
Transcriptional Regulation of the Huntingtin Gene
Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by a CAG trinucleotide expansion in the HTT gene, which encodes for an abnormal polyglutamine tract in the huntingtin protein (HTT). This review examines the known mechanisms of HTT gene regulation. We discuss HTT expression patterns, features of the HTT promoter, regulatory regions of the HTT promoter with functional significance, and HTT regulators located outside of the proximal promoter region. The factors that influence HTT expression in the brain and the mechanisms of HTT transcriptional regulation are currently poorly understood, despite continuing research. Expanding knowledge of HTT regulation will inform future studies investigating HTT function. Improving understanding of HTT expression and control may also uncover novel therapeutic approaches for HD through the development of methods to modulate mHTT levels.
INTRODUCTION
Huntington’s disease (HD) is caused by an expanded trinucleotide stretch of greater than 35 CAG repeats in the HTT gene that results in an abnormal polyglutamine tract in huntingtin [1]. HD is phenotypically characterized by progressive motor dysfunction, cognitive impairment, psychiatric symptoms, and personality changes [2, 3]. There is variation in the CAG repeat length mutation among HD patients. Larger CAG repeats are correlated with earlier HD onset [4]. Fully penetrant mutations have 40 or more CAG repeats. CAG repeats lengths of 35 or greater are classified as pathogenic, but mutations with 36 to 39 CAG repeats are incompletely penetrant and often cause later onset HD with slower progression and milder symptoms [5]. Wild type HTT has been shown to have anti-apoptotic effects [6] and plays a critical role in normal neuronal development, transcriptional regulation, cellular trafficking, and synaptic activity [7]. Mutant huntingtin (mHTT) forms intracellular oligomers and aggregates and is thought to have a toxic gain-of-function that interferes with many cellular and biological functions [2]. Previous work using mouse models of HD has shown that mHTT reduction positively influences behavioral and pathological HD symptom development and progression [8, 9]. In the first study of its kind, a regulatory single nucleotide polymorphism (rSNP) in an NF-κB binding site in the HTT promoter was associated with reduced, allele-specific HTT expression that bidirectionally influences HD age of onset [9]. Taken together, these studies support the hypothesis that modulation of HTT expression is a viable therapeutic avenue for HD. The huntingtin protein is ubiquitous in both the brain and peripheral tissues [10]. Peripherally, huntingtin is most abundant in the testes and is present in many other tissues, including liver and lung [11]. HTT expression patterns within the brain are inconsistent with HD pathophysiology. Expression is highest in the cortex, cerebellum and hippocampus, and is detectable (but not robust) in the striatum [11, 12]. The highest levels of HTT transcript and HTT in the brain are found in large neurons and in regions with dense neuronal populations [13]. Regions of reduced HTT within the striatum are positively correlated with regions of neuronal loss in HD brain, indicating that reduced HTT in these areas is the result of neuronal depletion rather than altered HTT expression [11, 12].
Various cell-based and animal models of HD have shown that the polyglutamine expansion in mHTT causes accelerated neurodegeneration and neuronal death [14, 15]. Conversely, over-expression of wild type HTT has a modest neuroprotective effect in a variety of model systems. In mice, elevated wild type HTT protects against apoptosis following external toxin exposure [16] and against NMDA receptor-mediated excitotoxicity [17]. In a mouse model of HD, wild type huntingtinover-expression had minimal effect on the HD phenotype, but did reduce the striatal neuronal atrophy caused by mutant huntingtin [18], while depletion of wild type huntingtin levels from conception worsens many aspects of the phenotype in HD mice [6, 19] and increases toxicity in HD cell culture models [20]. In heterozygous HD patients, HTT is expressed from both wild type and mutant alleles [13]. Homozygous HD patients express two mutant alleles in the absence of wild type huntingtin and develop normally with a similar age of HD onset to heterozygotes, but may have a more rapid progression of disease [21]. Finally, the effects of a transcription-lowering rSNP affecting NF-κB binding located in the promoter of the HTT gene was associated with a significant delay in age of onset when present on the mutant allele, but the same SNP on the normal HTT allele had only a modest effect in accelerating the age of onset [9].Together, these effects suggest that while HD is clearly caused by mHTT expression, the relative proportions of wild type HTT and mHTT may also be important modifying factors in HD.
