
The Role of Hypothalamic Pathology for Non-Motor Features of Huntington’s Disease
Abstract
Huntington’s disease (HD) is a fatal genetic neurodegenerative disorder. It has mainly been considered a movement disorder with cognitive symptoms and these features have been associated with pathology of the striatum and cerebral cortex. Importantly, individuals with the mutant huntingtin gene suffer from a spectrum of non-motor features often decades before the motor disorder manifests. These symptoms and signs include a range of psychiatric symptoms, sleep problems and metabolic changes with weight loss particularly in later stages. A higher body mass index at diagnosis is associated with slower disease progression. The common psychiatric symptom of apathy progresses with the disease. The fact that non-motor features are present early in the disease and that they show an association to disease progression suggest that unravelling the underlying neurobiological mechanisms may uncover novel targets for early disease intervention and better symptomatic treatment. The hypothalamus and the limbic system are important brain regions that regulate emotion, social cognition, sleep and metabolism. A number of studies using neuroimaging, postmortem human tissue and genetic manipulation in animal models of the disease has collectively shown that the hypothalamus and the limbic system are affected in HD. These findings include the loss of neuropeptide-expressing neurons such as orexin (hypocretin), oxytocin, vasopressin, somatostatin and VIP, and increased levels of SIRT1 in distinct nuclei of the hypothalamus. This review provides a summary of the results obtained so far and highlights the potential importance of these changes for the understanding of non-motor features in HD.
NON-MOTOR FEATURES OF HUNTINGTON’S DISEASE
Huntington’s disease (HD) is more than a movement disorder. It is always caused by an expanded CAG repeat in the huntingtin (HTT) gene. A positive gene test in combination with unequivocal signs of motor disturbances are required for the clinical diagnosis of HD today (i.e., manifest HD) [1]. However, a large number of clinical studies have demonstrated the presence of non-motor features decades before the manifestation of the movement disorder and alterations using neuroimaging techniques in individuals who carry the mutant HTT gene (i.e., premanifest HD [1–4]. The motor disturbances in HD have been associated with progressive and pronounced dysfunction and pathology in the striatum of the basal ganglia and the cerebral cortex, the most affected areas in HD. Cognitive changes are common and constitute deficits in executive function with reduced capacity for concentration, attention, mental flexibility and organization. These cognitive changes have also been associated with pathology in the striatum and the cerebral cortex. Other non-motor features of HD include a wide range of psychiatric symptoms such as apathy, depression, anxiety, irritability and altered social cognition [5, 6]. Reduced recognition of facial expression of emotions has also been detected in pre-manifest HD [7–10]. Reduced emotional recognition has recently been found to be associated with apathy, which is the psychiatric symptom that most clearly increases with disease progression [11, 12]. Sleep problems and an altered circadian rhythm occur in many individuals with HD [13–18]. Metabolic alterations include increased appetite with a higher caloric intake being necessary to counterbalance weight loss in HD [19, 20]. A high body mass index (BMI) at motor onset has been associated with a slower disease progression [21]. The underlying neurobiological mechanisms for these non-motor features are not known. As their occurrence is common early in the disease process and have associations to disease progression, further understanding of how they arise and develop may lead to important insight into early disease mechanisms and uncover new targets for disease modification.
The regulation of emotion, sleep and metabolism is governed by the hypothalamus and the limbic system. The hypothalamus is made up of interconnected nuclei that receive inputs both from the periphery, e.g., thyroid hormones, leptin, ghrelin and insulin, and the central nervous system (CNS). Besides regulating the endocrine axes of the body, its many different neuropeptide-expressing neuronal populations project within the region and to other areas of the brain to regulate emotion, sleep and metabolism. The hypothalamus is part of the larger limbic system that includes the hippocampus, gyrus cinguli, prefrontal cortex, insula, septal nuclei, amygdala, ventral striatum, ventral tegmental area and raphe nucleus [22, 23]. In light of the presence of non-motor features in HD, this system has gained increasing interest for investigations of changes in clinical material and experimental models of HD (previously reviewed in [24–27]). These authors reviewed the state of knowledge of this area in HD in 2012 in this journal [28] and provides here an updated overview of the major findings made in the hypothalamus and the limbic system in HD. The review is based on a literature search on the PubMed database up to 2019 with the search terms ‘Huntington disease’ ‘huntingtin’, ‘hypothalamus’, ‘limbic system’, ‘orexin’, ‘hypocretin’, ‘oxytocin’, and ‘vasopressin’.
HYPOTHALAMIC CHANGES IN CLINICAL HD DETECTED BY NEUROIMAGING STUDIES
A combination of neuroimaging studies and postmortem analyses of human hypothalamic tissue have identified alterations in the hypothalamic region in HD (Fig. 1, Table 1). Studies of the hypothalamic region in general are challenged by practical factors such as the scarcity of such tissue in brain banks and by intrinsic factors such as the difficulties to define the whole structure or specific nuclei within the hypothalamus due to the lack of clear anatomical borders. Nevertheless, structural analyses using voxel-based morphometry and mathematical modelling based on grey matter signals in the hypothalamic region have detected significant differences between premanifest HD and age- and sex-matched controls in several studies [18, 29–31]. Studies using positron emission tomography (PET) have found increased microglia activation and reductions in dopamine D2 receptors also in premanifest HD [32, 33]. Attempts to estimate the hypothalamic volume have not revealed any significant differences between HD and controls [34]. Nevertheless, these studies indicate that the hypothalamic region is affected early in clinical HD.
Fig. 1
Overview of main hypothalamic changes in Huntington’s disease. Altered immunoreactivity and gene expression levels of cell populations detected in specific hypothalamic nuclei. Changes that result in upregulation and downregulation are indicated by the arrows. The location of specific hypothalamic nuclei is indicated by the dashed arrows on the cross-sectional human hypothalamic section stained for Cresyl violet and Luxol fast blue. Hypothalamic changes are also detected in the blood and cerebrospinal fluid as well as from different imaging paradigms. These changes are thought to be a key contributor to the clinical features in Huntington’s disease. CART, cocaine and amphetamine regulated transcript; CREB-1, cyclic AMP-responsive element-binding protein 1; D2R, dopamine D2 receptor; FOXO3, Forkhead box O3; HPA, hypothalamic–pituitary–adrenal; LHA, lateral hypothalamic area; MCH, melanin-concentrating hormone; NTL, nucleus tuberalis lateralis; NKX2-1, NK2 homeobox 1; OX2R, Orexin 2 receptor; PVN, paraventricular nucleus; PDYN, prodynorphin, SIRT1, sirtuin 1; VIP, vasoactive intestinal peptide; VMH, ventromedial hypothalamus.
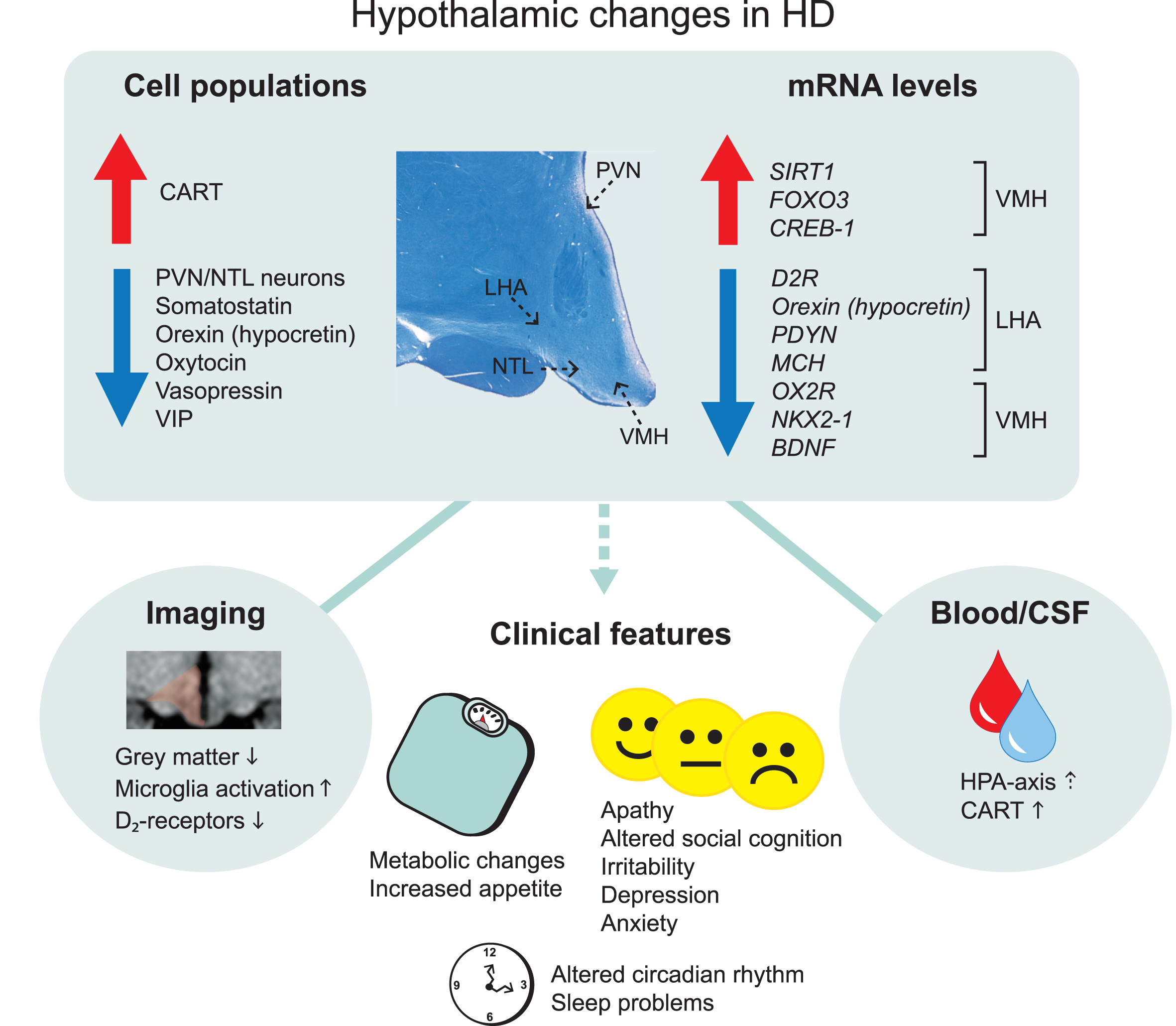
Fig. 2
Limbic system changes in Huntington’s disease. Schematic representation of the structures of the limbic system and overview of the main upregulated and downregulated changes (indicated by the arrows). A, amygdala; CC, cingulate cortex; F, fornix; fMRI, functional magnetic resonance imaging; HIPP, hippocampus; HYP, hypothalamus; PFC, prefrontal cortex; R, raphe nucleus; V, ventral tegmental area; VS, ventral striatum.
