
Progressive Polyglutamine Repeat Expansion in Peripheral Blood Cells and Sperm of Transgenic Huntington’s Disease Monkeys
Abstract
The expanded CAG repeat results in somatic mosaicism and genetic anticipation in Huntington’s disease (HD). Here we report a longitudinal study examining CAG repeat instability in lymphocytes and sperm of three HD monkeys throughout their whole life-span that encompass the prodromal to symptomatic stages of HD. We demonstrate a progressive increase in CAG repeat length in lymphocytes and sperm as the animals aged. We also examined the impact of CAG repeat length on expansion rate, which showed a clear linear correlation up to 62Q, and high instability after. Our findings stress the importance of further investigation in CAG instability in peripheral blood cells longitudinally.
Huntington’s disease (HD) is a monogenic, autosomal dominant disease characterized by progressive neurodegeneration that causes behavioral, cognitive, and motor dysfunction [1, 2]. HD symptoms generally arise in a range of ages but typically around 40 years of age, with death following 15 years after symptoms manifest. The pathological mutation responsible for HD is the expansion of CAG trinucleotide repeats (TNRs) in the exon 1 of the huntingtin (HTT; IT15) gene that results in an expanded polyglutamine (polyQ) residue at the N-terminus of the HTT protein [2, 3]. In humans, the general population has a CAG repeat length below 35 repeats, the intermediate length is between 36 and 39 repeats, and the full-mutation is above 40 repeats [2–7]. Due to a large variation in the development of neurological symptoms, the intermediate group can develop motor, cognitive, and behavioral abnormalities, and asymptomatic patients can pass on the full-mutation to its offspring in a process called genetic anticipation through a paternal lineage [8–10]. HTT is expressed ubiquitously, although the highest levels are found in the brain and testes [11]. HD primarily affects the brain, with striatal medium spiny neurons being the most vulnerable [2, 4, 7, 12].
CAG repeat length is inversely correlated with the age of onset, where adult-onset patients have between 50 and 60 repeats, and juvenile-onset patients have over 70 repeats [1, 2, 4–7, 13, 14]. However, as patient ages, CAG repeat stability varies among cell types, leading to somatic mosaicism [15]. Among different brain regions, the striatum has the highest instability, which is consistent with motor and cognitive-behavioral symptoms [15]. In fact, high CAG repeat instability observed in sperm has been correlated with the genomic instability of the male germ cells during spermatogenesis, which accounts for the genetic anticipation reported in paternally inherited HD [15–19].
A longitudinal study on CAG repeat instability in human HD patient peripheral blood is very limited. One study reported that HD patients’ leukocytes were less heterogeneous than sperm, and increased instability in sperm was positively correlated with the increase of CAG repeat length [15]. A longitudinal study in a Venezuelan cohort reported similar findings, with very little instability in HD patients’ lymphoblastoid cells over ten years or in sperm over two years [18]. Here, we investigated CAG repeat instability by measuring CAG repeat length in lymphocytes and sperm collected throughout the nine-year lifespan of a group of transgenic HD monkeys, which has been shown to resemble human disease progression based on clinical measurements including behavioral and cognitive declines and progressive anatomical changes including regional brain atrophy and alterations in white matter [20–27].
Lymphocyte samples were collected every two years after birth (rHD6-8) while sperm samples were collected approximately every two years, starting at the age of four when the monkeys reached puberty (rHD6, 8). The sperm of rHD7 was not available because of the manifestation of HD and was euthanized at five years of age. DNA isolated from each sample was used for PCR amplification of the polyQ domain of both endogenous HTT and mutant HTT (mHTT) genes. All Genescan electrograms are displayed in Supplementary Figures 1 and 2.