There are currently are several approaches to post-transcriptional lowering of HTT expression in development for the treatment of HD, including small molecules, ASOs, and small interfering RNA (siRNA), as summarized in several recent review articles [22, 23]. Most mHTT-lowering therapies do not distinguish between HTT alleles, and studies of HTT-reducing interventions in HD mouse models show clear phenotypic benefits [24]. Characterization of the regulatory mechanisms that control HTT gene expression will help expand our understanding of both normal huntingtin function and HD pathogenesis. The manipulation of HTT expression has enormous potential as a novel therapeutic approach for HD.
CONSERVED AND UNIQUE HTT PROMOTER REGULATORY ELEMENTS
The HTT promoter (Fig. 1) has high GC content and lacks TATA and CCAAT regulatory elements. There is a highly-conserved region between the human HTT promoter and the promoter of the mouse homologue, Hdh. The sequences in this region, located at positions –206 to –56 relative to the HTT translation start site (+1 site), have a 78.81% shared sequence identity. The shared sequence identities of the surrounding regions are lower: approximately 50% [25]. Multiple putative regulatory regions exist within the highly-conserved region. Some elements are shared, while others are specific to the promoter of a single species [25]. Unique features of the HTT promoter are: two 20 bp direct repeats, two 17 bp direct repeats (surrounded by identical 7 bp sequences that are identical to the first 7 bp of the full-length 17 bp repeats) and one full Alu element and one Alu element 3’ fragment, both of which are in reverse orientation to the direction of HTT transcription (Table 1, Supplementary Figure 1) [25].
Fig. 1.
Previously characterized HTT transcription factor binding sites and regulatory regions. Locations of TFBSs in the HTT proximal promoter (upper) and gene body (center) are shown. In parallel, uncharacterized regulatory regions in the HTT proximal promoter are indicated. TFs/UCRRs shown to upregulate (green) or downregulate (red) HTT transcription are pictured in addition to TFs with unknown effects on HTT transcription (grey).
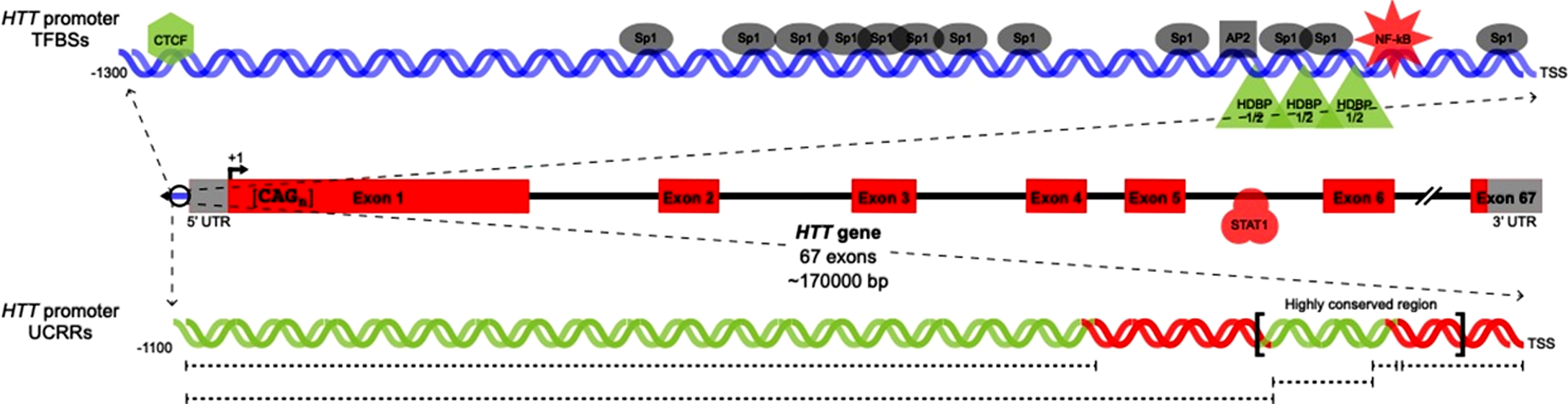
Table 1
HTT regulatory elements and features
| Regulatory feature/element | Binding site location relative to HTT +1 site | Location relative to +1 site (other species) | In vitro binding activity (method) | In vitro regulatory activity | Regulatory assay cell type (species) | Effect on HTT expression | Element type |
| 20 bp direct repeats25 | –213 to –194 | None | Not assessed | Yes | Non-neuronal (human) | Positive regulator | Predicted* |
| –193 to –174 | Neuronal (human) | ||||||
| 17 bp direct repeats25 | –518 to –502 | None | Not assessed | Not assessed | N/A | Unknown | Predicted* |
| –501 to –485 | |||||||
| UCRR 128 | –126 to –16 | N/A | Not assessed | Yes | Non-neuronal (human) | Positive regulator | Predicted* |
| UCRR 228 | –141 to –126 | N/A | Not assessed | Yes | Non-neuronal (human) | Positive regulator | Predicted* |