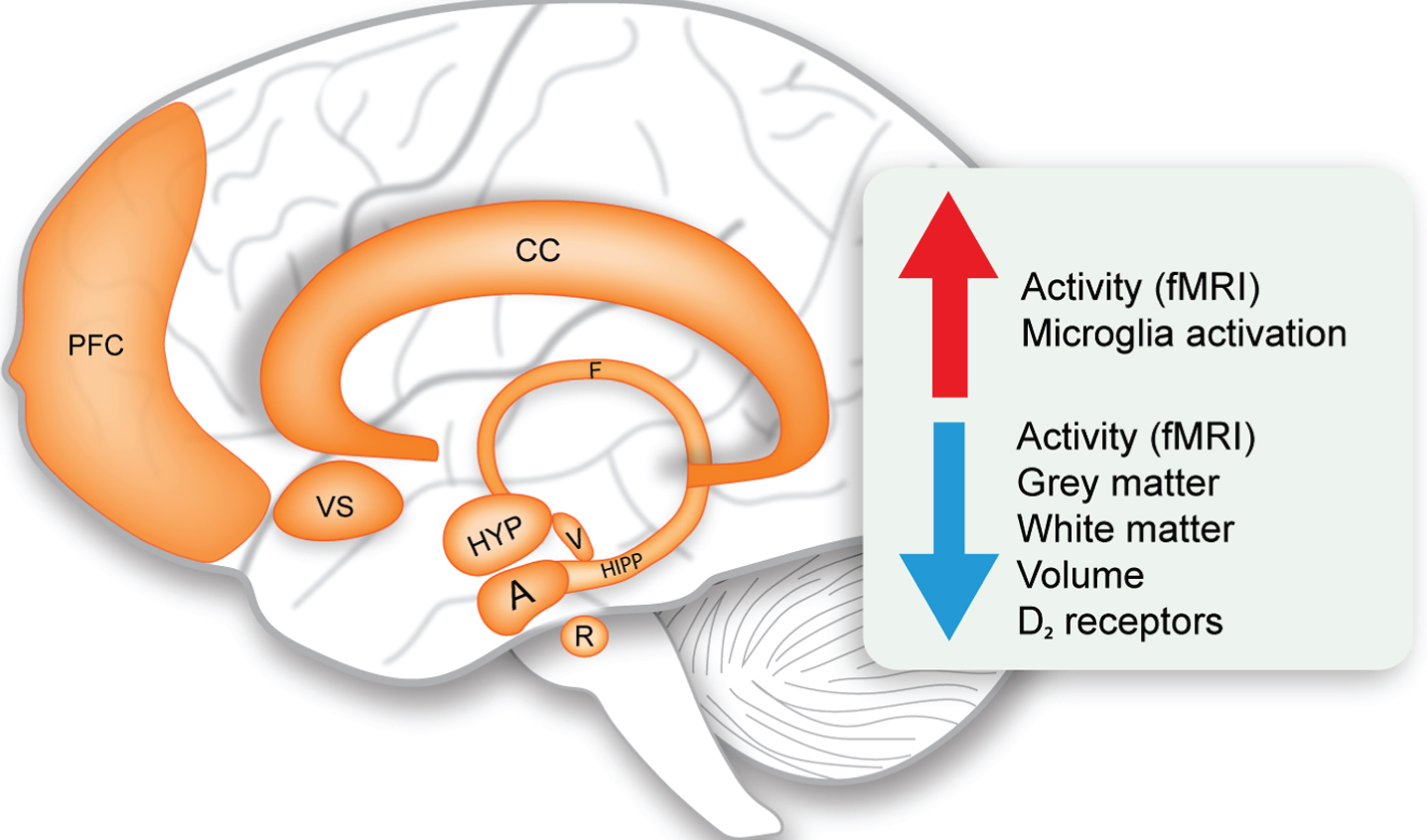
Table 1
Summary of main findings in the hypothalamus in clinical HD
| Pathology | HD stage | References | |
| Atrophy | ↑ | Premanifest and manifest (VBM) | [29, 31, 121] |
| Grey matter volume | ↓ | Premanifest and manifest (VBM) | [30, 31, 122] |
| = | Premanifest and manifest | [34] | |
| White matter volume | ↓ | Premanifest and manifest (DTI) | [122, 123] |
| Fornix demyelination | ↑ | Premanifest and manifest (DTI,VBM) | [124] |
| NTL volume/neurons | ↓ | Cell counts and volume (HD grade 2–4) | [35–38] |
| PVN neurons | ↓ | Cell counts (HD grade 2–4) | [35] |
| Microglial activation | ↑ | Premanifest and manifest (PET) | [32, 33] |
| Functioning of HPA axis | ↑ | Premanifest and manifest | [107, 109, 125, 126] |
| Circadian rhythm dysfunction | ↑ | Premanifest and manifest | [127] |
| AgRP | = | Immunoreactivity (HD grade 1–4) | [128] |
| BDNF | ↓ | mRNA levels (HD grade 1–4) | [52] |
| CART | ↑ | Immunoreactivity (HD grade 2–4) | [35] |
| = | Immunoreactivity, mRNA levels (HD grade 0, HD grade 1–4) | [40, 52, 128, 129] | |
| CREB-1 | ↑ | mRNA (HD grade 1–4) | [52] |
| CRH | = | Immunoreactivity (HD grade 1–4) | [129] |
| ↑ | mRNA (HD grade 1–4) | [129] | |
| Dopamine D2 receptor | ↓ | Premanifest and manifest (PET), mRNA levels (HD grade 1–4) | [32, 52] |
| FOXO3 | ↑ | mRNA (HD grade 1–4) | [52] |
| MCH | = | Immunoreactivity (HD grade 2–4) | [51, 52] |
| ↓ | mRNA levels (HD grade 1–4) | [52] | |
| NPY | = | Immunoreactivity (HD grade 2–4) | [35] |
| ↓ | Immunoreactivity (HD grade 1–4) | [128] | |
| ↑ | mRNA (HD grade 1–4) | [128] | |
| Orexin | = | Immunoreactivity (HD grade 0) | [40] |
| ↓ | Immunoreactivity, mRNA levels (HD grade 1–4) | [35, 51, 52, 130] | |
| Orexin 2 receptor | ↓ | mRNA levels (HD grade 1–4) | [52] |
| Oxytocin | ↓ | Immunoreactivity (HD grade 0) | [40] |
| ↓ | Immunoreactivity (HD grade 2–4) | [35] | |
| = | Immunoreactivity (HD grade 1–4) | [129] | |
| Prodynorphin | ↓ | mRNA levels (HD grade 1–4) | [52] |
| SIRT1 | ↑ | mRNA (HD grade 1–4) | [52] |
| Somatostatin | ↓ | Immunoreactivity (mid-late HD) | [39] |
| TRH | = | mRNA levels (HD grade 1–4) | [52, 129] |
| Tyrosine hydroxylase | ↓ | mRNA levels (HD grade 1–4) | [52] |
| Vasopressin | ↓ | Immunoreactivity (HD grade 0) | [40] |
| ↓ | Immunoreactivity (HD grade 1–4) | [35, 57] | |
| = | Immunoreactivity (HD grade 1–4) | [129] | |
| = | mRNA levels (HD grade 1–4) | [57] | |
| VIP | ↓ | Immunoreactivity (HD grade 1–4) | [57] |
| = | mRNA (HD grade 1–4) | [57] |
AgRP, agouti-related protein; BDNF, brain-derived neurotrophic factor; CART, cocaine and amphetamine regulated transcript; CREB-1, cyclic AMP-responsive element-binding protein 1; CRH, corticotrophin releasing hormone; DTI, diffusion tensor imaging; FOXO3, forkhead box O3; MCH, melanin-concentrating hormone; NPY, neuropeptide Y; PET, positron emission tomography; SIRT1, sirtuin 1; TRH, thyrotropin-releasing hormone; VBM, voxel based morphometry; VIP, vasoactive intestinal peptide.
NEUROPATHOLOGICAL CHANGES IN THE HYPOTHALAMUS IN CLINICAL HD
Postmortem analyses of hypothalami from individuals with HD have revealed a number of changes in different nuclei of this region (Fig. 1, Table 1). The hypothalamus constitutes several nuclei such as the paraventricular nucleus (PVN), supraoptic nucleus (SON), infundibular nucleus, ventromedial nucleus of the hypothalamus (VMH), suprachiasmatic nucleus (SCN), nucleus tuberalis lateralis (NTL), lateral hypothalamic area (LHA) and mammillary bodies. The frequency of huntingtin inclusions as assessed using the EM48 antibody has been estimated to be between 0.5–2.8% in the different nuclei [35]. Although stereological estimation for the total number of neurons and cells in the whole hypothalamus in sections stained for the Nissl stain cresyl violet did not reveal any significant differences between HD and control cases, a specific loss of neurons has been detected in the PVN by 23% and in the NTL by 32% in HD tissue [35, 36]. Early analyses have also indicated a reduced number of somatostatin neurons in the NTL [37–39]. The function of this nucleus is still not well understood. Another study found a reduced number of neurons expressing oxytocin by 45% and vasopressin by 24% in HD cases compared to controls [35]. The number of oxytocin and vasopressin immunopositive neurons were reduced in a case of premanifest HD with Vonsattel grade 0, suggesting that loss of oxytocin and vasopressin may occur early in HD [40]. Oxytocin has been implicated in the ability to recognize facial expression of emotion, which has been found to be reduced already in premanifest HD [41, 42]. Acute intranasal administration of oxytocin to HD gene carriers normalized their altered brain activation pattern in response to recognizing facial expression of emotional disgust, indicating a role for oxytocin in altered emotion recognition in HD [43]. As for vasopressin, it is not known whether vasopressin levels or their activity are altered in HD. The vasopressin system has been linked to aggression in animals where aggressive behavior can be inhibited by vasopressin V1a receptor antagonists [44, 45]. There is an ongoing Phase 2 clinical trial with Azevan Pharmaceuticals using a vasopressin receptor antagonist (SRX246) against irritability in manifest HD (https://clinicaltrials.gov/ct2/show/NCT02507284).
Several studies have investigated the LHA in HD. The LHA controls several functions that are disturbed in HD and pathology in this area could therefore be a contributing if not causative factor for part of the clinical manifestation of the disease. Experimental lesions of the LHA established the “lateral hypothalamic syndrome” characterized by hypophagia, adipsia, hypoactivity and weight loss [46–48]. Later studies established an important role for the LHA in the regulation of sleep, energy balance, reward and motivated behaviors [49]. In HD, there is a 38% reduction in the number of orexin (hypocretin) immunopositive-neurons in HD and mRNA levels are also diminished compared to control cases [35, 50–52]. Orexin is a neuropeptide that is only expressed in the LHA and it plays an important role in the regulation of sleep, emotion and metabolism [53, 54]. Melanin-concentrating hormone (MCH) is another emotion-regulating neuropeptide that is expressed in neighboring neurons within the LHA [55]. A recent study showed significant reduction of MCH mRNA levels in HD cases compared to controls although the number of MCH-immunopositive neurons is not affected [52]. Analyses of mRNA levels of other factors in the LHA in HD cases indicated that the dopamine D2-receptor (D2R) levels are also reduced [52]. mRNA levels of D2R are not reduced in other hypothalamic areas suggesting that the signal of D2R reductions detected using PET may arise from the LHA specifically.
Sleep problems and altered circadian rhythm are common in patients with HD [13–18, 56]. The SCN is the key brain region for regulating sleep and circadian rhythm. Postmortem analyses of the SCN from patients with HD have shown that numbers of vasoactive intestinal peptide (VIP)- and vasopressin-immunoreactive neurons were reduced by 85% and 33% respectively in HD cases compared to controls [57]. This occurred in the presence of normal mRNA levels of these genes as well as unaffected number of neurons expressing melatonin receptors in this area [57]. Hence, there is specific pathology in the SCN in HD that may contribute to alterations in sleep and circadian rhythm.
HYPOTHALAMIC GENE EXPRESSION CHANGES IN CLINICAL HD
Alterations in specific hypothalamic genes have also been reported in HD (Fig. 1, Table 1). A recent study showed reduced expression of mRNA levels of brain-derived neurotrophic factor (BDNF) in the VMH but not in the LHA, PVN or SON of HD cases compared to controls [52]. The VMH constitutes a group of cells involved in the regulation of energy homeostasis as well as aggression [58–64]. BDNF is expressed in the VMH [65] and reduced expression of BDNF in mice has been associated with aggressiveness, hyperphagia and obesity [66, 67]. BDNF is well known to be reduced in the cerebral cortex in clinical HD and has been implicated in the pathogenesis in HD [52, 68]. Hence, the effects of the BDNF system specifically in the VMH may also play a role in HD.
The metabolic regulators and energy sensors sirtuins (SIRT) have been implicated in HD. Neuroprotective effects of SIRT1 inhibitors have been shown in drosophila, mammalian cells and mouse models of HD [69]. One initial clinical trial to evaluate safety has been conducted using the SIRT1 inhibitor, selisistat, in HD patients [70]. SIRT1 acts in the hypothalamus to control metabolism and longevity partly through the regulation of maturation of hypothalamic peptide hormones [71–74]. A recent study showed an increase in mRNA levels and immunoreactivity of SIRT1 in both the LHA and VMH apart from other affected brain regions in HD such as the striatum and cerebral cortex [52]. No changes were found in the less affected cerebellum. Analyses of mRNA levels of downstream targets of SIRT1 such as FOXO3 indicated increased levels in the LHA and VMH in HD. FOXO3 has also been shown to be upregulated in the striatum in HD [75, 76]. This data indicate that the SIRT1 system is affected in sensitive brain regions in HD and that the hypothalamic effects of these factors need to be considered in future studies targeting this system in HD.
