
Possible Role of the Polyglutamine Elongation in Evolution of Amyloid-Related Evolvability
Abstract
The polyglutamine (polyQ) diseases, such as Huntington’s disease and the spinocerebellar ataxias, are characterized by the accumulation of elongated polyQ sequences (epolyQ) and mostly occur during midlife. Considering that polyQ disorders have not been selected out in evolution, there might be important physiological functions of epolyQ during development and/or reproduction. In a similar context, the physiological functions of neurodegeneration-associated amyloidogenic proteins (APs), such as β-amyloid in Alzheimer’s disease and α-synuclein in Parkinson’s disease, remain elusive. In this regard, we recently proposed that evolvability for coping with diverse stressors in the brain, which is beneficial for offspring, might be relevant to the physiological functions of APs. Given analogous properties of APs and epolyQ in terms of neurotoxic amyloid-fibril formation, the objective of this paper is to determine whether evolvability could also be applied to the physiological functions of epolyQ. Indeed, APs and epolyQ are similar in many ways, including functional redundancy of non-amyloidogenic homologues, hormesis conferred by the heterogeneity of the stress-induced protein aggregates, the transgenerational prion-like transmission of the protein aggregates via germ cells, and the antagonistic pleiotropy relationship between evolvability and neurodegenerative disease. Given that epolyQ is widely expressed from microorganisms to human brain, whereas APs are only identified in vertebrates, evolvability of epolyQ is considered to be much more primitive compared to those of APs during evolution. Collectively, epolyQ may be not only be important in the pathophysiology of polyQ diseases, but also in the evolution of amyloid-related evolvability.
ABBREVIATIONS
polyQ polyglutamine
HD Huntington’s disease
SBMA bulbar muscular atrophy
CAG cytosine-adenine-guanine
epolyQ elongated polyQ
APs amyloidogenic proteins
Aβ β-amyloid
AD Alzheimer’s disease
αS α-synuclein
PD Parkinson’s disease
APP amyloid precursor protein
HTT Huntingtin
mHTT-tg mutant HTT-transgenic
PQBP1 polyQ binding protein 1
INTRODUCTION
Polyglutamine (polyQ) diseases are autosomal dominant late-onset genetic disorders clinically characterized by progressive neurodegeneration leading to behavioral decline and motor impairments. At present, the polyQ diseases include five spinocerebellar ataxias types 1, 2, 6, 7, 17, Machado-Joseph disease, Huntington’s disease (HD), dentatorubral-pallidoluysian atrophy and spinal and bulbar muscular atrophy (SBMA) [1, 2]. The molecular pathogenesis of polyQ diseases is attributed to aggregation of elongated disease-specific polyQ proteins caused by cytosine-adenine-guanine (CAG) tri-nucleotide repeat expansion encoding for abnormal polyglutamine stretches [3]. It is unclear why specific neuronal populations undergo selective degeneration depending on the specific polyQ disease despite the ubiquitous expression of polyQ-containing proteins throughout the body.
Considering that elongated polyQ (epolyQ) peptides with neurotoxic properties have not been eliminated by the process of natural selection, such molecules may indeed play pivotal roles that are poorly understood. For instance, it is clear that amyloidogenesis is not necessarily biologically detrimental, and it is noteworthy that yeast prion evolvability has been characterized as a protein-based genetic switch that confers multiple phenotypes for yeast survival in a fluctuating environment [4]. Similar to yeast, human brains are also exposed to diverse stressors, such as oxidative stress, hyperthermia, hypoxia and inflammation. In this context, we recently proposed a hypothetical view suggesting that evolvability of amyloidogenic proteins (APs) relevant to aging-associated neurodegenerative diseases, including β-amyloid (Aβ) in Alzheimer’s disease (AD) and α-synuclein (αS) in Parkinson’s disease (PD), might be physiologically important for coping with diverse brain stressors [5]. Briefly, the heterogeneous structure of APs might correspond to each of the varied stressors, conferring hormesis on the brain. Furthermore, the transgenerational transmission of stress-specific APs presumably via the germ line, might lead to a preconditioning against forthcoming stressors in the brains of offspring. Because such epigenetic properties of APs are hereditable and are clearly advantageous for neuronal survival in brains of offspring, this might explain why APs have been perpetuated against the pressures of natural selection despite their neurotoxic properties.
Analogous to evolvability in APs, the main objective of this paper is to explore whether evolvability in epolyQ could be applicable to polyQ diseases as well. EpolyQ may exemplify evolvability in the presence of the functionally redundant wild-type homologue. Furthermore, the heterogeneity of epolyQ may correspond to diverse stressors, resulting in hormesis in the brain, and that the transgenerational transmission of epolyQ might lead to a preconditioning against similar stressors in the brains of offspring. Moreover, the integration of evolvability with the concept of the antagonistic pleiotropy might allow us to “overlook” the entire natural history of polyQ diseases. These properties associated with the evolvability of epolyQ are similar to those previously described for APs [5, 6]. Here, we further discuss the evolvability of epolyQ as playing an important role in the evolution of amyloid-related evolvability.
DISCUSSION
Previous reports relevant to the physiology of epolyQ
Indeed, there are previous reports making reference to the physiological role of the polyQ disease-related proteins. Muhlau and colleagues previously described that the structure of gray matter was increased with increasing long CAG repeat in HD [7]. More recently, Lee and colleagues found that the number of CAG repeats in huntingtin (HTT) gene, below disease threshold (15∼34) conferred advantageous changes in brain structure and general intelligence [8]. The similar effects were confirmed for the investigation of entire spectrum of CAG repeat (15∼58) [9]. Consistent with these findings, it was suggested that the expansion of the CAG repeat in HTT might be involved in DNA damage repair [10]. Although these results are interesting, the mechanisms are still obscure. It also is unclear whether the findings obtained in HD are applicable to other polyQ diseases.
Evolvability as a possible function of epolyQ in human brain
Provided that epolyQ and other APs both similarly exert neurotoxicity that in turn promotes neurodegeneration, it is possible that the concept of evolvability could also apply to epolyQ similar to APs.
Acquisition of a novel function by polyQ expansion
It is well known that gene duplication may afford the opportunity for evolutionary innovation in which a novel phenotype is obtained from one gene in the presence of its functionally redundant gene copy, the latter escaping selective pressures [11]. Based on genomic sequences, therefore, it is predicted that evolvability in APs might arise as a uniquely new function while non-amyloidogenic homologous counterparts are physiologically redundant during gene duplication [5]. For instance, the non-amyloidogenic homologues of APs, such as amyloid precursor protein (APP)-like protein 2 (APLP2) and β-synuclein (βS), may be physiologically redundant compared with their respective amyloidogenic homologues, APP and αS [5].
