LBH589, A Hydroxamic Acid-Derived HDAC Inhibitor, is Neuroprotective in Mouse Models of Huntington’s Disease
Abstract
Background: Modulation of gene transcription by HDAC inhibitors has been shown repeatedly to be neuroprotective in cellular, invertebrate, and rodent models of Huntington’s disease (HD). It has been difficult to translate these treatments to the clinic, however, because existing compounds have limited potency or brain bioavailability.
Objective: In the present study, we assessed the therapeutic potential of LBH589, an orally bioavailable hydroxamic acid-derived nonselective HDAC inhibitor in mouse models of HD.
Method: The efficacy of LBH589 is tested in two HD mouse models using various biochemical, behavioral and neuropathological outcome measures.
Results: We show that LBH589 crosses the blood brain barrier; induces histone hyperacetylation and prevents striatal neuronal shrinkage in R6/2 HD mice. In full-length knock-in HD mice LBH589-treatment improves motor performance and reduces neuronal atrophy.
Conclusions: Our efficacious results of LBH589 in fragment and full-length mouse models of HD suggest that LBH589 is a promising candidate for clinical assessment in HD patients and provides confirmation that non-selective HDAC inhibitors can be viable clinical candidates.
INTRODUCTION
Huntington’s disease (HD) is a progressive autosomal neurodegenerative disease characterized by selective neuronal loss that is especially prominent in the striatum and cerebral cortex. Although the exact causes of neuronal death in HD remain unclear, it seems likely that multiple molecular derangements culminate gradually in degenerative changes and cell death [1] and there is evidence for the involvement of a number of pathogenic mechanisms such as transcriptional dysregulation, mitochondrial dysfunction, excitotoxicity, oxidative stress and impaired proteolysis [2]. Numerous studies have implicated direct and indirect alterations in transcription due to the presence of mutant huntingtin as well as the therapeutic potential of modulating transcription [3, 4]. Histone deacetylase (HDAC) inhibitors, which promote histone hyperacetylation and selective gene transcription, including suberoylanilide hydroxamic acid (SAHA), sodium valporate, sodium butyrate, phenyl butyrate, trichostatin A (TSA), selisistat, HDACi 4b have all been shown to be neuroprotective in cellular, drosophila, and/or mouse models of HD [5– 12]. Since these compounds also enable acetylation of proteins uninvolved in epigenetic regulation, they may have more pleiotropic benefits as well. Among the principal challenges for developing these potential treatments for HD patients have been limited potency and poor brain bioavailability.
Although the majority of these HDAC inhibitors work broadly on all HDAC isoforms (pan inhibitors), it is not yet clear whether selective inhibitors would offer any advantages compared to the pan-inhibitors. Significant efforts are currently underway on the pharmaceutical development of isoform specific HDAC inhibitors and genetic validation studies in which HDAC KO mice are crossed with HD models to assess the effect of each HDAC isoform on HD phenotype. While pharmacological targeting of HDAC1/ HDAC3 has been shown to ameliorate disease phenotype in HD models, genetic depletion of HDAC3, HDAC5, HDAC6, HDAC7, and HDAC9 had no effect on an HD phenotype [10, 11, 13– 15]. So far, HDAC4 is the only HDAC whose genetic knock down improves behavioral and neuropathological phenotypes in HD mice and development of potent, selective small-molecule inhibitors of HDAC4 is underway [16, 17]. While progress has been made to create selective inhibitors, many challenges exist towards designing isoform selective HDAC inhibitors with favorable CNS characteristics.
LBH589 (Panobinostat), is a potent, orally bioavailable hydroxamic acid based deacetylase inhibitor which inhibits all Class I, II and IV HDAC enzymes at low nanomolar concentrations [18]. It has been under development, either alone or in combination with other compounds for oncological, inflammatory and viral diseases, including, Cutaneous T Cell lymphoma (CTCL), Hodgkin’s lymphoma, acute myeloid leukemia (AML), breast cancer, prostate cancer, diffuse intrinsic pontine gliomas (DIPG) and HIV [19– 23]. Panobinostat was recently approved by the FDA as a combination therapy with bortezomib and dexamethasone for patients with recurrent multiple myelomas [24]. In this report we assessed its potential for HD using transgenic R6/2 and full-length CAG140 knock-in mouse models of HD. Our results demonstrated that LBH589-treatment prevented neuronal shrinkage in R6/2 mice and significantly ameliorated the behavioral and neuropathological phenotypes in CAG140 knock-in mice.
