
What do we know about Late Onset Huntington’s Disease?
Abstract
Background: Although the typical age of onset for Huntington’s disease (HD) is in the fourth decade, between 4.4–11.5% of individuals with HD have a late onset (over 60 years of age). Diagnosis of Late onset HD (LoHD) can be missed, due to the perceived low likelihood of HD in the over 60-year-olds.
Objective: To review the epidemiology, genotype and phenotype of LoHD.
Methods: We systematically searched MEDLINE, EMBASE and Web of Science (inception-November 2016). Web of Science was then used to search for papers citing identified studies. Content experts were consulted for any additional studies. We included all studies reporting the clinical phenotype of LoHD for more than one participant.
Results: 20 studies were identified from a potential list of 1243. Among Caucasian HD cohorts, 4.4–11.5% of individuals have LoHD, and this proportion may be increasing. Proportion of LoHD without a positive family history ranges from 3–68%. 94.4% of reported cases of LoHD had CAG repeat lengths of ≤44. Motor manifestations are the commonest initial presentation, although 29.2% presented with non-motor manifestations as the first clinical feature in one case series. Individuals with LoHD may have slower progression of illness. Cognitive impairment rather than chorea may be the major source of disability in this group.
Conclusions: LoHD represents a substantial proportion of new diagnoses of HD and has some unique features. Further characterization of this population will aid clinicians in diagnosis.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease characterized by motor and movement disorder, cognitive impairment and behavioral abnormalities. HD is caused by a pathologic cytosine-adenine-guanine (CAG) repeat expansion, on the 5’ end of the Huntingtin gene [1]. The typical age of onset of HD is in the fourth decade, with a median survival of fifteen years from motor onset [2]. Recent prevalence studies, however, drew attention to a group of individuals with late-onset HD (LoHD). Some authors define LoHD as after 50 years [3–5], but more recent studies define LoHD as onset after 60 years [6–10]. Between 4.4–11.5% of individuals with HD have an onset age of over 60 [8, 10, 11]. Reported presentation of LoHD varies, and the natural history and prognosis of LoHD remains unclear. Diagnosis of LoHD can be missed due to the perceived low likelihood in the over 60-year-olds [12], and has substantial implications for family members. We aimed to search systematically for, and review the epidemiology, genotype, phenotype, diagnosis, progression and prognosis for people with LoHD.
METHODS
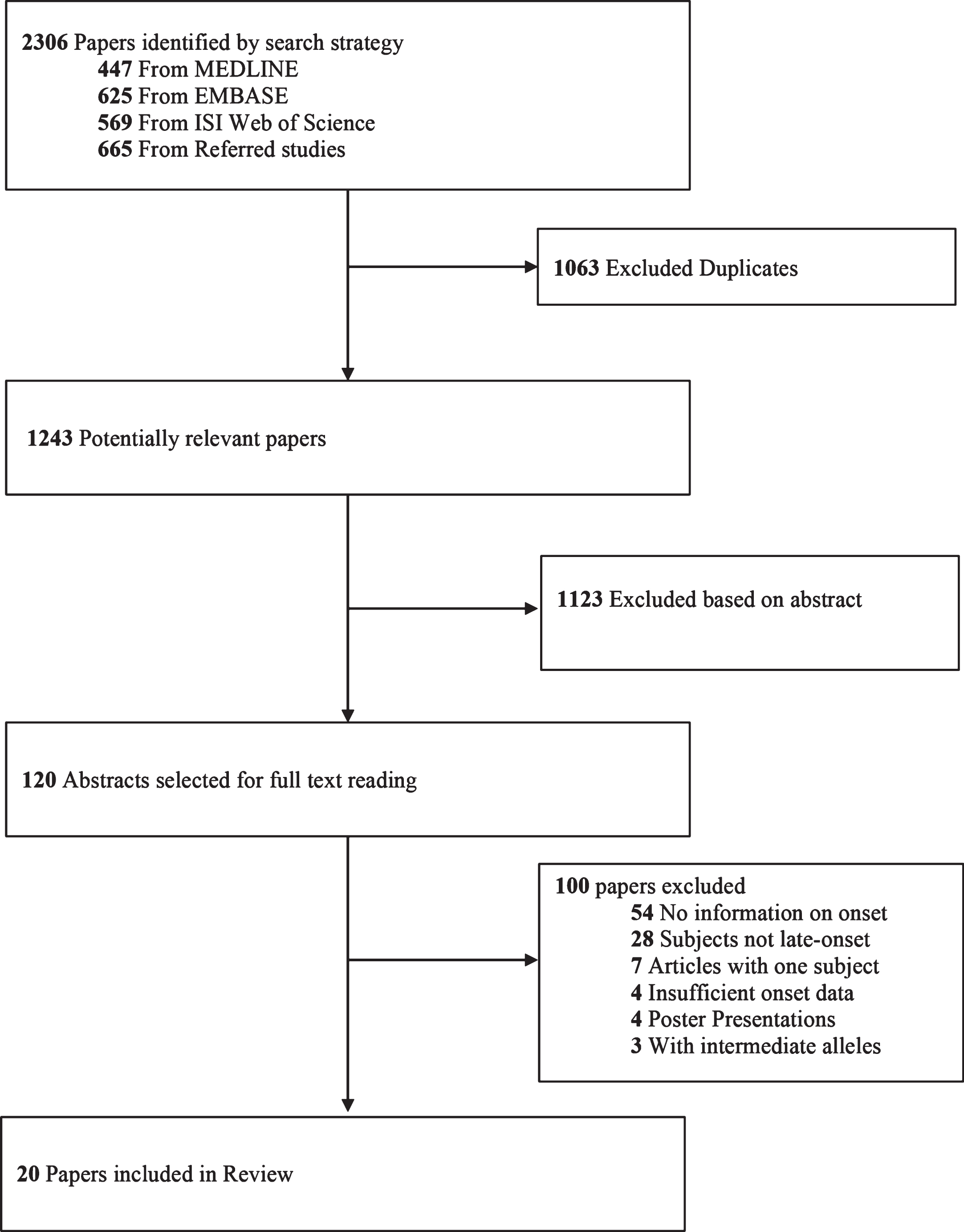