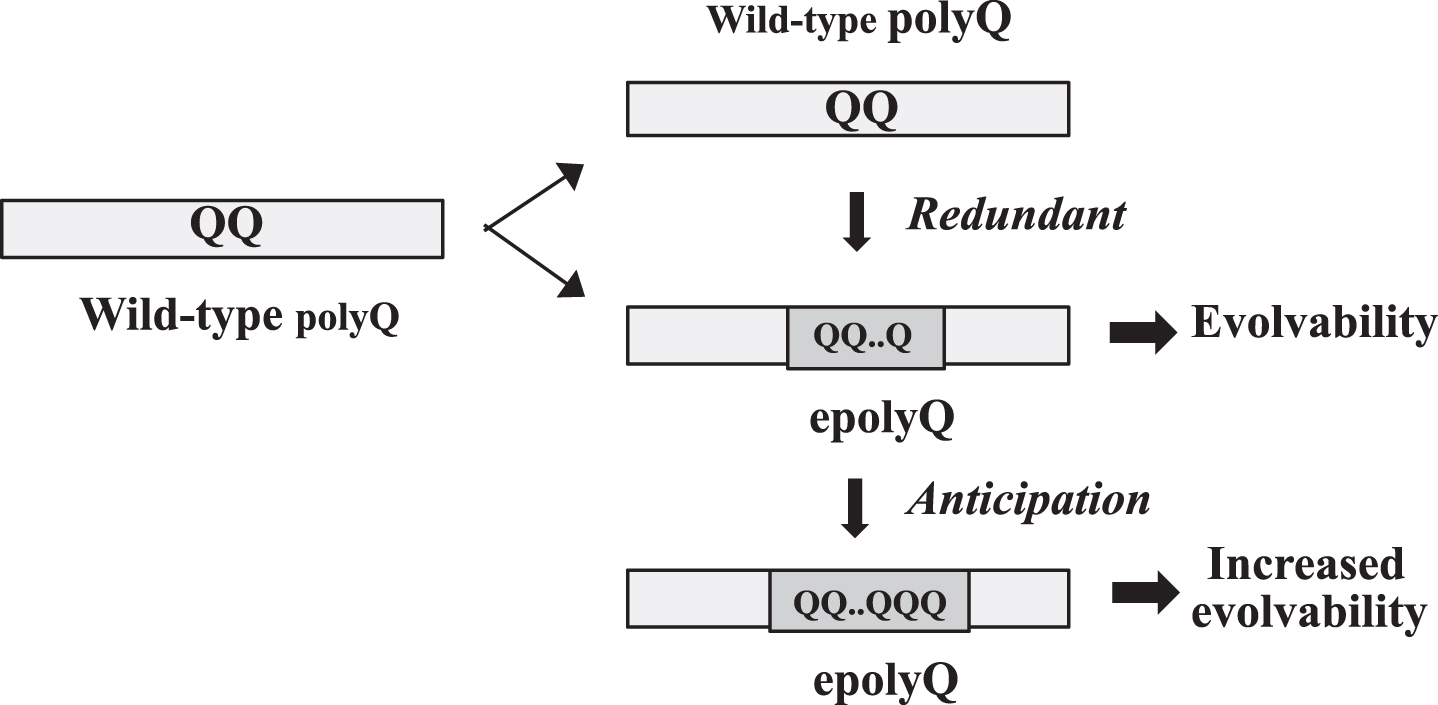
Although gene duplication is not directly applicable to the generation of epolyQ, a parallel concept to gene duplication might be relevant. Mechanistically, many polyQ proteins are encoded by tandem repeats of the codon CAG that are likely to mutate primarily by replication slippage due to the instability of DNA [12–15] (Fig. 1). In this regard, it is controversial whether slippage is just an artificial phenomena or is induced to occur by natural selection [16]. However, given that polyQ proteins are composed of mixed populations of half epolyQ and half wild-type polyQ proteins, it is possible to assume that the wild-type polyQ protein might be a functionally redundant counterpart for epolyQ (Fig. 1). The length of epolyQ may be further elongated through anticipation that may occur through slippage [17, 18].
Fig. 1.
Acquisition of evolvability as a novel function by polyQ expansion. Elongation of polyQ may occur through slippage presumably due to unstable DNA during gene replication. Since the polyQ proteins are composed of mixed populations of epolyQ with elongated stretches of polyQ and wild-type polyQ of normal-length, wild-type polyQ could be functionally redundant. The length of epolyQ may further be elongated by anticipation, resulting in the increased evolvability.
Relevance to stress resistance
Accumulating evidence suggests that neurons are equipped with adaptive mechanisms, including the unfolded protein response, ubiquitin-proteasome system and autophagy, which can be stimulated by preconditioning treatments that confer resistance to a subsequent toxic challenge, a phenomenon known as hormesis [19] According to recent studies demonstrating that APs such as Aβ and αS are composed of structurally heterogeneous populations [20–22], such structural heterogeneity of aggregated APs may confer hormesis, corresponding to the diverse brain stressors associated with neurodegenerative and neurological conditions.
Given that epolyQ peptides exhibit structurally heterogeneous amyloid-like fibrils [23, 24], it is possible that heterogeneity of epolyQ may be relevant to conferring hormesis in the brain (Fig. 2a). Also, because polyQ diseases occur preferentially in specific brain regions, such as the basal ganglia, cerebellum and spinal cord, it is predicted that structurally heterogeneous amyloid-like fibrils may interact with tissue-specific factors, leading to hormesis and the development of polyQ disorders.
Fig. 2.
A hypothetical mechanism of evolvability of erolyQ. a. Given that epolyQ proteins exhibit structurally heterogeneous amyloid-like fibrils, the heterogeneity of epolyQ may be relevant for hormesis in specific neuronal populations, conferring resistance against diverse stresses in the brain. b. The stress-responsive forms of epolyQ may go through the blood-brain barrier and enter bloodstream, reaching the reproductive system, thereby passing through the blood-testis (or blood-ovary) barrier. Subsequently, endogenous epolyQ in germ cells (sperm, oocytes) may be converted and transmitted to the embryo like a prion-like manner. Thus, transgenerational transmission of epolyQ may lead to preconditioning in the brain of offspring, which may occur during reproduction, followed by the onset of polyQ disorders.