| Neuronal (human) | |||||||
| UCRR 328 | –242 to –171 | N/A | Not assessed | Yes | Non-neuronal (human) | Positive regulator | Predicted* |
| Neuronal (human) | |||||||
| UCRR 431 | –1031 to –221 (w/ HCR) | N/A | Not assessed | Yes | Non-neuronal (hamster) | Positive regulator | Predicted* |
| Neuronal (mouse) | |||||||
| UCRR 528 | –1032 to –324 (w/ HCR) | N/A | Not assessed | Yes | Neuronal (human) | Negative regulator | Predicted* |
| Alu element (full)25 | –2100 to –1812 | None | Not assessed | Not assessed | N/A | Unknown | Predicted* |
| Alu element (partial)25 | –1724 to –1598 | None | Not assessed | Not assessed | N/A | Unknown | Predicted* |
| cAMP26 | None | –180 to –174 (mouse) | Yes (EMSA) | Yes | Non-neuronal (rat) | None (Positive regulator of rhd) | Putative† |
| –175 to –169 (rat) | Neuronal (rat) | ||||||
| AP225,28 | –249 to –242 | –270 to –262 (mouse) | Yes (EMSA) | Yes | Non-neuronal (human) | None detected | Putative† |
| Neuronal (human) | |||||||
| Sp125,28,29,30 | –15 to –9 | –299 to –293 (mouse) | Yes (EMSA) | Inconclusive | Non-neuronal (human) | Inconclusive | Putative† |
| –191 to –186 | –318 to –312 (mouse) | Neuronal (human) | |||||
| –211 to –206 | –374 to –368 (mouse) | ||||||
| –285 to –279 | –379 to –373 (mouse) | ||||||
| –461 to –455 | –427 to –421 (mouse) | ||||||
| –549 to –543 | |||||||
| –579 to –573 | |||||||
| –595 to –589 | |||||||
| –599 to –593 | |||||||
| –646 to –640 | |||||||
| –714 to –708 | |||||||
| –807 to –813 | |||||||
| HDBP1/232 | –221 to –215 | None | Yes (EMSA) | Yes | Neuronal (human) | Positive regulator | Putative† |
| –201 to –195 | |||||||
| –181 to –175 | |||||||
| p5329,33,34 | Promoter; intron 2; intron 3 | Promoter (partial; mouse) | Yes (ChIP/EMSA) | Inconclusive | Non-neuronal (mouse) | Inconclusive | Putative† |
| Non-neuronal (human) | |||||||
| Neuronal (human) | |||||||
| CTCF35 | –1270 to –1288 | None | Yes (EMSA) | Yes | Non-neuronal (human) | Positive regulator | Putative† |
| NF-κB9 | –139 to –148 | None | Yes (EMSA) | Yes | Non-neuronal (human) | Negative regulator | Putative† |
| STAT129 | HTT intron 5 | None | Yes (ChIP) | Yes | Non-neuronal (human) | Negative regulator | Putative† |
*Predicted = No binding activity or functional effect of the regulatory element on HTT expression has been investigated or detected. † Putative = A single group has detected binding activity between the regulatory element and the specified region and observed a functional effect of the regulatory element on HTT expression. This classification also applies to sites and elements for which reported binding interactions and/or functional effects have varied between publications. ‡ Independently Validated = Multiple groups have successfully detected binding activity between the regulatory element and the specified region, and observed a functional effect of the regulatory element on HTT expression. Bolded site locations indicate conserved sites. Italicized site locations indicate sites with experimentally confirmed regulatory element binding activity.