We searched MEDLINE and EMBASE from inception date to November 12, 2016 using the search terms (Huntington disease OR Huntington’s disease OR Huntington Chorea OR Huntington’s Chorea).mp AND (Late OR Delayed).mp AND (onset).mp. Web of Science was also queried using the same search terms. We included all English LoHD studies, which described the clinical features of more than one participant. We used Web of Science to complete the search, by searching for articles that cite the included studies. Content experts in the area were also consulted. The search yielded 1243 potentially relevant articles of which 120 were identified for full text reading based on the abstract. We included twenty studies in this review (Fig. 1).
Fig.1
Selection of included studies.
RESULTS
Epidemiology
A recent review by Rawlins et al. found the average prevalence of HD to be 7.33/100000 in North America and 6.68/100000 in the United Kingdom (UK) [13]. Rawlins et al. also pointed to an increase in HD prevalence in Australia, the Americas and the UK over the last few decades. A study from the UK suggested a number of reasons for the increased prevalence, including more accurate diagnosis (particularly with the advent of the definitive genetic test), improved supportive care, and general increase in life expectancy [14]. Impact of the baby boomer generation who are now in their 60 s, is also thought to contribute to the rise in prevalence [15].
The DNA diagnostic test for the mutation in the Huntingtin gene has improved our ability to diagnose HD in older people, in people with atypical features and in people without a family history [16, 17]. Evans et al. compared HD prevalence from 1990–1996 to 2004–2010, and found the greatest increase in prevalent cases, was in the 60–69 age group [14]. However, a recent study of the same database did not show increased incidence of LoHD cases in the same period, possibly indicating that people with younger onset HD are surviving longer [18]. In recent studies with patients’ diagnoses confirmed by genetic testing, the proportion of individuals with LoHD was between 4.4–11.5% [8, 10, 11]. The proportion of LoHD in older studies (1995 or earlier) was 4.3–28.0% [3, 6, 19, 20]. These older studies had varying definitions for LoHD, with three defining late age of onset as ≥ 60 and one ≥ 50 years.
Incidence studies for HD found individuals with LoHD make up an important proportion of new diagnoses. In Spain, Ramos-Arroyo et al. estimated that 16.1% of cases had LoHD between 1994–2002 [21]. Two analyses from British Columbia (from 1996–1999, and 1993–2000) found LoHD accounting for 17.7% and 19.6% respectively, of new diagnoses of HD [22]. Individuals with LoHD were more likely to have a negative family history and could result from repeat expansion from an unaffected parent [21]. This finding has important consequences for the person with LoHD and their extended families. Careful counselling needs to be offered, as this may be the first diagnosis of HD in the family.
