
Modulating Neurotrophin Receptor Signaling as a Therapeutic Strategy for Huntington’s Disease
Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by CAG repeat expansions in the IT15 gene which encodes the huntingtin (HTT) protein. Currently, no treatments capable of preventing or slowing disease progression exist. Disease modifying therapeutics for HD would be expected to target a comprehensive set of degenerative processes given the diverse mechanisms contributing to HD pathogenesis including neuroinflammation, excitotoxicity, and transcription dysregulation. A major contributor to HD-related degeneration is mutant HTT-induced loss of neurotrophic support. Thus, neurotrophin (NT) receptors have emerged as therapeutic targets in HD. The considerable overlap between NT signaling networks and those dysregulated by mutant HTT provides strong theoretical support for this approach. This review will focus on the contributions of disrupted NT signaling in HD-related neurodegeneration and how targeting NT receptors to augment pro-survival signaling and/or to inhibit degenerative signaling may combat HD pathologies. Therapeutic strategies involving NT delivery, peptidomimetics, and the targeting of specific NT receptors (e.g., Trks or p75NTR), particularly with small molecule ligands, are discussed.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominantly inherited neurodegenerative disorder caused by an expansion of CAG repeats in exon 1 of the IT15 gene which encodes an extended polyglutamine stretch in the huntingtin (HTT) protein [1–3]. Although progressive motor dysfunction is the primary clinical manifestation of HD, cognitive and psychiatric impairments are prevalent and often occur before motor symptoms manifest [3, 4]. Neuropathological hallmarks of HD include preferential loss of medium spiny projection neurons (MSNs) in the striatum and nuclear HTT aggregates [3, 5]. Synaptic dysfunction and ultimately dendritic and neuronal degeneration extends beyond the striatum to other brain areas such as the cortex and hippocampus. The mechanisms that give rise to these pathologies are numerous and diverse including inflammatory processes, excitotoxicity, metabolic disturbances, and dysregulated proteolysis and transcription [6]. Nevertheless, mutant HTT-induced loss of neurotrophic support is thought to have a major contributory role to HD-related degeneration [6, 7]. Neurotrophins (NTs) including nerve growth factor (NGF), brain derived neurotrophic factor (BDNF), NT-3, and NT-4/5 promote synaptic function and survival of neuronal populations particularly affected in HD including striatal MSNs and cortical projection neurons [8]. Moreover, each NT has been shown to be neuroprotective against HD-related pathology, with BDNF the most extensively studied [7, 9–11]. NTs bind to two types of receptors, namely tropomyosin receptor-kinases (Trks) and the 75 kDa pan-NT receptor (p75NTR) which are classically thought to regulate pro-survival and degenerative signaling, respectively. The intracellular signaling systems regulated by both Trks and p75NTR largely intersect with those contributing to HD pathogenesis (see Fig. 1). A majority of studies report that levels of NTs and/or their Trk receptors decrease while p75NTR levels increase in the HD brain [7, 11–15]. The ensuing loss of trophic signaling and attendant gain in degenerative signaling is thought to causally contribute to fundamental HD pathologies [11, 13]. Given the mechanistic complexity of HD and the large overlap between NT signaling networks and those affected by mutant HTT, targeting NT signaling has great potential to combat multiplex symptoms. This review will focus on mutant HTT-induced impairments in NT receptor signaling and how targeting these signaling pathways with NT administration or up-regulation and/or by pharmacological means may be an effective therapeutic strategy.
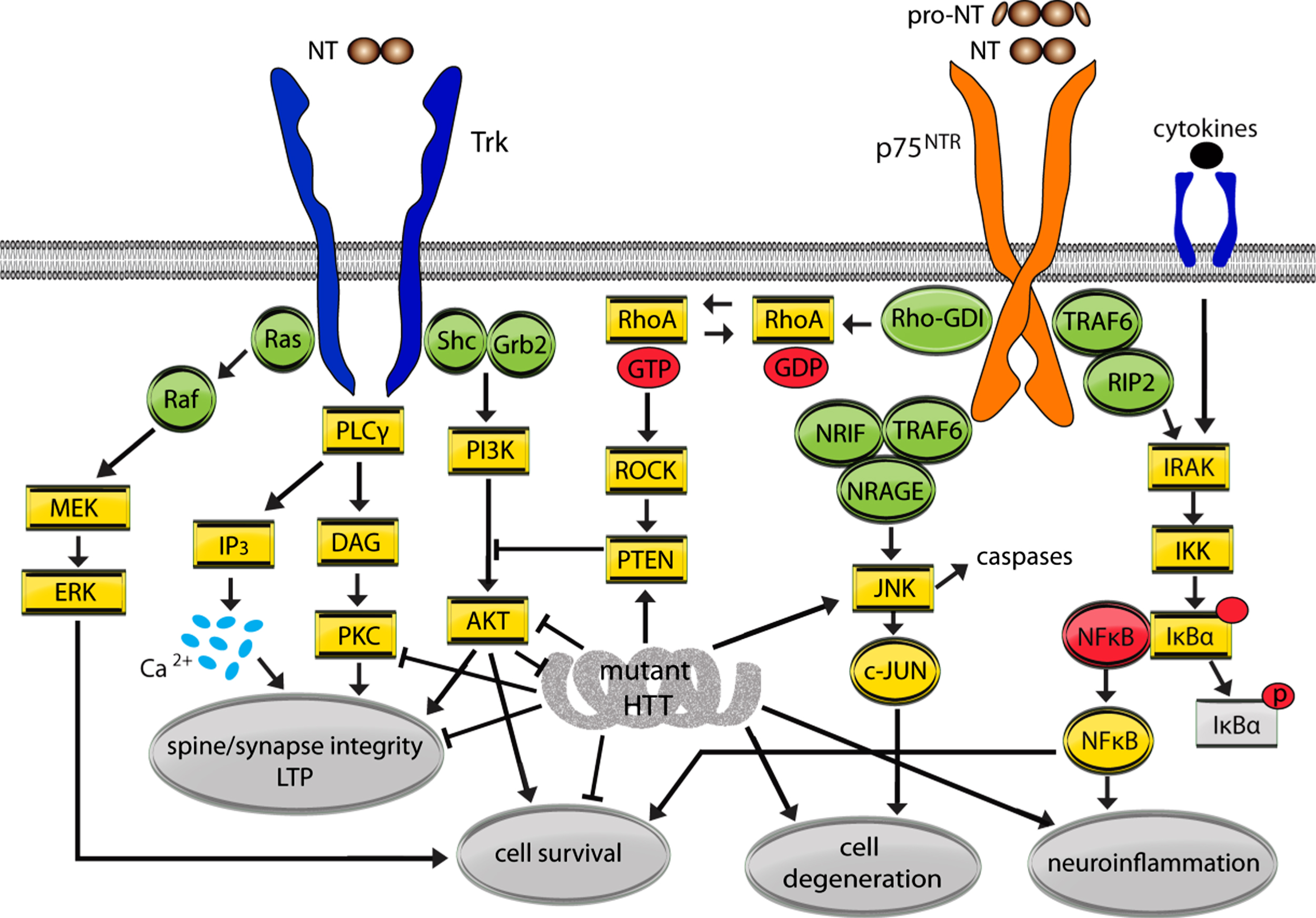
Fig.1
Mutant huntingtin (HTT) effects on Trk and p75NTR signaling pathways. This simplified schematic diagram depicts the signaling pathways regulated by the neurotrophin (NT) receptors, Trk and p75NTR, and their high integration with those dysregulated by mutant HTT. Arrows indicate up-regulation or promotion while ⊢ indicates inhibition or dysregulation. NTs signal through preformed or induced receptor dimers. Mature NT binding to Trks recruits adaptor proteins Shc and growth factor receptor-binding protein 2 (Grb2) and activate kinases resulting in further autophosphorylation and signaling via the PLCγ/PKC, MAPK (Ras/Raf/MEK/ERK) and PI3K/AKT pathways. p75NTR lacks intrinsic catalytic activity and regulates cellular processes by modulating Trk signaling and/or recruiting cell survival or cell death associated adaptor molecules including tumor necrosis factor receptor-associated factor 1–6 (TRAF1-6), NT receptor-interacting MAGE homolog (NRAGE), NT receptor-interacting factor (NRIF), and receptor-interacting protein 2 (RIP2). Depending on the cellular context and receptors present, mature NT binding to p75NTR may potentiate NT binding to Trk receptors and/or reinforce Trk signaling to promote cell survival. Mature NT or pro-NT binding to p75NTR can cause activation of the RhoA and JNK signaling pathways leading to dendritic spine loss, caspase release, and/or cell death.
2NEUROTROPHIN RECEPTOR SIGNALING
NTs are a family of secreted proteins comprised of NGF, BDNF, NT-3, and NT-4/5 that promote neuronal survival and synaptic function [16–18]. NTs are produced as a pro-protein that is proteolytically cleaved into a mature form prior to release from diverse cell types including neurons and glia. Both NT forms bind to two classes of cell surface receptors: Trks and p75NTR. Mature NTs principally bind to one of three Trk isoforms to promote cell survival or differentiation with NGF preferentially binding to TrkA, BDNF and NT-4/5 to TrkB, and NT-3 to TrkC. Some heterologous binding also occurs for example NT-3 has been shown to activate TrkB [18, 19]. All mature NTs also bind unselectively and with low affinity to p75NTR thus activating trophic signaling by potentiating Trk function or degenerative signaling if cognate Trks are absent or low [20–22]. Pro-NTs preferentially bind with high affinity to p75NTR and a protein dimer receptor complex of p75NTR-sortilin to initiate cell death signaling pathways. p75NTR also acts as a co-receptor for neurite outgrowth inhibitors with the NOGO receptor/LINGO-1 complex to regulate axonal regeneration and plasticity [23, 24].
2.1Trk signaling
Mature NTs have a central region that is the main Trk binding site and three hairpin β-turn loops (loops 1, 2, and 4) that vary amongst the NTs and confer receptor specificity. Thus, these loops have been exploited for the structure-based development of small molecule ligands for specific Trks, as discussed in the NT- and Trk-based Therapeutic Strategies section [20]. NTs signal through preformed or induced Trk dimers and trigger autophosphorylation at multiple tyrosine residues in the Trk cytoplasmic domain [18, 25, 26]. When the tyrosine residues are phosphorylated, they form cores of binding sites for adaptor proteins [e.g., Src, Shc, and growth factor receptor-binding protein 2 (Grb2)] leading to the activation of canonical downstream signaling pathways: MAPK (Ras/Raf/MEK/ERK), PI3K/AKT, and PLCγ/PKC [16, 18, 23] (Fig. 2). PLCγ/PKC signaling is initiated by binding to phosphorylated Y785 on TrkA and analogous sites on TrkB and TrkC (Y817/Y820), PI3K/AKT and MAPK/ERK activation involves Y490/Y515/Y516 [18, 27]. The effects of NT-Trk signaling via these pathways are typically related to cell survival and differentiation.
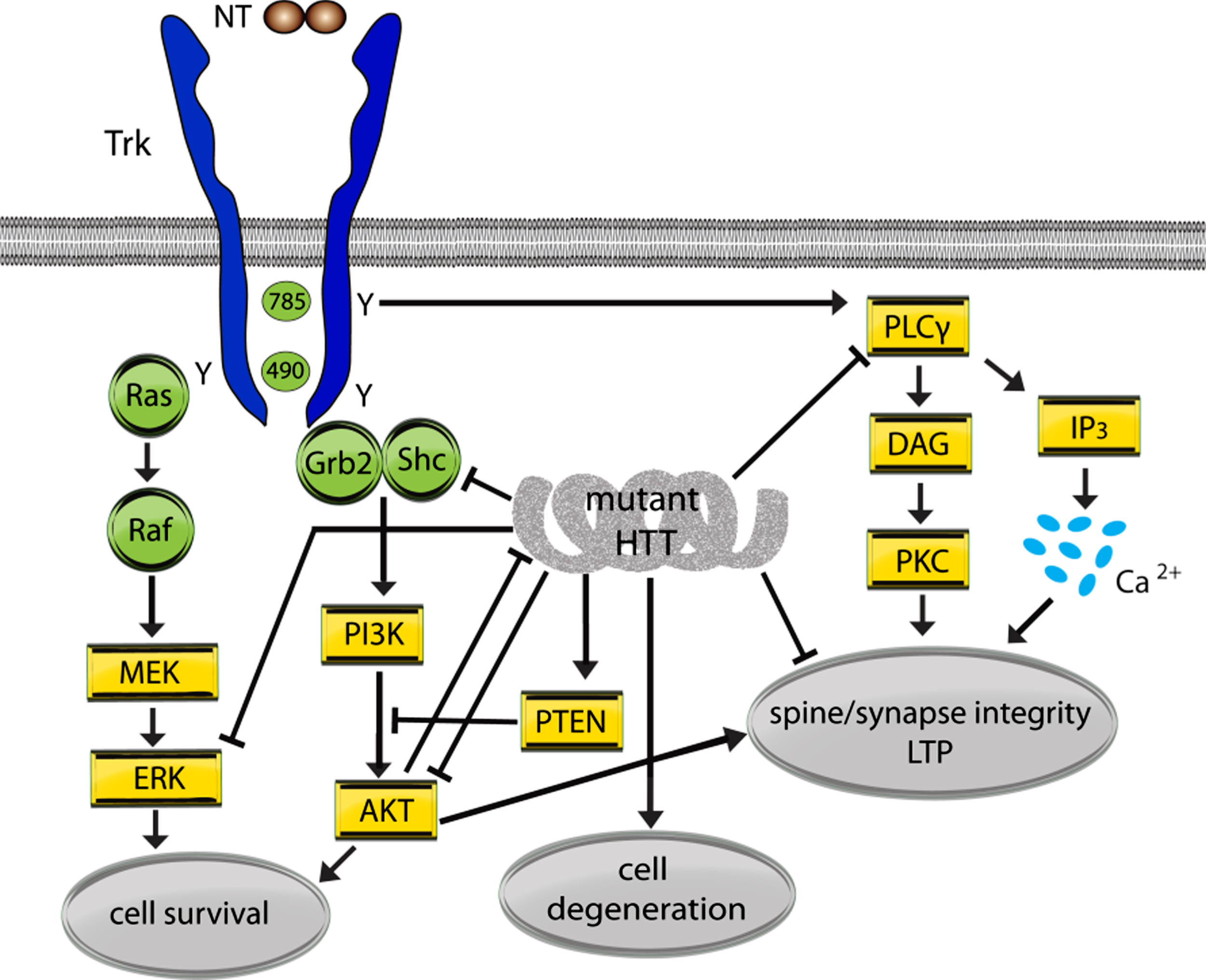
Fig.2
Mutant huntingtin (HTT) effects on Trk signaling pathways. A simplified schematic diagram showing the interactions between mutant HTT and Trk signaling pathways. Arrows indicate up-regulation or promotion while⊢ indicates inhibition or dysregulation. NTs signal through preformed or induced receptor dimers. Mature NT binding to Trks causes phosphorylation of intracellular domain tyrosine (Y) residues that form cores of binding sites for adaptor proteins Shc and growth factor receptor-binding protein 2 (Grb2) and activate kinase activity. Phosphorylation at Y785 (for TrkA; Y817/Y820 for Trks B and C) initiates signaling via the PLCγ/PKC pathway and Y490 (for TrkA; Y515/Y516 for Trks B and C) initiates signaling via the MAPK (Ras/Raf/MEK/ERK) and PI3K/AKT pathways. The effects of NT-Trk signaling via these pathways are typically related to synapse and dendritic spine integrity, long-term potentiation (LTP) and cell survival.
p75NTR signaling
p75NTR contains an extracellular domain with four cysteine-rich domains, two of which bind to NTs (domains 2 and 3), and a cell death domain in the intracellular region [28]. Pro- or mature NTs form homodimers that bind to dimeric p75NTR, which is either preformed or created with NT binding [29]. p75NTR lacks intrinsic enzymatic activity but regulates multifunctional cellular processes by modulating Trk signaling and recruiting adaptor molecules that act as bifunctional toggles for cell survival or death signaling. Interactions between p75NTR and adaptor molecules [e.g., tumor necrosis factor receptor-associated factor 1–6 (TRAF1-6), NT receptor-interacting MAGE homolog (NRAGE), NT receptor-interacting factor (NRIF), and receptor-interacting protein 2 (RIP2)] are ligand-dependent and, along with the co-receptors present, determine the signaling pathways activated [18, 23]. High concentrations of NTs and/or low Trk levels promote p75NTR interactions with adaptor proteins TRAF6, NRAGE, and NRIF leading to activation of the JNK pathway and cell death [23, 30] (Fig. 3). p75NTR also triggers JNK signaling via interfaces with NRAGE to activate caspases or by interacting with ceramide via sphingomyelinase activation [31, 32]. Moreover, binding of pro-NTs to p75NTR-sortilin also mediates cell death via JNK signaling and c-Jun activation after recruitment of adaptor molecules specific for certain cell types [23, 33]. For instance, in hippocampal neurons, pro-BDNF and pro-NGF binding induce p75NTR recruitment of NRAGE and/or NRIF to mediate apoptosis [23, 34].