Fig. 1
Longitudinal assessment of CAG trinucleotide repeats expansion in lymphocytes and sperm. A) CAG repeat expansion in lymphocyte samples collected throughout the life-span of the HD monkeys showing the gradual increase in the CAG trinucleotide repeat size. B) CAG repeat expansion in sperm samples collected throughout the life-span of the HD monkeys showing the gradual increase in the CAG trinucleotide repeat size. Blue lines represent electrogram data from the capillary electrophoresis, green lines represent individual curve, and red lines representthe overall curve-fit result
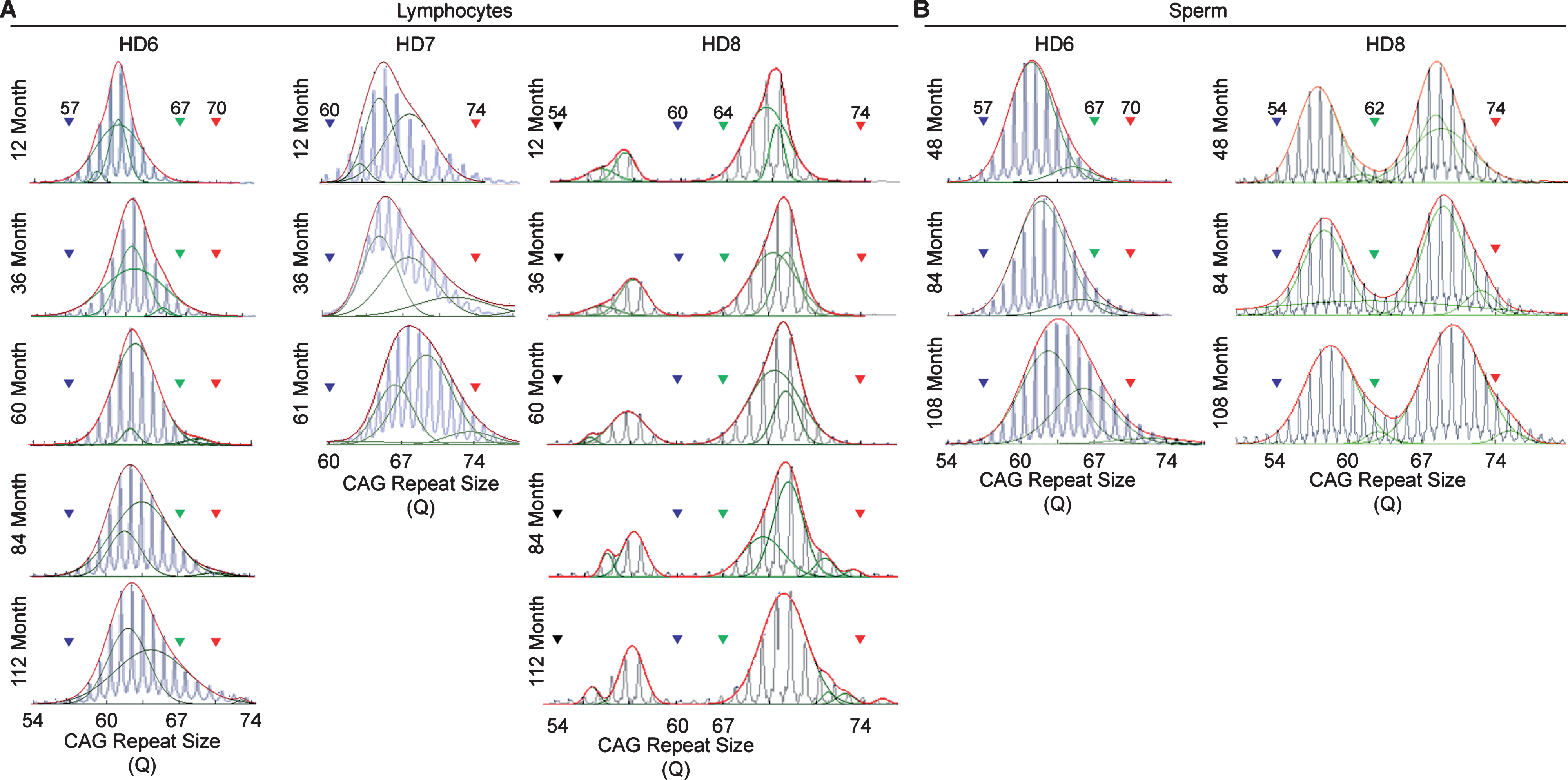
Fig. 2
The relationship between age of the HD monkeys and the repeat size. A) The age and CAG repeat size show a positive linear relationship in all three monkeys. B) The age and CAG repeat size in sperm show a positive linear relationship. Both lymphocytes and sperm showed larger allele with greater positive linear relationship than the smaller alleles. Curve 1-to-6 are the individual green curve shown in Figure 1.