One cAMP response element (CRE) in the Hdh promoter is absent from the human HTT promoter, but is present in the rat homologue, rhd [14, 25, 26]. The rhd CRE is located within the same region of high conservation shared between HTT and Hdh. Deletion of this region significantly reduces rhd promoter activity in rat neuronal and non-neuronal cells, indicating the presence of regulatory elements necessary for rhd expression in this region [26].
A single AP2 binding site is conserved between the HTT promoter and the Hdh promoter. There are 11 predicted Sp1 binding sites in the HTT promoter, and five predicted Sp1 binding sites in the Hdh promoter. Only one of these sites is conserved [25]. Four polymorphic sites are present in the region 303 bp upstream of the HTT +1 site. One of these sites is predicted to be in the HTT 5’ untranslated region (UTR). At two of the polymorphic sites, only single base pair substitutions occur. These substitutions are not predicted to be functionally significant because they do not disrupt the HTT promoter consensus sequence. The other two polymorphic sites contain one or two copies of a 6 bp repeat sequence or one, two, or three copies of a 20 bp repeat sequence [25, 27]. A single copy of both the 6 bp and 20 bp repeats is present in chimpanzee and gorilla, while the most common human allele contains one 6 bp sequence and a direct repeat of the 20 bp sequence [27].
The differences in regulatory elements between promoters from different species suggest that there may be unique transcriptional regulators for each gene homologue [25]. Conversely, regulatory elements in the HTT gene promoter that are shared between species are of interest because their conservation implies functional significance. These gene regions are of high priority for more detailed regulatory characterization.
*Predicted =No binding activity or functional effect of the regulatory element on HTT expression has been investigated or detected. †Putative =A single group has detected binding activity between the regulatory element and the specified region and observed a functional effect of the regulatory element on HTT expression. This classification also applies to sites and elements for which reported binding interactions and/or functional effects have varied between publications. ‡Independently Validated =Multiple groups have successfully detected binding activity between the regulatory element and the specified region, and observed a functional effect of the regulatory element on HTT expression. Bolded site locations indicate conserved sites. Italicized site locations indicate sites with experimentally confirmed regulatory element binding activity.
FUNCTIONALLY SIGNIFICANT HTT PROMOTER REGULATORY ELEMENTS
Specific in vitro binding interactions have been identified at one Sp1 site and one AP2 site in the HTT promoter [28]. In vitro binding interactions also occur between Sp1 and the putative 5’ UTR binding site and between Sp1 and the 6 bp polymorphic region. There is no functional effect of duplication of the 6 bp polymorphic region. Deletion of one copy of the 20 bp direct repeat significantly reduces HTT expression in both human neuronal and non-neuronal cells. The effect caused by deletion of one copy of the 20 bp direct repeat region shows the Sp1 site located within the 20 bp region is responsible for increased HTT expression, and that the observed change in HTT expression is not caused by the creation of a novel transcription factor binding site following duplication [28]. In human non-neuronal cells only, deletion of the HTT promoter region spanning –126 to –16 causes a decrease in HTT expression. In both human neuronal and non-neuronal cells, a total loss of HTT expression results from deletion of the region spanning –242 to –171, which is indicative of the presence of one or more positive cis-regulatory elements in this region [28].
In vitro binding interactions occur between Sp1 and AP2 at the binding sites conserved between human and mouse, but deletion of these sites has no significant effect on HTT expression in both human neuronal and non-neuronal cells [28]. Deletion of the –141 to –126 region causes a reduction in HTT expression, which may be the result of either the removal of the 3’ transcription start site (TSS) or of an unidentified cis-regulatory sequence. Removal of this region causes a more dramatic reduction of expression in human non-neuronal cells than in human neuronal cells [28]. De Souza and Kosior et al. showed siRNA-mediated knockdown of Sp1 in human non-neuronal cells modestly increases HTT expression [29]. In both human non-neuronal and neuronal cells, Wang et al. showed that over-expression of Sp1 increases HTT expression. Inhibition of Sp1 signaling in the same models decreased HTT expression [30]. These contradictory reports mean that the regulatory effect of Sp1 on HTT transcription is ambiguous.