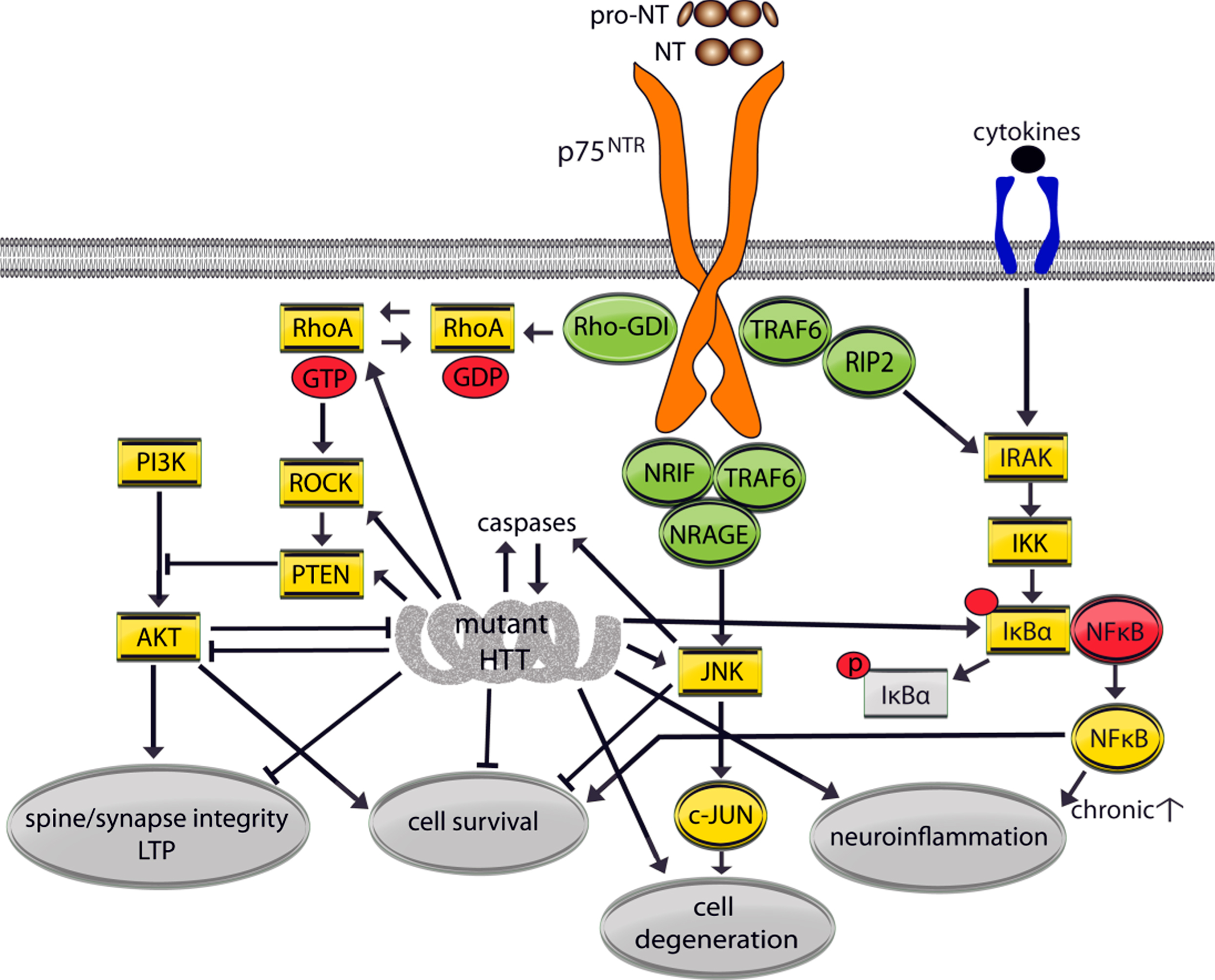
Fig.3
Mutant huntingtin (HTT) effects on p75NTR signaling pathways. p75NTR lacks intrinsic catalytic activity and regulates cellular processes by modulating Trk signaling and recruiting adaptor molecules including TRAF1-6, NRAGE, NRIF, and RIP2 which determine the signaling pathways activated. Regulated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling supports cell survival but when chronically activated, as in the presence of cytokines, inhibitor of kappa beta kinase (IKK)/NFκβ signaling mediates neuroinflammation. Inhibitor of nuclear factor κB (IκBα) sequesters NFκβ however when IκBα is phospohorylated (p) NFκβ is free to enter the nucleus to activate genes that promote inflammation. Mature NT or pro-NT binding to p75NTR can cause activation of the RhoA/Rho associated kinase (ROCK) and JNK signaling pathways. p75NTR interacts with Rho-GDI which activates RhoA small GTPase and ROCK signaling. ROCK then activates PTEN, which dephosphorylates PI3K thereby suppressing survival signaling and the promotion of synaptic plasticity mediated by TrkB. p75NTR can recruit TRAF6, NRAGE, and NRIF leading to activation of the JNK pathway and caspases, and cell death. Pro-NT binding to the sortilin/p75NTR complex (not shown) can also activate PTEN directly, which acts like toggle between survival and degenerative signaling, and the JNK pathway.
Conversely, TRK-p75NTR interactions enhance NT binding affinity to promote pro-survival signaling via MAPK/ERK, PI3K/AKT, and PLCγ/PKC pathways [35, 36]. Regulated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling also supports cell survival in the absence of Trks by NT-p75NTR binding, particularly NGF, and recruitment of RIP2 and TRAF6 [37, 38]. When chronically activated as in the presence of cytokines, inhibitor of kappa beta kinase (IKK)/NFκβ signaling mediates neuroinflammation [39]. NT-p75NTR binding or pro-NT binding with the sortilin/p75NTR complex can activate the phosphatase and tension homolog (PTEN), which acts like a switch between trophic and degenerative signaling. p75NTR interacts with Rho-GDI which activates RhoA/Rho associated kinase (ROCK) signaling. ROCK then activates PTEN, which dephosphorylates PI3K thereby suppressing survival signaling and synaptic plasticity/long-term potentiation (LTP) mediated by TrkB [40, 41] (Figs. 1 and 3). Finally, when NOGO or myelin-derived proteins bind to the p75NTR-NOGO receptor/LINGO1 complex, RhoA is activated and RAC is inhibited which negatively modulates the cytoskeletal system including deregulation of dendritic spine dynamics [42, 43].
3TRK SIGNALING IN HD
TrkA signaling in HD
Few studies have investigated the effects of mutant HTT on NGF or TrkA levels. In HD patients, mRNA levels of NGF in cortex and TrkA in hippocampus were unchanged [44, 45]; striatal levels have yet to be examined. NGF levels were unaltered in the striatum, cortex, and hippocampus at early symptomatic disease stages of the R6/1 transgenic mouse model of HD, but were reduced in hippocampus at later stages [46, 47]. Moreover, TrkA levels were decreased in striatal and cortical synapses of the CAG140 full-length HTT knock-in mouse model at symptomatic stages [48].
Disrupted NGF-TrkA signaling may contribute to HD-associated cholinergic deficits of striatal interneurons and hippocampal neurons. TrkA is absent from striatal MSNs but is present in cholinergic interneurons, which have synaptic plasticity deficits and abnormal morphologies in HD mouse models and/or patients [49–55]. Mutant HTT reduced striatal TrkA activation, as evidenced by decreased TrkA phosphorylation at Y791, in the R6/2 striatum (Simmons et al., unpublished results; [11]) and interferes with TrkA signaling contributing to neurite outgrowth [56, 57]. In vitro studies showed that mutant HTT interacts abnormally with huntingtin interacting protein 1 to reduce plasma membrane levels of TrkA and with Grb2 to disrupt TrkA signaling via the PI3K/AKT, MAPK, and JNK pathways that regulate neurite outgrowth [56, 57]. Finally, cholinergic neurochemical deficits in the R6/1 hippocampus were ameliorated by intracerebroventricular delivery of NGF which was also accompanied by increased neurogenesis and improved hippocampal-dependent memory [46]. In all, although NGF and TrkA have limited overlap with areas and cell types affected in HD, they may contribute considerably to specific aspects of HD degeneration.
TrkC signaling in HD
NT-3 has been shown to provide functional and trophic support to peripheral proprioceptive and motor neurons and to promote synapse formation and plasticity [58–62]. NT-3-containing neurons are located in brain regions affected in HD, including the striatum, cortex and hippocampus [63, 64]. In the cortex, NT-3 mRNA levels were greatly reduced in HD patients [45] as were NT-3 protein levels in R6/2 striatum (Simmons et al., unpublished results; [11]). NT-3’s receptor TrkC is expressed by MSNs, primarily in the D1a dopamine receptor-containing neurons of the direct pathway (dSPNs) of the basal ganglia that are involved in movement initiation [51, 63–65]. Interestingly, TrkB is predominately expressed in D2-containing MSNs of the opposing indirect pathway (iSPNs) [66, 67]. In R6/2 mice, striatal TrkC levels and activation are decreased at a mid-symptomatic stage (Simmons et al., unpublished; [11]) and this could contribute to the dysfunction of dSPNs seen in later stages of HD as their survival is compromised with diminished NT-3-TrkC signaling [66, 67]. Finally, NT-3-TrkC signaling has been shown to inhibit excitotoxicity-induced death of MSNs and ameliorate cortical neuron dysfunction [9, 10], and these effects are attributed to the PI3K/AKT, not ERK, pathway [68].
TrkB signaling in HD
The native ligands for TrkB are BDNF and NT-4/5. NT-4/5 is expressed throughout the CNS, including the striatum, and regulates the chemical phenotype of striatal neurons [9, 10, 69]. Little is known about the role of NT-4/5 in HD except that neurodegeneration was ameliorated by NT-4/5-secreting cells transplanted into the striatum of an excitotoxic rodent model of HD [9, 10]. Thus, this section will concentrate on discussing the contribution of BDNF-TrkB signaling disturbances to HD pathogenesis.
BDNF is involved in diverse cellular processes including development, synaptic plasticity, and neurotransmission. It is well known as a potent neuroprotective agent and promotes the survival of cortical and striatal neurons in vitro and prevents cell death in vivo following axotomy, ischemia, 3-nitroproionic acid toxicity and excitotoxic insults [70–76]. Pro- and mature forms of BDNF are present in the hippocampus and cortex while the primary source of mature BDNF for the striatum is anterograde transport via cortical projection neurons as well as dopaminergic and thalamic afferents [15, 63, 77–80]. BDNF’s receptor TrkB is widely expressed in the CNS including specific neuronal types affected in HD including cortical projection neurons, dopamine-containing nigrostriatal neurons, and striatal MSNs, particularly those of the indirect pathway (iSPNs) containing D2 dopamine receptors and enkephalin [63, 66, 67, 81]. TrkB is also located on striatal dendritic spines and is thus situated to form and stabilize these structures [82–87]. Accordingly, BDNF-TrkB signaling has been shown to be critical for memory processes including maintaining synapses and stabilizing LTP in hippocampus and striatum [82, 87–90].
Much evidence suggests that BDNF and TrkB mRNA and/or protein levels are markedly reduced in the cortex, striatum and hippocampus of HD patients and mouse models [7, 12, 13, 15, 77, 91–102]. Hippocampal and/or cortical pro-BDNF levels were also shown to decrease in early symptomatic HdhQ111 mice and zQ175 [77]. However, there have been reports that levels of cortical BDNF expression and striatal TrkB were unaltered in HD patients and/or in early to mid-symptomatic HD mice (for reviews see [15, 40]). These differing reports may be due to different HD mouse models used, technical limitations, and/or methodological differences including the use of ELISA which does not differentiate mature and pro-forms of BDNF, normalization to housekeeping genes that undergo disease-related changes, and/or BDNF antibodies with low specificity/sensitivity [11, 15, 40, 84]. To address these technical challenges, Ma et al. (2015) measured pro- and mature BDNF levels in zQ175 mice knocked-in with BDNF containing an HA epitope tag which allows highly specific HA antibodies to be used to accurately quantify BDNF [15]. This technique revealed that mature BDNF levels were reduced by ∼50% in the striatum of zQ175 mice at early symptomatic stages and in the cortex at later stages. Hippocampal and cortical pro-BDNF levels were also decreased in early symptomatic zQ175 mice [15].
Beyond the reported reductions in BDNF and TrkB expression, BDNF-TrkB signaling is disrupted by mutant HTT impaired axonal transport of BDNF from cortical neurons to striatum and compromised motility of vesicular BDNF in striatal and hippocampal neurons [80, 103–106]. TrkB cell surface expression is reduced in HD knock-in striatal cells (STHdhQ111) which may also hinder BDNF signaling [107]. Moreover, TrkB signal transduction is negatively impacted by deleterious effects of mutant HTT on scaffolding proteins, receptor phosphorylation, and downstream signaling intermediates. BDNF-TrkB signaling via the ERK pathway was markedly decreased in STHdhQ111 cells due to diminished Shc adaptor protein levels while TrkB phosphorylation was not affected [107]. On the other hand, striatal TrkB phosphorylation was reduced in R6/1 and R6/2 mice at tyrosine residues Y705 or Y515 but remained unchanged at Y817 in R6/2 and YAC128 mouse models of HD [91, 98, 100, 108]. These TrkB phosphorylation deficits were accompanied by decreased AKT, ERK1/2, and PLCγ signaling in the R6/1 and R6/2 striatum and occur at pre-symptomatic stages in the latter mouse model [91, 100, 108]. Depolarization-induced TrkB phosphorylation (Y515) in striatum was unaltered in the full-length BACHD mouse model of HD at mid-symptomatic stages while AKT signaling was attenuated suggesting mutant HTT effects on downstream signaling elements [40]. Thus, mutant HTT adversely affects BDNF-TrkB signaling at multiple levels: BDNF and TrkB expression, as well as signal transduction and downstream signaling stages. These deficits result in a severe loss of trophic support to neuronal populations that undergo degeneration in HD.