MATERIALS AND METHODS
Animals
This study was performed using female R6/2 and CAG 140 mice. R6/2 and CAG 140 KI males from stable colonies maintained at MGH facility were bred with females from their background C57BL/6×CBA F1. The progeny were genotyped by PCR using DNA extracted from tail tips [25] and were housed five per cage under standard conditions with ad-libitum access to water and food. The CAG repeat length of R6/2 mice was 125– 132. To ensure homogeneity of experimental cohorts mice from the same F generation were systematically assigned to experimental groups such that age, weight and CAG repeat lengths were balanced. All animal experiments were carried out in accordance with the NIH Guide for the Care and Use of Laboratory animals and were approved by institutional animal care and use committee at MGH.
Pharmacokinetics
Plasma and brain levels of LBH589 were determined by HPLC/MS method at Novartis Institute for Biomedical Research (Basel).
Drug treatment
LBH589 and the vehicle were obtained from the Novartis Institute for Biomedical Research (Basel). Mice were administered ip injections of LBH589 at the dose rate of 10 or 30 mg/kg every other day. Drug suspension was made fresh daily. Treatment was started from four weeks of age in R6/2 mice and from 6 weeks of age in CAG 140 mice. Body weights were recorded weekly at the same time of day. Treatment with LBH589 continued until death or euthanasia when triggered by pre-specified clinical criteria approved by the facility veterinarian, which was defined by the inability of the mice to right themselves thrice, within 20 seconds of being pushed onto one side.
Behavioral analyses
Motor performance was assessed using an accelerating rotarod (Stoelting, Ugo Basile, Italy) at 5, 8 and 11 weeks of age in R6/2 mice and 2,4,6,8 and 10 months of age in CAG 140 KI mice. At the beginning of each week, mice (n = 14-15) were trained for 30-seconds at a slow speed of 4.5 rpm. Subsequently, three trials were performed for three consecutive days. In each trial, mice were placed onto the rotarod at a constant speed of 4.5 rpm for 5 seconds, which then accelerates at a constant rate until a terminal angular velocity of 45 rpm was reached. The latency to fall from the rotarod was recorded for each mouse and the average of three trials was used for statistical analysis.
Histology
Histopathological analyses were performed at 90 days of age in R6/2 mice and at 10 months of age in CAG 140 mice. Mice were deeply anesthetized, then transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were post-fixed with perfusant for 2 days, cryoprotected in a graded series of 10% and 20% glycerol/2% DMSO, and serially sectioned at 50μm using a Leica SM 2000R freezing microtome (Leica, Wetzlar, Germany). Every eighth section was stained with thionin for striatal volume and neuronal volume analysis. Data was analyzed by ANOVA using the GraphPad Prism software. Results were considered statistically significant when p < 0.05.
Stereology
All analyses were performed blind using unbiased stereological approaches, StereoInvestigator software (MicroBrightField, Williston, VT, USA), and a Leica DMLB microscope with a motorized stage (Leica, Wetzlar, Germany). Striatal volumes were estimated on every 8th coronal section using the Cavalieri method. Stereological counts of neuronal cell body were obtained from the neostriatum at the level of the anterior commissure using the nucleator as described previously [26]. Data was analyzed by ANOVA.