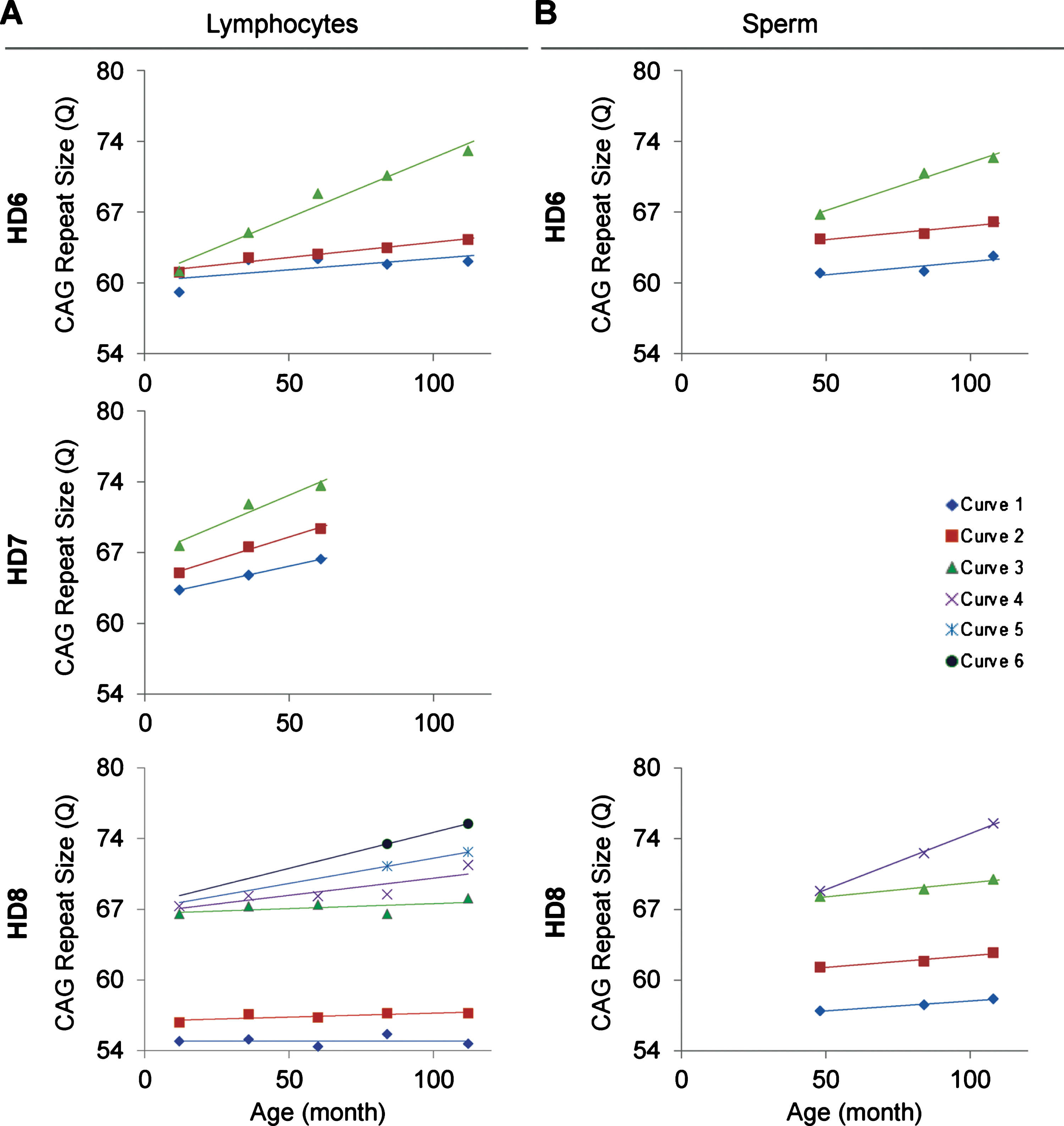
In order to resolve expanded alleles from the stable alleles, we used curve-fit method to capture multiple expansion episodes in our samples [28]. To resolve any overlapping CAG alleles that were evolved from the original mutant CAG allele, a normal distribution curves (Gaussian) were fitted to the overall data. The best fit for the overall data was determined based on the error and goodness of fit (R2). Individual curves represent alleles with different CAG repeat sizes derived from the original mutant HTT alleles [28]. Both the raw capillary electrophoresis electrogram data and curve-fit analyses showed a gradual increase of CAG repeat size in both lymphocytes (Fig. 1A) and sperm (Fig. 1B) throughout their lifespan. From the age of 12-to 112-month, rHD6 showed an addition of 2 CAG repeats (Qs; Fig. 2A top-blue diamond) in the allele with the smallest rate of CAG expansion to 11 Qs (Fig. 2A top-green triangle) in the allele with the highest rate of CAG expansion in lymphocytes (Fig. 2A, top). From the age of 12-to 61-month, rHD7 showed an addition of 2 (Fig. 2A middle-blue diamond) to 5 (Fig. 2A middle-green triangle) Qs in lymphocytes (Fig. 2A middle). Unlike rHD6 and rHD7, rHD8 had two independent insertions of the mutant HTT transgene that carry different CAG repeat sizes and expanded at different rate over time. From the age of 12-to 112-month, rHD8 showed an addition of 1 to 4 Qs in the lymphocytes (Fig. 2A bottom). From 84 month, two additional alleles with different CAG repeat size were emerged in rHD8 lymphocytes (Figs. 1A and Fig. 2A bottom). In sperm samples, rHD6 showed an addition of 2 (Fig. 2B top-blue diamond) to 5 (Fig. 2B top-green triangle) Qs from the age of 48-to 112-month (Fig. 2B top). Similar to rHD6, rHD8 showed an addition of 1 (Fig. 2B bottom-blue diamond) to 7 (Fig. 2B bottom-purple cross) Qs in the same time period as HD6 (Fig. 2B bottom). Both lymphocytes and sperm samples showed a positive correlation between CAG repeat size and animals’ age. In order to demonstrate the impact of CAG repeat size in TNR instability, we further investigated the correlation between the expansion rates (slopes of the lines shown in Fig. 2) and the initial allele sizes at the earliest time point (Supplementary Table 1 