Genotype
CAG repeat length is an important determinant for the age of onset. It correlates negatively with the age of onset, and accounts for approximately 70% of the variation in age of onset in HD [23]. However, the strength of correlation between CAG repeat length and age of onset decreases as the age of onset increases [24]. Langbehn et al. predict that more than 90% of individuals with repeat sizes of ≥44 would present before the age of 60 [25].
Among studies investigating LoHD, CAG repeat length was reported in 180/238 patients (Table 1). In studies where age of onset ≥60, mean CAG repeat length was calculated to be 40.9 (range 36–47, n = 145) with an average age of onset of 65.5. Across all studies, 170/180 reported cases of LoHD had a CAG repeat length of ≤44. It is interesting that repeat sizes as large as 47 have been recorded, where one would expect earlier onset [25]. This may relate to inaccurate determination of onset but in some instances, genetic modifiers or environmental factors could play a role in delaying expected onset for that repeat length.
Table 1
Characteristics and clinical features at presentation, of individuals with Late Onset Huntington Disease
| Study identifier | Sample size | Family History (%) | Mean onset (range) | Mean CAG (range) | Motor (%) | Chorea (%) | Cognitive (%) | Psychiatric (%) | Longitudinal study | Comments |
| Koutsis et al. [8] | 41 | 70.70 | 64.3 (60–73) | 40.5 (36–43) | 70.8 | 53.7 | 14.6 | 14.6 | Documentation when disease duration on reached severe stage | Mode of onset reported in mutually exclusive manner |
| Lipe et al. [7] | 34 | 32.4 | 66 (60–79) | 41.0 (38–44) | 100 | 85.3 | 29.4 | 32.4 | Progress reported and UHDRS available for 9 people. | |
| James et al. [6] | 33 (14 formally diagnosed) | 97.0 | 65 (median) (60–77) | 38.6 (38–39) n = 10 | 97 | 93.9 | 30.3 | 48.5 | Progress reported. | |
| Cornejo-Olivas et al. [10] | 31 | 78.6 (28 people) | 64.1 (60–73) | 42.5 (39–48) | 92.3 | 92.3 | 0 | 7.7 | No progress reported (NPR) | Mode of onset reported in mutually exclusive manner. |
| 26 people with onset data. | ||||||||||
| Myers et al. [3] | 25 | Unclear | 57.5 (50–70) | NA | 100 | 100 | 100 | 76.0 | Progress Reported | Onset defined as onset of chorea. Clinical features reported at examination. |
| Gomez-Tortosa et al. [5] | 13 | 100 | 57.8 (51–65) | 41.7 (39–47) | Not Reported | Not reported | Not reported | Not reported | NPR | |
| Britton et al. [38] | 10 | 100 | ≥50 | 45 (44–46) n = 4 | 100 | 100 | 0 | 0 | Progress reported | One person had onset at 40 and was excluded. |
| Davis et al. [51] | 10 | Unclear | 66.4 (60–74) | 40.6 (39–44) | 80 | 70 | 0 | 20 | NPR | Mode of onset reported in mutually exclusive manner. |
| Nance, Westphal and Nugent [52] | 6 | Selected to have no family history | 63.8 (60–70) | 39.8 (39–41) | 66.7 | 66.7 | 66.7 | 50.0 | NPR | |
| Yoshida et al. [9] | 5 | 40 (probable) | 64.4 (60–69) | 39.8 (39–40) | 100 | 100 | 0 | 0 | Progress Reported | Mode of onset reported in mutually exclusive manner. |
| Macmillan, Davies and Harper [53] | 4 | 50.0 | 65.0 (60–75) | 39.0 (38–41) | 100 | 100 | 50 | 25 | NPR | Age at presentation and clinical features at presentation reported. |
| Warren et al. [54] | 4 | 50 (probable) | 78.5 (65–87) | 39.8 (39–40) | 100 | 100 | 25 | Not Reported | NPR | Features reported at presentation. |
| Faught et al. [55] | 4 | 100 | 64.8 (63–68) | NA | 100 | 100 | 0 | 0 | Progress Reported | Clinical features available for 4 individuals |
| Panegyres and Goh [56] | 3 | 100 | 70.0 (66–76) | 38.3 (38–39) | 66.7 | 33.3 | 33.3 | 33.3 | Progress Reported | 3 people appear to satisfy criteria for HD. People were selected to have reduced penetrance alleles. |
| Ruiz et al. [57] | 3 | Selected to have no family history | 63.7 (63–64) | 39.0 (38–40) | 100 | 100 | 0 | 0 | Progress Reported | |
| Reuter et al. [40] | 3 | Selected from Parkinson disease mimics | 60.7 (58–64) | 43.0 (42–44) | 100 | Unclear | 0 | 0 | Progress Reported | One person in study had onset at 45 and was excluded. |
| Appollonio et al. [12] | 3 | 100 (probable) | 57.7 (53–61) | 40.7 (38–43) | 100 | 100 | 33.3 | 0 | Progress Reported | |