Disturbed BDNF/TrkB signaling has been shown to play a major role in the development of many key pathophysiological features of HD, including motor and cognitive deficits. Motor dysfunction in HD mice is exacerbated by genetic manipulations that decrease BDNF levels, as are dopaminergic deficits and degeneration of enkephalinergic striatal projection neurons [80, 81, 109, 110]. On the other hand, these pathologies are alleviated and motor performance improved by over-expressing BDNF in HD mice potentially by normalizing AKT and ERK signaling [98, 100]. Loss of BDNF via cortical afferents to striatal iSPNs is thought to contribute to HD motor symptoms, including chorea [6, 40]. Striatal iSPNs are preferentially affected in HD and suppress involuntary movement while dSPNs promote voluntary actions. BDNF-TrkB signaling via the AKT pathway is decreased in iSPNs, but not dSPNs, as was LTP [40]. Given that deficient BDNF-TrkB signaling in iSPNs causes hyperactivity in normal mice, these deficits may contribute directly to HD motor symptoms [111].
Mutant HTT-induced synaptic dysfunction, dendritic spine loss, and memory deficits have also been associated with perturbed BDNF-TrkB signaling (Fig. 2). As mentioned above, BDNF-TrkB signaling potently modulates the structural and functional aspects of synaptic plasticity, including dendritic spine formation and morphology as well as LTP [7, 89, 112–116]. Applying exogenous BDNF in vitro, up-regulating endogenous BDNF, or overexpressing the NT prevents dendritic spine loss and decreased cortico-striatal connectivity, and/or reverses hippocampal LTP deficits in HD mice [77, 80, 93, 98, 117–119]. This BDNF-induced preservation of synaptic integrity was accompanied by elevated AKT, PLCγ, and/or CREB signaling and preserved memory [93, 98, 117, 118].
Finally, BDNF-TrkB signaling may aid in mutant HTT clearance since it facilitates ubiquitin-proteosome processing which is one way that pathological protein aggregates are degraded [112]. Reduced BDNF-TrkB signaling via the AKT pathway may contribute to mutant HTT pathology as AKT has been shown to phosphorylate HTT or increase its degradation by the proteasome thus decreasing mutant HTT aggregation and toxicity [120–122]. In support of this suggestion, overexpressing BDNF or TrkB in HD cell lines and mouse models increases AKT signaling and/or reduces striatal HTT huntingtin aggregates and cell death [98, 100, 123].
Taken together these studies show the complex effects of mutant HTT on BDNF-TrkB signaling which likely involve early deficits in signal transduction including mitigated TrkB activation as well as reduced adaptor protein levels and downstream signaling. These deficits may be exacerbated at mid- to late symptomatic stages by declining levels of BDNF and TrkB. Ultimately, these severe BDNF-TrkB signaling disruptions have strong causal links to many of the hallmark HD pathologies.
NT- AND TRK-BASED THERAPEUTIC STRATEGIES FOR HD
The above section stressed that NT-Trk signaling networks are highly integrated with pathways responsible for synaptic function and neuronal health/survival and those that are dysregulated by mutant HTT (Figs. 1 and 2). Thus, targeting NT-Trk signaling could offer mechanism-based therapeutics to counteract HD degenerative processes by offsetting deficiencies in NT/Trk levels and/or by inducing Trk downstream signaling. This idea has prompted the development of NT- and Trk-based therapeutics for HD using two main approaches: 1) delivery of exogenous recombinant NTs or endogenous up-regulation of the native Trk ligands, particularly BDNF, and 2) ligands (e.g., antibodies or small molecules) that bind to and activate Trks in the CNS.
Each of the NT- and Trk-based therapeutic approaches has been shown to have beneficial effects against neurodegeneration in preclinical HD studies and, in clinical trials for other neurodegenerative disorders, cellular NT delivery and NT up-regulation via electrical stimulation have produced promising results. However, each approach also has general and/or HD-related technical limitations and logistic considerations that need addressing to achieve therapeutic efficacy with minimal or no unwanted pleiotropic effects. One major technical obstacle for NT administration and the currently available Trk ligands is low brain bioavailability which, for the former, requires invasive delivery and the latter requires better formulations. A consideration for NT-based therapeutic strategies is that mature NTs can produce deleterious effects in conditions in which Trk levels are low or absent by initiating p75NTR degenerative signaling and can be particularly detrimental if p75NTR levels are elevated [13, 124, 125]. This state occurs in HD patients and mouse models as TrkB levels are reduced while p75NTR levels are increased ([13, 95]; see more detailed discussions in the p75NTR signaling in HD section). In the context of this HD-related p75NTR/TrkB imbalance, BDNF-mediated neuroprotection is reduced by activating JNK signaling via p75NTR [13]. Thus, the therapeutic window for effective use of NT-based HD treatments would start before the altered p75NTR-TrkB ratio is prominent (i.e., prior to when TrkB levels are reduced to the extent that downstream signaling of existing receptors is inadequate to provide neuroprotection and JNK signaling via p75NTR induces degeneration). A similar concern arises for the use of TrkB ligands as HD therapeutics reduced levels of the target and signal transduction deficits may render them unable to activate trophic signaling. Therefore, TrkB ligands would have to be administered before severe TrkB loss or be sufficiently potent to activate the remaining receptors to induce trophism. Thus, the use of NT-/Trk-based treatment strategies requires careful consideration of the therapeutic window given that the target levels are altered in HD. These NT- and Trk-based therapeutic strategies will be discussed further in the following sections, as will their advantages and limitations, with emphasis on BDNF- and TrkB-based strategies as these are the most prevalent and relevant to HD.
NT-based therapy
The rationale for delivery of exogenous NTs is partially based on rodent studies demonstrating that each of the NTs have neuroprotective effects against striatal neurodegeneration and/or HD-related pathologies when they are administered intraventricularly, via systemic delivery of the recombinant protein, or by cellular delivery methods [8–10, 46, 97, 126–128]. While these preclinical studies demonstrate the efficacy of NT administration against HD pathologies, the delivery methods used have limited clinical feasibility as NTs have low stability with brief serum half-lives, low bioavailability via oral administration routes, and restricted blood brain barrier permeability. The low brain penetration of the macromolecular NTs dictates invasive delivery methods that still may result in limited diffusion throughout the brain and would necessitate chronically administering the NTs due to the progressive nature of the neurodegeneration in HD [129, 130]. Furthermore, the NTs could produce adverse pleiotropic effects, including pain, by activating multiple receptors (e.g., p75NTR). In attestation to these limitations, clinical trials involving intraventricular infusion of NGF in Alzheimer’s disease (AD) patients and intrathecal injection of recombinant BDNF in amyotrophic lateral sclerosis (ALS) patients showed limited CNS bioavailability of the NTs accompanied by minimal or no improvement in outcome measures and undesirable side effects including increased innervation of cerebral blood vessels, weight loss, disrupted sleep, and/or neuropathic pain [131–136]. Thus, efforts to develop NT-based therapies began to focus on alternate delivery methods that are less invasive and would increase levels and functional duration of the NTs in the brain.
Although invasive, intracranial implantation of NT-producing mesenchymal stem cells (MSCs) may be an effective delivery strategy as these cells may provide a sustained supply of trophic factors to degenerating neurons. MSCs have been shown to up-regulate BDNF expression but can also be engineered to deliver NTs and other factors that potentiate neuroprotective effects (e.g., BDNF and noggin) [137]. They migrate to regions of injured tissue and can be transplanted without eliciting immune rejection by the host, in fact, they have been shown to reduce inflammation [138]. Bone marrow-derived MSCs transplanted into the striata of R6/2 mice increased BDNF mRNA expression, improved motor and cognitive ability, and reduced brain volume loss [139]. When murine or human MSCs genetically engineered to overexpress BDNF or NGF were transplanted into striatum of HD mouse models, BDNF-MSCs improved motor performance, increased neurogenesis, and/or reduced anxiety-like behavior and striatal atrophy, while NGF-MSCs transiently alleviated motor deficits and did not affect survival of striatal neurons [128, 140]. Phase I and II clinical studies have demonstrated the biosafety of MSCs in diseases such as AD and ALS [141–143], but MSCs have yet to be tested in HD patients. A clinical trial for intrastriatal implants of BDNF-MSCs in HD patients is planned and has been accruing subjects via an observational study (PRE-CELL; ClinicalTrials.gov: NCT01937923) [127]. A relatively non-invasive and cost-effective delivery method has been developed in which MSCs are intranasally administered and travel extracellularly along axons of the olfactory pathway to bypass the blood brain barrier [144, 145]. This approach has been used to ameliorate behavioral symptoms and neurodegeneration in rodent models of AD and Parkinson’s disease (PD) but clinical application of this approach has not been attempted [145–148]. Thus, MSC delivery of NTs may circumvent some of the obstacles faced with the infusion approach in offering a more sustained and targeted delivery of NTs with more controlled dosing and less unwanted side effects.
Another approach to delivering NTs to subcortical brain regions is administering a viral vector engineered to encode the NT. Again this method is invasive as it involves injection of the virus into the target brain area, however, it may require fewer injections than the MSC approach since the virus would affect a stable change in the neuron’s ability to produce the NT [143, 149]. Preclinical studies employing this technique have been described in HD rodent models. Adenoviral BDNF and noggin, which reduces glial production, intraventricularly applied in R6/2 mice increased neurogenesis in the striatum and improved survival but did not affect motor function [150]. However, in this study, adenoviral expression was transient thus, the same group showed similar neurogenesis effects in rats using adeno-associated virus serotype 4 (AAV4) vectors to express BDNF and noggin in striatal neurons which produce prolonged changes, up to 2 and 6 years in rodents and primates respectively [149]. Similarly, an AAV1/2 vector containing BDNF injected bilaterally into the striatum of HD transgenic rats increased neurogenesis, ameliorated striatal atrophy, and improved motor and cognitive ability [151]. However, unlike MSCs, AAV vectors provoke immunogenic responses which limit their clinical use and clinical trials are needed to show safety and efficacy.
Instead of introducing the NTs themselves or using cell- or viral vector-mediated gene delivery approaches, many HD preclinical studies aimed at augmenting neuroprotective signaling have successfully increased endogenous levels of BDNF via behavioral modifications (e.g., exercise, environmental enrichment, and dietary restriction) or by pharmacological means including anti-depressants/psychotics (e.g., sertraline, NP03), anti-oxidants (e.g., B401), or glutamate receptor modulators/agonists (e.g., ampakines, LY379268) [47, 92, 93, 152–158]. These studies demonstrate that BDNF up-regulation produces neuroprotective effects and support pursuing BDNF- and Trk-based therapies for HD [for reviews, see [6, 7, 11]]. However, these behavioral strategies and compounds do not specifically target BDNF or TrkB and may have undesirable off-target effects.
BDNF expression has also been elevated in vivo via deep brain stimulation (DBS) or electroconvulsive therapy (ECT). ECT delivered chronically (over 10 days) increased BDNF mRNA and protein in the rat hippocampus and cortex for up to 48 hours and decreased levels of TrkB while increasing its activation (phosphorylation at site Y705) [159, 160]. ECT and DBS have been used clinically to ameliorate motor, cognitive, and/or behavioral symptoms (e.g., depression and agitation) in PD, AD and HD patients [161–165]. However, the procedure is invasive, select effects were transient, and in some patients adverse effects were noted including weight gain as well as impaired speech, gait, and cognition [166]. Moreover, whether any of these effects can be attributed to BDNF elevations are unknown. Further safety data and optimized stimulation paradigms for DBS are needed and will be addressed in an ongoing clinical trial examining the effects of pallidal-DBS in HD patients (ClinicalTrials.gov: NCT02535884) [164].
TrkB receptors as therapeutic targets
Despite the progress of the approaches described above, the prevailing limitations of NTs for clinical use, including invasive delivery, suboptimal pharmacological properties and pleiotropic activities via activation of multiple receptors (e.g., Trks and p75NTR), have led to development of alternative therapeutic strategies targeting specific NT receptors. Attempts to increase neurotrophic signaling via TrkB activation have revealed agonistic monoclonal TrkB antibodies that activate the receptor. Other efforts used a structure-based design approach to: 1) synthesize peptidomimetics that act as TrkB agonists, or 2) screen compound libraries, using in silico or cell-based methods, for small molecule TrkB activators.
The initial studies along these lines manufactured dimeric peptides containing the loop 2 sequence of BDNF, a region that confers TrkB binding and activation [167]. These peptides had BDNF-like activity in that they promoted the survival of embryonic chick sensory neurons in vitro [168] but have yet to be tested in vivo and their ability to activate TrkB was not investigated. Moreover, the therapeutic relevance of these reported peptide-based TrkB agonists is questionable considering these compounds have some of the same unfavorable pharmacological characteristics as the NTs themselves, primarily low oral bioavailability and in vivo instability, less options for optimized delivery methods, and effects at multiple receptors [169].
Monoclonal antibodies targeted to human and mouse TrkB have been generated that activate the receptor and/or support neurite outgrowth and cell survival similar to BDNF [170–172]. These bivalent agonist antibodies induced phosphorylation of TrkB at multiple sites (Y490, Y516, Y706/707, Y816), elevated intracellular Ca2 + release, and/or activated the MAPK and AKT signaling pathways in several cell-based assay systems. Two of these TrkB antibodies, 29D7 and 38B8, provided protection against mutant HTT-induced degeneration of striatal, not cortical, neurons in primary cortico-striatal co-cultures [173]. To date, functional monoclonal TrkB antibodies have not been tested in HD animal models or patients and therapeutic delivery will be a major challenge.
To circumvent delivery obstacles structurally inherent to macromolecular NTs and monoclonal antibodies, small molecule ligands selective for TrkB have been identified by cell-based or in silico screening methods that penetrate the blood brain barrier, activate TrkB and its downstream signaling, and provide neuroprotection. These small molecule TrkB ligands act as partial agonists as they phosphorylate differential patterns of tyrosine residues on the receptor and activate signaling pathways that are distinct from BDNF.
A cell-based apoptotic screen identified gedunin and flavone derivatives as TrkB agonists [174–177]. Deoxygedunin, a natural product from the Indian neem tree, prevented apoptosis in oxygen/glucose-deprived hippocampal neurons and, after oral or intraperitoneal delivery, phosphorylated TrkB (Y817) and initiated MAPK/ERK and P13K/AKT signaling while improving motor performance and reducing neurodegeneration in rodent models of PD [174, 177, 178]. Deoxygedunin has not been tested in HD animal models and, as its water solubility and brain permeability are low, derivatives of the compound are being pursued [177]. Finally, it is unknown whether deoxygedunin interacts with other Trks or p75NTR which could produce pleiotropic effects.