OTHER LIMBIC SYSTEM CHANGES IN CLINICAL HD
Although it is beyond the scope of this review to discuss in detail the alterations of the limbic system in HD, it is important to consider the interactions with other limbic structures which form widely distributed networks for a proper perspective. While the definition of the limbic system is not always clear, the anatomical structures generally accepted today and included in this review comprises of the hippocampus, amygdala, ventral striatum, nucleus accumbens, raphe nucleus, ventral tegmental area, cingulate cortex and prefrontal cortex [22, 23]. Table 2 summarizes the main alterations detected to date in the limbic system in clinical HD and in rodent models. Imaging studies using voxel based morphometry (VBM) in magnetic resonance images (MRI) have indicated increased atrophy and grey matter loss in these limbic structures already in premanifest HD [77–86]. Whereas histopathological studies on the limbic structures in HD are scarce, there is evidence for cell loss in the dorsal raphe nucleus, anterior cingulate cortex and prefrontal cortex in symptomatic HD cases with Vonsattel grade 1–4 [83, 87, 88]. Functional MRI (fMRI) studies have reinforced our understanding of limbic system dysfunction in HD. Reduced functional connectivity, network integrity and activity was found in the HD hippocampus, amygdala, ventral striatum, cingulate cortex and prefrontal cortex using different fMRI paradigms assessing verbal working memory, emotional processing, interference/conflict resolution and attention/alertness [89–99]. Such alterations in activity patterns produce multidimensional maps that to some extent reflect internal states of brain processing in response to a task. These changes are already present in the premanifest stage suggesting that early changes in the limbic structures might contribute in part to disease pathogenesis. As cognition and emotional functions are derived from multiple circuit connections involving cortical and subcortical regions of the limbic system, it seems indisputable that limbic system dysfunction can be ascribed to non-motor features of HD. Indeed, volumetric abnormalities in the amygdala have been found to be related to the clinical profile of HD with high level of anxiety and cognitive symptoms occurring before the onset of motor symptoms [77]. This prevailing view of limbic system involvement in HD is also supported by findings from rodent HD models with alterations in volume, reduction in BDNF and serotonin levels reported in several rat and mouse models (see Table 2 for details). The full extent of limbic system pathology in HD is not well understood and more systematic neuropathological postmortem analyses of these structures are warranted.
Table 2
Summary of main findings in the limbic system in clinical HD and rodent HD models
| Region/pathology | HD stage | HD rodent models | References | ||
| Hippocampus | |||||
| Atrophy | ↑ | Premanifest | Not assessed | [81] | |
| Network integrity | ↓ | Manifest | Not assessed | [99] | |
| Volume | = | Premanifest | = | VBM (R6/2) | [82, 131, 132] |
| ↓ | Premanifest and manifest | [78–80] | |||
| Cerebral blood flow | ↑ | Premanifest | Not assessed | [91] | |
| Neurogenesis | Not assessed | ↓ | R6/2, R6/1, N171-82Q, YAC128 | [133–138] | |
| Androgen receptor | Not assessed | ↓ | Protein levels (R6/1) | [139] | |
| BDNF | Not assessed | ↓ | mRNA and protein levels (R6/1), HdhQ111/Q111, zQ175 | [140–143] | |
| Dendritic spine density | Not assessed | ↓ | R6/2 | [144] | |
| Ferritin | Not assessed | ↑ | Immunoreactive neurons (R6/2) | [145] | |
| PSA-NCAM | Not assessed | ↓ | R6/1, R6/2 | [146] | |
| Serotonin | Not assessed | ↓ | R6/2 | [147] | |
| Serotonin receptors | Not assessed | ↓ | R6/1 | [148] | |
| Amygdala | |||||
| Atrophy | ↑ | Premanifest | tgHDrat | [81, 149] | |
| Volume | = | Premanifest | = | VBM (R6/2) | [82, 132] |
| ↓ | Premanifest and manifest | [77] | |||
| Functional connectivity | ↓ | Premanifest | Not assessed | [98] | |
| Dopamine D2 receptor | ↓ | Manifest | Not assessed | [150] | |
| TRH | ↑ | Peptide levels (manifest) | Not assessed | [151] | |
| Ventral striatum/nucleus accumbens | |||||
| Volume | ↓ | Premanifest and manifest (HD stage 1-2) | Not assessed | [78, 80, 82, 152] | |
| Functional connectivity | Not assessed | ↓ | BACHD rats | [153] | |
| Activity | ↓ | fMRI (premanifest) | Not assessed | [96, 97] | |
| BDNF | Not assessed | ↓ | mRNA levels (BACHD rats) | [154] | |
| Serotonin | Not assessed | ↓ | Immunoreactivity (tgHD rat) | [155] | |
| Somatostatin | ↑ | Peptide levels, immunoreactivity (manifest) | Not assessed | [151, 156] | |
| Tyrosine hydroxylase | Not assessed | ↑ | Immunoreactivity (tgHD rat) | [155] | |
| Raphe nucleus | |||||
| Volume | ↓ | Manifest | Not assessed | [83] | |
| Neuronal counts | ↓ | Manifest | Not assessed | [83] | |
| Serotonin | Not assessed | ↓ | Immunoreactivity (tgHD rat) | [155] | |
| Tyrosine hydroxylase | Not assessed | ↑ | Immunoreactivity (tgHD rat) | [155] | |
| Ventral tegmental area | |||||
| Dopaminergic cell fate | Not assessed | ↑ | Gene expression (tgHD rat) | [157] | |
| Tyrosine hydroxylase | Not assessed | ↑ | Immunoreactivity (tgHD rat) | [158] | |
| Cingulate cortex | |||||
| Volume | ↓ | Premanifest and manifest (VBM) | ↓ | R6/1 | [80, 84, 85, 159] |
| Functional connectivity | ↓ | Premanifest and manifest (fMRI) | Not assessed | [92] | |
| Activity | ↓ | Premanifest and manifest (fMRI) | Not assessed | [93–95, 160, 161] | |
| ↑ | Premanifest and manifest (fMRI) | [162–164] | |||
| Neuronal count | ↓ | Immunoreactivity (manifest) (HD grade 1–3) | Not assessed | [87] | |
| Prefrontal cortex | |||||
| Volume | ↓ | Premanifest (VBM) | ↓ | HdhQ250 | [86, 165] |
| = | Manifest (VBM) | [166] | |||
| White matter degeneration | ↑ | Premanifest (DWI) | Not assessed | [167, 168] | |
| Functional connectivity | ↓ | Premanifest (fMRI) | ↓ | BACHD rats | [89, 90, 153] |
| Activity | ↓ | Premanifest (fMRI) | ↓ | R6/2, tgHDrat | [91, 93, 169–171] |
| Neuronal size | ↓ | Immunoreactivity (manifest) | Not assessed | [88] | |
| Glial expression | ↑ | Immunoreactivity (manifest) | Not assessed | [88] | |
| Synaptic integrity | ↓ | Immunoreactivity (premanifest and manifest) | Not assessed | [172] | |
| Axonal transport | Not assessed | ↑ | Immunoreactivity (Q175KI) | [173] | |
| Glutamate uptake | ↓ | Manifest | Not assessed | [174] | |
| BDNF | Not assessed | ↓ | mRNA levels (R6/2, R6/1) | [142, 175] | |
| Orexin | ↓ | Immunoreactivity (HD grade 1–4) | Not assessed | [51] | |
| Serotonin | Not assessed | ↓ | Immunoreactivity (tgHD rat) | [155] |
BDNF, brain-derived neurotrophic factor; PSA-NCAM, polysialylated neural cell adhesion molecule; TRH, thyrotropin-releasing hormone; VBM, voxel-based morphometry; fMRI, functional magnetic resonance imaging; DWI, diffusion weighted imaging.
EFFECTS ON CIRCULATING FACTORS RELATED TO THE HYPOTHALAMUS IN CLINICAL HD
As the hypothalamus is targeted by and regulates circulating (neuro-)endocrine factors in the blood and cerebrospinal fluid (CSF), it became interesting to investigate whether measurements of such circulating factors would be altered in HD as an indication of hypothalamic dysfunction and potentially disease progression. Several studies have been performed using both CSF and blood to measure neuroendocrine factors with often contradicting or negative results [16, 100–104]. The major results are summarized in Table 3. Out of the published results, the increase of cocaine and amphetamine regulated transcript (CART) levels in the CSF is interesting as it may reflect the increased number of CART-immunopositive neurons in the hypothalamus in HD [35, 105]. Also, a subtle change in the hypothalamic-pituitary-adrenal (HPA) axis may be present [106–109]. With the development of more sensitive assays and a better standardization of collected material, it is possible that future studies will reveal other hypothalamic-related factors with relevance for the non-motor features of HD.
Table 3
Summary of main neuropeptide levels in the circulation in both clinical HD and rodent HD models
| Neuropeptide | HD stage | HD rodent models | References | ||
| Output from the hypothalamus | |||||
| ACTH | = | Serum, plasma levels (premanifest, HD stage 2-3) | ↑ | Serum levels (R6/2) | [104, 106, 176] |
| ↑ | Plasma levels (HD stage 1–4) | [126] | |||
| AgRP | ↑ | Plasma levels (premanifest) | Not assessed | [177] | |
| ↓ | Plasma levels (manifest) | [177] | |||
| CART | ↑ | CSF levels (early-mid stage HD) | Not assessed | [105] | |
| Cortisol/corticosterone | ↑ | Plasma, serum levels (HD stage 1–4) | ↑ | Serum levels (R6/2) | [106, 107, 126, 176, 178–180] |
| = | Serum levels (premanifest, HD stage 2-3) | = | Serum levels (BACHD) | [104, 116] | |
| Estradiol | = | Serum levels (premanifest, HD stage 2-3) | = | Serum levels (R6/1) | [104, 181] |
| FSH | = | Serum, plasma levels (premanifest, HD stage 2-3) | Not assessed | [104, 176] | |
| GHRF | = | Serum levels (premanifest, HD stage 2-3) | Not assessed | [104] | |
| Growth hormone | = | Serum and plasma levels (early HD, manifest) | Not assessed | [182, 183] | |
| ↑ | Plasma levels (manifest) | [176] | |||
| ↓ | Plasma levels (manifest) | [177] | |||
| IGF-1 | = | Plasma levels (manifest) | ↑ | Plasma levels (BACHD, YAC128) | [183, 184] |
| LH | = | Serum, plasma levels (premanifest, HD stage 2-3) | Not assessed | [104, 176] | |
| NPY | ↑ | CSF levels (early-mid stage HD) | Not assessed | [185] | |
| Orexin | = | Ventricular CSF (HD grade 1–4) | = | CSF levels (YAC128) | [51, 100, 101, 103, 130] |
| Prolactin | = | Serum, plasma levels (premanifest, HD stage 2-3) | Not assessed | [104, 176, 186, 187] | |
| ↓ | Plasma (premanifest and manifest) | [177] | |||
| Testosterone | = | Serum, plasma levels (premanifest, HD stage 2-3) | ↓ | Serum levels (R6/1, R6/2) | [104, 139, 176, 181, 188, 189] |
| ↓ | Plasma levels (manifest) | ↑ | Serum levels (tgHD rat) | [190, 191] | |
| TSH | = | Serum, plasma levels (premanifest, HD stage 2-3) | Not assessed | [104, 176, 186] | |
| Vasopressin | = | Serum levels (premanifest, HD stage 2-3) | = | Serum (BACHD) | [104, 116] |
| Input to the hypothalamus | |||||
| Adiponectin | = | Plasma levels (early HD) | ↓ | Serum levels (R6/2,N171-82Q) | [177, 182, 192] |
| Amylin | ↑ | Plasma (premanifest and manifest) | Not assessed | [177] | |
| Ghrelin | = | Plasma levels (early HD) | ↓ | Serum levels (R6/2,N171-82Q) | [110, 182, 192] |
| ↑ | Plasma (manifest) | [177, 183] | |||
| GIP | = | Plasma (premanifest and manifest) | Not assessed | [177] | |
| Glucagon | ↓ | Plasma (manifest) | Not assessed | [177] | |
| Glucose | = | Plasma levels (manifest) | ↑ | Serum levels (R6/2, R6/1, N171-82Q) | [106, 183, 192–195] |
| Insulin | = | Plasma levels (HD grade 1-2, manifest) | ↑ | Serum levels (R6/2) | [106, 177, 183, 193, 194, 196] |
| ↓ | Serum levels (R6/2, N171-82Q) | [192] | |||
| Leptin | = | Plasma levels (early HD) | ↑ | BACHD, BAC-225Q, YAC128 | [114, 116, 197–199] |
| ↓ | Plasma (manifest) | ↓ | Serum levels (R6/2,N171-82Q) | [177, 183, 192, 200] | |
| PP | = | Plasma (premanifest and manifest) | Not assessed | [177] | |
| PYY | = | Plasma (premanifest and manifest) | Not assessed | [177] | |
| Resistin | = | Plasma levels (early HD) | Not assessed | [197] |
ACTH, adrenocorticotropic hormone; AgRP, agouti-related protein; CART, cocaine and amphetamine regulated transcript; FSH, follicle-stimulating hormone; GHRF, growth hormone-releasing factor; IGF-1, insulin-like growth factor 1; LH, luteinizing hormone; NPY, neuropeptide Y; TSH, thyroid-stimulating hormone; GIP, gastric inhibitory polypeptide; PP, pancreatic polypeptide; PYY, peptide YY.