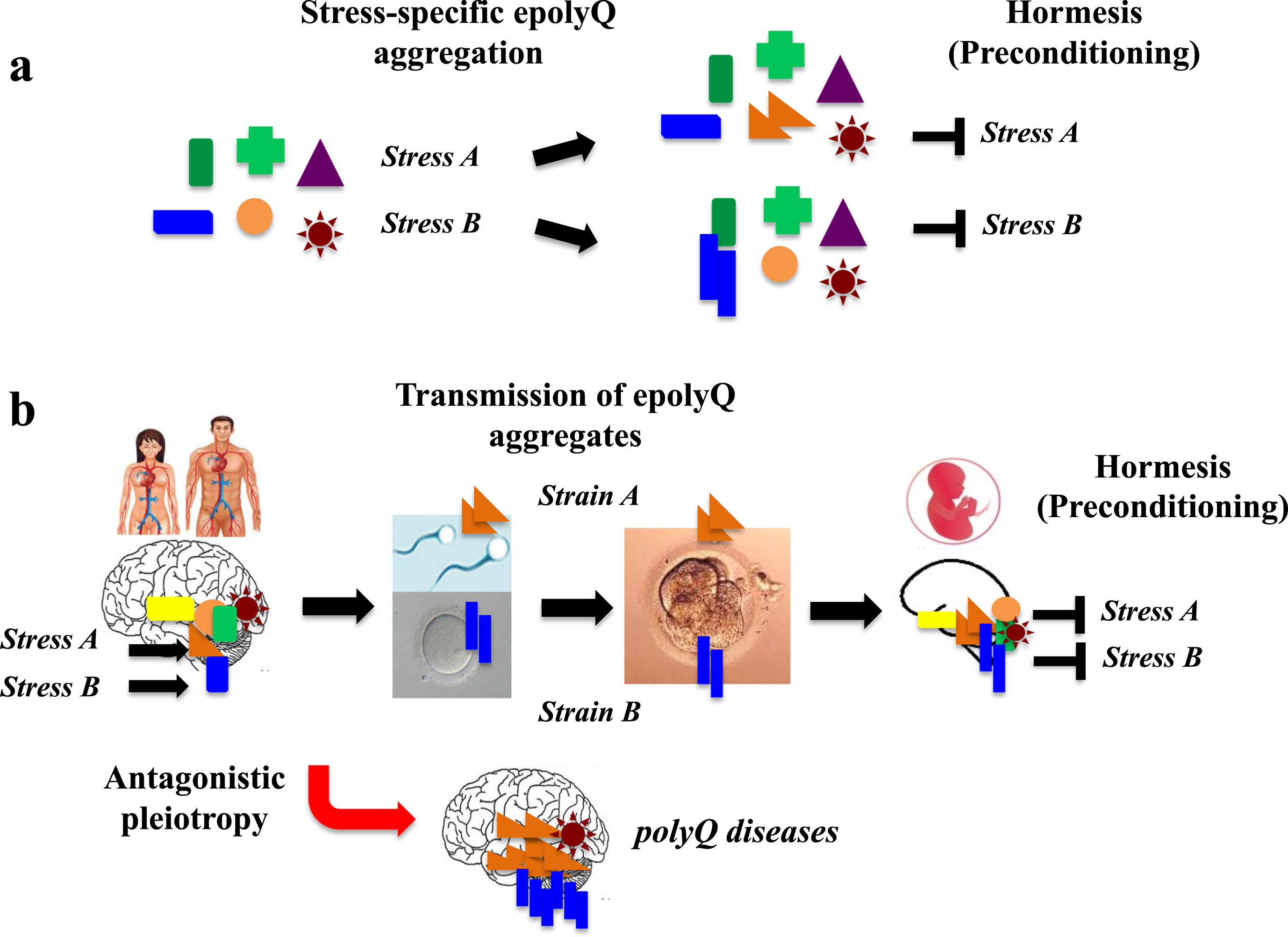
Transgenerational transmission of epolyQ
Evolvability is defined as the capacity of a population of organisms not merely to generate genetic diversity, but also to generate adaptive genetic diversity that is hereditary [25]. With this in mind, transgenerational transmission of APs may play a central role in AP evolvability. Indeed, a recent study suggests that transmission of APs could occur from parents to offspring via the germline, even at the animal level [26]. More precisely, APs may pass through the blood-brain barrier, propagating into the reproductive system, and then pass through the blood/testis (or blood/ovary) barrier, with conversion of endogenous APs to misfolded forms that are transmitted from the gametes to embryo in a prion-like fashion (Fig. 2b). Supporting this, APs, such as Aβ and αS, are abundantly expressed in serum as well as in gonadal tissues, including Sertoli cells and follicles [27].
Recent study suggests that epolyQ proteins are characterized by a prion-like transmission from cell to cell. For example, in striatal graft experiments, mutant HTT aggregates have been identified in donor cells, indicating a transmission of the mutant protein from the host tissue into the graft [28]. Furthermore, it was observed that full-length mutant HTT can be exocytosed via a non-conventional pathway in cell culture experiments [29]. Moreover, mutant HTT was s detected in the cerebrospinal fluid of HD patients [30].
In this context, it is reasonable to predict that similarly to APs, including Aβ and αS, the transgenerational transmission of epolyQ via the germline may also occur, conveying pathological stress information about specific neuronal populations encoded in the APs, altered structures of parental epolyQ, which could then be delivered to offspring. Such a mechanism would confer preconditioning against forthcoming stresses in the brains of offspring, which should be beneficial for their neuronal survival. If verified, evolvability in epolyQ could then be regarded as a significant physiological function.
Evolvablity and the inheritance of acquired characteristics
It is important to recognize that evolvability of amyloidogenic proteins in yeast prion as well as of neurodegenerative disease-related molecules, including APs and epolyQ, represents an epigenetic phenomenon. This is consistent with a recent paradigm that transgenerational epigenetics relevant to environmental stresses may play a major role in the inheritance of acquired characteristics, which historically, has remained a critical issue that still unsettled in the field of biology [31, 32].
Modulators of evolvability in epolyQ in human brain
If transgenerational transmission of the epolyQ occurs through the germ line in a prion-like fashion, it may be that epolyQ aggregates, especially non-fibrillar oligomeric forms, may be important [33]. As such, findings from pathological research into polyQ disease could be applied to a physiological understanding of epolyQ.
Post-translational modifications
In addition to a number of cellular factors related to protein aggregation including ubiquitin-proteasome system and autophagy, it has been shown that various post-translational modifications, such as phosphorylation, arginine methylation, palmitoylation, acetylation, SUMOylation, are involved in the pathogenesis of polyQ disease [5, 34]. Furthermore, increasing attention has recently been paid to the role of the FKBP12 protein in neurodegeneration. FKBP12 is a peptidyl-prolyl cis/trans isomerase and belong to the FK506-binding protein family [35]. Expression of FKBP12 was accumulated in the neurofibrillary tangles in patients with AD [36] and was markedly reduced in the striatum of HD mice [37], suggesting that the loss of FKBP12 may contribute to the pathogenesis of neurodegenerative diseases. Collectively, it is possible that some of these factors might be involved in the regulation of evolvability in epolyQ. In the similar context, it is of note that recent study suggests that exosomes may also play an important role in the transgenerational transmission of APs [5, 32, 38].
Role of wild-type homologues of PolyQ
During the process of gene duplication, many APs give rise to their non-amyloidogenic homologs. More specifically, APs, including APP, αS, tau, SOD1 and prion protein, are homologous with APLP2, βS, microtubule associated protein 2c, copper chaperone protein for superoxide dimustase and Doppel, respectively [39]. Interestingly, one property of non-amyloidogenic homologs may be to suppress neurodegeneration related to their respective APs. In a similar fashion, wild-type polyQ proteins may also function to suppress neurodegeneration induced by epolyQ proteins. Clearly, wild-type HTT reduces the cellular toxicity of mutant HTT in vivo [40]. Mechanistically, it was recently shown that the N-terminal fragments of wild type HTT have the capacity to inhibit aggregation of mutant HTT N-terminal fragments [41]. Thus, the functional redundancy of wild-type polyQ proteins may be critical to compensate for decreased functioning of epolyQ.
p53 in polyQ neurodegeneration and evolvability
p53 expression has been shown to be upregulated in the pathogenesis of polyQ disease, with possible relevance to neuronal cell death [42], and supporting this view, p53 levels were increased in the brains of mutant HTT-transgenic (mHTT-tg) mice [42]. On the molecular level, mHTT bound p53, upregulating levels of nuclear p53 as well as p53 transcriptional activity in neuronal cultures [42, 43]. Furthermore, neurodegeneration in mHTT-tg flies and neurobehavioral abnormalities in mHTT-tg mice were suppressed by p53 gene deletion [42]. Conversely, p53 was shown to regulate expression of HTT [44, 45]. This function of p53 might be related to the role of HTT in DNA damage repair, in which HTT and ATM co-localize at sites of DNA damage [10, 46].