and Fig. 3). A linear regression correlation was calculated using the rolling window until the linear repression correlation coefficient start to lose statistical significant. In lymphocytes, there was a strong statistically significant positive correlation between initial allele size and expansion rate up to 62Q (R2 = 0.9991, P = 0.0005. Figure 3A, blue line). However, no correlation and statistically significance were found after 62Q (R2 = 0.05256, P = 0.5529, Fig. 3A, red line). The overall trend showed a weak and no statistically significant correlation (R2 = 0.2320, P = 0.0956, Fig. 3A, black line). In sperm samples, no statistically significant correlation was found either after 62Q or in overall data (Fig. 3B). When both lymphocytes and sperm data were combined, a strong statistically significant positive correlation was found before 62Q (R2 = 0.9239, P = 0.002, Fig. 3C, blue line). However, no statistically significant correlation was found after 62Q (R2 = 0.006637, P = 0.9347, Fig. 3C, red line). Overall data showed a statistically significant but a weak positive correlation between the initial allele size and expansion rate (R2 = 0.2464, P = 0.0306, Fig. 3C, blackline).
Fig. 3
Relationship between the expansion rates to the initial allele size. A) CAG repeat expansion rate in lymphocytes showed a positive correlation up to 62Q (blue dotted line, R2 = 0.9991, P = 0.0005) while the alleles sized larger than 62 showed no clear correlation (red dotted line, R2 = 0.0526, P = 0.5529). Overall trend showed generally positive correlation, but no statistical significant was found (black dotted line, R2 = 0.232, P = 0.0956). B) CAG repeat expansion rate in sperm showed no clear correlation after 62Q (red dotted line, R2 = 0.3762, P = 0.2712). Overall trend showed generally positive correlation, but no statistical significant was found (black dotted line, R2 = 0.447, P = 0.1034). C) Combined samples of lymphocytes and sperm samples showed a strong statistically significant positive correlation up to 62Q (blue dotted line, R2 = 0.9239, P = 0.0022) while the alleles sized larger than 62 showed no clear correlation (red dotted line, R2 = 0.006637, P = 0.9347). Overall trend showed statistically significant positive correlation (black dotted line, R2 = 0.2464, P = 0.0306).