HTT transcription increases when expression of HTT is controlled by longer promoter constructs that contain the –1031 to –221 bp region, in addition to the highly-conserved region, upstream from the HTT +1 site [31]. Conversely, a decrease in transcriptional activity occurs when HTT expression is driven by the promoter containing the highly-conserved region and the –1032 to –324 bp region [28]. These results were reported by separate studies that used, respectively, mouse and human neuroblastoma cell lines to evaluate HTT promoter activity [28, 31]. The observed difference in HTT promoter activity may be attributed to physiological variation between these cell lines, or may be caused by an unidentified negative cis-regulatory element in the –323 to –222 region. Currently, the mechanisms driving the regulatory effects of these HTT promoter regions are unknown.
HD gene regulatory region-binding protein (HDBP) 1 and HDBP2 have conserved C-terminal regions that bind to 7 bp sequences separated by 13 bp “spacer” sequences next to the 20 bp direct repeats in the HTT promoter [32]. Disruption of the binding sequences destroys HDBP1/2 binding interactions in human neuronal cells. HTT promoter function is also reliant upon the preservation of these 7 bp consensus sequences [32]. There is a portion of a putative p53 response element upstream of the Hdh +1 site at position –1285. A similar, partial consensus sequence exists in the HTT promoter at –630 and –1200 relative to the HTT +1 site [33].
Binding interactions between p53 and three regions of the HTT gene (in the promoter, intron 2 and intron 3) have been detected in vitro with a lower binding affinity than functionally validated p53 response elements at other locations in the genome (for example, within GADD45) [34].
In response to γ irradiation-induced DNA damage and resultant p53 pathway activation, an increase in HTT expression has been reported in human non-neuronal cells. In murine non-neuronal cells, HTT levels increase following p53 pathway activation by γ irradiation. The recapitulation of this phenotype in neuronal cells has not been reported. The effect of p53 activation on HTT expression suggests that p53 is a putative trans-activator of HTT.
There is a positive correlation between levels of p53 and murine htt. An analysis of htt levels in mice with zero, one, or two copies of the mutant Hdh allele and zero, one or two copies of the functional p53 allele showed that a larger number of functional p53 alleles increased levels of both wild type and mutant htt [33]. This effect was reported to be most significant in tissue from the brain and testes, and was not observed in peripheral tissues [33]. p53 has the same dose-dependent effect on HTT expression in a murine cell model. Transfection of murine fibroblasts with a reporter construct containing an HTT promoter fragment driving luciferase gene expression generated a fluorescent signal following p53 over-expression in a dose-dependent manner [33]. In contrast to the Ryan et al. and Feng et al. studies, De Souza and Kosior et al. showed that siRNA-mediated knockdown of p53 in human non-neuronal cells has no effect on HTT expression [29]. Currently, it is unclear if p53 is a transcriptional regulator of HTT.
Previous analyses of the regulation of HTT expression have largely focused on the HTT proximal promoter, and overlooked the regions within and surrounding the HTT gene. A computational analysis of the HTT gene locus (including the entire HTT gene, portions the genes immediately upstream and downstream of HTT, and intergenic sequences) identified a STAT1 binding site in intron 5 of HTT [29].
The HTT locus was scanned and scored for putative regulatory markers using publicly available datasets. High scoring regions were probed for putative transcription factor binding sites using ChIP-seq and transcription factor binding profile datasets, and a list of putative HTT transcription factors was generated. Analysis of protein-protein interactions and cell type specific DNase hypersensitivity marks prioritized these candidate transcription factors for further investigation. STAT1 was selected for functional characterization [29].
siRNA-mediated knockdown of STAT1 in human non-neuronal cells significantly increased HTT expression. STAT1 binding enrichment within HTT intron 5 was detected using ChIP-qPCR, confirming the direct interaction of STAT1 with the predicted binding site within HTT. The identification of STAT1 as a novel regulator of HTT shows that HTT transcription can be modulated by regulatory sites outside the proximal promoter region, and within the HTT gene body [29].