Moreover, it was shown that p53 activation increased Bax expression in cerebellar and pontine nuclei neurons transfected with ataxin-3 [47]. Taken together, these findings implicating p53 in the pathogenesis of polyQ-related neurodegeneration may reflect the stimulatory effect of p53 on polyQ evolvability. Indeed, it is of particular note that p53 is prone to form amyloid-fibrils [48]. Given the central role of p53 in the pathogenesis of cancer and possible involvement of p53 in other neurodegenerative disorders, including AD and PD [49], p53 may be a common enhancer of evolvability in these major diseases.
PolyQ binding protein 1 (PQBP1)
To date, myriads of molecules have been shown to interact with the polyQ proteins [50]. Among them, PQBP1 is of particular interest because this molecule was discovered through its affinity for polyQ sequences of normal length [51], suggesting that PQBP1 might be a physiological regulator of polyQ proteins. PQBP1 was subsequently characterized as a highly conserved nuclear protein expressed ubiquitously, which binds to intrinsically disordered/denatured proteins that interact with splicing-related factors to regulate post-transcriptional gene expression [51]. Subsequently, a loss-of-function of PQBP1 due to frame-shift mutations in the PQBP1 gene was shown to prevent its interaction with the spliceosomal protein U5-15 kD, causing X-linked mental retardation [52]. Taken together, these results implicate the physiological role of PQBP1 as a possible regulator of the evolvability of epolyQ proteins.
Recently, Langfelder et al. analyzed the HD knock-in mice using integrated genomics and proteomics and found that 22 striatal module genes were validated as modifiers of toxicities of mouse HTT in CAG length-dependent manners thereby [53], suggesting that a number of polyQ interacting proteins might be involved in the regulation of the evolvability of epolyQ proteins.
Antagonistic pleiotropy and epolyQ evolvability
One unanswered question is how evolvability in epolyQ, which occurs during reproductive stage, is related to polyQ diseases that generally occur in midlife. Recently, we proposed that evolvability during the reproductive stage and neurodegenerative diseases, such as AD and PD, occurring with aging, may be in an antagonistic pleiotropy relationship [6]. Since polyQ diseases are generally manifested post-reproductively, a similar concept may be applicable to epolyQ as well.
Consistent with this notion, it has been previously suggested that antagonistic pleiotropy is applicable to a number of diseases, including HD, cystic fibrosis, sickle cell anemia β thalassemia, and cancer [54]. Conventionally, the mechanism underlying antagonistic pleiotropy in HD has yielded two potential benefits. First, HD might be linked to an increase in fecundity [54]. Supporting this, it was shown that HD patients have significantly greater fecundity when compared to unaffected individuals. For instance, a Canadian study of the relative fitness of affected and unaffected individuals comparing the fitness values of HD patients (n = 157) and non-affected individuals (n = 170), found that the mean number of offspring for HD patients was 39% higher than their unaffected siblings and 18% higher than a set of unrelated, age and gender-matched control individuals [55]. Second, epolyQ may reduce the risk of cancer [54, 56]. It was reported that a higher number of CAG repeats within the HTT allele is associated with increased p53 activity. Because p53 is a tumor suppressor protein with a role in inducing apoptosis, elevated p53 may also protect against tumor growth and cancer development [57]. Therefore, increased CAG repeat length in HD, and perhaps elevated p53, may be one explanation for the reduced incidence of cancer observed in a large HD registry. However, even though long CAG repeats correlate with a lower incidence of cancer, they may enhance the severity of specific subtypes of cancer once it is initiated, suggesting that the mechanism by which HD regulates cancer might be complicated [58].
Indeed, our proposed viewpoint does not necessarily exclude conventional explanations for the benefits of antagonistic pleiotropy in HD. For instance, it is possible that evolvability of epolyQ against stressors, which may benefit the development of offspring, might be related to the increased fecundity previously observed in HD [54]. Also, since p53 is also a self-aggregating protein [59], it is tempting to speculate that upregulation of p53 may be related to promotion of evolvability as well as the polyQ diseases. In light of this, it is notable that possible involvement of p53 in antagonistic pleiotropy has been previously described [60].
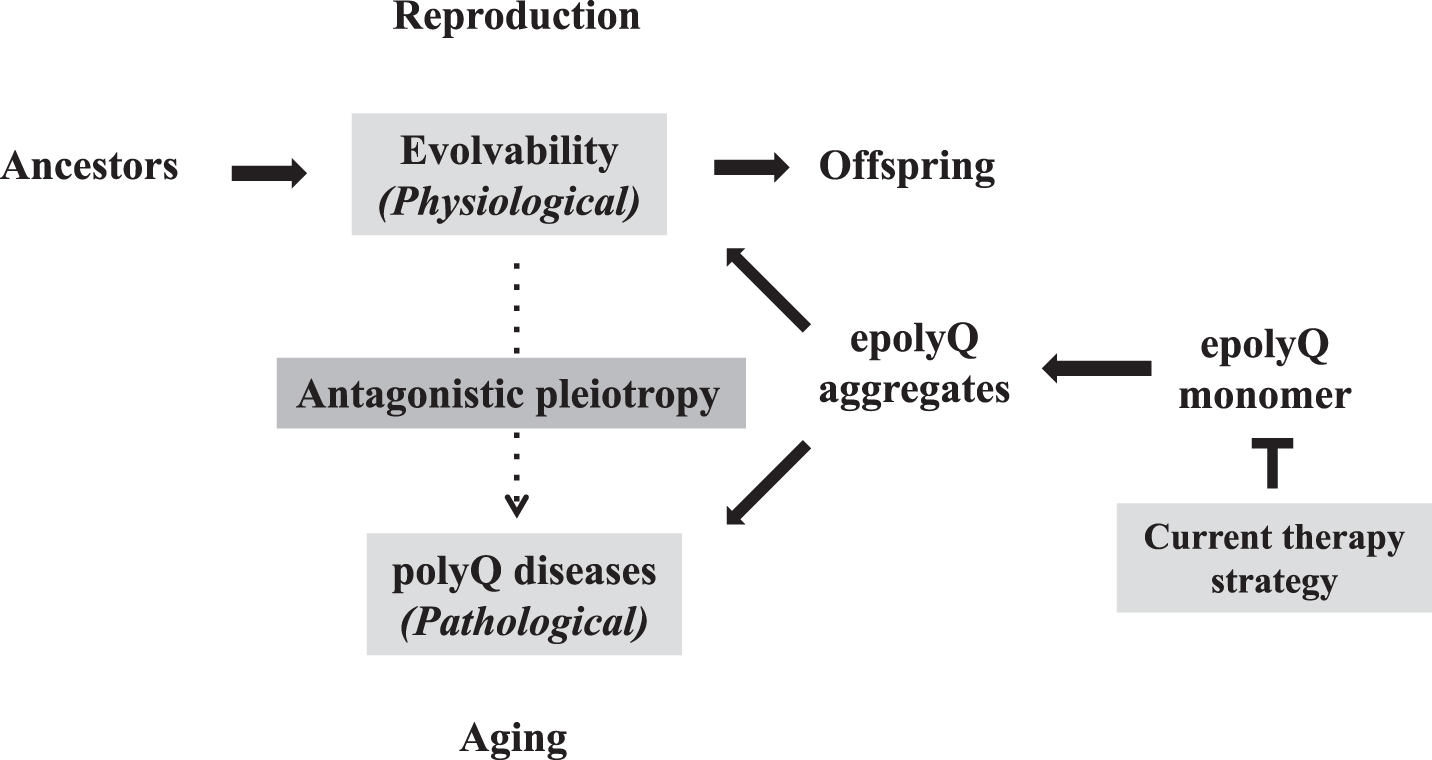
As described earlier, the increasing the CAG repeat-length was shown to stimulate human intelligence [8, 9]. Thus, aside from an increase of fecundity and cancer suppression, further investigations are warranted to characterize the benefits of the increasing the CAG repeat-length in the development/reproduction stages. Collectively, evolvability, a novel function of epolyQ during reproduction, could exist in an antagonistic pleiotropy with the polyQ diseases, similar to aging-related neurodegeneration (Fig. 3). In both phenomena, the aggregation of epolyQ may be a critical factor. Overall, our proposed view thus enables us to “overlook” the whole course of polyQ diseases. Naturally, one may predict that suppressing the antagonistic pleiotropy mechanism may represent a therapeutic strategy for polyQ diseases, especially from the viewpoint of evolution. This reinforces that the current goal for therapy of polyQ diseases, namely suppression of the expression level of soluble epolyQ, is likely the most effective strategy (Fig. 3). Consistent with this notion, it was previously shown that inclusion body formation reduced the risk of death in primary neuronal cultures, suggesting that the polyQ aggregates are protective [61]. Furthermore, it was recently described that soluble mutant HTT exon 1 can trigger apoptosis, and inclusions that form from the mutant HTT exon 1 can deactivate apoptosis to allow cells to survive longer, the cells with aggregates eventually die via a delayed necrosis mechanism [62]. Thus, the more effective therapeutic strategy would be to reduce the soluble mutant HTT expression. Indeed, recently encouraging are dosage-reduction strategies against the polyQ diseases, including antisense oligonucleotides and molecules directed at transcription dysregulation [63, 64]. Furthermore, it is expected that the results obtained in the polyQ diseases could be applied to aging-associated neurodegenerative diseases, such as AD and PD.