LESSONS LEARNT FROM EXPERIMENTAL STUDIES IN HD
There are several animal models expressing different variants of the human mutant huntingtin gene. Several of these transgenic mice display hypothalamic pathology (Table 4). The R6/2 mouse model shows the highest degree of hypothalamic pathology with reduced number of neurons expressing orexin, oxytocin, vasopressin and VIP [50, 106, 110–113]. Experimental studies provide a complementary approach to the analyses of clinical material as they provide the possibility to study and establish causative relationships. As the hypothalamus receives major input both from the periphery and the rest of the CNS, the question arises as to what extent are the changes detected in this region a direct consequence to the expression of mutant huntingtin in hypothalamic cells. Experiments using recombinant adeno-associated viral (rAAV) vector mediated delivery of different fragments of mutant huntingtin specifically to the hypothalamus have been conducted to address this point. These studies have shown a direct and early effect on the neuronal populations expressing orexin, oxytocin, vasopressin, MCH and BDNF in the hypothalamus, suggesting that the findings made in human postmortem hypothalamic tissue may be directly related to the effects of mutant huntingtin in this area [52, 114, 115]. Other studies have been performed in order to investigate whether the inactivation of mutant huntingtin in the floxed BACHD mouse model using delivery of Cre-recombinase by rAAV vectors or different breeding strategies. rAAV-vector mediated delivery of Cre-recombinase into the hypothalamus has shown a causative link between hypothalamic dysfunction and non-motor phenotypes such as metabolic dysfunction and depressive-like behavior [114, 116]. However, experiments to inactivate mutant huntingtin in specific nuclei or cell populations in the hypothalamus using breeding of the BACHD mice with mice expressing Cre-recombinase under the VMH-specific promoter SF-1, under the PVN-specific promoter Sim-1, or the leptin receptor promoter, did not show any beneficial effects on the psychiatric or metabolic phenotype [117–119]. It would be useful to investigate the involvement of specific hypothalamic circuitries in the development of the early HD phenotype. Further studies using cell specific expression or inactivation of mutant huntingtin in combination with modulating the activity of specific circuitries using chemo- or opto-genetic techniques with analyses of behavioral outputs are likely to give further insight into the causal links between huntingtin-mediated hypothalamic dysfunction and non-motor features of HD.
Table 4
Summary of main hypothalamic findings in HD rodent models
| Pathology | HD rodent models | References | |
| Atrophy | ↑ | R6/2 | [110, 131, 201] |
| Neuronal count | ↓ | R6/2, N171-82Q | [50, 202] |
| Upregulation of HPA axis | ↑ | R6/2 | [106] |
| Circadian rhythm dysfunction | ↑ | R6/2 (CAG250, CAG 140), BACHD, Q175 | [203–207] |
| Androgen receptor | ↓ | protein (R6/1) | [139] |
| Brn2 | ↓ | R6/2 | [112] |
| CART | ↓ | peptide, immunoreactivity, mRNA (R6/2, HD190QG) | [110, 208] |
| ↑ | mRNA (BACHD) | [116] | |
| Clock genes (mPer2, mBmal1, Cry1, Dbp) | ↓ | R6/2 | [13, 209, 210] |
| CRH | ↓ | peptide (R6/2) | [106, 110, 112] |
| = | mRNA (BACHD) | [116] | |
| GnRH | ↓ | immunoreactivity (BACHD, R6/2) | [119, 189] |
| ↓ | mRNA (R6/1) | [181] | |
| HAP1 levels | ↓ | N171-82Q | [202] |
| MCH | ↓ | peptide, immunoreactivity, mRNA (R6/2, AAV-hypo) | [52, 110] |
| = | mRNA (BACHD) | [116] | |
| NPY | ↓ | mRNA (R6/2, HD190QG, BACHD) | [116, 208] |
| Orexin | ↓ | protein, immunoreactivity, mRNA (R6/2, YAC128, AAV-hypo) | [50, 52, 100, 111, 114, 211] |
| ↑ | immunoreactivity (BACHD) | [116] | |
| Orexin 2 receptor | ↓ | mRNA (BACHD) | [116] |
| Oxytocin | ↓ | immunoreactivity, mRNA (R6/2, HD190QG, AAV-hypo) | [112, 114, 208] |
| = | immunoreactivity, mRNA (BACHD) | [116] | |
| Preprosomatostatin | ↓ | mRNA (R6/2, HD190QG) | [208] |
| POMC | ↓ | peptide, immunoreactivity (R6/2) | [110] |
| Prodynorphin | ↓ | mRNA (AAV-hypo) | [52] |
| Tachykinin receptor 3 | ↓ | mRNA (BACHD) | [116] |
| TSH releasing hormone | ↓ | mRNA (R6/2, HD190QG) | [208] |
| Tyrosine hydroxylase | ↓ | mRNA (AAV-hypo) | [52] |
| Vasopressin | ↓ | mRNA, immunoreactivity (R6/2, HD190QG, AAV-hypo) | [111, 112, 114, 208] |
| = | immunoreactivity, mRNA (BACHD) | [116] | |
| VIP | ↓ | Immunoreactivity, mRNA (R6/2) | [113] |
| VIP receptor | ↓ | mRNA (R6/2) | [113] |
HPA, hypothalamic–pituitary–adrenal; CART, cocaine and amphetamine regulated transcript; CRH, corticotrophin releasing hormone; GnRH, gonadotropin-releasing hormone; HAP1, huntingtin-associated protein 1; MCH, melanin-concentrating hormone; NPY, neuropeptide Y; POMC, proopiomelanocortin; TSH, thyroid stimulating hormone, VIP, vasoactive intestinal peptide.
CLINICAL TRIALS IN HD: FROM A HYPOTHALAMIC PATHOLOGY PERSPECTIVE
According to the clinical trials database (https://clinicaltrials.gov/), a total of 124 HD clinical trials have been completed to date worldwide with an additional 46 currently active or in the process of recruiting. These trials have largely focused on restoring motor or cognitive function and very few studies have focused on the non-motor disturbances. Gene therapy approaches to silence the disease-causing mutant HTT protein are currently at the forefront of treatment strategies for HD with the successful completion of the Phase 1/2a IONIS-HTTRx clinical trial (Ionis Pharmaceuticals) using intrathecal administration of antisense oligonucleotides (ASOs) to reduce both wild-type and mutant HTT [120]. A Phase 3 trial in adult patients with manifest HD (RG6042, Roche Pharmaceuticals) began in April 2019. The hypothalamus is located in a prime position anatomically, surrounding the third ventricle. Although it is likely that intrathecally-delivered ASOs can also target the hypothalamus, preclinical studies using different intrathecally-delivered HTT-lowering agents have not focused on whether there is target engagement in the hypothalamus. With the results of the ongoing HTT-lowering clinical trials, it will be imperative to determine whether intrathecally delivered ASOs reach the hypothalamus and if so, the extent to which this improves the non-motor symptoms and signs. In preparation for future preclinical and clinical trials with HTT-lowering agents, it is worth considering that hypothalamic pathology exists alongside or perhaps even before striatal and cortical pathology. It will be important to further investigate what brain regions need to be targeted to impact upon non-motor features of HD and to pinpoint a treatment timeframe during the life of an affected individual where it is still possible to prevent or reverse symptoms and eventually halt disease progression.
CONCLUSION
The hypothalamus and other areas of the limbic system are part of the brain regions affected in HD. The changes detected mainly in the hypothalamus include effects on factors that regulate motivated behavior, emotions, social cognition, sleep and/or metabolism with reduced levels of orexin, oxytocin, vasopressin, VIP and somatostatin and increased levels of SIRT1 in specific hypothalamic nuclei. Experimental studies in mice have begun to establish causative relationships between expression of mutant huntingtin in the hypothalamus and its effects on neuropeptide expression as well as in the development of depressive-like behavior and metabolic disturbances. Hence, pathology in the hypothalamus may have relevance for the development of non-motor features in HD. Further studies are still needed to investigate the overall extent of these changes in the limbic system and the potential of modulating affected targets for symptomatic effects and/or disease modification in HD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
RYC is supported by the Swedish Society for Medical Research and NEURO Sweden. SG is supported by the Swedish Brain Foundation. ÅP is supported by research grants from the Swedish Research Council, the Swedish Brain Foundation and the Region of Scania grants (ALF).