The flavone product 7,8-dihydroxyflavone (7,8-DHF) and its synthetic derivative 4’-dimethylamino-7,8-DHF (4’DMA-7,8-DHF) have been shown to activate TrkB, not TrkA or TrkC, and to provide neuroprotection in animal models of neurodegenerative disease, including Rett syndrome, AD, Down’s syndrome, and PD [175, 179–183]. Moreover, two preclinical studies have been conducted in HD mice showing that chronic oral administration of 7,8-DHF and/or 4’DMA-7,8-DHF activates TrkB and its downstream signaling and protects against neurodegeneration [184, 185]. In the N171-82Q mouse model of HD, 7,8-DHF and 4’DMA-7,8-DHF phosphorylated TrkB (Y706) and MAPK in the striatum, ameliorated brain volume loss, increased striatal DARPP-32 levels and neurogenesis, improved motor function, and prolonged survival [184]. A more recent study showed that 7,8-DHF prevented the reduction of TrkB phosphorylation at residue Y816, not Y515, increased PLCγ signaling, and reduced p75NTR levels in the striatum of R6/1 mice [185]. This increased TrkB signaling was associated with improved cognition and ameliorated HD-related neuropathologies in striatum including: HTT aggregate number and size, reduced enkephalin levels, striatal atrophy, and elevated inducible nitric oxide synthase, a neuroinflammation indicator [185]. The neuroprotective effects of 7,8-DHF may be partially attributed to TrkB activation, however, TrkB-independent effects also contribute as in vitro studies show that in the presence of the Trk inhibitor K252a, 7,8-DHF still prevents cell death and it acts as an antioxidant to protect cells that are devoid of TrkB [185, 186]. Finally, it is unknown whether 7,8-DHF activates p75NTR and it decreases p75NTR levels which could contribute to its beneficial effects as a p75NTR/Trk imbalance plays a role in HD pathogenesis [13, 185].
In silico screening of compound libraries was used by the Longo and Massa laboratories to identify small molecule non-peptide TrkB ligands that have structural similarities to loop 2 of BDNF and selectively activate TrkB, but not the other Trks or p75NTR in in vitro studies [20, 187]. One of these ligands, LM22A-4, has been shown to phosphorylate TrkB to recruit the Shc, GRB2 and SOS1 adaptor proteins and initiate signaling via all the major Trk signaling routes to promote neuronal survival in in vitro assays [20, 187–189]. LM22A-4 also demonstrated neuroprotective effects in mouse models of numerous neurodegenerative conditions, including Rett syndrome, stroke, and traumatic brain and spinal cord injury [187, 189–192]. LM22A-4 has also been shown to be effective against HD pathology. In R6/2 mice, systemic LM22A-4 administration activated TrkB (phosphorylation at Y705) as well as downstream signaling via AKT, PLCγ, and CREB [91]. It also decreased intranuclear HTT aggregates in striatum and cortex, reduced striatal microglial activation, and prevented degeneration of two main types of striatal neurons affected in HD: parvalbumin-containing interneurons and DARRP-32-containing medium-spiny projection neurons. LM22A-4 also prevented dendritic spine loss of medium spiny neurons and improved motor performance in both R6/2 and BACHD mice [91]. Taken together, the above findings suggest that targeting TrkB can combat HD degenerative mechanisms and improve motor performance and is therefore a viable therapeutic strategy for HD.
TrkA and TrkC as HD therapeutic targets
TrkA- and TrkC-based therapeutic strategies for HD have received much less attention than those involving TrkB but have followed similar developmental paths in that receptor structure has been used to design peptidomimetics or to identify small molecules that activate or potentiate the effects of the receptors’ signaling. Peptidomimetic TrkA agonists with structural similarity to NGF loop 4 have been shown to activate AKT and ERK signaling and provide neuroprotection in cell culture systems [193–195]. One of these peptide-based selective TrkA agonists, D3, delivered intraventricularly to an AD mouse model increased hippocampal and cortical AKT signaling and improved memory in AD mice but disrupted it in wild-types [196, 197]. Peptidomimetic TrkC agonists have also been identified that have trophic activity in in vitro assays but have not been tested in vivo [193]. To date, neither peptide-based TrkA nor TrkC agonists have been tested in HD models. Again, peptide-based therapeutic strategies are limited by potential pleiotropic effects caused by multi-receptor binding and sub-optimal pharmacological characteristics, including poor brain penetration and in vivo instability [169].
Small molecule non-peptide TrkA receptor agonists have been identified by cell-based apoptotic assays (e.g., gambogic amide) and in silico screening (e.g., MT2). Gambogic amide binds TrkA but not the other Trks and stabilizes TrkA dimers thus potentiating NGF activity. It activates TrkA by phosphorylation at Y490, Y751, and Y494 as well as PI3K/AKT and MAPK/ERK signaling in primary hippocampal neurons and blocks neuroexcitotoxicity in vivo [169, 177]. However, gambogic amide also binds to transferrin receptors and can induce apoptosis. MT2 activates TrkA by phosphorylation at Y490, Y674/675, and Y785 to initiate AKT and ERK signaling and decreased amyloidogenesis and neuronal death in NGF-deprived rat hippocampal cultures [198]. Neither of these TrkA ligands have been tested in in vitro or in vivo models of HD. However, potentiating NGF effects in R6/2 mouse model of HD with a substituted pyrimidine, KP544 (also known as CXB-909), shown to induce MAPK signaling in vitro, ameliorated motor deficits and prevented enlarged ventricles [199, 200]. These results suggest the therapeutic potential of small molecule modulation of TrkA signaling in HD.
A TrkB/TrkC co-activator, LM22B-10, was identified by an expanded in silico screening of small molecule libraries using the pharmacophore based on BDNF loop 2 that identified the TrkB ligand LM22A-4 [201]. LM22B-10 is a non-peptide small molecule that binds to and phosphorylates TrkB (Y515, Y705, Y817) and TrkC (Y516, Y820) but not TrkA or p75NTR. It recruits the Shc adaptor protein and triggers ERK and AKT signaling in hippocampus and striatum as well as PLCγ signaling in in vitro assays. Furthermore, LM22B-10 activated synaptic TrkB and TrkC and alleviated deficits in pre- and post-synaptic proteins as well as dendritic spine loss in aged mice [201]. Analysis of LM22B-10/Trk kinetics revealed that LM22B-10 produced signaling patterns that were similar to those of NT-3/TrkC and differed from BDNF/TrkB. The LM22B-10-induced TrkB/TrkC signaling may be synergistic as LM22B-10’s neurotrophic effects on hippocampal neurons exceeded that of the native NTs in that it promoted survival and neurite outgrowth to a greater extent than BDNF or NT-3 [201]. This functional synergy afforded by multi-target ligands could potentially combat diverse pathological mechanisms present in complex diseases such as HD [202]. Accordingly, initial examinations of LM22B-10 in R6/2 mice revealed that increasing TrkB and TrkC signaling reduced HTT aggregates and ameliorated DARPP-32 and motor deficits (Simmons et al., unpublished). These preliminary results are proof-of-concept that simultaneous targeting of multiple NT receptors may be a feasible and effective therapeutic strategy for HD and other neurodegenerative diseases. Current investigations are focused on developing TrkB/C ligands with improved pharmacological properties as brain permeability after oral administration of LM22B-10 is low and the ligand has a sub-optimal metabolic profile.
Testing of TrkA- and TrkC-based treatments in HD has received much less attention than those involving TrkB, however the data that does exists along these lines suggests that they may be effective therapeutic strategies. However, all the Trk ligands developed to date share the limitation of poor brain penetrance necessitating the development of novel derivatives with better bioavailability particularly via oral administration routes.
Challenges in developing small molecule Trk ligands
Identifying Trk ligands that can activate large multi-subunit receptors can be challenging especially concerning monomeric and monovalent small molecules [20, 203]. Bivalency is typically considered a requirement for receptor dimerization and activation, however, monovalent small molecules can bind and activate pre-formed dimeric receptors and may activate Trks in this way as TrkA and most TrkB receptors exist as homodimers [20, 25, 26, 197, 203–205]. Alternatively, small molecules can cause dimerization by binding to monomeric receptors and changing their conformation or by allosteric mechanisms [20]. Partial agonism is typically the result of monomeric/monovalent small molecule binding to Trks initiating patterns of autophosphorylation and signaling that can differ from the native NTs which are dimerically bound [91, 184, 187, 201]. This point is demonstrated by the differing patterns of Trk phosphorylation of 7,8-DHF, LM22A-4 and LM22B-10 from each other and the native NT, as described in the above sections. These differences and the less robust effects produced by the small molecule Trk ligands compared to the full agonism of the NTs makes establishing target engagement by assessing activation of TrkB and its downstream signaling difficult. In fact, two recent reports failed to discern an effect of 7,8-DHF or LM22A-4 on TrkB activation and AKT and ERK signaling in several in vitro assays [173, 206]. These findings are in sharp contrast to the positive results on these endpoints from several other independent laboratories testing these compounds in similar in vitro paradigms [20, 187–189]. These conflicting results are most likely due to the use of variable cellular contexts and experimental conditions [206]. These variables include insufficient assay sensitivity and replications to detect the less pronounced biological effects, particularly Trk phosphorylation, produced by the small molecule ligands relative to the native NTs. For example, to detect LM22A-4 activation of TrkB (Y490) in hippocampal neurons by Western blotting 8–11 replications were performed from 4-5 independent cultures [187]. Moreover, in vitro systems undeniably differ from the in vivo milieu and may not accurately recapitulate the environment needed for receptor activation (e.g., presence of pre-formed Trk homodimers or transactivation of Trks by G protein-coupled receptors) [23, 206]. Accordingly, in in vivo studies from multiple independent research groups, both LM22A-4 and 7,8-DHF have been shown to activate TrkB and its downstream signaling to provide neuroprotection in numerous pre-clinical animal models of diverse neurodegenerative conditions, as mentioned above [LM22A-4:[91, 187, 189–191, 207, 208]; 7,8-DHF: [175, 179–185]]. Thus, despite discrepant in vitro results, developing small molecule Trk ligands for neurodegenerative diseases, including HD, remains a viable therapeutic strategy especially given the in vivo success of the currently available ligands and that the eventual goal of developing these compounds is in vivo activity and utility. However, the lack of reliable assays that can detect Trk activation and that are translatable between in vitro and in vivo systems remains an obstacle in identifying novel small molecule Trk activators [20, 206].
P75NTR SIGNALING IN HD
p75NTR signaling mediates diverse cellular processes including neurite outgrowth, synaptic plasticity, and inflammation as well as cell death or survival depending on its modulation of other receptors (e.g., Trks and sortilin) and the intracellular signaling adaptors it recruits [20, 23, 209]. In the adult brain, p75NTR is predominantly present in basal forebrain cholinergic neurons, but it is also expressed in neurons of brain areas primarily affected in HD including striatum, cortex, and hippocampus as well as other cell types (e.g., glial cells) [13, 210–215]. Within the striatum, p75NTR is located on many of the specific cell types vulnerable to mutant HTT including DARPP32-containing MSNs, cholinergic and GABAergic interneurons, and oligodendrocytes [13, 15, 40, 55, 215].
In HD patients, p75NTR expression is up-regulated in the hippocampus and striatum, but not in the cortex, and is elevated in all three of these brain areas in HD mouse models at mid- to late symptomatic stages [13, 55, 95, 215, 216]. Interestingly, p75NTR increases were larger in the rostral than caudal regions of the R6/1 striatum suggesting a role of p75NTR in striatal atrophy which follows a rostro-caudal gradient [1, 13]. However, in the zQ175 HD mouse model, p75NTR levels were unaltered in striatum at pre-symptomatic stages (5-6 months of age) and associated with survival signaling via the AKT and NFκβ pathways [217] but were decreased in striatum and cortex at symptomatic stages (12 months of age) [15]. Levels of sortilin, p75NTR’s cell death co-receptor, are increased in the striatum, but not the cortex of 12 month-old zQ175 mice [15]. While p75NTR levels were generally decreased in zQ175 striatum, they were increased in striatal immature oligodendrocytes, as were sortilin levels, which may contribute to the myelin deficiencies seen in HD patients and mouse models [15, 218, 219].
The elevated p75NTR levels and reduced levels of TrkB and/or its signaling intermediates seen in HD patients and most mouse models generates an imbalance in the ratio of p75NTR to TrkB which likely contributes greatly to HD pathogenesis [13, 40, 55, 91, 95, 216]. This p75NTR/TrkB imbalance has been linked to increased pro-apoptotic JNK signaling and reduced pro-survival signaling by inhibiting BDNF/TrkB activation of the AKT pathway in mutant HTT-expressing cells [13]. It has also been reported to reduce the resistance of striatal cells to mutant HTT-induced excitotoxic injury [13]. The effects of this imbalance can be exacerbated by a paucity of NTs, as seen with reduced BDNF and NT-3 in the HD striatum and/or cortex, which can lead to a default mode of p75NTR signaling involving degenerative pathways including JNK [20, 220]. Increased JNK signaling, as induced by mutant HTT [13, 55, 101, 215, 221, 222], has been linked to multiple HD-related pathologies, including excessive tau phosphorylation. Recent evidence suggests that HD may be amongst a large group of neurodegenerative diseases categorized as tauopathies [223]. Tau is phosphorylated directly by numerous kinases including JNK and toxic forms of the protein, including hyperphosphorylated and oligomeric tau, are present in the brains of HD patients and mouse models [223].
p75NTR signaling networks are highly integrated with those affected by mutant HTT and those that are involved in synaptic plasticity and neuronal survival (Fig. 3). Along these lines, several recent studies have shown that dysregulated p75NTR signaling contributes substantially to fundamental HD pathologies, particularly structural and functional synaptic plasticity deficits. BDNF binding to p75NTR has been shown to regulate hippocampal long-term depression [17]. p75NTR is located on dendritic spines of hippocampal CA1 pyramidal neurons as well as dentate gyrus neurons and is a negative regulator of dendrite complexity and spine formation by activating RhoA signaling [43, 224]. In the hippocampus of the HdhQ7/Q111 mouse model of HD, post-synaptic levels of p75NTR and RhoA activation are up-regulated [215]. These increases likely contribute to impaired synaptic function as normalizing p75NTR levels and RhoA activity by genetic knockdown of p75NTR in HdhQ7/Q111 mice prevented decreases in spine density and synaptic proteins, including PSD-95, CREB-binding protein (CBP), and phosphorylated GluA1 [210, 215]. This leveling of hippocampal p75NTR was also associated with improved LTP and cognition in HD mice [215].
Aberrant p75NTR signaling also contributes significantly to HD-related corticostriatal synaptic plasticity deficits. BDNF/TrkB-dependent LTP was selectively impaired in striatal projection neurons of the indirect (iSPN), not direct (dSPN), pathway of basal ganglia circuits at an age coinciding with the emergence of motor symptoms in BACHD and Q175 HD mouse models [40]. The LTP deficit was correlated with dendritic spine loss and reduced post-synaptic AMPA receptors on the iSPNs. Knockdown of p75NTR expression rescued iSPN LTP in BACHD mice. The proposed mechanism underlying this effect involves p75NTR-induced inhibition of TrkB signaling via RhoA/ROCK activation of PTEN, which blocks PI3K/AKT signaling necessary for TrkB-induced potentiation. Expression of PTEN is increased in striatum of HD mice [40, 55], specifically in iSPNs which could lead to diminished LTP induction in these cells [40]. LTP deficits were prevented by pharmacological blocking of multiple intermediates in the p75NTR-RhoA signaling pathway, including Rho-GDI and ROCK, lending support for this mechanism [40]. Although p75NTR signaling was associated with synaptic plasticity dysfunction in iSPNs, which are involved in motor suppression, striatal knockdown of p75NTR did not significantly ameliorate deficits in rotarod motor performance [215].