Epigenetic modifications also contribute to HTT expression variability. A CTCF TFBS in the HTT promoter has unique methylation occupancy in liver and cortex tissues. siRNA-mediated silencing of CTCF reduces HTT expression, implicating CTCF as a regulator of HTT. Differential methylation of the CTCF binding site may be an additional HTT expression modifier that operates in a tissue-specific manner [35].
siRNA-mediated knockdown of NF-κB in human non-neuronal cells significantly increases HTT expression [29]. There is a regulatory single nucleotide polymorphism (rSNP) in a NF-κB binding site within the HTT promoter. This site changes HTT expression by modulating the relative proportions of wild type and mutant HTT. The rSNP impairs NF-κB binding and decreases HTT expression. Later age of onset of HD is observed in patients with the rSNP on the mutant HTT allele, while a modest effect with an earlier age of onset is observed in patients with the rSNP on the wild type HTT allele [9]. The implications of this result are hugely significant, as they show NF-κBbinding to the HTT promoter alters HTT expression and that this SNP is a bidirectional modifier of HD age of onset. These results are also the first human evidence that modulation of HTT levels alters the course of HD, and supports the development of agents that decrease HTT levels as therapeutic strategies for HD.
OTHER REGULATORS OF HTT EXPRESSION
A recently identified HTT antisense transcript that contains the HD CAG repeat tract expansion and has a 5’ cap, 3’ polyA tail and three exons is detectable in frontal cortex tissues from healthy and HD brains [36]. This antisense transcript is alternatively spliced into two smaller transcripts, one of which contains the CAG repeat tract expansion. An increased number of CAG repeats in HTT is correlated with reduced expression of the antisense transcript [36]. Over-expression of the transcript is correlated with decreased endogenous HTT mRNA, and siRNA-mediated knockdown of the transcript causes an increase in HTT mRNA [36].
CONCLUSION
To date, there have been various attempts to characterize the regulation of HTT expression that have yielded inconsistent and often contradictory results. For example, studies have reported different numbers and locations of Sp1 binding sites in the HTT promoter [25, 35]. The sequence of the published binding site recognized by Sp1 is a consensus sequence [37], so variability in the query sequence used to predict the locations of Sp1 binding sites may contribute to the inconsistencies between these studies. The same concept can also be applied to functional assays, because transcription factor binding affinities are dependent on the sequences of their binding sites. The relative binding affinities of Sp1 to putative binding sites with different variations of the Sp1 consensus sequence has not been evaluated in the HTT promoter. As a result, the actual locations at which Sp1 binding occurs, and the effects of these binding interactions on HTT expression, are uncertain.
No studies have successfully reproduced the regulatory effects of transcription factors on HTT expression or confirmed the findings reported by other groups. For example, separate investigations have reported that p53 both influences or has no effect on HTT expression [33, 34, 36]. According to the criteria we have developed and now propose to classify types of regulatory elements in the HTT promoter (Table 1), there are no independently validated regulatory elements that influence HTT expression in the literature.
STAT1 is a recently characterized novel regulator of HTT expression that binds HTT intron 5 [36]. The discovery of this intragenic TFBS indicates that a thorough analysis of HTT transcriptional control should probe the entire HTT gene locus, rather than focusing only on TF activity at the HTT proximal promoter.
There are also no published studies that explore the combinatorial action of transcription factors on the modulation of HTT expression. Because many of the regulatory elements previously associated with HTT expression are nonspecific transcription factors, it is possible that the specificity of HTT expression is conferred by the synergistic action of these factors. This type of regulation may also explain why some studies report no functional effect of transcription factor binding site disruption, even though binding interactions between transcription factors and these sites have been validated using in vitro methods in the same studies.
More generally, inconsistencies in the literature may also be attributed to the methods of investigation used(for example, cell type) or to the length of the regulatory regions examined. Regardless of the source of the variability, it is apparent that, to date, no comprehensive evaluation of the regulation of HTT expression has yet been conducted. A thorough, functionally-validated assessment of the regulators of HTT expression, and how HTT expression changes in response to external stimuli, will be important for the continued development of novel therapeutic strategies aimed at reducing mHTT levels.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: 10.3233/JHD-180331.