REFERENCES
[1] | Ross CA , Aylward EH , Wild EJ , Langbehn DR , Long JD , Warner JH , et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. (2014) ;10: (4):204–16. |
[2] | Duff K , Paulsen JS , Beglinger LJ , Langbehn DR , Stout JC , Predict-HD Investigators of the Huntington Study Group. Psychiatric symptoms in Huntington’s disease before diagnosis: The predict-HD study. Biol Psychiatry. (2007) ;62: (12):1341–6. |
[3] | Stout JC , Paulsen JS , Queller S , Solomon AC , Whitlock KB , Campbell JC , et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology. (2011) ;25: (1):1–14. |
[4] | Tabrizi SJ , Scahill RI , Durr A , Roos RA , Leavitt BR , Jones R , et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: The 12-month longitudinal analysis. Lancet Neurol. (2011) ;10: (1):31–42. |
[5] | van Duijn E , Kingma EM , van der Mast RC . Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. (2007) ;19: (4):441–8. |
[6] | Anderson KE , van Duijn E , Craufurd D , Drazinic C , Edmondson M , Goodman N , et al. Clinical management of neuropsychiatric symptoms of Huntington disease: Expert-based consensus guidelines on agitation, anxiety, apathy, psychosis and sleep disorders. J Huntingtons Dis. (2018) ;7: (3):355–66. |
[7] | Sprengelmeyer R , Young AW , Calder AJ , Karnat A , Lange H , Homberg V , et al. Loss of disgust. Perception of faces and emotions in Huntington’s disease. Brain. (1996) ;119: (Pt 5):1647–65. |
[8] | Sprengelmeyer R , Schroeder U , Young AW , Epplen JT . Disgust in pre-clinical Huntington’s disease: A longitudinal study. Neuropsychologia. (2006) ;44: (4):518–33. |
[9] | Johnson SA , Stout JC , Solomon AC , Langbehn DR , Aylward EH , Cruce CB , et al. Beyond disgust: Impaired recognition of negative emotions prior to diagnosis in Huntington’s disease. Brain. (2007) ;130: (Pt 7):1732–44. |
[10] | Henley SM , Wild EJ , Hobbs NZ , Warren JD , Frost C , Scahill RI , et al. Defective emotion recognition in early HD is neuropsychologically and anatomically generic. Neuropsychologia. (2008) ;46: (8):2152–60. |
[11] | Kempnich CL , Andrews SC , Fisher F , Wong D , Georgiou-Karistianis N , Stout JC . Emotion recognition correlates with social-neuropsychiatric dysfunction in Huntington’s disease. J Int Neuropsychol Soc. (2018) ;24: (5):417–23. |
[12] | van Duijn E , Craufurd D , Hubers AA , Giltay EJ , Bonelli R , Rickards H , et al. Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry. (2014) ;85: (12):1411–8. |
[13] | Morton AJ , Wood NI , Hastings MH , Hurelbrink C , Barker RA , Maywood ES . Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci. (2005) ;25: (1):157–63. |
[14] | Arnulf I , Nielsen J , Lohmann E , Schiefer J , Wild E , Jennum P , et al. Rapid eye movement sleep disturbances in Huntington disease. Arch Neurol. (2008) ;65: (4):482–8. |
[15] | Goodman AO , Morton AJ , Barker RA . Identifying sleep disturbances in Huntington’s disease using a simple disease-focused questionnaire. PLoS Curr. (2010) ;2: , RRN1189. |
[16] | Lazar AS , Panin F , Goodman AO , Lazic SE , Lazar ZI , Mason SL , et al. Sleep deficits but no metabolic deficits in premanifest Huntington’s disease. Ann Neurol. (2015) ;78: (4):630–48. |
[17] | Piano C , Losurdo A , Della Marca G , Solito M , Calandra-Buonaura G , Provini F , et al. Polysomnographic findings and clinical correlates in Huntington disease: A cross-sectional cohort study. Sleep. (2015) ;38: (9):1489–95. |
[18] | Bartlett DM , Dominguez DJ , Reyes A , Zaenker P , Feindel KW , Newton RU , et al. Investigating the relationships between hypothalamic volume and measures of circadian rhythm and habitual sleep in premanifest Huntington’s disease. Neurobiol Sleep Circadian Rhythms. (2019) ;6: , 1–8. |
[19] | Marder K , Zhao H , Eberly S , Tanner CM , Oakes D , Shoulson I , et al. Dietary intake in adults at risk for Huntington disease: Analysis of PHAROS research participants. Neurology. (2009) ;73: (5):385–92. |
[20] | Djousse L , Knowlton B , Cupples LA , Marder K , Shoulson I , Myers RH , et al. Weight loss in early stage of Huntington’s disease. Neurology. (2002) ;59: (9):1325–30. |
[21] | van der Burg JMM , Gardiner SL , Ludolph AC , Landwehrmeyer GB , Roos RAC , Aziz NA . Body weight is a robust predictor of clinical progression in Huntington disease. Ann Neurol. (2017) ;82: (3):479–83. |
[22] | Morgane PJ , Mokler DJ . The limbic brain: Continuing resolution. Neurosci Biobehav Rev. (2006) ;30: (2):119–25. |
[23] | Morgane PJ , Galler JR , Mokler DJ . A review of systems and networks of the limbic forebrain/limbic midbrain. Prog Neurobiol. (2005) ;75: (2):143–60. |
[24] | Petersen A , Bjorkqvist M . Hypothalamic-endocrine aspects in Huntington’s disease. Eur J Neurosci. (2006) ;24: (4):961–7. |
[25] | Hult S , Schultz K , Soylu R , Petersen A . Hypothalamic and neuroendocrine changes in Huntington’s disease. Curr Drug Targets. (2010) ;11: (10):1237–49. |
[26] | van Wamelen DJ , Aziz NA , Roos RA , Swaab DF . Hypothalamic alterations in Huntington’s disease patients: Comparison with genetic rodent models. J Neuroendocrinol. (2014) ;26: (11):761–75. |
[27] | Bartlett DM , Cruickshank TM , Hannan AJ , Eastwood PR , Lazar AS , Ziman MR . Neuroendocrine and neurotrophic signaling in Huntington’s disease: Implications for pathogenic mechanisms and treatment strategies. Neurosci Biobehav Rev. (2016) ;71: :444–54. |
[28] | Petersen A , Gabery S . Hypothalamic and limbic system changes in Huntington’s disease. J Huntingtons Dis. (2012) ;1: :5–16. |
[29] | Kassubek J , Juengling FD , Kioschies T , Henkel K , Karitzky J , Kramer B , et al. Topography of cerebral atrophy in early Huntington’s disease: A voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry. (2004) ;75: (2):213–20. |
[30] | Douaud G , Gaura V , Ribeiro MJ , Lethimonnier F , Maroy R , Verny C , et al. Distribution of grey matter atrophy in Huntington’s disease patients: A combined ROI-based and voxel-based morphometric study. Neuroimage. (2006) ;32: (4):1562–75. |
[31] | Soneson C , Fontes M , Zhou Y , Denisov V , Paulsen JS , Kirik D , et al. Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol Dis. (2010) ;40: (3):531–43. |
[32] | Politis M , Pavese N , Tai YF , Tabrizi SJ , Barker RA , Piccini P . Hypothalamic involvement in Huntington’s disease: An in vivo PET study. Brain. (2008) ;131: (Pt 11):2860–9. |
[33] | Politis M , Pavese N , Tai YF , Kiferle L , Mason SL , Brooks DJ , et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: A multimodal imaging study. Hum Brain Mapp. (2011) ;32: (2):258–70. |
[34] | Gabery S , Georgiou-Karistianis N , Lundh SH , Cheong RY , Churchyard A , Chua P , et al. Volumetric analysis of the hypothalamus in Huntington disease using 3T MRI: The IMAGE-HD Study. PLoS One. (2015) ;10: (2):e0117593. |
[35] | Gabery S , Murphy K , Schultz K , Loy CT , McCusker E , Kirik D , et al. Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol. (2010) ;120: (6):777–88. |
[36] | Kremer HP , Roos RA , Dingjan GM , Bots GT , Bruyn GW , Hofman MA . The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington’s disease. Neurosci Lett. (1991) ;132: (1):101–4. |
[37] | Kremer HP , Roos RA , Dingjan G , Marani E , Bots GT . Atrophy of the hypothalamic lateral tuberal nucleus in Huntington’s disease. J Neuropathol Exp Neurol. (1990) ;49: (4):371–82. |
[38] | Kremer HP . The hypothalamic lateral tuberal nucleus: Normal anatomy and changes in neurological diseases. Prog Brain Res. (1992) ;93: :249–61. |
[39] | Timmers HJ , Swaab DF , van de Nes JA , Kremer HP . Somatostatin 1-12 immunoreactivity is decreased in the hypothalamic lateral tuberal nucleus of Huntington’s disease patients. Brain Res. (1996) ;728: (2):141–8. |
[40] | Gabery S , Halliday G , Kirik D , Englund E , Petersen A . Selective loss of oxytocin and vasopressin in the hypothalamus in early Huntington disease: A case study. Neuropathol Appl Neurobiol. (2015) ;41: (6):843–8. |
[41] | Lopatina OL , Komleva YK , Gorina YV , Higashida H , Salmina AB . Neurobiological aspects of face recognition: The role of oxytocin. Front Behav Neurosci. (2018) ;12: :195. |
[42] | Labuschagne I , Jones R , Callaghan J , Whitehead D , Dumas EM , Say MJ , et al. Emotional face recognition deficits and medication effects in pre-manifest through stage-II Huntington’s disease. Psychiatr Res. (2013) ;207: (1-2):118–26. |
[43] | Labuschagne I , Poudel G , Kordsachia C , Wu Q , Thomson H , Georgiou-Karistianis N , et al. Oxytocin selectively modulates brain processing of disgust in Huntington’s disease gene carriers. Prog Neuropsychopharmacol Biol Psychiatry. (2018) ;81: :11–6. |
[44] | Ferris CF , Melloni RH Jr , Koppel G , Perry KW , Fuller RW , Delville Y . Vasopressin/serotonin interactions in the anterior hypothalamus control aggressive behavior in golden hamsters. J Neurosci. (1997) ;17: (11):4331–40. |
[45] | Ferris CF , Lu SF , Messenger T , Guillon CD , Heindel N , Miller M , et al. Orally active vasopressin V1a receptor antagonist, SRX251, selectively blocks aggressive behavior. Pharmacol Biochem Behav. (2006) ;83: (2):169–74. |
[46] | Anand BK , Brobeck JR . Hypothalamic control of food intake in rats and cats. Yale J Biol Med. (1951) ;24: (2):123–40. |
[47] | Teitelbaum P , Epstein AN . The lateral hypothalamic syndrome: Recovery of feeding and drinking after lateral hypothalamic lesions. Psychol Rev. (1962) ;69: :74–90. |
[48] | Boyle PC , Keesey RE . Chronically reduced body weight in rats sustaining lesions of the lateral hypothalamus and maintained on palatable diets and drinking solutions. J Comp Physiol Psychol. (1975) ;88: (1):218–23. |
[49] | Stuber GD , Wise RA . Lateral hypothalamic circuits for feeding and reward. Nat Neurosci. (2016) ;19: (2):198–205. |
[50] | Petersen A , Gil J , Maat-Schieman ML , Bjorkqvist M , Tanila H , Araujo IM , et al. Orexin loss in Huntington’s disease. Hum Mol Genet. (2005) ;14: (1):39–47. |
[51] | Aziz A , Fronczek R , Maat-Schieman M , Unmehopa U , Roelandse F , Overeem S , et al. Hypocretin and melanin-concentrating hormone in patients with Huntington disease. Brain Pathol. (2008) ;18: (4):474–83. |
[52] | Baldo B , Gabery S , Soylu-Kucharz R , Cheong RY , Henningsen JB , Englund E , et al. SIRT1 is increased in affected brain regions and hypothalamic metabolic pathways are altered in Huntington disease. Neuropathol Appl Neurobiol. (2019) ;45: (4):361–79. |
[53] | Tyree SM , Borniger JC , de Lecea L . Hypocretin as a hub for arousal and motivation. Front Neurol. (2018) ;9: :413. |
[54] | Haghparast A , Fatahi Z , Arezoomandan R , Karimi S , Taslimi Z , Zarrabian S . Functional roles of orexin/hypo-cretin receptors in reward circuit. Prog Brain Res. (2017) ;235: :139–54. |
[55] | Diniz GB , Bittencourt JC . The melanin-concentrating hormone as an integrative peptide driving motivated behaviors. Front Syst Neurosci. (2017) ;11. |
[56] | Park S , Colwell CS . Do disruptions in the circadian timing system contribute to autonomic dysfunction in Huntington’s disease? Yale J Biol Med. (2019) ;92: (2):291–303. |