| Bird and Steinbart [58] | 2 | 100 (probable) | 77 (75–79) | 39.5 (39–40) | 50 | 50 | 50 | 0 | Progress Reported | |
| Galluci et al. [59] | 2 | Not Reported | 53.5 (53–54) | 40.5 (38–43) | 100 | 100 | 0 | 0 | Progress Reported | |
| Hindley et al. [60] | 2 | 100 (probable) | 66.5 (64–69) | 41 (40–42) | 100 | 100 | 100 | 100 | Progress Reported |
There is some debate in the literature about the lowest limit of CAG repeat length that can lead to HD. Clinical HD always develops in individuals with CAG repeat length of 40 or greater, and there is reduced penetrance among individuals with repeat lengths from 36–39. CAG repeat lengths of 27–35 have generally been described as intermediate alleles (IA), where the CAG repeat size can expand into the pathogenic HD range in the next generation, but is not thought to cause onset of HD in the carrier of the IA [26]. Some have suggested that individuals with IAs have subtle motor, cognitive and behavioral changes compared to normal controls, and could perhaps develop late-onset HD if they were to live long enough [27–29]. Kenney et al. report the case history of a person with a clinical picture suggestive of HD, supported by neuropathological changes, but only 29 CAG repeats [30]. However, we note that unlike other neurodegenerative disorders such as Alzheimer’s disease, neuropathological changes are not as definitive for the diagnosis of HD [31]. In addition, some genetically proven advanced cases show only minimal pathological changes [32].
Groen et al. reported two cases of possible LoHD with IAs (30 and 31 repeats), one of whom had children with clinical HD and CAG repeat length of 43 [33]. There are also other case reports of possible LoHD with IAs [34–36]. Oosterloo et al. reviewed the 10 reported cases of IAs with HD, and found that only four of these cases fit with the clinical presentation of HD [37]. The authors suggest that it is difficult to conclude that individuals with IAs can develop HD as even in the four cases identified, alternative explanations such as HD mimics and somatic mosaicism were not always excluded [37].
Phenotype
Table 1 summarizes the clinical phenotype of people with LoHD. Four papers compared LoHD with HD without late onset, and some longitudinal data was available for fourteen papers.
All studies reported motor manifestations, predominantly chorea, to be the most common clinical feature at presentation. Current understanding is that individuals with LoHD present with chorea as the chief manifestation of the disease [6–10, 38]. Lipe et al. reported that approximately 85% of people had chorea at onset [7]. When the first clinical feature is reported in a mutually exclusive fashion, Koutsis et al. found 53.7% of people with LoHD had chorea at onset, and 90.2% of people had chorea at gene test request [8]. 17.1% had unsteadiness at onset. James et al. also showed that gait abnormalities are prominent in early stages of LoHD [6, 8].
The motor abnormalities progress with increasing disease duration so that patients develop progressive chorea and have more falls [7], but there are no large studies so far in which the progression of LoHD is specifically followed.
Cognitive and psychiatric onset ranged from 0–100% in the studies shown (Table 1). A recent study found that 29.4% of people with LoHD present with cognitive onset [7]. Zero percent and 14.6% of people in studies from Peru and Greece respectively, had cognitive onset when the different symptom domains were measured in a mutually exclusive fashion [8, 10]. Despite not reporting cognitive onset, the Peruvian cohort (n = 31) scored poorly in cognitive testing. The authors found that individuals with LoHD have a predominance of executive and visuospatial dysfunction, as well as problems in attention, calculation and verbal fluency as measured by Mini-Mental State Examination (MMSE) and Montreal Cognitive Assessment. Thus it is possible that people with LoHD have subtle cognitive changes at presentation that may not be fully appreciated, and only motor features are reported as these are more evident. This could potentially underestimate the proportion with cognitive onset among people with LoHD, particularly in retrospective studies.
