038737. |
[65] | Dharshini SAP , Taguchi Y-H , Gromiha MM . Investigating the energy crisis in Alzheimer disease using transcriptome study. Sci Rep. (2019) ;9: :18509. |
[66] | Chen C , Turnbull DM , Reeve AK . Mitochondrial dysfunction in Parkinson’s disease-cause or consequence? Biology. (2019) ;8: :38. |
[67] | Fu R , Ceballos-Picot I , Torres RJ , Larovere LE , Yamada Y , Nguyen KV , et al. Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain. (2014) ;137: :1282–303. |
[68] | Göttle M , Prudente CN , Fu R , Sutcliffe D , Pang H , Cooper D , et al. Loss of dopamine phenotype among midbrain neurons in Lesch-Nyhan disease. Ann Neurol. (2014) ;76: :95–107. |
[69] | Cancel G , Gourfinkel-An I , Stevanin G , Didierjean O , Abbas N , Hirsch E , et al. Somatic mosaicism of the CAG repeat expansion in spinocerebellar ataxia type 3/Machado-Joseph disease. Hum Mutat. (1998) ;11: :23–7. |
[70] | Gao R , Matsuura T , Coolbaugh M , Zühlke C , Nakamura K , Rasmussen A , et al. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet. (2008) ;16: :215–22. |
[71] | Katagiri S , Hayashi T , Takeuchi T , Yamada H , Gekka T , Kawabe K , et al. Somatic instability of expanded CAG repeats of ATXN7 in Japanese patients with spinocerebellar ataxia type 7. Doc Ophthalmol. (2015) ;130: :189–95. |
[72] | Tanaka F , Sobue G , Doyu M , Ito Y , Yamamoto M , Shimada N , et al. Differential pattern in tissue-specific somatic mosaicism of expanded CAG trinucleotide repeat in dentatorubral-pallidoluysian atrophy, Machado-Joseph disease, and X-linked recessive spinal and bulbar muscular atrophy. J Neurol Sci. (1996) ;135: :43–50. |
[73] | Trang H , Stanley SY , Thorner P , Faghfoury H , Schulze A , Hawkins C , et al. Massive CAG repeat expansion and somatic instability in maternally transmitted infantile spinocerebellar ataxia type 7. JAMA Neurol. (2015) ;72: :219–23. |
[74] | Zühlke C , Hellenbroich Y , Schaaff F , Gehlken U , Wessel K , Schubert T , et al. CAG repeat analyses in frozen and formalin-fixed tissues following primer extension preamplification for evaluation of mitotic instability of expanded SCA1 alleles. Hum Genet. (1997) ;100: :339–44. |
[75] | Laffita-Mesa JM , Velázquez-Pérez LC , Santos Falcón N , Cruz-Mariño T , González Zaldívar Y , Vázquez Mojena Y , et al. Unexpanded and intermediate CAG polymorphisms at the SCA2 locus (ATXN2) in the Cuban population: Evidence about the origin of expanded SCA2 alleles. Eur J Hum Genet. (2012) ;20: :41–9. |
[76] | Chong SS , McCall AE , Cota J , Subramony SH , Orr HT , Hughes MR , et al. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. (1995) ;10: :344–50. |
[77] | Hashida H , Goto J , Kurisaki H , Mizusawa H , Kanazawa I . Brain regional differences in the expansion of a CAG repeat in the spinocerebellar ataxias: Dentatorubral-pallidoluysian atrophy, Machado-Joseph disease, and spinocerebellar ataxia type 1. Ann Neurol. (1997) ;41: :505–11. |
[78] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) ;6: :409–14. |
[79] | Lopes-Cendes I , Maciel P , Kish S , Gaspar C , Robitaille Y , Clark HB , et al. Somatic mosaicism in the central nervous system in spinocerebellar ataxia type 1 and Machado-Joseph disease. Ann Neurol. (1996) ;40: :199–206. |
[80] | Matsuura T , Sasaki H , Yabe I , Hamada K , Hamada T , Shitara M , et al. Mosaicism of unstable CAG repeats in the brain of spinocerebellar ataxia type 2. J Neurol. (1999) ;246: :835–9. |