[57] | van Wamelen DJ , Aziz NA , Anink JJ , van Steenhoven R , Angeloni D , Fraschini F , et al. Suprachiasmatic nucleus neuropeptide expression in patients with Huntington’s disease. Sleep. (2013) ;36: (1):117–25. |
[58] | Kim KW , Donato J Jr , Berglund ED , Choi YH , Kohno D , Elias CF , et al. FOXO1 in the ventromedial hypothalamus regulates energy balance. J Clin Invest. (2012) ;122: (7):2578–89. |
[59] | Lin D , Boyle MP , Dollar P , Lee H , Lein ES , Perona P , et al. Functional identification of an aggression locus in the mouse hypothalamus. Nature. (2013) ;470: (7333)221–6. |
[60] | Silva BA , Mattucci C , Krzywkowski P , Cuozzo R , Carbonari L , Gross CT . The ventromedial hypothalamus mediates predator fear memory. Eur J Neurosci. (2016) ;43: (11):1431–9. |
[61] | Silva BA , Mattucci C , Krzywkowski P , Murana E , Illarionova A , Grinevich V , et al. Independent hypothalamic circuits for social and predator fear. Nat Neurosci. (2013) ;16: (12):1731–3. |
[62] | Falkner AL , Dollar P , Perona P , Anderson DJ , Lin D . Decoding ventromedial hypothalamic neural activity during male mouse aggression. J Neurosci. (2014) ;34: (17):5971–84. |
[63] | Wang L , Chen IZ , Lin D . Collateral pathways from the ventromedial hypothalamus mediate defensive behaviors. Neuron. (2015) ;85: (6):1344–58. |
[64] | Kunwar PS , Zelikowsky M , Remedios R , Cai H , Yilmaz M , Meister M , et al. Ventromedial hypothalamic neurons control a defensive emotion state. Elife. (2015) ;4. |
[65] | Xu B , Goulding EH , Zang K , Cepoi D , Cone RD , Jones KR , et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. (2003) ;6: (7):736–42. |
[66] | Lyons WE , Mamounas LA , Ricaurte GA , Coppola V , Reid SW , Bora SH , et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. (1999) ;96: (26):15239–44. |
[67] | Unger TJ , Calderon GA , Bradley LC , Sena-Esteves M , Rios M . Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. (2007) ;27: (52):14265–74. |
[68] | Zuccato C , Ciammola A , Rigamonti D , Leavitt BR , Goffredo D , Conti L , et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. (2001) ;293: (5529)493–8. |
[69] | Smith MR , Syed A , Lukacsovich T , Purcell J , Barbaro BA , Worthge SA , et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum Mol Genet. (2014) ;23: (11):2995–3007. |
[70] | Sussmuth SD , Haider S , Landwehrmeyer GB , Farmer R , Frost C , Tripepi G , et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br J Clin Pharmacol. (2015) ;79: (3):465–76. |
[71] | Ramadori G , Lee CE , Bookout AL , Lee S , Williams KW , Anderson J , et al. Brain SIRT Anatomical distribution and regulation by energy availability. J Neurosci. (2008) ;28: (40):9989–96. |
[72] | Satoh A , Brace CS , Ben-Josef G , West T , Wozniak DF , Holtzman DM , et al. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci. (2010) ;30: (30):10220–32. |
[73] | Satoh A , Brace CS , Rensing N , Cliften P , Wozniak DF , Herzog ED , et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. (2013) ;18: (3):416–30. |
[74] | Nillni EA . The metabolic sensor Sirt1 and the hypothalamus: Interplay between peptide hormones and pro-hormone convertases. Mol Cell Endocrinol. (2016) ;438: :77–88. |
[75] | Kannike K , Sepp M , Zuccato C , Cattaneo E , Timmusk T . Forkhead transcription factor FOXO3a levels are increased in Huntington disease because of overactivated positive autofeedback loop. J Biol Chem. (2014) ;289: (47):32845–57. |
[76] | Scarpa JR , Jiang P , Losic B , Readhead B , Gao VD , Dudley JT , et al. Systems genetic analyses highlight a TGFbeta-FOXO3 dependent striatal astrocyte network conserved across species and associated with stress, sleep, and Huntington’s disease. PLoS Genet. (2016) ;12: (7):e1006137. |
[77] | Ahveninen LM , Stout JC , Georgiou-Karistianis N , Lorenzetti V , Glikmann-Johnston Y . Reduced amygdala volumes are related to motor and cognitive signs in Huntington’s disease: The IMAGE-HD study. Neuroimage Clin. (2018) ;18: :881–7. |
[78] | van den Bogaard SJ , Dumas EM , Acharya TP , Johnson H , Langbehn DR , Scahill RI , et al. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. (2011) ;258: (3):412–20. |
[79] | Rosas HD , Koroshetz WJ , Chen YI , Skeuse C , Vangel M , Cudkowicz ME , et al. Evidence for more widespread cerebral pathology in early HD: An MRI-based morphometric analysis. Neurology. (2003) ;60: (10):1615–20. |
[80] | Martinez-Horta S , Perez-Perez J , Sampedro F , Pagonabarraga J , Horta-Barba A , Carceller-Sindreu M , et al. Structural and metabolic brain correlates of apathy in Huntington’s disease. Mov Disord. (2018) ;33: (7):1151–9. |
[81] | Tang X , Ross CA , Johnson H , Paulsen JS , Younes L , Albin RL , et al. Regional subcortical shape analysis in premanifest Huntington’s disease. Hum Brain Mapp. (2019) ;40: (5):1419–33. |
[82] | Majid DS , Aron AR , Thompson W , Sheldon S , Hamza S , Stoffers D , et al. Basal ganglia atrophy in prodromal Huntington’s disease is detectable over one year using automated segmentation. Mov Disord. (2011) ;26: (14):2544–51. |
[83] | Zweig RM , Ross CA , Hedreen JC , Peyser C , Cardillo JE , Folstein SE , et al. Locus coeruleus involvement in Huntington’s disease. Arch Neurol. (1992) ;49: (2):152–6. |
[84] | Hobbs NZ , Pedrick AV , Say MJ , Frost C , Dar Santos R , Coleman A , et al. The structural involvement of the cingulate cortex in premanifest and early Huntington’s disease. Mov Disord. (2011) ;26: (9):1684–90. |
[85] | Ruocco HH , Bonilha L , Li LM , Lopes-Cendes I , Cendes F . Longitudinal analysis of regional grey matter loss in Huntington disease: Effects of the length of the expanded CAG repeat. J Neurol Neurosurg Psychiatry. (2008) ;79: (2):130–5. |
[86] | Gomez-Anson B , Alegret M , Munoz E , Monte GC , Alayrach E , Sanchez A , et al. Prefrontal cortex volume reduction on MRI in preclinical Huntington’s disease relates to visuomotor performance and CAG number. Parkinsonism Relat Disord. (2009) ;15: (3):213–9. |
[87] | Thu DC , Oorschot DE , Tippett LJ , Nana AL , Hogg VM , Synek BJ , et al. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain. (2010) ;133: (Pt 4):1094–110. |
[88] | Rajkowska G , Selemon LD , Goldman-Rakic PS . Neuronal and glial somal size in the prefrontal cortex: A postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. (1998) ;55: (3):215–24. |
[89] | Espinoza FA , Liu J , Ciarochi J , Turner JA , Vergara VM , Caprihan A , et al. Dynamic functional network connectivity in Huntington’s disease and its associations with motor and cognitive measures. Hum Brain Mapp. (2019) ;40: (6):1955–68. |
[90] | Wolf RC , Sambataro F , Vasic N , Schonfeldt-Lecuona C , Ecker D , Landwehrmeyer B . Altered frontostriatal coupling in pre-manifest Huntington’s disease: Effects of increasing cognitive load. Eur J Neurol. (2008) ;15: (11):1180–90. |
[91] | Wolf RC , Gron G , Sambataro F , Vasic N , Wolf ND , Thomann PA , et al. Brain activation and functional connectivity in premanifest Huntington’s disease during states of intrinsic and phasic alertness. Hum Brain Mapp. (2012) ;33: (9):2161–73. |
[92] | Thiruvady DR , Georgiou-Karistianis N , Egan GF , Ray S , Sritharan A , Farrow M , et al. Functional connectivity of the prefrontal cortex in Huntington’s disease. J Neurol Neurosurg Psychiatry. (2007) ;78: (2):127–33. |
[93] | Poudel GR , Driscoll S , Dominguez DJ , Stout JC , Churchyard A , Chua P , et al. Functional brain correlates of neuropsychiatric symptoms in presymptomatic Huntington’s disease: The IMAGE-HD Study. J Huntingtons Dis. (2015) ;4: (4):325–32. |
[94] | Henley SM , Wild EJ , Hobbs NZ , Frost C , MacManus DG , Barker RA , et al. Whole-brain atrophy as a measure of progression in premanifest and early Huntington’s disease. Mov Disord. (2009) ;24: (6):932–6. |
[95] | Henley SM , Wild EJ , Hobbs NZ , Scahill RI , Ridgway GR , Macmanus DG , et al. Relationship between CAG repeat length and brain volume in premanifest and early Huntington’s disease. J Neurol. (2009) ;256: (2):203–12. |
[96] | Enzi B , Edel MA , Lissek S , Peters S , Hoffmann R , Nicolas V , et al. Altered ventral striatal activation during reward and punishment processing in premanifest Huntington’s disease: A functional magnetic resonance study. Exp Neurol. (2012) ;235: (1):256–64. |
[97] | Malejko K , Weydt P , Sussmuth SD , Gron G , Landwehrmeyer BG , Abler B . Prodromal Huntington disease as a model for functional compensation of early neurodegeneration. PLoS One. (2014) ;9: (12). |
[98] | Mason SL , Zhang J , Begeti F , Guzman NV , Lazar AS , Rowe JB , et al. The role of the amygdala during emotional processing in Huntington’s disease: From pre-manifest to late stage disease. Neuropsychologia. (2015) ;70: :80–9. |
[99] | Coppen EM , van der Grond J , Hafkemeijer A , Rombouts SA , Roos RA . Early grey matter changes in structural covariance networks in Huntington’s disease. Neuroimage Clin. (2016) ;12: :806–14. |
[100] | Bjorkqvist M , Petersen A , Nielsen J , Ecker D , Mulder H , Hayden MR , et al. Cerebrospinal fluid levels of orexin-A are not a clinically useful biomarker for Huntington disease. Clin Genet. (2006) ;70: (1):78–9. |
[101] | Baumann CR , Hersberger M , Bassetti CL . Hypocretin-1 (orexin A) levels are normal in Huntington’s disease. J Neurol. (2006) ;253: (9):1232–3. |
[102] | Gaus SE , Lin L , Mignot E . CSF hypocretin levels are normal in Huntington’s disease patients. Sleep. (2005) ;28: (12):1607–8. |
[103] | Meier A , Mollenhauer B , Cohrs S , Rodenbeck A , Jordan W , Meller J , et al. Normal hypocretin-1 (orexin-A) levels in the cerebrospinal fluid of patients with Huntington’s disease. Brain Res. (2005) ;1063: (2):201–3. |
[104] | Kalliolia E , Silajdzic E , Nambron R , Costelloe SJ , Martin NG , Hill NR , et al. A 24-hour study of the hypothalamo-pituitary axes in Huntington’s disease. PLoS One. (2015) ;10: (10):e0138848. |
[105] | Bjorkqvist M , Leavitt BR , Nielsen JE , Landwehrmeyer B , Ecker D , Mulder H , et al. Cocaine- and amphetamine-regulated transcript is increased in Huntington disease. Mov Disord. (2007) ;22: (13):1952–4. |
[106] | Bjorkqvist M , Petersen A , Bacos K , Isaacs J , Norlen P , Gil J , et al. Progressive alterations in the hypothalamic-pituitary-adrenal axis in the R6/2 transgenic mouse model of Huntington’s disease. Hum Mol Genet. (2006) ;15: (10):1713–21. |
[107] | Aziz NA , Pijl H , Frolich M , van der Graaf AW , Roelfsema F , Roos RA . Increased hypothalamic-pituitary-adrenal axis activity in Huntington’s disease. J Clin Endocrinol Metab. (2009) ;94: (4):1223–8. |
[108] | Shirbin CA , Chua P , Churchyard A , Lowndes G , Hannan AJ , Pang TY , et al. Cortisol and depression in pre-diagnosed and early stage Huntington’s disease. Psychoneuroendocrinology. (2013) ;38: (11):2439–47. |
[109] | Hubers AA , van der Mast RC , Pereira AM , Roos RA , Veen LJ , Cobbaert CM , et al. Hypothalamic-pituitary-adrenal axis functioning in Huntington’s disease and its association with depressive symptoms and suicidality. J Neuroendocrinol. (2015) ;27: (3):234–44. |