The prevalence of psychiatric symptoms and depression is reported to be between 32% and 48% [6, 7]. When first manifestations were considered in a mutually exclusive fashion, 7.7–14.6% of individuals with LoHD had psychiatric onset [8, 10]. To date only two studies described the prevalence of psychiatric symptoms at onset such as depression (9–18%), apathy (9%) and psychosis (3%) [6, 7].
Comparing the mode of presentation between the different ages of onset would be very useful in characterizing HD. The frequency of chorea, cognitive and psychiatric manifestations as first presentations does not appear to be different between people with LoHD and HD [8, 10]. However, there is evidence to show that there may be subtle differences in the phenotype. In a small cohort (n = 13), Gomez-Tortosa et al. showed that individuals with LoHD (onset ≥50 years) had reduced cognitive scores in the visual domain, compared to other individuals with HD [5]. Differences in multiple domains were also observed between juvenile onset HD and HD. Koutsis et al. showed increased gait unsteadiness at diagnosis in LoHD compared to HD [8]. A comparison of HD with a cohort of people with very late onset HD (>75) may elicit more pronounced differences in clinical phenotype.
Diagnosis and family history
As noted, the diagnosis of LoHD may be difficult. Falush et al. found evidence for significant under-diagnosis of cases with late onset by measuring mutational flow. This method relies on mathematically extrapolating the expected genotype of the population (mutational flow) and comparing the expected value to what is observed [39]. Clinical assessment is complicated by the fact that there is considerable heterogeneity in the presentation of LoHD (Table 1), and chorea may not be the chief manifestation of the disease [40].
Family history may also be uninformative in LoHD. Between 32 and 97% of people with LoHD had a family history in large studies (Table 1). Koutsis et al. reported that 70.7% of people with LoHD had a family history in the largest case series to date (n = 41) [8]. A similar rate was reported in Peru [10]. Lipe et al. however, reported only 32.4% of people with LoHD had a family history [7]. These differences may be due to the differing methods of data acquisition. Koutsis et al. had access to all people with HD identified in Greece making it more likely that a positive family history was recorded. Differences in background life expectancy could also play a role, as it is possible that some expansion carriers may die from competing causes before disease onset.
Another important consideration is that LoHD can be under-reported due to patient factors. For instance, patients may consider the decline in cognitive and motor skills to be a part of ageing [9]. In addition, the cognitive impairment of HD is associated with unawareness of the disease manifestations, which is common at all ages of HD. James et al. showed that 91% (28 of 31) of people with late-onset presented because the signs and symptoms are first noticed by the carers rather than the people with LoHD themselves, suggesting a possible reason for under-diagnosis [6].
Progression
While chorea is the predominant manifestation, cognitive decline is often the major determinant of disability. Gomez-Tortosa et al. found that the MMSE correlated better than motor variables with Total Functional Capacity (a measure of every day function) among individuals with LoHD in a small cohort (n = 13) [5]. While the unwanted movements can be associated with gait abnormalities and weight loss, chorea may not be the major source of disability. It is unclear however, to what extent psychiatric symptoms contribute to morbidity. Myers et al. reported that the psychiatric symptoms do not contribute to disability but this study was conducted before the advent of the genetic test, and with a relatively small cohort (n = 25) [3].
It is generally thought that LoHD follows a milder course compared to HD. This is supported by the finding that individuals with a smaller CAG repeat experience a more benign course [41]. However, age may have a confounding effect, since disease progression and functional outcomes are better correlated with the CAG repeat length when adjusted for age of onset [42–44]. There is also evidence to show that the ageing process in itself is an important driver of institutionalization in individuals with HD [45].
Prognosis
There have been conflicting reports in the literature concerning the survival of individuals with LoHD. Whether LoHD is associated with the same [6, 46, 47] or worse [4, 7] prognosis in comparison with HD is not decided. There are a greater number of associated co-morbidities (such as cancer, cerebrovascular abnormalities and Alzheimer’s disease) in LoHD due to increasing age [4, 7]. Anecdotally, it has been reported that death in LoHD is a result of these comorbidities rather than HD [4, 7]. It is likely that the variability in duration of disease reflects differences in the relative co-morbidities in the different populations to date. A variety of genetic [48] and environmental factors may influence the age of onset [23]. Similarly, it is possible that a wide range of heritable as well as environmental factors could also impact survival.