[81] | Libby RT , Monckton DG , Fu Y-H , Martinez RA , McAbney JP , Lau R , et al. Genomic context drives SCA7 CAG repeat instability, while expressed SCA7 cDNAs are intergenerationally and somatically stable in transgenic mice. Hum Mol Genet. (2003) ;12: :41–50. |
[82] | Ito H , Fujita K , Tagawa K , Chen X , Homma H , Sasabe T , et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol Med. (2015) ;7: :78–101. |
[83] | Bosso Taniguchi J , Kondo K , Fujita K , Chen X , Homma H , Mao Y , et al. RpA1 ameliorates symptoms of mutant ataxin-1 knock-in mice and enhance DNA damage repair. J Neurol Sci. (2017) ;381: :299. |
[84] | Abraham KJ , Chan JNY , Salvi JS , Ho B , Hall A , Vidya E , et al. Intersection of calorie restriction and magnesium in the suppression of genome-destabilizing RNA-DNA hybrids. Nucleic Acids Res. (2016) ;44: :8870–84. |
[85] | Chakraborty A , Tapryal N , Venkova T , Mitra J , Vasquez V , Sarker AH , et al. Deficiency in classical nonhomologous end-joining-mediated repair of transcribed genes is linked to SCA3 pathogenesis. Proc Natl Acad Sci U S A. (2020) ;117: :8154–65. |
[86] | Gao R , Chakraborty A , Geater C , Pradhan S , Gordon KL , Snowden J , et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife. (2019) ;8: :e42988. |
[87] | Pfeiffer A , Luijsterburg MS , Acs K , Wiegant WW , Helfricht A , Herzog LK , et al. Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J. (2017) ;36: :1066–83. |
[88] | Torres-Ramos Y , Montoya-Estrada A , Cisneros B , Tercero-Pérez K , León-Reyes G , Leyva-García N , et al. Oxidative stress in spinocerebellar ataxia type 7 is associated with disease severity. Cerebellum. (2018) ;17: :601–9. |
[89] | Martinez E , Palhan VB , Tjernberg A , Lymar ES , Gamper AM , Kundu TK , et al. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo . Mol Cell Biol. (2001) ;21: :6782–95. |
[90] | Jung Y , Mikata Y , Lippard SJ . Kinetic studies of the TATA-binding protein interaction with cisplatin-modified DNA. J Biol Chem. (2001) ;276: :43589–96. |
[91] | Vichi P , Coin F , Renaud JP , Vermeulen W , Hoeijmakers JH , Moras D , et al. Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. EMBO J. (1997) ;16: :7444–56. |
[92] | Goodwin JF , Schiewer MJ , Dean JL , Schrecengost RS , de Leeuw R , Han S , et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. . (2013) ;3: :1254–71. |
[93] | Li L , Chang W , Yang G , Ren C , Park S , Karantanos T , et al. Targeting poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage response pathway in castration-resistant prostate cancer. Sci Signal. (2014) ;7: :ra47. |
[94] | Lloret A , Beal MF . PGC-1α, sirtuins and PARPs in Huntington’s disease and other neurodegenerative conditions: NAD+to rule them all. Neurochem Res. (2019) ;44: :2423–34. |
[95] | Hertz NT , Berthet A , Sos ML , Thorn KS , Burlingame AL , Nakamura K , et al. A neo-substrate that amplifies catalytic activity of Parkinson’s-disease-related kinase PINK1. Cell. (2013) ;154: :737–47. |
[96] | Houl JH , Ye Z , Brosey CA , Balapiti-Modarage LPF , Namjoshi S , Bacolla A , et al. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat Commun. (2019) ;10: :5654. |
[97] | Tereshchenko AV , Schultz JL , Bruss JE , Magnotta VA , Epping EA , Nopoulos PC . Abnormal development of cerebellar-striatal circuitry in Huntington disease. Neurology. (2020) ;94: :e1908–15. |