[110] | van der Burg JM , Bacos K , Wood NI , Lindqvist A , Wierup N , Woodman B , et al. Increased metabolism in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. (2008) ;29: (1):41–51. |
[111] | Wood NI , Goodman AO , van der Burg JM , Gazeau V , Brundin P , Bjorkqvist M , et al. Increased thirst and drinking in Huntington’s disease and the R6/2 mouse. Brain Res Bull. (2008) ;76: (1-2):70–9. |
[112] | Yamanaka T , Tosaki A , Miyazaki H , Kurosawa M , Furukawa Y , Yamada M , et al. Mutant huntingtin fragment selectively suppresses Brn-2 POU domain transcription factor to mediate hypothalamic cell dysfunction. Hum Mol Genet. (2010) ;19: (11):2099–112. |
[113] | Fahrenkrug J , Popovic N , Georg B , Brundin P , Hannibal J . Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J Mol Neurosci. (2007) ;31: (2):139–48. |
[114] | Hult S , Soylu R , Bjorklund T , Belgardt BF , Mauer J , Bruning JC , et al. Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab. (2011) ;13: (4):428–39. |
[115] | Soylu-Kucharz R , Adlesic N , Baldo B , Kirik D , Petersen A . Hypothalamic overexpression of mutant huntingtin causes dysregulation of brown adipose tissue. Sci Rep. (2015) ;5: :14598. |
[116] | Hult Lundh S , Nilsson N , Soylu R , Kirik D , Petersen A . Hypothalamic expression of mutant huntingtin contributes to the development of depressive-like behavior in the BAC transgenic mouse model of Huntington’s disease. Hum Mol Genet. (2013) ;22: :3485–97. |
[117] | Lundh SH , Soylu R , Petersen A . Expression of mutant huntingtin in leptin receptor-expressing neurons does not control the metabolic and psychiatric phenotype of the BACHD mouse. PLoS One. (2012) ;7: (12):e51168. |
[118] | Baldo B , Cheong RY , Petersen A . Effects of deletion of mutant huntingtin in steroidogenic factor 1 neurons on the psychiatric and metabolic phenotype in the BACHD mouse model of Huntington disease. PLoS One. (2014) ;9: (10):e107691. |
[119] | Soylu-Kucharz R , Baldo B , Petersen A . Metabolic and behavioral effects of mutant huntingtin deletion in Sim1 neurons in the BACHD mouse model of Huntington’s disease. Sci Rep. (2016) ;6: :28322. |
[120] | Tabrizi SJ , Leavitt BR , Landwehrmeyer GB , Wild EJ , Saft C , Barker RA , et al. Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med. (2019) ;380: (24):2307–16. |
[121] | Kassubek J , Gaus W , Landwehrmeyer GB . Evidence for more widespread cerebral pathology in early HD: An MRI-based morphometric analysis. Neurology. (2004) ;62: (3):523–4; author reply 4. |
[122] | Peinemann A , Schuller S , Pohl C , Jahn T , Weindl A , Kassubek J . Executive dysfunction in early stages of Huntington’s disease is associated with striatal and insular atrophy: A neuropsychological and voxel-based morphometric study. J Neurol Sci. (2005) ;239: (1):11–9. |
[123] | Devon RS , Orban PC , Gerrow K , Barbieri MA , Schwab C , Cao LP , et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc Natl Acad Sci U S A. (2006) ;103: (25):9595–600. |
[124] | Bohanna I , Georgiou-Karistianis N , Egan GF . Connecti-vity-based segmentation of the striatum in Huntington’s disease: Vulnerability of motor pathways. Neurobiol Dis. (2011) ;42: (3):475–81. |
[125] | van Duijn E , Selis MA , Giltay EJ , Zitman FG , Roos RA , van Pelt H , et al. Hypothalamic-pituitary-adrenal axis functioning in Huntington’s disease mutation carriers compared with mutation-negative first-degree controls. Brain Res Bull. (2010) ;83: (5):232–7. |
[126] | Heuser IJ , Chase TN , Mouradian MM . The limbic-hypothalamic-pituitary-adrenal axis in Huntington’s disease. Biol Psychiatry. (1991) ;30: (9):943–52. |
[127] | Bellosta Diago E , Perez Perez J , Santos Lasaosa S , Viloria Alebesque A , Martinez Horta S , Kulisevsky J , et al. Circadian rhythm and autonomic dysfunction in presymptomatic and early Huntington’s disease. Parkinsonism Relat Disord. (2017) ;44: :95–100. |
[128] | van Wamelen DJ , Aziz NA , Anink JJ , Roos RA , Swaab DF . Neuropeptide alterations in the infundibular nucleus of Huntington’s disease patients. J Neuroendocrinol. (2013) ;25: (2):198–205. |
[129] | van Wamelen DJ , Aziz NA , Anink JJ , Roos RA , Swaab DF . Paraventricular nucleus neuropeptide expression in Huntington’s disease patients. Brain Pathol. (2012) ;22: (5):654–61. |
[130] | Roos RA , Aziz NA . Hypocretin-1 and secondary signs in Huntington’s disease. Parkinsonism Relat Disord. (2007) ;13: (Suppl 3):S387–90. |
[131] | Sawiak SJ , Wood NI , Williams GB , Morton AJ , Carpenter TA . Voxel-based morphometry in the R6/2 transgenic mouse reveals differences between genotypes not seen with manual 2D morphometry. Neurobiol Dis. (2009) ;33: (1):20–7. |
[132] | Sawiak SJ , Wood NI , Williams GB , Morton AJ , Carpenter TA . Use of magnetic resonance imaging for anatomical phenotyping of the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. (2009) ;33: (1):12–9. |
[133] | Duan W , Peng Q , Masuda N , Ford E , Tryggestad E , Ladenheim B , et al. Sertraline slows disease progression and increases neurogenesis in N171-82Q mouse model of Huntington’s disease. Neurobiol Dis. (2008) ;30: (3):312–22. |
[134] | Gil JM , Mohapel P , Araujo IM , Popovic N , Li JY , Brundin P , et al. Reduced hippocampal neurogenesis in R6/2 transgenic Huntington’s disease mice. Neurobiol Dis. (2005) ;20: (3):744–51. |
[135] | Lazic SE , Grote H , Armstrong RJ , Blakemore C , Hannan AJ , van Dellen A , et al. Decreased hippocampal cell proliferation in R6/1 Huntington’s mice. Neuroreport. (2004) ;15: (5):811–3. |
[136] | Peng Q , Masuda N , Jiang M , Li Q , Zhao M , Ross CA , et al. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp Neurol. (2008) ;210: (1):154–63. |
[137] | Simpson JM , Gil-Mohapel J , Pouladi MA , Ghilan M , Xie Y , Hayden MR , et al. Altered adult hippocampal neurogenesis in the YAC128 transgenic mouse model of Huntington disease. Neurobiol Dis. (2011) ;41: (2):249–60. |
[138] | Fedele V , Roybon L , Nordstrom U , Li JY , Brundin P . Neurogenesis in the R6/2 mouse model of Huntington’s disease is impaired at the level of NeuroD1. Neuroscience. (2011) ;173: :76–81. |
[139] | Hannan AJ , Ransome MI . Deficits in spermatogenesis but not neurogenesis are alleviated by chronic testosterone therapy in R6/1 Huntington’s disease mice. J Neuroendocrinol. (2012) ;24: (2):341–56. |
[140] | Zajac MS , Pang TY , Wong N , Weinrich B , Leang LS , Craig JM , et al. Wheel running and environmental enrichment differentially modify exon-specific BDNF expression in the hippocampus of wild-type and pre-motor symptomatic male and female Huntington’s disease mice. Hippocampus. (2010) ;20: (5):621–36. |
[141] | Zhang H , Zhang C , Vincent J , Zala D , Benstaali C , Sainlos M , et al. Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington’s disease models. Nat Commun. (2018) ;9: (1):4272. |
[142] | Pang TY , Stam NC , Nithianantharajah J , Howard ML , Hannan AJ . Differential effects of voluntary physical exercise on behavioral and brain-derived neurotrophic factor expression deficits in Huntington’s disease transgenic mice. Neuroscience. (2006) ;141: (2):569–84. |
[143] | Ma Q , Yang J , Li T , Milner TA , Hempstead BL . Selective reduction of striatal mature BDNF without induction of proBDNF in the zQ175 mouse model of Huntington’s disease. Neurobiol Dis. (2015) ;82: :466–77. |
[144] | Bulley SJ , Drew CJ , Morton AJ . Direct visualisation of abnormal dendritic spine morphology in the hippocampus of the R6/2 transgenic mouse model of Huntington’s disease. J Huntingtons Dis. (2012) ;1: (2):267–73. |
[145] | Simmons DA , Casale M , Alcon B , Pham N , Narayan N , Lynch G . Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. (2007) ;55: (10):1074–84. |
[146] | van der Borght K , Brundin P . Reduced expression of PSA-NCAM in the hippocampus and piriform cortex of the R6/1 and R6/2 mouse models of Huntington’s disease. Exp Neurol. (2007) ;204: (1):473–8. |
[147] | Mochel F , Durant B , Durr A , Schiffmann R . Altered dopamine and serotonin metabolism in motorically asymptomatic R6/2 mice. PLoS One. (2011) ;6: (3):e18336. |
[148] | Pang TY , Du X , Zajac MS , Howard ML , Hannan AJ . Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington’s disease. Hum Mol Genet. (2009) ;18: (4):753–66. |
[149] | Faure A , Hohn S , Von Horsten S , Delatour B , Raber K , Le Blanc P , et al. Altered emotional and motivational processing in the transgenic rat model for Huntington’s disease. Neurobiol Learn Mem. (2011) ;95: (1):92–101. |
[150] | Pavese N , Andrews TC , Brooks DJ , Ho AK , Rosser AE , Barker RA , et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: A PET study. Brain. (2003) ;126: (Pt 5):1127–35. |
[151] | Nemeroff CB , Youngblood WW , Manberg PJ , Prange AJ Jr , Kizer JS . Regional brain concentrations of neuropeptides in Huntington’s chorea and schizophrenia. Science. (1983) ;221: (4614):972–5. |
[152] | Coppen EM , Jacobs M , van den Berg-Huysmans AA , van der Grond J , Roos RAC . Grey matter volume loss is associated with specific clinical motor signs in Huntington’s disease. Parkinsonism Relat Disord. (2018) ;46: :56–61. |
[153] | Zlebnik NE , Gildish I , Sesia T , Fitoussi A , Cole EA , Carson BP , et al. Motivational impairment is accompanied by corticoaccumbal dysfunction in the BACHD-Tg5 rat model of Huntington’s disease. Cereb Cortex. 2019; doi: 10.1093/cercor/bhz009 |
[154] | Manfre G , Novati A , Faccini I , Rossetti AC , Bosch K , Molteni R , et al. BACHD rats expressing full-length mutant huntingtin exhibit differences in social behavior compared to wild-type littermates. PLoS One. (2018) ;13: (2):e0192289. |
[155] | Jahanshahi A , Vlamings R , van Roon-Mom WM , Faull RL , Waldvogel HJ , Janssen ML , et al. Changes in brainstem serotonergic and dopaminergic cell populations in experimental and clinical Huntington’s disease. Neuroscience. (2013) ;238: :71–81. |
[156] | Beal MF , Bird ED , Langlais PJ , Martin JB . Somatostatin is increased in the nucleus accumbens in Huntington’s disease. Neurology. (1984) ;34: (5):663–6. |
[157] | Huguet G , Temel Y , Kadar E , Pol S , Casaca-Carreira J , Segura-Torres P , et al. Altered expression of dopaminergic cell fate regulating genes prior to manifestation of symptoms in a transgenic rat model of Huntington’s disease. Brain Res. (2019) ;1712: :101–8. |
[158] | Jahanshahi A , Vlamings R , Kaya AH , Lim LW , Janssen ML , Tan S , et al. Hyperdopaminergic status in experimental Huntington disease. J Neuropathol Exp Neurol. (2010) ;69: (9):910–7. |
[159] | van Dellen A , Cordery PM , Spires TL , Blakemore C , Hannan AJ . Wheel running from a juvenile age delays onset of specific motor deficits but does not alter protein aggregate density in a mouse model of Huntington’s disease. BMC Neurosci. (2008) ;9: :34. |
[160] | Reading SA , Dziorny AC , Peroutka LA , Schreiber M , Gourley LM , Yallapragada V , et al. Functional brain changes in presymptomatic Huntington’s disease. Ann Neurol. (2004) ;55: (6):879–83. |