REFERENCES
[1] | The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: (6):971–83. |
[2] | Vonsattel JP , DiFiglia M . Huntington disease. J Neuropathol Exp Neurol. (1998) ;57: (5):369. |
[3] | Ambrose CM , Duyao MP , Barnes G , Bates GP , Lin CS , Srinidhi J , Baxendale S , Hummerich H , Lehrach H , Altherr M , Wasmuth J . Structure and expression of the Hunting-ton's disease gene: Evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet. (1994) ;20: (1):27–38. |
[4] | Duyao M , Ambrose C , Myers R , Novelletto A , Persichetti F , Frontali M , Folstein S , Ross C , Franz M , Abbott M , Gray J . Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. (1993) ;4: (4):387–92. |
[5] | Andrew SE , Goldberg YP , Kremer B , Telenius H , Theil-mann J , Adam S , Starr E , Squitieri F , Lin B , Kalchman MA , Graham RK . The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. (1993) ;4: :398–403. |
[6] | Leavitt BR , Guttman JA , Hodgson JG , Kimel GH , Singaraja R , Vogl AW , Hayden MR . Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. (2001) ;68: (2):313–24. |
[7] | Zuccato C , Valenza M , Cattaneo E . Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. (2010) ;90: (3):905–81. |
[8] | Yamamoto A , Lucas JJ , Hen R . Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. (2000) ;101: (1):57–66. |
[9] | Bečanović K , Nørremølle A , Neal SJ , Kay C , Collins JA , Arenillas D , Lilja T , Gaudenzi G , Manoharan S , Doty CN , Beck J . A SNP in the HTT promoter alters NF-[kappa] B binding and isa bidirectional genetic modifier of Huntington disease. Nat Neurosci. (2015) ;18: (6):807–16. |
[10] | Sharp AH , Loev SJ , Schilling G , Li SH , Li XJ , Bao J , Wagster MV , Kotzuk JA , Steiner JP , Lo A , Hedreen J . Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. (1995) ;14: (5):1065–74. |
[11] | Li SH , Schilling G , Young Iii WS , Margolis RL , Stine OC , Wagster MV , Abbott MH , Franz ML , Ranen NG , Fol-stein SE , Hedreen JC . Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron. (1993) ;11: (5):985–93. |
[12] | Strong TV , Tagle DA , Valdes JM , Elmer LW , Boehm K , Swaroop M , Kaatz KW , Collins FS , Albin RL . Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat Genet. (1993) ;5: (3):259–65. |
[13] | Gutekunst CA , Levey AI , Heilman CJ , Whaley WL , Yi H , Nash NR , Rees HD , Madden JJ , Hersch SM . Identification and localization of huntingtin in brain and human lym-phoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci USA. (1995) ;92: (19):8710–4. |
[14] | Saudou F , Finkbeiner S , Devys D , Greenberg ME . Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. (1998) ;95: (1):55–66. |
[15] | De Almeida LP , Ross CA , Zala D , Aebischer P , Deglon N . Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J Neurosci. (2002) ;22: (9):3473–83. |
[16] | Rigamonti D , Bauer JH , De-Fraja C , Conti L , Sipione S , Sciorati C , Clementi E , Hackam A , Hayden MR , Li Y , Cooper JK . Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. (2000) ;20: (10):3705–13. |
[17] | Leavitt BR , van Raamsdonk JM , Shehadeh J , Fernandes H , Murphy Z , Graham RK , Wellington CL , Raymond LA , Hayden MR . Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. (2006) ;96: (4):1121–9. |
[18] | Van Raamsdonk JM , Pearson J , Murphy Z , Hayden MR , Leavitt BR . Wild-type huntingtin ameliorates striatal neuronal atrophy but does not prevent other abnormalities in the YAC128 mouse model of Huntington disease. BMC Neurosci. (2006) ;7: (1):80. |
[19] | Van Raamsdonk JM , Pearson J , Rogers DA , Bissada N , Vogl AW , Hayden MR , Leavitt BR . Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. (2005) ;14: (10):1379–92. |