Fig. 3.
Antagonistic pleiotropy of polyQ-related pathophysiology Evolvability is proposed to be a physiological phenomenon during reproduction, whereas the polyQ diseases, such as HD and spinocerebellar ataxias, manifest as pathological phenomena during the post-reproductive senescent period. Both are derived from the soluble epolyQ protofibrils and participate in an antagonistic pleiotropic relationship as illustrated. Current therapy strategy targets the dose-reduction of soluble epolyQ.
Possible role of epolyQ in the evolution of amyloid-related evolvability
Next, provided that evolvability is a physiological function associated with both APs and epolyQ, how then does evolvability in epolyQ differ from that in other APs?
Previously, it was shown that polyQ-containing proteins exist in both fauna and flora [65]. Moreover, epolyQ has also been identified in lower organisms such as amoeba (Dictyostelium discoideum), that possess epolyQ up to several hundred in repeat length [66]. Particularly interesting, triplet repeats have been associated with phenotypic variation in filamentous fungi such as Neurospora crassa [67] as well as in yeast [68] (Fig. 4).
Fig. 4.
Evolution of amyloid-related evolvability. Synthesis of yeast prion is observed in microorganisms such as yeast, whereas expression of APs, including Aβ and αS, has not been observed in invertebrates, suggesting that APs are vertebrate-specific proteins. Notably, polyQ-containing proteins are ubiquitously expressed from microorganisms, such as yeast and fungi, to animals and plants. Thus, epolyQ might be regarded as an evolution of amyloid-related evolvability between yeast prion and APs.
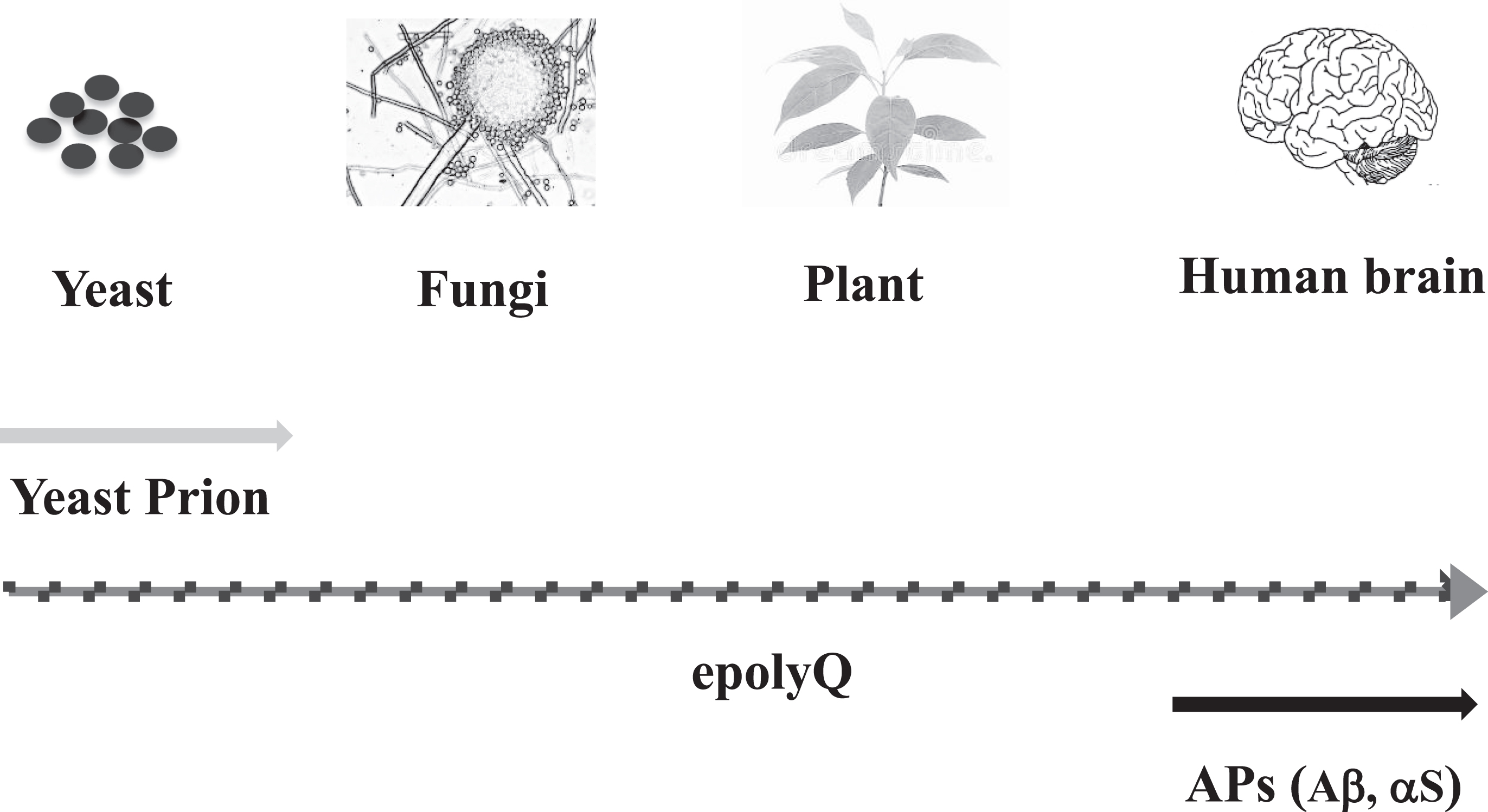
Distinct from evolvability in epolyQ, that related to APs may be a relatively new phenomenon across evolution. For instance, the APP ancestral gene is expressed in lower animals such as Drosophila, in which no homologous proteins are expressed [69, 70]. Furthermore, though the αS ancestral gene was identified in Torpedo, the complete set of αS family members has only been identified in mammals and birds, and no homologous αS proteins have been found in invertebrates, suggesting that they are vertebrate-specific proteins [71]. In particular, the pairs of APs and their non-amyloidogenic homologues, including Aβ, αS, and other APs, such as tau, SOD1 and prion protein, are only observed in vertebrate brain [39], and such amyloidogenic and non-amyloidogenic pairs may be important for evolvability [5].