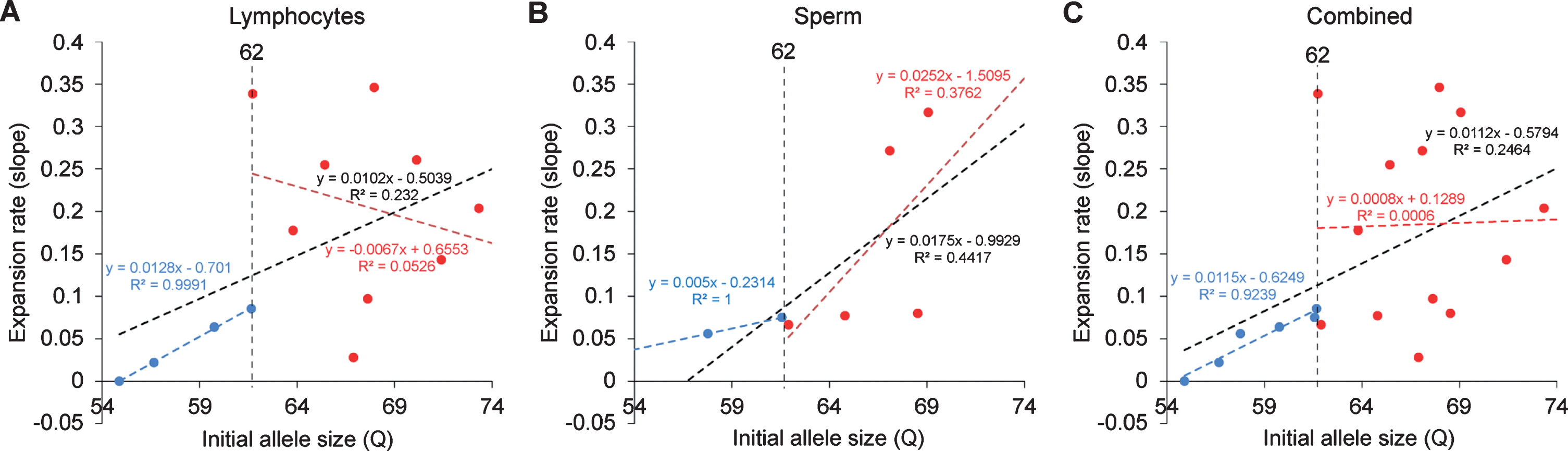
Fig. 4
Instability model based on the data provided in this article.
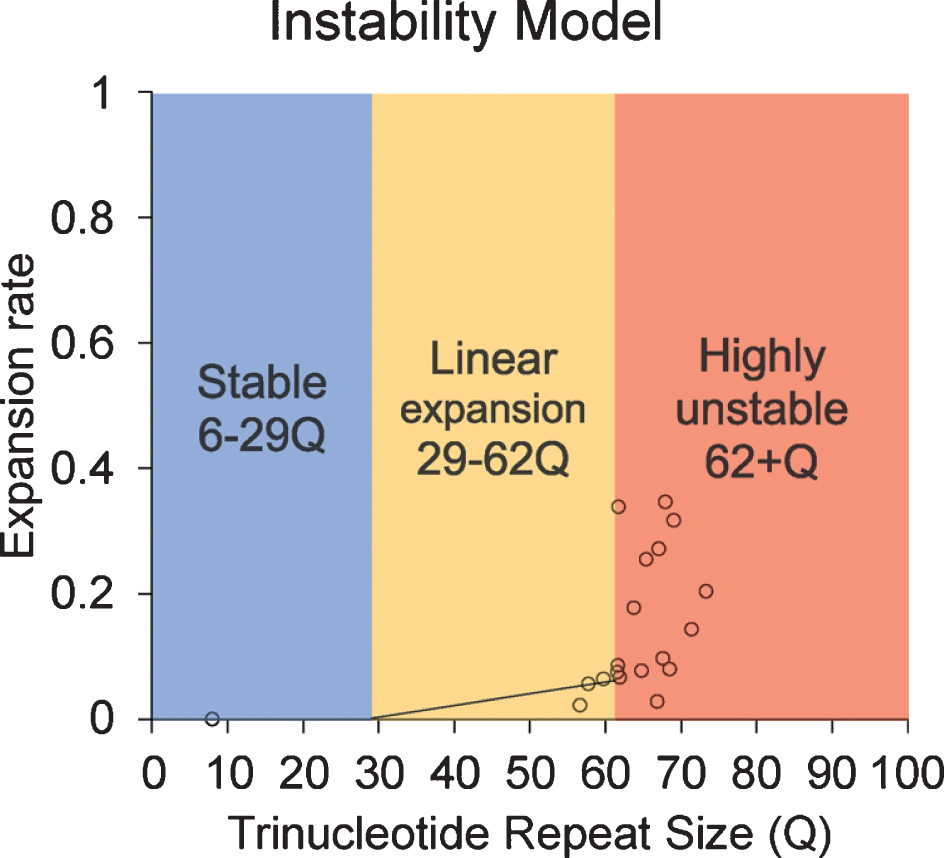
Although the current study is limited by sample size and the limitation in statistical analysis, our results reveal an increasing trend in CAG repeat length in both lymphocytes and sperm of HD monkeys as they age. This is the first study to longitudinally monitor CAG repeat instability in lymphocytes and sperm during the lifespan of a monkey model of HD. Our results reveal that TNR instability in HD monkey sperm increased with age. Although a previous publication suggested the paternal age does not affect TNR instability [18], our study shows a clear positive correlation between the CAG repeat size as disease progress as the animal age [20, 25, 26]. Although all assessments were conducted at approximately five years of age to minimize stress induced self-injury behaviors, progressive cognitive behavioral decline and anatomical changes were clearly shown in HD monkeys [20, 25, 26]. We also observed the increase of CAG repeat length in lymphocytes over time similar to that of sperm, which has not been reported in prior reports [29]. Additionally, the discrepancy between studies in rodent models and human HD patients, especially in sperm, could be due to the fundamental differences in spermatogenesis that impact CAG repeat instability in HD [16, 19, 30–32]. Rodent goes through many developmental stages (19 steps) with multiple replication steps resulting in 10 times more sperm production compared to humans, while human only undergoes 6 developmental stages [30]. Our data suggest that there might be a threshold of CAG repeat size where dramatic increase of instability occurred. Here, we propose a model based on the current data in HD monkeys (Fig. 4). Our data suggest that 62Q might be the threshold where a larger CAG repeat expansion starts to occur. In a human study, CAG repeat seems to start to expand rapidly after 50Q in sperm samples [18]. Although it is unlikely that a person who carries more than 60Q would not have developed a neurological symptom before the reproductive age, the neurological symptom onset age has a considerable variation [9], and it would be advisable to inform the patient about the risk.
Our findings stress the importance of further investigation in CAG instability in peripheral blood cells longitudinally. The physiological and genetic similarities between rhesus macaques and humans suggest that HD monkeys could replicate human conditions, including CAG instability. Thus, CAG instability in blood cells could potentially be a good indicator of somatic cell stability, while HD monkeys’ sperm could be a useful model for investigating genetic anticipation in HD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank the YNPRC veterinarians and animal care staff. Special thanks to the veterinarian staff, primate enrichment