When chronically activated, p75NTR signaling via the inhibitor of kappa beta kinase (IKK)/NFκβ pathway may contribute to the neuroinflammatory responses shown to integrally contribute to HD pathogenesis [225]. IKK/NFκB signaling mediates glial activation and inflammatory processes and is excessively activated in HD patients and mouse models [39, 226, 227]. The increased NFκβ signaling could be caused by mutant HTT-induced decreases in levels of inhibitor of nuclear factor κB (Iκβα) [228–230]. Iκβα prevents NFκβ from translocating to the nucleus to activate genes that promote inflammation and its levels are reduced in the R6/1 hippocampus [216] and R6/2 striatum [55]. Independent of its NFκβ activating effects, IKK has been shown to phosphorylate and acetylate HTT thus tagging it for clearance via autophagy and/or the ubiquitin-proteasome pathways and up-regulating IKK-related gene expression with an HDAC inhibitor decreased HTT aggregates in the cortex and/or striatum of HD mice [231, 232].
Thus, dysregulated p75NTR signaling promotes HD-related neuroinflammation and compromises hippocampal and corticostriatal synaptic plasticity contributing to memory problems and motor deficits [11, 13, 40, 216]. Given how interconnected p75NTR signaling networks are with those affected by mutant HTT, therapeutic targeting of p75NTR has the potential to combat a broad array of HD pathologies, especially if its degenerative signaling could be blocked while concurrently augmenting its trophic signaling.
P75NTR -BASED THERAPEUTIC STRATEGIES FOR HD
In developing p75NTR-based HD therapeutics, two distinct strategies have been utilized to normalize disrupted p75NTR signaling. The first involves a pharmacological modulation that removes the imbalance in the ratio of p75NTR to TrkB by lowering p75NTR levels [216]. The second entails using small molecule ligands that selectively bind to and activate p75NTR to increase survival signaling while reducing degenerative signaling [55].
Addressing the first strategy, the p75NTR/TrkB imbalance in the hippocampus of R6/1 mice was eradicated by chronic treatment with a modulator of sphinosine-1 phosphatase receptor, fingolimod [216]. This effect of fingolimod may be due to its inhibition of TNFα, a cytokine that was shown to increase p75NTR in R6/1 hippocampal cultures conceivably via NF-κB signaling [216, 233]. Chronically treating pre-symptomatic R6/1 mice with fingolimod ameliorated the loss of PSD-95 and dendritic spines in hippocampus and improved long-term memory possibly by increasing BDNF expression and cAMP/CREB signaling while reducing RhoA activity [216]. Fingolimod also reduced astrocyte-mediated inflammation potentially by reducing signaling via the NF-κB pathway. These beneficial effects of fingolimod on cognition and structural synaptic plasticity in hippocampus of R6/1 mice is likely due, in part, to its prevention of p75NTR up-regulation as signaling associated with the receptor was affected, including the RhoA and NF-κB pathways. However, its immunomodulatory properties and elevation of BDNF would also be expected to contribute significantly to its efficacy in ameliorating the HD phenotype.
The other approach to normalizing p75NTR signaling in HD involves direct targeting of the receptor using potent and selective small molecule p75NTR ligands. The Longo and Massa laboratories used in silico screening to identify non-peptide small molecules corresponding to NGF’s loop 1 β-turn domain which interacts with p75NTR [234]. These compounds are specific p75NTR ligands in that they do not bind Trk receptors and were negative when screened for binding to >50 common G-protein coupled and ion channel receptors [234, 235]. In in vitro studies, the p75NTR ligands prevented neuronal death and promoted survival signaling while blocking degenerative signaling by pro-NGF-independent mechanisms or by competing with pro-NGF for p75NTR binding [234–236]. One p75NTR ligand identified, LM11A-31, has the favorable characteristics of a drug lead, including suitable oral bioavailability and BBB penetration with a brain-to-plasma ratio >1, and has shown therapeutic efficacy in in vitro and in vivo models of multiple neurodegenerative disorders including AD and traumatic brain injury [20, 234–242].
LM11A-31 has also been examined for its effects against HD pathology in multiple mouse models of the disease. LM11A-31 restored p75NTR-associated trophic signaling via the AKT pathway and inhibited degenerative signaling via the JNK pathway in the R6/2 and BACHD striatum [55]. It also prevented the increases in striatal RhoA/ROCK and NF-κB signaling seen in R6/2 mice [55, 243]. LM11A-31’s normalizing effects on p75NTR signaling could be the mechanism by which the ligand ameliorates several fundamental HD pathologies. For example, intranuclear HTT aggregates of striatal, cortical and hippocampal neurons in R6/2 mice were reduced by the p75NTR ligand potentially via its actions on AKT and/or RhoA/ROCK signaling [55], as activation of the former and inhibition of the latter have been shown to reduce mutant HTT aggregation and toxicity [106, 120, 121, 244, 245]. LM11A-31 prevented dendritic spine loss of striatal MSNs and hippocampal CA1 pyramidal neurons of R6/2 and BACHD mice most likely by triggering AKT and PAK signaling [55], which positively regulates spine formation and function [115, 246], while inhibiting RhoA signaling, which negatively affects spines [243, 247–250]. Microglial activation was also reduced by LM11A-31 possibly by alleviating deficits in IκB-α [55], which isolates NF-κB thereby blocking its ability to induce neuroinflammation [228–230]. Moreover, LM11A-31 elevated DARRP-32 levels and prevented neurite degeneration of MSNs and cholinergic interneurons in the striatum which may involve its effects on multiple signaling pathways including JNK, RhoA, and AKT [251–253]. Importantly, LM11A-31 prolonged the survival of R6/2 mice and improved cognition and motor function in both R6/2 and BACHD mice. In vivo target engagement of LM11A-31 was suggested by elevated striatal levels of p75NTR cleavage products [55], which are proteolytically formed with ligand binding [254, 255]. Thus, the effects of LM11A-31 are most likely due to its direct interactions with p75NTR. Taken together these results indicate that targeting p75NTR signaling may be an effective therapeutic strategy for HD. Notably, LM11A-31 is in Phase IIa clinical trials for AD (ClinicalTrials.gov: NCT03069014) and is therefore a feasible candidate for HD clinical testing.
CONCLUSIONS AND FUTURE DIRECTIONS
Mutant HTT produces a pervasive set of degenerative effects that warrants the development of disease modifying therapeutics capable of counteracting broad and fundamental disease mechanisms. Loss of neurotrophic signaling and up-regulation of degenerative signaling are key mechanisms underlying HD-related neuronal dysfunction and death. The substantial overlap between NT signaling pathways and those dysregulated by mutant HTT provides strong rationale for targeting NT receptors as a therapeutic strategy for HD. Support for this approach is provided by preclinical studies showing that small molecule ligands for both types of NT receptors, Trks and p75NTR, reduce degenerative signaling and/or increase trophic signaling to prevent HD-related pathology in mouse models of the disease. Regarding Trk ligands, future studies will determine if novel small molecules with optimized bioavailability can be identified that also prevent HD pathogenesis to improve cognition and motor ability. Since the p75NTR ligand, LM11A-31, is already in Phase IIa clinical trials for AD (ClinicalTrials.gov: NCT03069014), current work is focused on preparing for HD clinical trials, including development of non-invasive biomarkers capable of detecting the therapeutic efficacy of LM11A-31.
CONFLICT OF INTEREST
The author does not have any conflicts of interest.
REFERENCES
[1] | Vonsattel J , Myers R , Stevens T , Ferrante R , Bird E , Richardson EJ . Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. (1985) ;44: (6):559–77. |
[2] | Group HsDCR. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: (6):971–83. |
[3] | Vonsattel J , DiFiglia M . Huntington disease. J Neuropathol Exp Neurol. (1998) ;57: (5):369–84. |
[4] | Paulsen JS , Long JD . Onset of Huntington’s disease: Can it be purely cognitive? Mov Disord. (2014) ;29: (11):1342–50. |
[5] | Walker FO . Huntington’s disease. Lancet. (2007) ;369: (9557):218–28. |
[6] | Zuccato C , Valenza M , Cattaneo E . Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. (2010) ;90: (3):905–81. |
[7] | Zuccato C , Cattaneo E . Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. (2007) ;81: (5-6):294–330. |
[8] | Ventimiglia R , Mather PE , Jones BE , Lindsay RM . The neurotrophins BDNF, NT-3 and NT-4/5 promote survival and morphological and biochemical differentiation of striatal neurons in vitro. Eur J Neurosci. (1995) ;7: (2):213–22. |
[9] | Perez-Navarro E , Canudas A , Akerund P , Alberch J , Arenas E . Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington’s disease. J Neurochem. (2000) ;75: :2190–9. |
[10] | Perez-Navarro E , Alberch J , Neveu I , Arenas E . Brain-derived neurotrophic factor, neurotrophin-3 and neurotrophin-4/5 differentially regulate the phenotype and prevent degenerative changes in striatal projection neurons after excitotoxicity in vivo. Neuroscience. (1999) ;91: (4):1257–64. |
[11] | Simmons DA , Longo FM , Massa SM . Neurotrophin receptor signaling as a therapeutic target for Huntington’s disease. CNS Neurol Disord Drug Targets. (2017) ;16: (3):291–302. |
[12] | Gines S , Bosch M , Marco S , Gavalda N , Diaz-Hernandez M , Lucas JJ , et al. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. Eur J Neurosci. (2006) ;23: (3):649–58. |
[13] | Brito V , Puigdellivol M , Giralt A , Del Toro D , Alberch J , Gines S . Imbalance of p75(NTR)/TrkB protein expression in Huntington’s disease: Implication for neuroprotective therapies. Cell Death Dis. (2013) ;4: :e595. |
[14] | Zuccato C , Cattaneo E . Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. (2009) ;5: (6):311–22. |
[15] | Ma Q , Yang J , Li T , Milner TA , Hempstead BL . Selective reduction of striatal mature BDNF without induction of proBDNF in the zQ175 mouse model of Huntington’s disease. Neurobiol Dis. (2015) ;82: :466–77. |
[16] | Chao MV . Neurotrophins and their receptors: A convergence point for many signalling pathways. Nature reviews Neuroscience. (2003) ;4: (4):299–309. |
[17] | Lu B , Chow A . Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. (1999) ;58: :76–87. |
[18] | Reichardt LF . Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. (2006) ;361: (1473):1545–64. |
[19] | Barbacid M . Structural and functional properties of the TRK family of neurotrophin receptors. Ann N Y Acad Sci. (1995) ;766: :442–58. |
[20] | Longo FM , Massa SM . Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat Rev Drug Discov. (2013) ;12: (7):507–25. |
[21] | Bamji SX , Majdan M , Pozniak CD , Belliveau DJ , Aloyz R , Kohn J , et al. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. (1998) ;140: (4):911–23. |
[22] | Casaccia-Bonnefil P , Carter BD , Dobrowsky RT , Chao MV . Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. (1996) ;383: (6602):716–9. |
[23] | Pramanik S , Sulistio YA , Heese K . Neurotrophin signaling and stem cells-implications for neurodegenerative diseases and stem cell therapy. Mol Neurobiol. (2017) ;54: :7401–59. |
[24] | Mi S , Lee X , Shao Z , Thill G , Ji B , Relton J , et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. (2004) ;7: (3):221–8. |
[25] | Shen J , Maruyama IN . Nerve growth factor receptor TrkA exists as a preformed, yet inactive, dimer in living cells. FEBS Lett. (2011) ;585: (2):295–9. |
[26] | Shen J , Maruyama IN . Brain-derived neurotrophic factor receptor TrkB exists as a preformed dimer in living cells. J Mol Signal. (2012) ;7: (1):2. |
[27] | Huang EJ , Reichardt LF . Neurotrophins: Roles in neuronal development and function. Annu Rev Neurosci. (2001) ;24: :677–736. |
[28] | Gong Y , Cao P , Yu HJ , Jiang T . Crystal structure of the neurotrophin-3 and p75NTR symmetrical complex. Nature. (2008) ;454: (7205):789–93. |
[29] | Vilar M , Charalampopoulos I , Kenchappa RS , Reversi A , Klos-Applequist JM , Karaca E , et al. Ligand-independent signaling by disulfide-crosslinked dimers of the p75 neurotrophin receptor. J Cell Sci. (2009) ;122: (Pt 18):3351–7. |
[30] | Gentry JJ , Barker PA , Carter BD . The p75 neurotrophin receptor: Multiple interactors and numerous functions. Prog Brain Res. (2004) ;146: :25–39. |
[31] | Dobrowsky RT , Werner MH , Castellino AM , Chao MV , Hannun YA . Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science. (1994) ;265: (5178):1596–9. |
[32] | Salehi AH , Xanthoudakis S , Barker PA . NRAGE, a p75 neurotrophin receptor-interacting protein, induces caspase activation and cell death through a JNK-dependent mitochondrial pathway. J Biol Chem. (2002) ;277: (50):48043–50. |
[33] | Kraemer BR , Yoon SO , Carter BD . The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb Exp Pharmacol. (2014) ;220: :121–64. |
[34] | Volosin M , Trotter C , Cragnolini A , Kenchappa RS , Light M , Hempstead BL , et al. Induction of proneurotrophins and activation of p75NTR-mediated apoptosis via neurotrophin receptor-interacting factor in hippocampal neurons after seizures. J Neurosci. (2008) ;28: (39):9870–9. |
[35] | Hempstead BL , Martin-Zanca D , Kaplan DR , Parada LF , Chao MV . High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. (1991) ;350: (6320):678–83. |
[36] | Meeker R , Williams K . Dynamic nature of the p75 neurotrophin receptor in response to injury and disease. J Neuroimmune Pharmacol. (2014) ;9: (5):615–28. |
[37] | Carter BD , Kaltschmidt C , Kaltschmidt B , Offenhauser N , Bohm-Matthaei R , Baeuerle PA , et al. Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science. (1996) ;272: (5261):542–5. |
[38] | Khursigara G , Orlinick JR , Chao MV . Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem. (1999) ;274: (5):2597–600. |