As such, it is predicted that evolutionarily, evolvability in other APs may have surpassed that in epolyQ (Fig. 4). The mechanistic reasons for this difference between evolvability in epolyQ and in APs such as Aβ and αS, remains elusive. Yet, one possibility is that differences in transmission efficiency for these two protein types might account for this. To elaborate further, evolvability of epolyQ in primitive microorganisms should be simple, due to direct transmission of epolyQ protofibrils to offspring in concert with division/proliferation. In contrast, evolvability in higher organisms may be more complex because of the requirement for both somatic and gonadal transmission. Provided that the majority of epolyQ are transcription factors [50], whereas other APs are either cytoplasmic chaperone-like proteins or secreted trans-membrane proteins, the latter may likely be more suited for the transgenerational mode of transmission. Thus, it is suggested that the evolvability of epolyQ might be primitive in evolution compared to those of APs (Fig. 4). The difference in neurotoxicity between epolyQ and APs is less likely an explanation for this, since both polyQ disorders and other neurodegenerative diseases occur in the post-reproductive senescent period, which is not evolutionarily important. Thus, evolvability in epolyQ might have been less prominent, while those of APs have emerged in vertebrate organisms, leading to rapid evolution of the brain.
Advantage and disadvantage of the present hypothesis
Our hypothesis could be applied to some important issues in the field of polyQ research. For instance, it is noteworthy that Hayden and associates have described that the reduced penetrance of the HD alleles occur at high frequency in the general population compared to the HD patients, although the mechanism is obscure [72, 73]. In this regard, our hypothesis may raise a possibility that various negative regulators of the epolyQ evolvability, including post-translational modifications and wild-type homologues of polyQ, might be increased in the normal population. Alternatively, the mechanisms of the antagonistic pleiotropy of the epolyQ evolvability might be different between patients and the normal population. Of course, our hypothesis is still subject to scrutiny, and the various proposals, including the transgenerational transmission of amyloid fibrils, must still be experimentally demonstrated. If the species of epolyQ transmissible from parent to offspring consists of protofibrils, their detection may prove extremely difficult. Conceivably, further technical innovations will be required to detect the intermediate species of protein aggregates to demonstrate our hypothesis. On this point, however, it was recently shown that semen contains multiple types of amyloid fibrils in the absence of disease [74]. Further investigations are warranted to validate our hypothesis.
Another issue with our hypothesis regards disorders related to non-coding microsatellite expansion. Although the polyQ diseases are caused by epolyQ aggregation, some trinucleotide repeat disorder are caused by a non-coding microsatellite expansion, such as myotonic dystrophy type 1, featuring a CTG expansion located in the 3’ untranslated region of the dystrophia myotonica-protein kinase (DMPK) gene and myotonic dystrophy type 2, associated with an untranslated CCTG expansion [75]. Similar pathogenic repeat expansions in RNAs occur in spinocerebellar ataxia types 8, 10 and 12, and HD-like type 2 (HDL2) [76]. It is apparent that our present concept based on the amyloidogenesis of APs cannot be applied to these disorders lacking epolyQ aggregation. Curiously, it was shown that antisense transcripts from the JPH3 locus encoded a toxic polypeptide with an expanded polyQ stretch in BAC transgenic mouse model of HDL2 [77].
It is also worth noting that the expansion of the GGGGCC hexanucleotide repeat located within the first intron of the C9orf72 gene, is associated with the most prevalent form of familial amyotrophic lateral sclerosis, as well as frontotemporal dementia [78]. Various possibilities have been put forth, including impediments to expression of the C9orf72 gene itself, expression of putatively toxic sense or anti-sense transcripts of the repeats, and the expression of putatively toxic repeat-associated, non-ATG translation products [78]. Collectively, the relevance of these possibilities to our hypothesis certainly merits further investigation.
CONCLUSION
Much progress in our understanding of the mechanism of polyQ disease has been made since the discovery of the polyQ expansion in various polyQ diseases, such as SBMA and HD in the early 1990 s [79, 80]. Although it has become clear that the neurotoxicity of epolyQ is disease-causative, the biological significance of epolyQ remain poorly understood. Given the analogous properties of both epolyQ and APs in terms of redundancy through non-amyloidogenic homologues, hormesis, and transgenerational transmission, evolvability could be regarded as a physiological function of epolyQ. Furthermore, evolvability during the reproductive stage and neurodegenerative disorders during post-reproductive senescent life may exist in an antagonistic pleiotropic relationship in both polyQ diseases and other neurodegenerative conditions. Moreover, evolvability in epolyQ might be more primitive than that of APs across evolution. Given the vast separation between yeast prion and APs in evolution, it is plausible that epolyQ evolvability might be situated between them, playing a critical role in the evolution of amyloid-related evolvability. Collectively, further investigation is required to clarify both the pathological and physiological actions of epolyQ, which may provide clues leading to effective therapies for the polyQ diseases. On a final note, speculation might arise as to the possibility that AP-related evolvability might eventually be replaced by evolvability with other molecules or other regulatory mechanisms in the distant future, induced for instance by radical environmental changes.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHOR’S CONTRIBUTIONS
MH conceived the study and MH and GH wrote the paper. Both authors have read and approved the manuscript.
ACKNOWLEDGMENTS
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (University of Chicago). This paper is supported by neither public nor private grants.