team, and animal care personnel for providing superior medical and daily care to HD monkeys as disease progressed. We also thank current and past members of the Chan Lab team who contributed to the development of the HD monkey model. YNPRC is supported by the Office of Research and Infrastructure Program (ORIP)/OD P51OD11132. The Transgenic Huntington’s Disease Monkey Resource “THDMR” and this study were supported by a grant awarded by the ORIP/NIH (OD010930) to AWSC.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JHD-190359.
REFERENCES
[1] | van Dijk JG , van der Velde EA , Roos RA , Bruyn GW . Juvenile Huntington disease. Hum Genet. (1986) ;73: (3):235–9. |
[2] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Primers. (2015) ;1: :15005. |
[3] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: :971–83. |
[4] | Crook ZR , Housman DE . Surveying the landscape of Huntington’s disease mechanisms, measurements, and medicines. J Huntingtons Dis. (2013) ;2: (4):405–36. |
[5] | Gil JM , Rego AC . Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. (2008) ;27: (11):2803–20. |
[6] | Langbehn DR , Hayden MR , Paulsen JS . CAG-repeat length and the age of onset in Huntington disease (HD): A review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. (2010) ;153B: (2):397–408. |
[7] | Ross CA , Aylward EH , Wild EJ , Langbehn DR , Long JD , Warner JH , et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. (2014) ;10: (4):204–16. |
[8] | Ha AD , Beck CA , Jankovic J . Intermediate CAG repeats in Huntington’s disease: Analysis of COHORT. Tremor Other Hyperkinet Mov (N Y). (2012) ;2: :tre-02-64-287-4. |
[9] | Gusella JF , MacDonald ME . Huntington’s disease: The case for genetic modifiers. Genome Med. (2009) ;1: (8):80. |
[10] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Primers. (2015) :15005. |
[11] | Chong SS , Almqvist E , Telenius H , LaTray L , Nichol K , Bourdelat-Parks B , et al. Contribution of DNA sequence and CAG size to mutation frequencies of intermediate alleles for Huntington disease: Evidence from single sperm analyses. Hum Mol Genet. (1997) ;6: (2):301–9. |
[12] | Reiner A , Albin RL , Anderson KD , D'Amato CJ , Penney JB , Young AB . Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. (1988) ;85: (15):5733–7. |
[13] | Aziz NA , van Belzen MJ , Coops ID , Belfroid RD , Roos RA . Parent-of-origin differences of mutant HTT CAG repeat instability in Huntington’s disease. Eur J Med Genet. (2011) ;54: (4):e413–8. |
[14] | Butterworth NJ , Williams L , Bullock JY , Love DR , Faull RL , Dragunow M . Trinucleotide (CAG) repeat length is positively correlated with the degree of DNA fragmentation in Huntington’s disease striatum. Neuroscience. (1998) ;87: (1):49–53. |
[15] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) ;6: (4):409–14. |
[16] | Kovtun IV , McMurray CT . Features of trinucleotide repeat instability in vivo. Cell Res. (2008) ;18: (1):198–213. |