Conclusions
Age of onset for clinical HD can be difficult to determine, due to its slowly progressive nature, unawareness of manifestations among people with HD, and considerable variation in clinical presentation [49]. Diagnosing LoHD is sometimes delayed because of the heterogeneity in the presentation of the disease, and a lack of family history. Converging epidemiologic evidence suggests that approximately 10% of people with Huntington Disease have late onset. People with LoHD are most likely to present with motor manifestations, although cognitive and psychiatric features are also common. Cognitive impairment rather than chorea may be the major source of disability in this group- particularly as suppression of chorea does not appear to improve function [50]. The mortality in people with LoHD is possibly determined by associated co-morbidities and disease duration may not differ from HD. Identification of genetic modifiers (other than CAG repeat length) that influence onset, as well as environmental modifiers, could be valuable in the quest to delay onset of HD.
CONFLICTS OF INTEREST
The authors declare that there is no conflict of interest in publishing the paper.
ACKNOWLEDGMENTS
Funding Source: CTL is supported by an Australian National Health and Medical Research Council- Australian Research Council Dementia Research Development Fellowship (APP1107657).
REFERENCES
[1] | The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: (6):971–83. |
[2] | Keum JW , Shin A , Gillis T , Mysore JS , Elneel KA , Lucente D , et al. The HTT CAG-expansion mutation determines age at death but not disease duration in Huntington disease. Am J Hum Genet. (2016) ;98: (2):287–98. |
[3] | Myers R , Sax D , Schoenfeld M , Bird E , Wolf P , Vonsattel J , et al. Late onset of Huntington’s disease. J Neurol Neurosurg Psychiatry. (1985) ;48: (6):530–4. |
[4] | Foroud T , Gray J , Ivashina J , Conneally PM . Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatry. (1999) ;66: (1):52–6. |
[5] | Gómez-Tortosa E , del Barrio A , Ruiz PJG , Pernaute RS , Benítez J , Barroso A , et al. Severity of cognitive impairment in juvenile and late-onset Huntington disease. Arch Neurol. (1998) ;55: (6):835–43. |
[6] | James C , Houlihan G , Snell R , Cheadle J , Harper P . Late-onset Huntington’s disease: A clinical and molecular study. Age Ageing. (1994) ;23: (6):445–8. |
[7] | Lipe H , Bird T . Late onset Huntington Disease: Clinical and genetic characteristics of 34 cases. J Neurol Sci. (2009) ;276: (1):159–62. |
[8] | Koutsis G , Karadima G , Kladi A , Panas M . Late-onset Huntington’s disease: Diagnostic and prognostic considerations. Parkinsonism Relat Disord. (2014) ;20: (7):726–30. |
[9] | Yoshida K , Yanagawa S , Tsuchiya A , Nakajima T , Fukushima Y , Ikeda Si . Huntington’s disease with onset ages greater than 60 years. Geriatr Gerontol Int. (2007) ;7: (1):80–2. |
[10] | Cornejo-Olivas MR , Inca-Martinez MA , Espinoza-Huertas K , Veliz-Otani D , Velit-Salazar MR , Marca V , et al. Clinical and molecular features of late onset Huntington disease in a Peruvian cohort. J Huntingtons Dis. (2015) ;4: (1):99–105. |
[11] | Alonso ME , Ochoa A , Boll MC , Sosa AL , Yescas P , López M , et al. Clinical and genetic characteristics of Mexican Huntington’s disease patients. Mov Disord. (2009) ;24: (13):2012–5. |
[12] | Appollonio I , Frisoni GB , Curtò N , Trabucchi M , Frattola L . Which diagnostic procedures in the elderly? The case of late-onset Huntington’s disease. J Geriatr Psychiatry Neurol. (1997) ;10: (1):39–46. |
[13] | Rawlins MD , Wexler NS , Wexler AR , Tabrizi SJ , Douglas I , Evans SJ , et al. The prevalence of Huntington’s disease. Neuroepidemiology. (2016) ;46: (2):144–53. |
[14] | Evans SJ , Douglas I , Rawlins MD , Wexler NS , Tabrizi SJ , Smeeth L . Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry. (2013) ;84: (10):1156–60. |
[15] | Loy CT , Lownie A , McCusker E . Huntington’s disease. Lancet. (2010) ;376(9751):1463. |