[39] | Mattson MP , Meffert MK . Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. (2006) ;13: (5):852–60. |
[40] | Plotkin JL , Day M , Peterson JD , Xie Z , Kress GJ , Rafalovich I , et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron. (2014) ;83: (1):178–88. |
[41] | Song W , Volosin M , Cragnolini AB , Hempstead BL , Friedman WJ . ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J Neurosci. (2010) ;30: (46):15608–15. |
[42] | Meeker RB , Williams KS . The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen Res. (2015) ;10: (5):721–5. |
[43] | Yamashita T , Tucker KL , Barde YA . Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. (1999) ;24: (3):585–93. |
[44] | Connor B , Young D , Lawlor P , Gai W , Waldvogel H , Faull RL , et al. Trk receptor alterations in Alzheimer’s disease. Brain Res Mol Brain Res. (1996) ;42: (1):1–17. |
[45] | Zuccato C , Ciammola A , Rigamonti D , Leavitt B , Goffredo D , Conti L , et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. (2001) ;293: (5529):493–8. |
[46] | Zhang H , Petit GH , Gaughwin PM , Hansen C , Ranganathan S , Zuo X , et al. NGF rescues hippocampal cholinergic neuronal markers, restores neurogenesis, and improves the spatial working memory in a mouse model of Huntington’s Disease. J Huntingtons Dis. (2013) ;2: (1):69–82. |
[47] | Spires TL , Grote HE , Varshney NK , Cordery PM , van Dellen A , Blakemore C , et al. Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J Neurosci. (2004) ;24: (9):2270–6. |
[48] | Valencia A , Sapp E , Kimm JS , McClory H , Ansong KA , Yohrling G , et al. Striatal synaptosomes from Hdh140Q/140Q knock-in mice have altered protein levels, novel sites of methionine oxidation, and excess glutamate release after stimulation. J Huntingtons Dis. (2013) ;2: (4):459–75. |
[49] | Holtzman DM , Kilbridge J , Li Y , Cunningham ET Jr , Lenn NJ, Clary DO, et al. TrkA expression in the CNS: Evidence for the existence of several novel NGF-responsive CNS neurons. J Neurosci. (1995) ;15: (2):1567–76. |
[50] | Sobreviela T , Clary DO , Reichardt LF , Brandabur MM , Kordower JH , Mufson EJ . TrkA-immunoreactive profiles in the central nervous system: Colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. (1994) ;350: (4):587–611. |
[51] | Merlio JP , Ernfors P , Jaber M , Persson H . Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. (1992) ;51: (3):513–32. |
[52] | Deng YP , Reiner A . Cholinergic interneurons in the Q140 knock-in mouse model of Huntington’s disease: Reductions in dendritic branching and thalamostriatal input. J Comp Neurol. (2006) ;524: (17):3518–29. |
[53] | Smith R , Chung H , Rundquist S , Maat-Schieman ML , Colgan L , Englund E , et al. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum Mol Genet. (2006) ;15: (21):3119–31. |
[54] | Vetter JM , Jehle T , Heinemeyer J , Franz P , Behrens PF , Jackisch R , et al. Mice transgenic for exon 1 of Huntington’s disease: Properties of cholinergic and dopaminergic pre-synaptic function in the striatum. J Neurochem. (2003) ;85: (4):1054–63. |
[55] | Simmons DA , Belichenko NP , Ford EC , Semaan S , Monbureau M , Aiyaswamy S , et al. A small molecule p75NTR ligand normalizes signalling and reduces Huntington’s disease phenotypes in R6/2 and BACHD mice. Hum Mol Genet. (2016) ;25: (22):4920–4938. |
[56] | Song C , Perides G , Liu YF . Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem. (2002) ;277: (8):6703–7. |
[57] | Rong J , McGuire JR , Fang ZH , Sheng G , Shin JY , Li SH , et al. Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J Neurosci. (2006) ;26: (22):6019–30. |
[58] | Fang H , Liu C , Yang M , Li H , Zhang F , Zhang W , et al. Neurotrophic factor and Trk signaling mechanisms underlying the promotion of motor recovery after acute spinal cord injury in rats. Exp Ther Med. (2017) ;14: (1):652–6. |
[59] | Zhou XF , Rush RA . Functional roles of neurotrophin 3 in the developing and mature sympathetic nervous system. Mol Neurobiol. (1996) ;13: (3):185–97. |
[60] | Liu Y , Kelamangalath L , Kim H , Han SB , Tang X , Zhai J , et al. NT-3 promotes proprioceptive axon regeneration when combined with activation of the mTor intrinsic growth pathway but not with reduction of myelin extrinsic inhibitors. Exp Neurol. (2016) ;283: (Pt A):73–84. |
[61] | Mendoza E , Miranda-Barrientos JA , Vazquez-Roque RA , Morales-Herrera E , Ruelas A , De la Rosa G , et al. In vivo mitochondrial inhibition alters corticostriatal synaptic function and the modulatory effects of neurotrophins. Neuroscience. (2014) ;280: :156–70. |
[62] | Ramos-Languren LE , Escobar ML . Plasticity and metaplasticity of adult rat hippocampal mossy fibers induced by neurotrophin-3. Eur J Neurosci. (2013) ;37: (8):1248–59. |
[63] | Zermeno V , Espindola S , Mendoza E , Hernandez-Echeagaray E . Differential expression of neurotrophins in postnatal C57BL/6 mice striatum. Int J Biol Sci. (2009) ;5: (2):118–27. |
[64] | Checa N , Canals JM , Alberch J . Developmental regulation of BDNF and NT-3 expression by quinolinic acid in the striatum and its main connections. Exp Neurol. (2000) ;165: (1):118–24. |
[65] | Freeman AY , Soghomonian JJ , Pierce RC . Tyrosine kinase B and C receptors in the neostriatum and nucleus accumbens are co-localized in enkephalin-positive and enkephalin-negative neuronal profiles and their expression is influenced by cocaine. Neuroscience. (2003) ;117: (1):147–56. |
[66] | Baydyuk M , Xu B . BDNF signaling and survival of striatal neurons. Front Cell Neurosci. (2014) ;8: :254. |
[67] | Chen JY , Wang EA , Cepeda C , Levine MS . Dopamine imbalance in Huntington’s disease: A mechanism for the lack of behavioral flexibility. Front Neurosci. (2013) ;7: :114. |
[68] | Liot G , Gabriel C , Cacquevel M , Ali C , MacKenzie ET , Buisson A , et al. Neurotrophin-3-induced PI-3 kinase/Akt signaling rescues cortical neurons from apoptosis. Exp Neurol. (2004) ;187: (1):38–46. |
[69] | Alexi T , Venero JL , Hefti F . Protective effects of neurotrophin-4/5 and transforming growth factor-alpha on striatal neuronal phenotypic degeneration after excitotoxic lesioning with quinolinic acid. Neuroscience. (1997) ;78: (1):73–86. |
[70] | Alderson RF , Alterman AL , Barde YA , Lindsay RM . Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron. (1990) ;5: (3):297–306. |
[71] | Knusel B , Beck KD , Winslow JW , Rosenthal A , Burton LE , Widmer HR , et al. Brain-derived neurotrophic factor administration protects basal forebrain cholinergic but not nigral dopaminergic neurons from degenerative changes after axotomy in the adult rat brain. J Neurosci. (1992) ;12: (11):4391–402. |
[72] | Sawai H , Clarke DB , Kittlerova P , Bray GM , Aguayo AJ . Brain-derived neurotrophic factor and neurotrophin-4/5 stimulate growth of axonal branches from regenerating retinal ganglion cells. J Neurosci. (1996) ;16: (12):3887–94. |
[73] | Beck T , Lindholm D , Castren E , Wree A . Brain-derived neurotrophic factor protects against ischemic cell damage in rat hippocampus. J Cereb Blood Flow Metab. (1994) ;14: (4):689–92. |
[74] | Burke MA , Mobley WC , Cho J , Wiegand SJ , Lindsay RM , Mufson EJ , et al. Loss of developing cholinergic basal forebrain neurons following excitotoxic lesions of the hippocampus: Rescue by neurotrophins. Exp Neurol. (1994) ;130: (2):178–95. |
[75] | Lindholm D . Role of neurotrophins in preventing glutamate induced neuronal cell death. J Neurol. (1994) ;242: (1 Suppl 1):S16–8. |
[76] | Wu CL , Hwang CS , Chen SD , Yin JH , Yang DI . Neuroprotective mechanisms of brain-derived neurotrophic factor against 3-nitropropionic acid toxicity: Therapeutic implications for Huntington’s disease. Ann N Y Acad Sci. (2010) ;1201: :8–12. |
[77] | Lynch G , Kramar EA , Rex CS , Jia Y , Chappas D , Gall CM , et al. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington’s disease. J Neurosci. (2007) ;27: (16):4424–34. |
[78] | Schmidt-Kastner R , Wetmore C , Olson L . Comparative study of brain-derived neurotrophic factor messenger RNA and protein at the cellular level suggests multiple roles in hippocampus, striatum and cortex. Neuroscience. (1996) ;74: (1):161–83. |
[79] | Altar CA , Cai N , Bliven T , Juhasz M , Conner JM , Acheson AL , et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. (1997) ;389: (6653):856–60. |
[80] | Baquet ZC , Gorski JA , Jones KR . Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. (2004) ;24: (17):4250–8. |
[81] | Canals JM , Pineda JR , Torres-Peraza JF , Bosch M , Martin-Ibanez R , Munoz MT , et al. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci. (2004) ;24: (35):7727–39. |
[82] | Yoshii A , Constantine-Paton M . Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. (2010) ;70: (5):304–22. |
[83] | Drake CT , Milner TA , Patterson SL . Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci. (1999) ;19: (18):8009–26. |
[84] | Ma B , Savas JN , Chao MV , Tanese N . Quantitative analysis of BDNF/TrkB protein and mRNA in cortical and striatal neurons using alpha-tubulin as a normalization factor. Cytometry A. (2012) ;81: (8):704–17. |
[85] | Wu K , Xu JL , Suen PC , Levine E , Huang YY , Mount HT , et al. Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Brain Res Mol Brain Res. (1996) ;43: (1-2):286–90. |
[86] | Lynch G , Rex CS , Chen LY , Gall CM . The substrates of memory: Defects, treatments, and enhancement. Eur J Pharmacol. (2008) ;585: (1):2–13. |
[87] | Rex CS , Lin CY , Kramar EA , Chen LY , Gall CM , Lynch G . Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. (2007) ;27: (11):3017–29. |
[88] | Jia Y , Gall CM , Lynch G . Presynaptic BDNF promotes postsynaptic long-term potentiation in the dorsal striatum. J Neurosci. (2010) ;30: (43):14440–5. |
[89] | Bramham CR , Messaoudi E . BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog Neurobiol. (2005) ;76: (2):99–125. |
[90] | Yamada K , Nabeshima T . Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharmacol Sci. (2003) ;91: (4):267–70. |
[91] | Simmons DA , Belichenko NP , Yang T , Condon C , Monbureau M , Shamloo M , et al. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J Neurosci. (2013) ;33: (48):18712–27. |
[92] | Simmons DA , Mehta RA , Lauterborn JC , Gall CM , Lynch G . Brief ampakine treatments slow the progression of Huntington’s disease phenotypes in R6/2 mice. Neurobiol Dis. (2011) ;41: (2):436–44. |
[93] | Simmons DA , Rex CS , Palmer L , Pandyarajan V , Fedulov V , Gall CM , et al. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc Natl Acad Sci U S A. (2009) ;106: (12):4906–11. |
[94] | Conforti P , Mas Monteys A , Zuccato C , Buckley NJ , Davidson B , Cattaneo E . In vivo delivery of DN:REST improves transcriptional changes of REST-regulated genes in HD mice. Gene Therapy. (2013) ;20: (6):678–85. |
[95] | Zuccato C , Marullo M , Conforti P , MacDonald ME , Tartari M , Cattaneo E . Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. (2008) ;18: (2):225–38. |
[96] | Duan W , Peng Q , Masuda N , Ford E , Tryggestad E , Ladenheim B , et al. Sertraline slows disease progression and increases neurogenesis in N171-82Q mouse model of Huntington’s disease. Neurobiol Dis. (2008) ;30: (3):312–22. |
[97] | Giampa C , Montagna E , Dato C , Melone MA , Bernardi G , Fusco FR . Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS One. (2013) ;8: (5):e64037. |
[98] | Xie Y , Hayden MR , Xu B . BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J Neurosci. (2010) ;30: (44):14708–18. |
[99] | Gray M , Shirasaki DI , Cepeda C , Andre VM , Wilburn B , Lu XH , et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. (2008) ;28: (24):6182–95. |
[100] | Gharami K , Xie Y , An JJ , Tonegawa S , Xu B . Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J Neurochem. (2008) ;105: (2):369–79. |
[101] | Apostol BL , Simmons DA , Zuccato C , Illes K , Pallos J , Casale M , et al. CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol Cell Neurosci. (2008) ;39: (1):8–20. |
[102] | Samadi P , Boutet A , Rymar VV , Rawal K , Maheux J , Kvann JC , et al. Relationship between BDNF expression in major striatal afferents, striatum morphology and motor behavior in the R6/2 mouse model of Huntington’s disease. Genes Brain Behav. (2013) ;12: (1):108–24. |
[103] | Gauthier LR , Charrin BC , Borrell-Pages M , Dompierre JP , Rangone H , Cordelieres FP , et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. (2004) ;118: (1):127–38. |
[104] | Colin E , Zala D , Liot G , Rangone H , Borrell-Pages M , Li XJ , et al. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. (2008) ;27: (15):2124–34. |
[105] | Liot G , Zala D , Pla P , Mottet G , Piel M , Saudou F . Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J Neurosci. (2013) ;33: (15):6298–309. |
[106] | Zala D , Colin E , Rangone H , Liot G , Humbert S , Saudou F . Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. (2008) ;17: (24):3837–46. |
[107] | Gines S , Paoletti P , Alberch J . Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. J Biol Chem. (2010) ;285: (28):21537–48. |
[108] | Nguyen KQ , Rymar VV , Sadikot AF . Impaired TrkB signaling underlies reduced BDNF-mediated trophic support of striatal neurons in the R6/2 mouse model of Huntington’s disease. Front Cell Neurosci. (2016) ;10: :37. |
[109] | Strand AD , Baquet ZC , Aragaki AK , Holmans P , Yang L , Cleren C , et al. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. (2007) ;27: (43):11758–68. |
[110] | Pineda JR , Canals JM , Bosch M , Adell A , Mengod G , Artigas F , et al. Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington’s disease. J Neurochem. (2005) ;93: (5):1057–68. |
[111] | Besusso D , Geibel M , Kramer D , Schneider T , Pendolino V , Picconi B , et al. BDNF-TrkB signaling in striatopallidal neurons controls inhibition of locomotor behavior. Nat Commun. (2013) ;4: :2031. |
[112] | Jia JM , Chen Q , Zhou Y , Miao S , Zheng J , Zhang C , et al. Brain-derived neurotrophic factor-tropomyosin-related kinase B signaling contributes to activity-dependent changes in synaptic proteins. J Biol Chem. (2008) ;283: (30):21242–50. |