[20] | Ho LW , Brown R , Maxwell M , Wyttenbach A , Rubinsztein DC . Wild type Huntingtin reduces the cellular toxicity of mutant Huntingtin in mammalian cell models of Huntington’s disease. J Med Genet. (2001) ;38: (7):450–2. |
[21] | Squitieri F , Gellera C , Cannella M , Mariotti C , Cislaghi G , Rubinsztein DC , Almqvist EW , Turner D , Bachoud-Levi AC , Simpson SA , Delatycki M . Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. (2003) ;126: (4):946–55. |
[22] | Miniarikova J , Evers M , Konstantinova P . Translation of microRNA-based huntingtin lowering therapies from pre-clinical studies to the clinic. Mol Ther. (2018) ;26: (4):947–62. |
[23] | Keiser MS , Kordasiewicz HB , McBride JL . Gene suppression strategies for dominantly inherited neurodegenerative diseases: Lessons from Huntington’s disease and spinocerebellar ataxia. Hum Mol Genet. (2015) ;25: (R1):R53–64. |
[24] | Kordasiewicz HB , Stanek LM , Wancewicz EV , Mazur C , McAlonis MM , Pytel KA , Artates JW , Weiss A , Cheng SH , Shihabuddin LS , Hung G . Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. (2012) ;74: (6):1031–44. |
[25] | Lin B , Nasir J , Kalchman MA , Mcdonald H , Zeisler J , Goldberg YP , Hayden MR . Structural analysis of the 5′ region of mouse and human Huntington disease genes reveals conservation of putative promoter region and di-and trinucleotide polymorphisms. Genomics. (1995) ;25: (3):707–15. |
[26] | Holzmann C , Maueler W , Petersohn D , Schmidt T , Thiel G , Epplen JT , Riess O . Isolation and characterization of the rat huntingtin promoter. Biochem J. (1998) ;336: (1):227–34. |
[27] | Coles R , Leggo J , Rubinsztein DC . Analysis of the 5’upstream sequence of the Huntington’s disease (HD) gene shows six new rare alleles which are unrelated to the age at onset of HD. J Med Genet. (1997) ;34: (5):371–4. |
[28] | Coles R , Caswell R , Rubinsztein DC . Functional analysis of the Huntington’s disease (HD) gene promoter. HumMol Genet. (1998) ;7: (5):791–800. |
[29] | De Souza RA , Kosior N , Thomson SB , Mathelier A , Zhang AW , Becanovic K , Wasserman WW , Leavitt BR . Computational analysis of transcriptional regulation sites at the HTT gene locus. J Huntingtons Dis. (2018) ;7: (3):223–37. |
[30] | Wang R , Luo Y , Ly PT , Cai F , Zhou W , Zou H , Song W . Sp1 regulates human huntingtin gene expression. J Mol Neurosci. (2012) ;47: (2):311–21. |
[31] | Holzmann C , Schmidt T , Thiel G , Epplen JT , Riess O . Functional characterization of the human Huntington’s disease gene promoter. Mol Brain Res. (2001) ;92: (1):85–97. |
[32] | Tanaka K , Shouguchi-Miyata J , Miyamoto N , Ikeda JE . Novel nuclear shuttle proteins, HDBP1 and HDBP2, bind to neuronal cell-specific cis-regulatory element in the promoter for the human Huntington’s disease gene. J Biol Chem. (2004) ;279: (8):7275–86. |
[33] | Ryan AB , Zeitlin SO , Scrable H . Genetic interaction between expanded murine Hdh alleles and p53 reveal deleterious effects of p53 on Huntington’s disease pathogenesis. Neurobiol Dis. (2006) ;24: (2):419–27. |
[34] | Feng Z , Jin S , Zupnick A , Hoh J , De Stanchina E , Lowe S , Prives C , Levine AJ . p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene. (2006) ;25: (1):1–7. |
[35] | De Souza RA , Islam SA , McEwen LM , Mathelier A , Hill A , Mah SM , Wasserman WW , Kobor MS , Leavitt BR . DNA methylation profiling in human Huntington’s disease brain. Hum Mol Genet. (2016) ;25: (10):2013–30. |
[36] | Chung DW , Rudnicki DD , Yu L , Margolis RL . A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum Mol Genet. (2011) ;20: (17):3467–77. |
[37] | Briggs MR , Kadonaga JT , Bell SP , Tjian R . Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science. (1986) :47–52. |