[16] | Nance MA , Westphal B . Diagnosis of patients presenting to a Huntington’s disease clinic without a family history of Huntington’s disease. Neurology. (1996) ;46: (2 Suppl.):A251-A. |
[17] | Davis MB , Bateman D , Quinn NP , Marsden CD , Harding AE . Mutation analysis in patients with possible but apparently sporadic Huntington’s disease. Lancet. (1994) ;344: (8924):714–7. |
[18] | Wexler NS , Collett L , Wexler AR , Rawlins MD , Tabrizi SJ , Douglas I , et al. Incidence of adult Huntington’s disease in the UK: A UK-based primary care study and a systematic review. BMJ Open. (2016) ;6: (2):e009070. |
[19] | Morrison P , Johnston W , Nevin N . The epidemiology of Huntington’s disease in Northern Ireland. J Med Genet. (1995) ;32: (7):524–30. |
[20] | Adams P , Falek A , Arnold J . Huntington disease in Georgia: Age at onset. Am J Hum Genet. (1988) ;43: (5):695. |
[21] | Ramos-Arroyo M , Moreno S , Valiente A . Incidence and mutation rates of Huntington’s disease in Spain: Experience of 9 years of direct genetic testing. J Neurol Neurosurg Psychiatry. (2005) ;76: (3):337–42. |
[22] | Almqvist E , Elterman D , MacLeod P , Hayden M . High incidence rate and absent family histories in one quarter of patients newly diagnosed with Huntington disease in British Columbia. Clin Genet. (2001) ;60: (3):198–205. |
[23] | Wexler NS . Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. (2004) ;101: (10):3498–503. |
[24] | Kremer B , Squitieri F , Telenius H , Andrew S , Theilmann J , Spence N , et al. Molecular analysis of late onset Huntington’s disease. J Med Genet. (1993) ;30: (12):991–5. |
[25] | Langbehn D , Brinkman R , Falush D , Paulsen J , Hayden M . A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. (2004) ;65: (4):267–77. |
[26] | Semaka A , Creighton S , Warby S , Hayden M . Predictive testing for Huntington disease: Interpretation and significance of intermediate alleles. Clin Genet. (2006) ;70: (4):283–94. |
[27] | Ha AD , Beck CA , Jankovic J . Intermediate CAG repeats in Huntington’s disease: Analysis of COHORT. Tremor Other Hyperkinet Mov (N Y). (2012) ;2. |
[28] | Killoran A , Biglan KM , Jankovic J , Eberly S , Kayson E , Oakes D , et al. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology. (2013) ;80: (22):2022–7. |
[29] | Cubo E , Ramos-Arroyo MA , Martinez-Horta S , Martínez-Descalls A , Calvo S , Gil-Polo C . Clinical manifestations of intermediate allele carriers in Huntington disease. Neurology. (2016) ;87: (6):571–8. |
[30] | Kenney C , Powell S , Jankovic J . Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov Disord. (2007) ;22: (1):127–30. |
[31] | Semaka A , Warby S , Leavitt BR , Hayden MR . Re: Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov Disord. (2008) ;23: (12):1794–5. author reply 3. |
[32] | Caramins M , Halliday G , McCusker E , Trent RJ . Genetically confirmed clinical Huntington’s disease with no observable cell loss. J Neurol Neurosurg Psychiatry. (2003) ;74: (7):968–70. |
[33] | Groen JL , De Bie RM , Foncke EM , Roos RA , Leenders KL , Tijssen MA . Late-onset Huntington disease with intermediate CAG repeats: True or false? J Neurol Neurosurg Psychiatry. (2010) ;81: (2):228–30. |
[34] | Ha AD , Jankovic J . Exploring the correlates of intermediate CAG repeats in Huntington disease. Postgrad Med. (2011) ;123: (5):116–21. |
[35] | Andrich J , Arning L , Wieczorek S , Kraus PH , Gold R , Saft C . Huntington’s disease as caused by 34 CAG repeats. Mov Disord. (2008) ;23: (6):879–81. |
[36] | Garcia-Ruiz PJ , Garcia-Caldentey J , Feliz C , del Val J , Herranz A , Martínez-Castrillo JC . Late onset Huntington’s disease with 29 CAG repeat expansion. J Neurol Sci. (2016) ;363: , 114–5. |
[37] | Oosterloo M , Van Belzen MJ , Bijlsma EK , Roos RA . Is there convincing evidence that intermediate repeats in the HTT gene cause Huntington’s disease? J Huntingtons Dis. (2015) ;4: (2):141–8. |
[38] | Britton J , Uitti R , Ahlskog J , Robinson R , Kremer B , Hayden M . Hereditary late-onset chorea without significant dementia genetic evidence for substantial phenotypic variation in Huntington’s disease. Neurology. (1995) ;45: (3):443–7. |