[113] | Kowianski P , Lietzau G , Czuba E , Waskow M , Steliga A , Morys J . BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. doi: 10.1007/s10571-017-05104 |
[114] | Cuesto G , Enriquez-Barreto L , Carames C , Cantarero M , Gasull X , Sandi C , et al. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J Neurosci. (2011) ;31: (8):2721–33. |
[115] | Majumdar D , Nebhan CA , Hu L , Anderson B , Webb DJ . An APPL1/Akt signaling complex regulates dendritic spine and synapse formation in hippocampal neurons. Mol Cell Neurosci. (2011) ;46: (3):633–44. |
[116] | Tebano MT , Martire A , Chiodi V , Ferrante A , Popoli P . Role of adenosine A(2A) receptors in modulating synaptic functions and brain levels of BDNF: A possible key mechanism in the pathophysiology of Huntington’s disease. Scientific World Journal. (2010) ;10: :1768–82. |
[117] | Giralt A , Rodrigo T , Martin ED , Gonzalez JR , Mila M , Cena V , et al. Brain-derived neurotrophic factor modulates the severity of cognitive alterations induced by mutant huntingtin: Involvement of phospholipaseCgamma activity and glutamate receptor expression. Neuroscience. (2009) ;158: (4):1234–50. |
[118] | Spires TL , Grote HE , Garry S , Cordery PM , Van Dellen A , Blakemore C , et al. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington’s disease transgenic mice. Eur J Neurosci. (2004) ;19: (10):2799–807. |
[119] | Simmons DA , Rex CS , Pandyarajan V , Lauterborn JC , Lynch G , Gall CM . Ampakines increase BDNF levels and rescue synaptic plasticity in Huntington’s Disease knock-in mice. Abstr Soc Neurosci. (2007) ;798.22. |
[120] | Rangone H , Pardo R , Colin E , Girault JA , Saudou F , Humbert S . Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J Biol Chem. (2005) ;280: (23):22021–8. |
[121] | Humbert S , Bryson EA , Cordelieres FP , Connors NC , Datta SR , Finkbeiner S , et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. (2002) ;2: (6):831–7. |
[122] | Colin E , Regulier E , Perrin V , Durr A , Brice A , Aebischer P , et al. Akt is altered in an animal model of Huntington’s disease and in patients. Eur J Neurosci. (2005) ;21: (6):1478–88. |
[123] | Silva A , Naia L , Dominguez A , Ribeiro M , Rodrigues J , Vieira OV , et al. Overexpression of BDNF and full-length TrkB receptor ameliorate striatal neural survival in Huntington’s disease. Neurodegener Dis. (2015) ;15: (4):207–18. |
[124] | Fortress AM , Buhusi M , Helke KL , Granholm AC . Cholinergic degeneration and alterations in the TrkA and p75NTR balance as a result of pro-NGF injection into aged rats. J Aging Res. (2011) ;2011: :460543. |
[125] | Yoon SO , Casaccia-Bonnefil P , Carter B , Chao MV . Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. (1998) ;18: (9):3273–81. |
[126] | Zuccato C , Liber D , Ramos C , Tarditi A , Rigamonti D , Tartari M , et al. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol Res. (2005) ;52: :133–9. |
[127] | Deng P , Torrest A , Pollock K , Dahlenburg H , Annett G , Nolta JA , et al. Clinical trial perspective for adult and juvenile Huntington’s disease using genetically-engineered mesenchymal stem cells. Neural Regen Res. (2016) ;11: (5):702–5. |
[128] | Pollock K , Dahlenburg H , Nelson H , Fink KD , Cary W , Hendrix K , et al. Human mesenchymal stem cells genetically engineered to overexpress brain-derived neurotrophic factor improve outcomes in Huntington’s disease mouse models. Mol Ther. (2016) ;24: (5):965–77. |
[129] | Poduslo JF , Curran GL . Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res. (1996) ;36: (2):280–6. |
[130] | Saltzman WM , Mak MW , Mahoney MJ , Duenas ET , Cleland JL . Intracranial delivery of recombinant nerve growth factor: Release kinetics and protein distribution for three delivery systems. Pharm Res. (1999) ;16: (2):232–40. |
[131] | Allen SJ , Watson JJ , Shoemark DK , Barua NU , Patel NK . GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Thera. (2013) ;138: (2):155–75. |
[132] | Beck M , Flachenecker P , Magnus T , Giess R , Reiners K , Toyka KV , et al. Autonomic dysfunction in ALS: A preliminary study on the effects of intrathecal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord. (2005) ;6: (2):100–3. |
[133] | Eriksdotter Jonhagen M , Nordberg A , Amberla K , Backman L , Ebendal T , Meyerson B , et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. (1998) ;9: (5):246–57. |
[134] | Isaacson LG , Saffran BN , Crutcher KA . Intracerebral NGF infusion induces hyperinnervation of cerebral blood vessels. Neurobiol Aging. (1990) ;11: (1):51–5. |
[135] | Levi-Montalcini R . The nerve growth factor 35 years later. Science. (1987) ;237: (4819):1154–62. |
[136] | Ochs G , Penn RD , York M , Giess R , Beck M , Tonn J , et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) ;1: (3):201–6. |
[137] | Olson SD , Pollock K , Kambal A , Cary W , Mitchell GM , Tempkin J , et al. Genetically engineered mesenchymal stem cells as a proposed therapeutic for Huntington’s disease. Mol Neurobiol. (2012) ;45: (1):87–98. |
[138] | Kerkis I , Haddad MS , Valverde CW , Glosman S . Neural and mesenchymal stem cells in animal models of Huntington’s disease: Past experiences and future challenges. Stem Cell Res Ther. (2015) ;6: :232. |
[139] | Rossignol J , Fink KD , Crane AT , Davis KK , Bombard MC , Clerc S , et al. Reductions in behavioral deficits and neuropathology in the R6/2 mouse model of Huntington’s disease following transplantation of bone-marrow-derived mesenchymal stem cells is dependent on passage number. Stem Cell Res Ther. (2015) ;6: :9. |
[140] | Dey ND , Bombard MC , Roland BP , Davidson S , Lu M , Rossignol J , et al. Genetically engineered mesenchymal stem cells reduce behavioral deficits in the YAC 128 mouse model of Huntington’s disease. Behav Brain Res. (2010) ;214: (2):193–200. |
[141] | Aleynik A , Gernavage KM , Mourad Y , Sherman LS , Liu K , Gubenko YA , et al. Stem cell delivery of therapies for brain disorders. Clin Transl Med. (2014) ;3: :24. |
[142] | Tuszynski MH , Yang JH , Barba D , U HS , Bakay RA , Pay MM , et al. Nerve growth factor gene therapy: Activation of neuronal responses in Alzheimer disease. JAMA Neurol. (2015) ;72: (10):1139–47. |
[143] | Weissmiller AM , Wu C . Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl Neurodegener. (2012) ;1: (1):14. |
[144] | Danielyan L , Schafer R , von Ameln-Mayerhofer A , Buadze M , Geisler J , Klopfer T , et al. Intranasal delivery of cells to the brain. Eur J Cell Biol. (2009) ;88: (6):315–24. |
[145] | Chapman CD , Frey 2nd WH , Craft S , Danielyan L , Hallschmid M , Schioth HB , et al. Intranasal treatment of central nervous system dysfunction in humans. harm Res. (2013) ;30: (10):2475–84. |
[146] | Danielyan L , Beer-Hammer S , Stolzing A , Schafer R , Siegel G , Fabian C , et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. (2014) ;23: (Suppl 1):S123–39. |
[147] | Danielyan L , Klein R , Hanson LR , Buadze M , Schwab M , Gleiter CH , et al. Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res. (2010) ;13: (2-3):195–201. |
[148] | Salama M , Sobh M , Emam M , Abdalla A , Sabry D , El-Gamal M , et al. Effect of intranasal stem cell administration on the nigrostriatal system in a mouse model of Parkinson’s disease. Exp Ther Med. (2017) ;13: (3):976–82. |
[149] | Benraiss A , Bruel-Jungerman E , Lu G , Economides AN , Davidson B , Goldman SA . Sustained induction of neuronal addition to the adult rat neostriatum by AAV4-delivered noggin and BDNF. Gene Therapy. (2012) ;19: (5):483–93. |
[150] | Cho SR , Benraiss A , Chmielnicki E , Samdani A , Economides A , Goldman SA . Induction of neostriatal neurogenesis slows disease progression in a transgenic murine model of Huntington disease. J Clin Invest. (2007) ;117: (10):2889–902. |
[151] | Connor B , Sun Y , von Hieber D , Tang SK , Jones KS , Maucksch C . AAV1/2-mediated BDNF gene therapy in a transgenic rat model of Huntington’s disease. Gene Therapy. (2016) ;23: (3):283–95. |
[152] | Duan W , Guo Z , Jiang H , Ware M , Li X , Mattson M . Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. (2003) ;100: :2911–6. |
[153] | Hockly E , Cordery PM , Woodman B , Mahal A , van Dellen A , Blakemore C , et al. Environmental enrichment slows disease progression in R6/2 Huntington’s disease mice. Ann Neurol. (2002) ;51: (2):235–42. |
[154] | Reiner A , Lafferty DC , Wang HB , Del Mar N , Deng YP . The group 2 metabotropic glutamate receptor agonist LY379268 rescues neuronal, neurochemical and motor abnormalities in R6/2 Huntington’s disease mice. Neurobiol Dis. (2012) ;47: (1):75–91. |
[155] | Reiner A , Wang HB , Del Mar N , Sakata K , Yoo W , Deng YP . BDNF may play a differential role in the protective effect of the mGluR2/3 agonist LY379268 on striatal projection neurons in R6/2 Huntington’s disease mice. Brain Res. (2012) ;1473: :161–72. |
[156] | Wang SE , Lin CL , Hsu CH , Sheu SJ , Chien CT , Wu CH . Treatment with a herbal formula B401 enhances neuroprotection and angiogenesis in the R6/2 mouse model of Huntington’s disease. Drug Des Devel Ther. (2015) ;9: :887–900. |
[157] | Pouladi MA , Brillaud E , Xie Y , Conforti P , Graham RK , Ehrnhoefer DE , et al. NP03, a novel low-dose lithium formulation, is neuroprotective in the YAC128 mouse model of Huntington disease. Neurobiol Dis. (2012) ;48: (3):282–9. |
[158] | Jeon J , Kim W , Jang J , Isacson O , Seo H . Gene therapy by proteasome activator, PA28gamma, improves motor coordination and proteasome function in Huntington’s disease YAC128 mice. Neuroscience. (2016) ;324: :20–8. |
[159] | Enomoto S , Shimizu K , Nibuya M , Suzuki E , Nagata K , Kondo T . Activated brain-derived neurotrophic factor/TrkB signaling in rat dorsal and ventral hippocampi following 10-day electroconvulsive seizure treatment. Neurosci Lett. (2017) ;660: :45–50. |
[160] | Zetterstrom TS , Pei Q , Grahame-Smith DG . Repeated electroconvulsive shock extends the duration of enhanced gene expression for BDNF in rat brain compared with a single administration. Brain Res Mol Brain Res. (1998) ;57: (1):106–10. |
[161] | Beste C , Muckschel M , Elben S , C JH , McIntyre CC , Saft C , et al. Behavioral and neurophysiological evidence for the enhancement of cognitive control under dorsal pallidal deep brain stimulation in Huntington’s disease. Brain Struct Funct. (2015) ;220: (4):2441–8. |
[162] | Gonzalez V , Cif L , Biolsi B , Garcia-Ptacek S , Seychelles A , Sanrey E , et al. Deep brain stimulation for Huntington’s disease: Long-term results of a prospective open-label study. J Neurosurg. (2014) ;121: (1):114–22. |
[163] | Wojtecki L , Groiss SJ , Ferrea S , Elben S , Hartmann CJ , Dunnett SB , et al. A prospective pilot trial for pallidal deep brain stimulation in Huntington’s disease. Front Neurol. (2015) ;6: :177. |
[164] | Wojtecki L , Groiss SJ , Hartmann CJ , Elben S , Omlor S , Schnitzler A , et al. Deep brain stimulation in Huntington’s disease-preliminary evidence on pathophysiology, efficacy and safety. Brain Sci. (2016) ;6: (3), pii: E38. |
[165] | Mitre M , Mariga A , Chao MV . Neurotrophin signalling: Novel insights into mechanisms and pathophysiology. Clin Sci. (2017) ;131: (1):13–23. |
[166] | Buhmann C , Huckhagel T , Engel K , Gulberti A , Hidding U , Poetter-Nerger M , et al. Adverse events in deep brain stimulation: A retrospective long-term analysis of neurological, psychiatric and other occurrences. PLoS One. (2017) ;12: (7):e0178984. |
[167] | Ibanez CF , Ilag LL , Murray-Rust J , Persson H . An extended surface of binding to Trk tyrosine kinase receptors in NGF and BDNF allows the engineering of a multifunctional pan-neurotrophin. EMBO J. (1993) ;12: (6):2281–93. |
[168] | O’Leary PD , Hughes RA . Design of potent peptide mimetics of brain-derived neurotrophic factor. J Biol Chem. (2003) ;278: (28):25738–44. |
[169] | Jang SW , Okada M , Sayeed I , Xiao G , Stein D , Jin P , et al. Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death. Proc Natl Acad Sci U S A. (2007) ;104: (41):16329–34. |
[170] | Qian MD , Zhang J , Tan XY , Wood A , Gill D , Cho S . Novel agonist monoclonal antibodies activate TrkB receptors and demonstrate potent neurotrophic activities. J Neurosci. (2006) ;26: (37):9394–403. |
[171] | Lin JC , Tsao D , Barras P , Bastarrachea RA , Boyd B , Chou J , et al. Appetite enhancement and weight gain by peripheral administration of TrkB agonists in non-human primates. PLoS One. (2008) ;3: (4):e1900. |
[172] | Cazorla M , Arrang JM , Premont J . Pharmacological characterization of six trkB antibodies reveals a novel class of functional agents for the study of the BDNF receptor. Br J Pharmacol. (2011) ;162: (4):947–60. |
[173] | Todd D , Gowers I , Dowler SJ , Wall MD , McAllister G , Fischer DF , et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS One. (2014) ;9: (2):e87923. |
[174] | Jang SW , Liu X , Chan CB , France SA , Sayeed I , Tang W , et al. Deoxygedunin, a natural product with potent neurotrophic activity in mice. PLoS One. (2010) ;5: (7):e11528. |
[175] | Jang SW , Liu X , Yepes M , Shepherd KR , Miller GW , Liu Y , et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. (2010) ;107: (6):2687–92. |
[176] | Liu X , Obianyo O , Chan CB , Huang J , Xue S , Yang JJ , et al. Biochemical and biophysical investigation of the brain-derived neurotrophic factor mimetic 7,8-dihydroxyflavone in the binding and activation of the TrkB receptor. J Biol Chem. (2014) ;289: (40):27571–84. |
[177] | Obianyo O , Ye K . Novel small molecule activators of the Trk family of receptor tyrosine kinases. Biochim Biophys Acta. (2013) ;1834: (10):2213–8. |
[178] | Nie S , Xu Y , Chen G , Ma K , Han C , Guo Z , et al. Small molecule TrkB agonist deoxygedunin protects nigrostriatal dopaminergic neurons from 6-OHDA and MPTP induced neurotoxicity in rodents. Neuropharmacology. (2015) ;99: :448–58. |
[179] | Stagni F , Giacomini A , Guidi S , Emili M , Uguagliati B , Salvalai ME , et al. A flavonoid agonist of the TrkB receptor for BDNF improves hippocampal neurogenesis and hippocampus-dependent memory in the Ts65Dn mouse model of DS. Exp Neurol. (2017) ;298: (Pt A):79–96. |