REFERENCES
[1] | Zoghbi HY OH . Glutamine repeats and neurodegeneration. Annu Rev Neurosci. (2000) ;23: :217–47. |
[2] | Di Prospero NA , Fischbeck KH . Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet. (2005) ;6: (10):756–65. |
[3] | La Spada AR , Paulson HL , Fischbeck KH . Trinucleotide repeat expansion in neurological disease. Ann Neurol. (1994) ;36: (6):814–22. |
[4] | Wickner RB . Yeast and fungal prions. Cold Spring Harb Perspect Biol. (2016) ;8: (9). |
[5] | Hashimoto M , Ho G , Sugama S , Takamatasu Y , Shimizu Y , Takenouchi T , et al. Evolvability of amyloidogenic proteins in human brain. J Alzheimers Dis. (2018) ;62: (1):73–83. |
[6] | Hashimoto M , Ho G , Takamatasu Y , Shimizu Y , Sugama S , Takenouchi T , et al. Evolvability and neurodegenerative disease: Antagonistic pleiotropy phenomena derived from amyloid aggregates. J Parkinsons Dis. (2018) ;8: (3):405–8. |
[7] | Muhlau M , Winkelmann J , Rujescu D , Giegling I , Kout-souleris N , Gaser C , et al. Variation within the Huntington’s disease gene influences normal brain structure. PLoS One. (2012) ;7: (1):e29809. |
[8] | Lee JK , Ding Y , Conrad AL , Cattaneo E , Epping E , Mathews K , et al. Sex-specific effects of the Huntington gene on normal neurodevelopment. J Neurosci Res. (2017) ;95: (1-2):398–408. |
[9] | Lee JK , Conrad A , Epping E , Mathews K , Magnotta V , Dawson JD , et al. Effect of trinucleotide repeats in the Huntington’s gene on intelligence. EBioMedicine. (2018) ;31: :47–53. |
[10] | Maiuri T , Mocle AJ , Hung CL , Xia J , van Roon-Mom WM , Truant R . Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. (2017) ;26: (2):395–406. |
[11] | Ohno S . Gene duplication and the uniqueness of vertebrate genomes circa 1970-1999. Semin Cell Dev Biol. (1999) ;10: (5):517–22. |
[12] | Chastain PD , Sinden RR . CTG repeats associated with human genetic disease are inherently flexible. J Mol Biol. (1998) ;275: (3):405–11. |
[13] | Pearson CE , Wang YH , Griffith JD , Sinden RR . Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n. (CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res. (1998) ;26: (3):816–23. |
[14] | Sinden RR , Potaman VN , Oussatcheva EA , Pearson CE , Lyubchenko YL , Shlyakhtenko LS . Triplet repeat DNA structures and human genetic disease: Dynamic mutations from dynamic DNA. J Biosci. (2002) ;27: (1 Suppl 1):53–65. |
[15] | Veeraraghavan J , Rossi ML , Bambara RA . Analysis of DNA replication intermediates suggests mechanisms of repeat sequence expansion. J Biol Chem. (2003) ;278: (44): 42854–66. |
[16] | Li H , Liu J , Wu K , Chen Y . Insight into role of selection in the evolution of polyglutamine tracts in humans. PLoS One. (2012) ;7: (7):e41167. |
[17] | Butland SL , Devon RS , Huang Y , Mead CL , Meynert AM , Neal SJ , et al. CAG-encoded polyglutamine length polymorphism in the human genome. BMC Genomics. (2007) ;8: :126. |
[18] | Kraus-Perrotta C , Lagalwar S . Expansion, mosaicism and interruption: Mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1. Cerebellum Ataxias. (2016) ;3: :20. |
[19] | Mollereau B , Rzechorzek NM , Roussel BD , Sedru M , Van den Brink DM , Bailly-Maitre B , et al. Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations. Brain Res. (2016) ;1648: (Pt B):603–16. |
[20] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. (2015) ;522: (7556):340–4. |
[21] | Qiang W , Yau WM , Lu JX , Collinge J , Tycko R . Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature. (2017) ;541: (7636):217–21. |
[22] | Theillet FX , Binolfi A , Bekei B , Martorana A , Rose HM , Stuiver M , et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. (2016) ;530: (7588):45–50. |
[23] | Buchanan LE , Carr JK , Fluitt AM , Hoganson AJ , Moran SD , de Pablo JJ , et al. Structural motif of polyglutamine amyloid fibrils discerned with mixed-isotope infrared spectroscopy. Proc Natl Acad Sci USA. (2014) ;111: (16):5796–801. |
[24] | Chen S , Berthelier V , Hamilton JB , O’Nuallain B , Wetzel R . Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. (2002) ;41: (23):7391–9. |
[25] | Kirschner M , Gerhart J . Evolvability. Proc Natl Acad Sci USA. (1998) ;95: (15):8420–7. |
[26] | Becker C . Like father, like son. eLife. (2017) ;6: . |
[27] | Beer J , Masters CL , Beyreuther K . Cells from peripheral tissues that exhibit high APP expression are characterized by their high membrane fusion activity. Neurodegeneration. (1995) ;4: (1):51–9. |
[28] | Cicchetti F , Lacroix S , Cisbani G , Vallieres N , Saint-Pierre M , St-Amour I , et al. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol. (2014) ;76: (1):31–42. |
[29] | Trajkovic K , Jeong H , Krainc D . Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J Neurosci. (2017) ;37: (37):9000–12. |
[30] | Fodale V , Boggio R , Daldin M , Cariulo C , Spiezia MC , Byrne LM , et al. Validation of ultrasensitive mutant huntingtin detection in human cerebrospinal fluid by single molecule counting immunoassay. J Huntingtons Dis. (2017) ;6: (4):349–61. |
[31] | Liu Y . Like father like son. A fresh review of the inheritance of acquired characteristics. EMBO Rep. (2007) ;8: (9):798–803. |
[32] | Sharma A . Transgenerational epigenetics: Integrating soma to germline communication with gametic inheritance. Mech Ageing Dev. (2017) ;163: :15–22. |
[33] | Silveira JR , Raymond GJ , Hughson AG , Race RE , Sim VL , Hayes SF , et al. The most infectious prion protein particles. Nature. (2005) ;437: (7056):257–61. |
[34] | Sambataro F , Pennuto M . Post-translational modifications and protein quality control in motor neuron and polyglu-tamine diseases. Front Mol Neurosci. (2017) ;10: :82. |
[35] | Aghdasi B , Ye K , Resnick A , Huang A , Ha HC , Guo X , etal. FKBP12, the 12-kDaFK506-binding protein, is aphys-iologic regulator of the cell cycle. Proc Natl Acad Sci USA. (2001) ;98: (5):2425–30. |
[36] | Sugata H , Matsuo K , Nakagawa T , Takahashi M , Mukai H , Ono Y , et al. A peptidyl-prolyl isomerase, FKBP12, accumulates in Alzheimer neurofibrillary tangles. Neurosci Lett. (2009) ;459: (2):96–9. |
[37] | Sun CS , Lee CC , Li YN , Yao-Chen Yang S , Lin CH , Chang YC , et al. Conformational switch of polyglutamine-expanded huntingtin into benign aggregates leads to neuroprotective effect. Sci Rep. (2015) ;5: :14992. |
[38] | Takeuchi T , Suzuki M , Fujikake N , Popiel HA , Kikuchi H , Futaki S , et al. Intercellular chaperone transmission via exosomes contributes to maintenance of protein home-ostasis at the organismal level. Proc Natl Acad Sci USA. (2015) ;112: (19):E2497–506. |
[39] | Fujita M , Wei J , Nakai M , Masliah E , Hashimoto M . Chaper-one and anti-chaperone: Two-faced synuclein as stimulator of synaptic evolution. Neuropathology. (2006) ;26: (5):383–92. |
[40] | Leavitt BR , Guttman JA , Hodgson JG , Kimel GH , Singaraja R , Vogl AW , et al. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. (2001) ;68: (2):313–24. |