[39] | Falush D , Almqvist EW , Brinkmann RR , Iwasa Y , Hayden MR . Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial underascertainment of late-onset cases. Am J Hum Genet. (2001) ;68: (2):373–85. |
[40] | Reuter I , Hu M , Andrews T , Brooks D , Clough C , Chaudhuri KR . Late onset levodopa responsive Huntington’s disease with minimal chorea masquerading as Parkinson plus syndrome. J Neurol Neurosurg Psychiatry. (2000) ;68: (2):238–41. |
[41] | Rosenblatt A , Liang K-Y , Zhou H , Abbott M , Gourley L , Margolis R , et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. (2006) ;66: (7):1016–20. |
[42] | Ross CA , Pantelyat A , Kogan J , Brandt J . Determinants of functional disability in Huntington’s disease: Role of cognitive and motor dysfunction. Mov Disord. (2014) ;29: (11):1351–8. |
[43] | Ravina B , Romer M , Constantinescu R , Biglan K , Brocht A , Kieburtz K , et al. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov Disord. (2008) ;23: (9):1223–7. |
[44] | Rosenblatt A , Kumar BV , Mo A , Welsh CS , Margolis RL , Ross CA . Age, CAG repeat length, and clinical progression in Huntington’s disease. Mov Disord. (2012) ;27: (2):272–6. |
[45] | Rosenblatt A , Kumar BV , Margolis RL , Welsh CS , Ross CA . Factors contributing to institutionalization in patients with Huntington’s disease. Mov Disord. (2011) ;26: (9):1711–6. |
[46] | Roos R , Hermans J , Vegter-Van Der Vlis M , Van Ommen G , Bruyn G . Duration of illness in Huntington’s disease is not related to age at onset. J Neurol Neurosurg Psychiatry. (1993) ;56: (1):98–100. |
[47] | Mahant N , McCusker E , Byth K , Graham S . Huntington’s disease Clinical correlates of disability and progression. Neurology. (2003) ;61: (8):1085–92. |
[48] | Lee J-M , Wheeler Vanessa C , Chao Michael J , Vonsattel Jean Paul G , Pinto Ricardo M , Lucente D , et al. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162: (3):516–26. |
[49] | Walker FO . Huntington’s disease. Lancet. (2007) ;369: (9557):218–28. |
[50] | FrankS . Tetrabenazine as anti-chorea therapy in Huntington disease: An open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol. (2009) ;9: , 62. |
[51] | Davis MB , Bateman D , Quinn NP , Marsden CD , Harding AE . Mutation analysis in patients with possible but apparently sporadic Huntington’s disease. Lancet. 344: (8924):714–7. |
[52] | Nance M , Westphal B , Nugent S . Diagnosis of patients presenting to a Huntington disease (HD) clinic without a family history of HD. Neurology. (1996) ;47: (6):1578–80. |
[53] | MacMillan J , Davies P , Harper P . Molecular diagnostic analysis for Huntington’s disease: A prospective evaluation. J Neurol Neurosurg Psychiatry. (1995) ;58: (4):496–8. |
[54] | Warren J , Firgaira F , Thompson E , Kneebone C , Blumbergs P , Thompson P . The causes of sporadic and ‘senile’ chorea. Aust N Z J Med. (1998) ;28: (4):429–31. |
[55] | Faught E , Falgout J , Leli D . Late-onset variant of Huntington’s chorea. South Med J. (1983) ;76: (10):1266–70. |
[56] | Panegyres PK , Goh JG . The neurology and natural history of patients with indeterminate CAG repeat length mutations of the Huntington disease gene. J Neurol Sci. (2011) ;301: (1):14–20. |
[57] | Garcia Ruiz P , Gomez-Tortosa E , Barrio Ad , Benitez J , Morales B , Vela L , et al. Senile chorea: A multicenter prospective study. Acta Neurol Scand. (1997) ;95: (3):180–3. |
[58] | Bird TD , Lipe HP , Steinbart EJ . Geriatric neurogenetics: Oxymoron or reality? Arch Neurol. (2008) ;65: (4):537–9. |
[59] | Gallucci DC , Frisoni GB , Trabucchi M , Appollonio IM . The relevance of genetic testing in late-onset Huntington’s disease. J Am Geriatr Soc. (1996) ;44: (5):609–11. |
[60] | Hindley N , Norbury G , Jobst K , Rosser E , Huson S , Pearce MJ , et al. Late onset Huntington’s disease as a cause of dementia: Where should the clinician’s index of suspicion lie? Int J Geriatr Psychiatry (1996) ;11: (8):729–33. |