[180] | Tan Y , Nie S , Zhu W , Liu F , Guo H , Chu J , et al. 7,8-dihydroxyflavone ameliorates cognitive impairment by inhibiting expression of tau pathology in ApoE-knockout mice. Front Aging Neurosci. (2016) ;8: :287. |
[181] | He J , Xiang Z , Zhu X , Ai Z , Shen J , Huang T , et al. Neuroprotective effects of 7, 8-dihydroxyflavone on midbrain dopaminergic neurons in MPP+-treated monkeys. Sci Rep. (2016) ;6: :34339. |
[182] | Gao L , Tian M , Zhao HY , Xu QQ , Huang YM , Si QC , et al. TrkB activation by 7, 8-dihydroxyflavone increases synapse AMPA subunits and ameliorates spatial memory deficits in a mouse model of Alzheimer’s disease. J Neurochem. (2016) ;136: (3):620–36. |
[183] | Johnson RA , Lam M , Punzo AM , Li H , Lin BR , Ye K , et al. 7,8-dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J Appl Physiol. (2012) ;112: (5):704–10. |
[184] | Jiang M , Peng Q , Liu X , Jin J , Hou Z , Zhang J , et al. Small molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington’s disease. Hum Mol Genet. (2013) ;22: :2462–70. |
[185] | Garcia-Diaz Barriga G , Giralt A , Anglada-Huguet M , Gaja-Capdevila N , Orlandi JG , Soriano J , et al. 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington’s disease mouse model through specific activation of the PLCgamma1 pathway. Hum Mol Genet. (2017) ;26: (16):3144–60. |
[186] | Chen J , Chua KW , Chua CC , Yu H , Pei A , Chua BH , et al. Antioxidant activity of 7,8-dihydroxyflavone provides neuroprotection against glutamate-induced toxicity. Neurosci Lett. (2011) ;499: (3):181–5. |
[187] | Massa SM , Yang T , Xie Y , Shi J , Bilgen M , Joyce JN , et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. (2010) ;120: (5):1774–85. |
[188] | Kajiya M , Takeshita K , Kittaka M , Matsuda S , Ouhara K , Takeda K , et al. BDNF mimetic compound LM22A-4 regulates cementoblast differentiation via the TrkB-ERK/Akt signaling cascade. Int Immunopharmacol. (2014) ;19: (2):245–52. |
[189] | Li W , Bellot-Saez A , Phillips ML , Yang T , Longo FM , Pozzo-Miller L . A small-molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis Model Mech. (2017) ;10: (7):837–45. |
[190] | Han J , Pollak J , Yang T , Siddiqui MR , Doyle KP , Taravosh-Lahn K , et al. Delayed administration of a small molecule tropomyosin-related kinase B ligand promotes recovery after hypoxic-ischemic stroke. Stroke. (2012) ;43: (7):1918–24. |
[191] | Schmid DA , Yang T , Ogier M , Adams I , Mirakhur Y , Wang Q , et al. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci. (2012) ;32: (5):1803–10. |
[192] | Yu G , Wang W . Protective effects of LM22A-4 on injured spinal cord nerves. Int J Clin Exp Pathol. (2015) ;8: (6):6526–32. |
[193] | Zaccaro MC , Lee HB , Pattarawarapan M , Xia Z , Caron A , L’Heureux PJ , et al. Selective small molecule peptidomimetic ligands of TrkC and TrkA receptors afford discrete or complete neurotrophic activities. Chem Biol. (2005) ;12: (9):1015–28. |
[194] | Peleshok J , Saragovi HU . Functional mimetics of neurotrophins and their receptors. Biochem Soc Trans. (2006) ;34: (Pt 4):612–7. |
[195] | Xie Y , Tisi MA , Yeo TT , Longo FM . Nerve growth factor (NGF) loop 4 dimeric mimetics activate ERK and AKT and promote NGF-like neurotrophic effects. J Biol Chem. (2000) ;275: (38):29868–74. |
[196] | Aboulkassim T , Tong XK , Tse YC , Wong TP , Woo SB , Neet KE , et al. Ligand-dependent TrkA activity in brain differentially affects spatial learning and long-term memory. Mol Pharmacol. (2011) ;80: (3):498–508. |
[197] | Maliartchouk S , Feng Y , Ivanisevic L , Debeir T , Cuello AC , Burgess K , et al. A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol Pharmacol. (2000) ;57: (2):385–91. |
[198] | Scarpi D , Cirelli D , Matrone C , Castronovo G , Rosini P , Occhiato EG , et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. (2012) ;3: :e389. |
[199] | Geist MA , Volbracht C , Podhorna J , Falsig J , Leist M . Wide spectrum modulation by KP-544 in models relevant for neuronal survival. Neuroreport. (2007) ;18: (6):571–5. |
[200] | Dey ND , Boersen AJ , Myers RA , York LR , Bombard MC , Lu M , et al. The novel substituted pyrimidine, KP544, reduces motor deficits in the R6/2 transgenic mouse model of Huntington’s disease. Restor Neurol Neurosci. (2007) ;25: (5-6):485–92. |
[201] | Yang T , Massa SM , Tran KC , Simmons DA , Rajadas J , Zeng AY , et al. A small molecule TrkB/TrkC neurotrophin receptor co-activator with distinctive effects on neuronal survival and process outgrowth. Neuropharmacology. (2016) ;110: (Pt A):343–361. |
[202] | Guzior N , Wieckowska A , Panek D , Malawska B . Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr Med Chem. (2015) ;22: (3):373–404. |
[203] | Maliartchouk S , Debeir T , Beglova N , Cuello AC , Gehring K , Saragovi HU . Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J Biol Chem. (2000) ;275: (14):9946–56. |
[204] | Jiang G , Hunter T . Receptor signaling: When dimerization is not enough. Curr Biol. (1999) ;9: (15):R568–71. |
[205] | Livnah O , Stura EA , Middleton SA , Johnson DL , Jolliffe LK , Wilson IA . Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. (1999) ;283: (5404):987–90. |
[206] | Boltaev U , Meyer Y , Tolibzoda F , Jacques T , Gassaway M , Xu Q , et al. Multiplex quantitative assays indicate a need for reevaluating reported small-molecule TrkB agonists. Sci Signal. 201;10: (493). pii: eaal1670. |
[207] | Warnault V , Darcq E , Morisot N , Phamluong K , Wilbrecht L , Massa SM , et al. The BDNF valine 68 to methionine polymorphism increases compulsive alcohol drinking in mice that is reversed by tropomyosin receptor kinase B activation. Biol Psychiatry. (2016) ;79: (6):463–73. |
[208] | Kron M , Lang M , Adams IT , Sceniak M , Longo F , Katz DM . A BDNF loop-domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis Model Mech. (2014) ;7: (9):1047–55. |
[209] | Mamidipudi V , Wooten MW . Dual role for p75(NTR) signaling in survival and cell death: Can intracellular mediators provide an explanation? J Neurosci Res. (2002) ;68: (4):373–84. |
[210] | Dougherty KD , Milner TA . p75NTR immunoreactivity in the rat dentate gyrus is mostly within presynaptic profiles but is also found in some astrocytic and postsynaptic profiles. J Comp Neurol. (1999) ;407: (1):77–91. |
[211] | Miller MW , Pitts FA . Neurotrophin receptors in the somatosensory cortex of the mature rat: Co-localization of p75, trk, isoforms and c-neu. Brain Res. (2000) ;852: (2):355–66. |
[212] | Cragnolini AB , Friedman WJ . The function of p75NTR in glia. Trends Neurosci. (2008) ;31: (2):99–104. |
[213] | Ozbas-Gerceker F , Gorter JA , Redeker S , Ramkema M , van der Valk P , Baayen JC , et al. Neurotrophin receptor immunoreactivity in the hippocampus of patients with mesial temporal lobe epilepsy. Neuropathology and applied neurobiology. (2004) ;30: (6):651–64. |
[214] | Cragnolini AB , Huang Y , Gokina P , Friedman WJ . Nerve growth factor attenuates proliferation of astrocytes via the p75 neurotrophin receptor. Glia. (2009) ;57: (13):1386–92. |
[215] | Brito V , Giralt A , Enriquez-Barreto L , Puigdellivol M , Suelves N , Zamora-Moratalla A , et al. Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J Clin Invest. (2014) ;124: (10):4411–28. |
[216] | Miguez A , Garcia-Diaz Barriga G , Brito V , Straccia M , Giralt A , Gines S , et al. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum Mol Genet. (2015) ;24: (17):4958–70. |
[217] | Wehner AB , Milen AA , Albin RL , Pierchala BA . The p75 neurotrophin receptor augments survival signaling in the striatum of pre-symptomatic Q175 mice. Neuroscience. (2016) ;324: :297–306. |
[218] | Bartzokis G , Lu PH , Tishler TA , Fong SM , Oluwadara B , Finn JP , et al. Myelin breakdown and iron changes in Huntington’s disease: Pathogenesis and treatment implications. Neurochem Res. (2007) ;32: (10):1655–64. |
[219] | Jin J , Peng Q , Hou Z , Jiang M , Wang X , Langseth AJ , et al. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum Mol Genet. (2015) ;24: (9):2508–27. |
[220] | Roux PP , Barker PA . Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol. (2002) ;67: (3):203–33. |
[221] | Apostol BL , Illes K , Pallos J , Bodai L , Wu J , Strand A , et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet. (2006) ;15: (2):273–85. |
[222] | Liu YF . Expression of polyglutamine-expanded Huntingtin activates the SEK1-JNK pathway and induces apoptosis in a hippocampal neuronal cell line. J Biol Chem. (1998) ;273: (44):28873–7. |
[223] | Gratuze M , Cisbani G , Cicchetti F , Planel E . Is Huntington’s disease a tauopathy? Brain. (2016) ;139: (Pt 4):1014–25. |
[224] | Zagrebelsky M , Holz A , Dechant G , Barde YA , Bonhoeffer T , Korte M . The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci. (2005) ;25: (43):9989–99. |
[225] | Moller T . Neuroinflammation in Huntington’s disease. J Neural Transm. (2010) ;117: (8):1001–8. |
[226] | Khoshnan A , Ko J , Watkin EE , Paige LA , Reinhart PH , Patterson PH . Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci. (2004) ;24: (37):7999–8008. |
[227] | Trager U , Andre R , Lahiri N , Magnusson-Lind A , Weiss A , Grueninger S , et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain. (2014) ;137: (Pt 3):819–33. |
[228] | Hayden MS , Ghosh S . NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. (2012) ;26: (3):203–34. |
[229] | Huxford T , Huang DB , Malek S , Ghosh G . The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. (1998) ;95: (6):759–70. |
[230] | Khoshnan A , Patterson PH . The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol Dis. (2011) ;43: (2):305–11. |
[231] | Jia H , Kast RJ , Steffan JS , Thomas EA . Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: Implications for the ubiquitin-proteasomal and autophagy systems. Hum Mol Genet. (2012) ;21: (24):5280–93. |
[232] | Thompson LM , Aiken CT , Kaltenbach LS , Agrawal N , Illes K , Khoshnan A , et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. (2009) ;187: (7):1083–99. |
[233] | Choi S , Friedman WJ . Inflammatory cytokines IL-1beta and TNF-alpha regulate p75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signalling mechanisms. ASN Neuro. (2009) ;1: (2), pii: e00010. |
[234] | Massa SM , Xie Y , Yang T , Harrington AW , Kim ML , Yoon SO , et al. Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J Neurosci. (2006) ;26: (20):5288–300. |
[235] | Yang T , Knowles JK , Lu Q , Zhang H , Arancio O , Moore LA , et al. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS One. (2008) ;3: (11):e3604. |
[236] | Tep C , Lim TH , Ko PO , Getahun S , Ryu JC , Goettl VM , et al. Oral administration of a small molecule targeted to block proNGF binding to p75 promotes myelin sparing and functional recovery after spinal cord injury. J Neurosci. (2013) ;33: (2):397–410. |
[237] | Knowles JK , Rajadas J , Nguyen TV , Yang T , LeMieux MC , Vander Griend L , et al. The p75 neurotrophin receptor promotes amyloid-beta(1-42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci. (2009) ;29: (34):10627–37. |
[238] | Simmons DA , Knowles JK , Belichenko NP , Banerjee G , Finkle C , Massa SM , et al. A small molecule p75NTR ligand, LM11A-31, reverses cholinergic neurite dystrophy in Alzheimer’s disease mouse models with mid- to late-stage disease progression. PLoS One. (2014) ;9: (8):e102136. |
[239] | Friesland A , Weng Z , Duenas M , Massa SM , Longo FM , Lu Q . Amelioration of cisplatin-induced experimental peripheral neuropathy by a small molecule targeting p75 NTR. Neurotoxicology. (2014) ;45: :81–90. |
[240] | Shi J , Longo FM , Massa SM . A small molecule p75(NTR) ligand protects neurogenesis after traumatic brain injury. Stem Cells. (2013) ;31: (11):2561–74. |
[241] | Longo FM , Massa SM . Small molecule modulation of p75 neurotrophin receptor functions. CNS Neurol Disord Drug Targets. (2008) ;7: (1):63–70. |
[242] | Meeker RB , Poulton W , Clary G , Schriver M , Longo FM . Novel p75 neurotrophin receptor ligand stabilizes neuronal calcium, preserves mitochondrial movement and protects against HIV associated neuropathogenesis. Exp Neurol. (2016) ;275: (Pt 1):182–98. |
[243] | Narayanan KL , Chopra V , Rosas HD , Malarick K , Hersch S . Rho kinase pathway alterations in the brain and leukocytes in Huntington’s disease. Mol Neurobiol. (2016) ;53: (4):2132–40. |
[244] | Bauer PO , Wong HK , Oyama F , Goswami A , Okuno M , Kino Y , et al. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J Biol Chem. (2009) ;284: (19):13153–64. |
[245] | Shao J , Welch WJ , Diprospero NA , Diamond MI . Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol Cell Biol. (2008) ;28: (17):5196–208. |
[246] | Bokoch GM . Biology of the p21-activated kinases. Annu Rev Biochem. (2003) ;72: :743–81. |
[247] | Nakayama AY , Harms MB , Luo L . Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. (2000) ;20: (14):5329–38. |
[248] | Penzes P , Cahill ME . Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton. (2012) ;69: (7):426–41. |
[249] | Penzes P , Rafalovich I . Regulation of the actin cytoskeleton in dendritic spines. Adv Exp Med Biol. (2012) ;970: :81–95. |
[250] | Tashiro A , Yuste R . Role of Rho GTPases in the morphogenesis and motility of dendritic spines. Methods Enzymol. (2008) ;439: :285–302. |
[251] | Bogush A , Pedrini S , Pelta-Heller J , Chan T , Yang Q , Mao Z , et al. AKT and CDK5/p35 mediate brain-derived neurotrophic factor induction of DARPP-32 in medium size spiny neurons in vitro. J Biol Chem. (2007) ;282: (10):7352–9. |
[252] | Govek E , Newey S , Van Aelst L . The role of the Rho GTPases in neuronal development. Genes Dev. (2005) ;19: :1–49. |
[253] | Perrin V , Dufour N , Raoul C , Hassig R , Brouillet E , Aebischer P , et al. Implication of the JNK pathway in a rat model of Huntington’s disease. Exp Neurol. (2009) ;215: (1):191–200. |
[254] | Ceni C , Kommaddi RP , Thomas R , Vereker E , Liu X , McPherson PS , et al. The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. Journal of cell science. (2010) ;123: (Pt 13):2299–307. |
[255] | Skeldal S , Matusica D , Nykjaer A , Coulson EJ . Proteolytic processing of the p75 neurotrophin receptor: A prerequisite for signalling?: Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR). Bioessays. (2011) ;33: (8):614–25. |