[161] | Zimbelman JL , Paulsen JS , Mikos A , Reynolds NC , Hoffmann RG , Rao SM . fMRI detection of early neural dysfunction in preclinical Huntington’s disease. J Int Neuropsychol Soc. (2007) ;13: (5):758–69. |
[162] | Paulsen JS , Zimbelman JL , Hinton SC , Langbehn DR , Leveroni CL , Benjamin ML , et al. fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington’s disease. AJNR Am J Neuroradiol. (2004) ;25: (10):1715–21. |
[163] | Georgiou-Karistianis N , Sritharan A , Farrow M , Cunnington R , Stout J , Bradshaw J , et al. Increased cortical recruitment in Huntington’s disease using a Simon task. Neuropsychologia. (2007) ;45: (8):1791–800. |
[164] | Georgiou-Karistianis N , Poudel GR , Dominguez DJ , Langmaid R , Gray MA , Churchyard A , et al. Functional and connectivity changes during working memory in Huntington’s disease: 18 month longitudinal data from the IMAGE-HD study. Brain Cogn. (2013) ;83: (1):80–91. |
[165] | Jin J , Peng Q , Hou Z , Jiang M , Wang X , Langseth AJ , et al. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum Mol Genet. (2015) ;24: (9):2508–27. |
[166] | Muhlau M , Weindl A , Wohlschlager AM , Gaser C , Stadtler M , Valet M , et al. Voxel-based morphometry indicates relative preservation of the limbic prefrontal cortex in early Huntington disease. J Neural Transm (Vienna). (2007) ;114: (3):367–72. |
[167] | Matsui JT , Vaidya JG , Wassermann D , Kim RE , Magnotta VA , Johnson HJ , et al. Prefrontal cortex white matter tracts in prodromal Huntington disease. Hum Brain Mapp. (2015) ;36: (10):3717–32. |
[168] | Bourbon-Teles J , Bells S , Jones DK , Coulthard E , Rosser A , Metzler-Baddeley C . Myelin breakdown in human Huntington’s disease: Multi-modal evidence from diffusion MRI and quantitative magnetization transfer. Neuroscience. (2019) ;403: :79–92. |
[169] | Wolf RC , Vasic N , Schonfeldt-Lecuona C , Landwehr-meyer GB , Ecker D . Dorsolateral prefrontal cortex dysfunction in presymptomatic Huntington’s disease: Evidence from event-related fMRI. Brain. (2007) ;130: (Pt 11):2845–57. |
[170] | Walker AG , Ummel JR , Rebec GV . Reduced expression of conditioned fear in the R6/2 mouse model of Huntington’s disease is related to abnormal activity in prelimbic cortex. Neurobiol Dis. (2011) ;43: (2):379–87. |
[171] | Hohn S , Dallerac G , Faure A , Urbach YK , Nguyen HP , Riess O , et al. Behavioral and in vivo electrophysiological evidence for presymptomatic alteration of prefrontostriatal processing in the transgenic rat model for huntington disease. J Neurosci. (2011) ;31: (24):8986–97. |
[172] | DiProspero NA , Chen EY , Charles V , Plomann M , Kordower JH , Tagle DA . Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. (2004) ;33: (5):517–33. |
[173] | Smith GA , Rocha EM , McLean JR , Hayes MA , Izen SC , Isacson O , et al. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum Mol Genet. (2014) ;23: (17):4510–27. |
[174] | Hassel B , Tessler S , Faull RL , Emson PC . Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem Res. (2008) ;33: (2):232–7. |
[175] | Zuccato C , Marullo M , Conforti P , MacDonald ME , Tartari M , Cattaneo E . Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. (2008) ;18: (2):225–38. |
[176] | Saleh N , Moutereau S , Durr A , Krystkowiak P , Azulay JP , Tranchant C , et al. Neuroendocrine disturbances in Huntington’s disease. PLoS One. (2009) ;4: (3):e4962. |
[177] | Wang R , Ross CA , Cai H , Cong WN , Daimon CM , Carlson OD , et al. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front Physiol. (2014) ;5: :231. |
[178] | Leblhuber F , Peichl M , Neubauer C , Reisecker F , Steinparz FX , Windhager E , et al. Serum dehydroepiandrosterone and cortisol measurements in Huntington’s chorea. J Neurol Sci. (1995) ;132: (1):76–9. |
[179] | Adamczak-Ratajczak A , Kupsz J , Owecki M , Zielonka D , Sowinska A , Checinska-Maciejewska Z , et al. Circadian rhythms of melatonin and cortisol in manifest Huntington’s disease and in acute cortical ischemic stroke. J Physiol Pharmacol. (2017) ;68: (4):539–46. |
[180] | Renoir T , Pang TY , Zajac MS , Chan G , Du X , Leang L , et al. Treatment of depressive-like behaviour in Huntington’s disease mice by chronic sertraline and exercise. Br J Pharmacol. (2012) ;165: (5):1375–89. |
[181] | Du X , Pang TY , Mo C , Renoir T , Wright DJ , Hannan AJ . The influence of the HPG axis on stress response and depressive-like behaviour in a transgenic mouse model of Huntington’s disease. Exp Neurol. (2014) ;263C: :63–71. |
[182] | Aziz NA , Pijl H , Frolich M , Schroder-van der Elst JP , van der Bent C , Roelfsema F , et al. Growth hormone and ghrelin secretion are associated with clinical severity in Huntington’s disease. Eur J Neurol. (2010) ;17: (2):280–8. |
[183] | Popovic V , Svetel M , Djurovic M , Petrovic S , Doknic M , Pekic S , et al. Circulating and cerebrospinal fluid ghrelin and leptin: Potential role in altered body weight in Huntington’s disease. Eur J Endocrinol. (2004) ;151: (4):451–5 |
[184] | Pouladi MA , Stanek LM , Xie Y , Franciosi S , Southwell AL , Deng Y , et al. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum Mol Genet. (2012) ;21: (10):2219–32. |
[185] | Wagner L , Bjorkqvist M , Lundh SH , Wolf R , Borgel A , Schlenzig D , et al. Neuropeptide Y (NPY) in cerebrospinal fluid from patients with Huntington’s disease: Increased NPY levels and differential degradation of the NPY1-30 fragment. J Neurochem. (2016) ;137: (5):820–37. |
[186] | Aziz NA , Pijl H , Frolich M , Roelfsema F , Roos RA . Altered thyrotropic and lactotropic axes regulation in Huntington’s disease. Clin Endocrinol (Oxf). (2010) ;73: (4):540–5. |
[187] | Markianos M , Panas M , Kalfakis N , Vassilopoulos D . Plasma homovanillic acid and prolactin in Huntington’s disease. Neurochem Res. (2009) ;34: (5):917–22. |
[188] | Markianos M , Panas M , Kalfakis N , Vassilopoulos D . Plasma testosterone, dehydroepiandrosterone sulfate, and cortisol in female patients with Huntington’s disease. Neuro Endocrinol Lett. (2007) ;28: (2):199–203. |
[189] | Papalexi E , Persson A , Bjorkqvist M , Petersen A , Woodman B , Bates GP , et al. Reduction of GnRH and infertility in the R6/2 mouse model of Huntington’s disease. Eur J Neurosci. (2005) ;22: (6):1541–6. |
[190] | Bode FJ , Stephan M , Suhling H , Pabst R , Straub RH , Raber KA , et al. Sex differences in a transgenic rat model of Huntington’s disease: Decreased 17beta-estradiol levels correlate with reduced numbers of DARPP32+ neurons in males. Hum Mol Genet. (2008) ;17: (17):2595–609. |
[191] | Markianos M , Panas M , Kalfakis N , Vassilopoulos D . Plasma testosterone in male patients with Huntington’s disease: Relations to severity of illness and dementia. Ann Neurol. (2005) ;57: (4):520–5. |
[192] | Martin B , Golden E , Carlson OD , Pistell P , Zhou J , Kim W , et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes. (2009) ;58: (2):318–28. |
[193] | Duarte AI , Sjogren M , Santos MS , Oliveira CR , Moreira PI , Bjorkqvist M . Dual therapy with liraglutide and ghrelin promotes brain and peripheral energy metabolism in the R6/2 mouse model of Huntington’s disease. Sci Rep. (2018) ;8: (1):8961. |
[194] | Sjogren M , Duarte AI , McCourt AC , Shcherbina L , Wierup N , Bjorkqvist M . Ghrelin rescues skeletal muscle catabolic profile in the R6/2 mouse model of Huntington’s disease. Sci Rep. (2017) ;7: (1):13896. |
[195] | Josefsen K , Nielsen MD , Jorgensen KH , Bock T , Norremolle A , Sorensen SA , et al. Impaired glucose tolerance in the R6/1 transgenic mouse model of Huntington’s disease. J Neuroendocrinol. (2008) ;20: (2):165–72. |
[196] | Aziz NA , Pijl H , Frolich M , Snel M , Streefland TC , Roelfsema F , et al. Systemic energy homeostasis in Huntington’s disease patients. J Neurol Neurosurg Psychiatry. (2010) ;81: (11):1233–7. |
[197] | Aziz NA , Pijl H , Frolich M , van der Graaf AW , Roelfsema F , Roos RA . Leptin secretion rate increases with higher CAG repeat number in Huntington’s disease patients. Clin Endocrinol (Oxf). (2010) ;73: (2):206–11. |
[198] | Kudwa AE , Menalled LB , Oakeshott S , Murphy C , Mushlin R , Fitzpatrick J , et al. Increased body weight of the BAC HD transgenic mouse model of Huntington’s disease accounts for some but not all of the observed HD-like motor deficits. PLoS Curr. (2013) ;5. |
[199] | Wegrzynowicz M , Bichell TJ , Soares BD , Loth MK , McGlothan JS , Mori S , et al. Novel BAC mouse model of Huntington’s disease with 225 CAG repeats exhibits an early widespread and stable degenerative phenotype. J Huntingtons Dis. (2015) ;4: (1):17–36. |
[200] | Phan J , Hickey MA , Zhang PX , Chesselet MF , Reue K . Adipose tissue dysfunction tracks disease progression in two Huntington’s disease mouse models. Hum Mol Genet. (2009) ;18: (6):1006–16. |
[201] | Zacharoff L , Tkac I , Song Q , Tang C , Bolan PJ , Mangia S , et al. Cortical metabolites as biomarkers in the R6/2 model of Huntington’s disease. J Cereb Blood Flow Metab. (2012) ;32: (3):502–14. |
[202] | Li SH , Yu ZX , Li CL , Nguyen HP , Zhou YX , Deng C , et al. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington’s disease. J Neurosci. (2003) ;23: (17):6956–64. |
[203] | Kudo T , Schroeder A , Loh DH , Kuljis D , Jordan MC , Roos KP , et al. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp Neurol. (2011) ;228: (1):80–90. |
[204] | Kuljis DA , Gad L , Loh DH , MacDowell Kaswan Z , Hitchcock ON , Ghiani CA , et al. Sex differences in circadian dysfunction in the BACHD mouse model of Huntington’s disease. PLoS One. (2016) ;11: (2):e0147583. |
[205] | Loh DH , Kudo T , Truong D , Wu YF , Colwell CS . The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS One. (2013) ;8: (7). |
[206] | Wang HB , Loh DH , Whittaker DS , Cutler T , Howland D , Colwell CS . Time-restricted feeding improves circadian dysfunction as well as motor symptoms in the Q175 mouse model of Huntington’s Disease. Eneuro. (2018) ;5: (1), ENEURO.0431-17.2017. |
[207] | Schroeder AM , Wang HB , Park S , Jordan MC , Gao F , Coppola G , et al. Cardiac dysfunction in the BACHD mouse model of Huntington’s disease. PLoS One. (2016) ;11: (1):e0147269. |
[208] | Kotliarova S , Jana NR , Sakamoto N , Kurosawa M , Miyazaki H , Nekooki M , et al. Decreased expression of hypothalamic neuropeptides in Huntington disease transgenic mice with expanded polyglutamine-EGFP fluorescent aggregates. J Neurochem. (2005) ;93: (3):641–53. |
[209] | Maywood ES , Fraenkel E , McAllister CJ , Wood N , Reddy AB , Hastings MH , et al. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci. (2010) ;30: (30):10199–204. |
[210] | Pallier PN , Maywood ES , Zheng Z , Chesham JE , Inyushkin AN , Dyball R , et al. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J Neurosci. (2007) ;27: (29):7869–78. |
[211] | Williams RH , Morton AJ , Burdakov D . Paradoxical function of orexin/hypocretin circuits in a mouse model of Huntington’s disease. Neurobiol Dis. (2011) ;42: (3):438–45. |