[41] | Sethi R , Tripathi N , Pallapati AR , Gaikar A , Bharatam PV , Roy I . Does N-terminal huntingtin function as a ‘holdase’ for inhibiting cellular protein aggregation? FEBS J. (2018) ;285: (10):1791–811. |
[42] | Bae BI , Xu H , Igarashi S , Fujimuro M , Agrawal N , Taya Y , et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. (2005) ;47: (1):29–41. |
[43] | Steffan JS , Kazantsev A , Spasic-Boskovic O , Greenwald M , Zhu YZ , Gohler H , et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. (2000) ;97: (12): 6763–8. |
[44] | Feng Z , Jin S , Zupnick A , Hoh J , de Stanchina E , Lowe S , et al. p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene. (2006) ;25: (1):1–7. |
[45] | Ryan AB , Zeitlin SO , Scrable H . Genetic interaction between expanded murine Hdh alleles and p53 reveal deleterious effects of p53 on Huntington’s disease pathogenesis. Neurobiol Dis. (2006) ;24: (2):419–27. |
[46] | Lu XH , Mattis VB , Wang N , Al-Ramahi I , van den Berg N , Fratantoni SA , et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Hunting-ton’s disease. Sci Transl Med. (2014) ;6: (268):268ra178. |
[47] | Chou AH , Lin AC , Hong KY , Hu SH , Chen YL , Chen JY , et al. p53 activation mediates polyglutamine-expanded ataxin-3 upregulation of Bax expression in cerebellar and pontine nuclei neurons. Neurochem Int. (2011) ;58: (2):145–52. |
[48] | Ano Bom AP , Rangel LP , Costa DC , de Oliveira GA , Sanches D , Braga CA , et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J Biol Chem. (2012) ;287: (33):28152–62. |
[49] | Checler F , Alves da Costa C . p53 in neurodegen-erative diseases and brain cancers. Pharmacol Ther. (2014) ;142: (1):99–113. |
[50] | Okazawa H . Polyglutamine diseases: A transcription disorder? Cell Mol Life Sci. (2003) ;60: (7):1427–39. |
[51] | Waragai M , Lammers CH , Takeuchi S , Imafuku I , Udagawa Y , Kanazawa I , et al. PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum Mol Genet. (1999) ;8: (6):977–87. |
[52] | Mizuguchi M , Obita T , Serita T , Kojima R , Nabeshima Y , Okazawa H . Mutations in the PQBP1 gene prevent its interaction with the spliceosomal protein U5-15 kD. Nat Commun. (2014) ;5: :3822. |
[53] | Langfelder P , Cantle JP , Chatzopoulou D , Wang N , Gao F , Al-Ramahi I , et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. (2016) ;19: (4):623–33. |
[54] | Carter AJ , Nguyen AQ . Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med Genet. (2011) ;12: :160. |
[55] | Shokeir MH . Investigation on Huntington’s disease in the Canadian Prairies. II. Fecundity and fitness. Clin Genet. (1975) ;7: (4):349–53. |
[56] | Eskenazi BR , Wilson-Rich NS , Starks PT . A Darwinian approach to Huntington’s disease: Subtle health benefits of a neurological disorder. Med Hypotheses. (2007) ;69: (6):1183–9. |
[57] | Chang JR , Ghafouri M , Mukerjee R , Bagashev A , Chabrashvili T , Sawaya BE . Role of p53 in neurodegen-erative diseases. Neurodegener Dis. (2012) ;9: (2):68–80. |
[58] | Thion MS , Humbert S . Cancer: From wild-type to mutant huntingtin. J Huntingtons Dis. (2018) ;7: (3):201–8. |
[59] | Costa DC , de Oliveira GA , Cino EA , Soares IN , Rangel LP , Silva JL . Aggregation and prion-like properties of misfolded tumor suppressors: Is cancer a prion disease? Cold Spring Harb Perspect Biol. (2016) ;8: (10). |
[60] | Ungewitter E , Scrable H . Antagonistic pleiotropy and p53. Mech Ageing Dev. (2009) ;130: (1-2):10–7. |
[61] | Arrasate M , Mitra S , Schweitzer ES , Segal MR , Finkbeiner S . Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. (2004) ;431: (7010):805–10. |
[62] | Ramdzan YM , Trubetskov MM , Ormsby AR , Newcombe EA , Sui X , Tobin MJ , et al. Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep. (2017) ;19: (5):919–27. |
[63] | Dickey AS , La Spada AR . Therapy development in Huntington disease: From current strategies to emerging opportunities. Am J Med Genet A. (2018) ;176: (4):842–61. |
[64] | Lane RM , Smith A , Baumann T , Gleichmann M , Norris D , Bennett CF , et al. Translating antisense technology into a treatment for Huntington’s disease. Methods Mol Biol. (2018) ;1780: :497–523. |
[65] | Lindqvist C , Laakkonen L , Albert VA . Polyglutamine variation in a flowering time protein correlates with island age in a Hawaiian plant radiation. BMC Evol Biol. (2007) ;7: :105. |
[66] | Scala C , Tian X , Mehdiabadi NJ , Smith MH , Saxer G , Stephens K , et al. Amino acid repeats cause extraordinary coding sequence variation in the social amoeba Dictyostelium discoideum. PLoS One. (2012) ;7: (9):e46150. |
[67] | Michael TP , Park S , Kim TS , Booth J , Byer A , Sun Q , et al. Simple sequence repeats provide a substrate for phenotypic variation in the Neurospora crassa circadian clock. PLoS One. (2007) ;2: (8):e795. |
[68] | Gemayel R , Chavali S , Pougach K , Legendre M , Zhu B , Boeynaems S , et al. Variable glutamine-rich repeats modulate transcription factor activity. Mol Cell. (2015) ;59: (4):615–27. |
[69] | Luo LQ , Martin-Morris LE , White K . Identification, secretion, and neural expression of APPL, a Drosophila protein similar to human amyloid protein precursor. J Neurosci. (1990) ;10: (12):3849–61. |
[70] | Coulson EJ , Paliga K , Beyreuther K , Masters CL . What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int. (2000) ;36: (3): 175–84. |
[71] | Toni M , Cioni C . Fish synucleins: An update. Mar Drugs. (2015) ;13: (11):6665–86. |
[72] | Semaka A , Kay C , Doty CN , Collins JA , Tam N , Hayden MR . High frequency of intermediate alleles on Huntington disease-associated haplotypes in British Columbia’s general population. Am J Med Genet B Neuropsychiatr Genet. (2013) ;162B: (8):864–71. |
[73] | Kay C , Collins JA , Miedzybrodzka Z , Madore SJ , Gordon ES , Gerry N , et al. Huntington disease reduced penetrance alleles occur at high frequency in the general population. Neurology. (2016) ;87: (3):282–8. |
[74] | Roan NR , Sandi-Monroy N , Kohgadai N , Usmani SM , Hamil KG , Neidleman J , et al. Semen amyloids participate in spermatozoa selection and clearance. eLife. (2017) ;6. |
[75] | Udd B , Krahe R . The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. (2012) ;11: (10):891–905. |
[76] | Ranum LP , Day JW . Dominantly inherited, non-coding microsatellite expansion disorders. Curr Opin Genet Dev. (2002) ;12: (3):266–71. |
[77] | Wilburn B , Rudnicki DD , Zhao J , Weitz TM , Cheng Y , Gu X , et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Hunt-ington’s disease-like 2 mice. Neuron. (2011) ;70: (3):427–40. |
[78] | Lin Y , Mori E , Kato M , Xiang S , Wu L , Kwon I , et al. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell. (2016) ;167: (3):789–802 e12. |
[79] | La Spada AR , Wilson EM , Lubahn DB , Harding AE , Fischbeck KH . Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. (1991) ;352: (6330):77–9. |
[80] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: (6):971–83. |