[17] | McMurray CT . Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. (2010) ;11: (11):786–99. |
[18] | Wheeler VC , Persichetti F , McNeil SM , Mysore JS , Mysore SS , MacDonald ME , et al. Factors associated with HD CAG repeat instability in Huntington disease. J Med Genet. (2007) ;44: (11):695–701. |
[19] | Yoon SR , Dubeau L , de Young M , Wexler NS , Arnheim N . Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc Natl Acad Sci U S A. (2003) ;100: (15):8834–8. |
[20] | Chan AW , Jiang J , Chen Y , Li C , Prucha MS , Hu Y , et al. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic Huntington disease monkey model from infancy to adulthood. PLoS One. (2015) ;10: (5):e0122335. |
[21] | Chan AW , Xu Y , Jiang J , Rahim T , Zhao D , Kocerha J , et al. A two years longitudinal study of a transgenic Huntington disease monkey. BMC Neurosci. (2014) ;15: :36. |
[22] | Kocerha J , Liu Y , Willoughby D , Chidamparam K , Benito J , Nelson K , et al. Longitudinal transcriptomic dysregulation in the peripheral blood of transgenic Huntington’s disease monkeys. BMC Neurosci. (2013) ;14: :88. |
[23] | Kocerha J , Xu Y , Prucha MS , Zhao D , Chan AW . microRNA-128a dysregulation in transgenic Huntington’s disease monkeys. Mol Brain. (2014) ;7: :46. |
[24] | Lallani S , Villalba RM , Chen Y , Jenkins S , Smith Y , Chan AWS . Striatal interneurons in transgenic nonhuman primate model of Huntington’s disease. Sci Rep. (2019) ;9: (1):3528. |
[25] | Meng Y , Jiang J , Bachevalier J , Zhang X , Chan AW . Developmental whole brain white matter alterations in transgenic Huntington’s disease monkey. Sci Rep. (2017) ;7: (1):379. |
[26] | Raper J , Bosinger S , Johnson Z , Tharp G , Moran SP , Chan AWS . Increased irritability, anxiety, and immune reactivity in transgenic Huntington’s disease monkeys. Brain Behav Immun. (2016) ;58: :181–90. |
[27] | Yang SH , Cheng PH , Banta H , Piotrowska-Nitsche K , Yang JJ , Cheng EC , et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature. (2008) ;453: (7197):921–4. |
[28] | Mollersen L , Rowe AD , Larsen E , Rognes T , Klungland A . Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. (2010) ;6: (12):e1001242. |
[29] | MacDonald ME , Barnes G , Srinidhi J , Duyao MP , Ambrose CM , Myers RH , et al. Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J Med Genet. (1993) ;30: (12):982–6. |
[30] | Ehmcke J , Wistuba J , Schlatt S . Spermatogonial stem cells: Questions, models and perspectives. Hum Reprod Update. (2006) ;12: (3):275–82. |
[31] | Hermann BP , Sukhwani M , Simorangkir DR , Chu T , Plant TM , Orwig KE . Molecular dissection of the male germ cell lineage identifies putative spermatogonial stem cells in rhesus macaques. Hum Reprod. (2009) ;24: (7):1704–16. |
[32] | Simard O , Gregoire MC , Arguin M , Brazeau MA , Leduc F , Marois I , et al. Instability of trinucleotidic repeats during chromatin remodeling in spermatids. Hum Mutat. (2014) ;35: (11):1280–4. |