Protein extraction and immunoblotting
Frozen murine cortical tissues were homogenized in PBS containing Complete EDTA-free Protease Inhibitor Cocktail (Roche Applied Science, USA) and 1 mM PMSF, using a Kontes Pellet Pestle (Kimble/Kontes, USA). Frozen muscle tissues were instead homogenized in the same buffer using an Omni Tissue Homogenizer (Omni, USA). Both homogeneates were then sonicated with a Branson Sonifier (Branson Ultrasonic Corp., USA) and lysed overnight at 4°C in 2X volume of 63 mM Tris buffer pH 6.8, 2% SDS, 10% glycerol, 1 mM DTT, Complete EDTA-free Protease Inhibitor Cocktail and 1 mM PMSF. Protein concentration was quantified with the BCA protein assay kit (Pierce, Thermo Scientific, USA) and analyzed by SDS-PAGE and western blot analysis using anti- acetyl histone H3 antibody (06– 599; Millipore, USA),anti- histone H3 antibody (9715;Cell Signaling Technology Inc., USA), anti-GADPH (MAB374; Millipore) and anti-actin (A2066; Sigma-Aldrich, USA). Proteins were visualized using an ECL detection substrate (Pierce, Thermo Scientific, USA).Protein levels were quantified by densitometry with the ImageJ software (NIH, USA), normalizing to actin or GAPDH levels.
Statistical analysis
Analyses were performed either using SAS software (version 9.1 Cary, NC) or using GraphPad Prism software (version 5.0, San Diego, USA). All the behavioral and body weight data with repeated measures was analyzed by SAS using a mixed model method that includes age by treatment effects. All Stereological data was analyzed by one-way analysis of variance (ANOVA) using GraphPad Prism. For Immunoblotting, two-tailed, unpaired Student t-test was used to determine the statistical significance between two groups. Results were considered statistically significant when p < 0.05.
RESULTS
Pharmacokinetic profiling of LBH589
Initial pharmacokinetic studies were performed using C57/BL6 mice. Mice were given a single ip injection of 10 mg/kg LBH589, sacrificed at 0.08, 0.5, 1, 2, 4, 8, and 24 hr post-dosing (n = 3 at each time point) and brain and plasma levels were analyzed by LC-MS/MS (LOQ = 1– 6 pmol/mL in plasma, 23 pmol/g in brain). The compound was rapidly eliminated from the systemic circulation (Fig. 1A). Although brain penetration was limited early, the brain to plasma ratio increased favorably with time, from 0.04 at five minutes to 0.59 at 2 hours to 3.66 at 24 hours post injection. The half-life of the compound was 7.6 hr in plasma and 15 hr in brain. Brain and plasma levels in R6/2 HD mice were comparable to wild-type mice. Two hours after injection, the average plasma and brain levels in R6/2 mice (n = 5) were 319±85 pmoles/ml and 110±51 pmoles/g respectively (Fig. 1B).
LBH589 increases histone acetylation in HD mice
We assessed histone H3 acetylation as a pharmacodynamic marker of HDAC inhibition in R6/2 mice. LBH589 has been previously demonstrated to rapidly induce histone acetylation in various tumor tissues [18]. R6/2 mice were treated with either vehicle or LBH589 at the dose rate of 20 mg/kg/three times per week for 3 weeks and the acetylation of histone H3 was determined in brain and muscle tissue by western blotting. When assessed in R6/2 mice sacrificed 2 hr after the last dose, total H3 acetylation significantly increased in brain (2.8 fold, t(7) = 3.57, p = 0.0091) and even more so in muscle (9.5 fold, t(7) = 3.36, p = 0.0120) (Fig. 2A-B).
LBH589 reduces striatal neuronal atrophy in R6/2 mice
We first tested the in vivo efficacy of LBH589 in the R6/2 mouse model of HD. These mice express an exon-1 fragment of human huntingtin and undergo rapid progression of disease with death occurring near three months of age [27]. To determine the optimal dose and dosing frequency for the efficacy studies, we first performed chronic and sub-chronic tolerability studies, in which groups of mice (n = 10– 15) were treated with different doses from 100 mg/kg to 10 mg/kg. The higher dose of 100 mg/kg three times per week resulted in a 12% loss of body weight within one week of initiating treatment. Doses of 60 mg/kg three times per week and 30 mg/kg/day were also poorly tolerated as they resulted in 18% and 14% loss in body weight respectively after 2 week of treatment. However, no significant loss in body weight in R6/2 mice was observed with 10 or 20 mg/kg three times per week after 4 week of treatment (Supplementary Table 1).
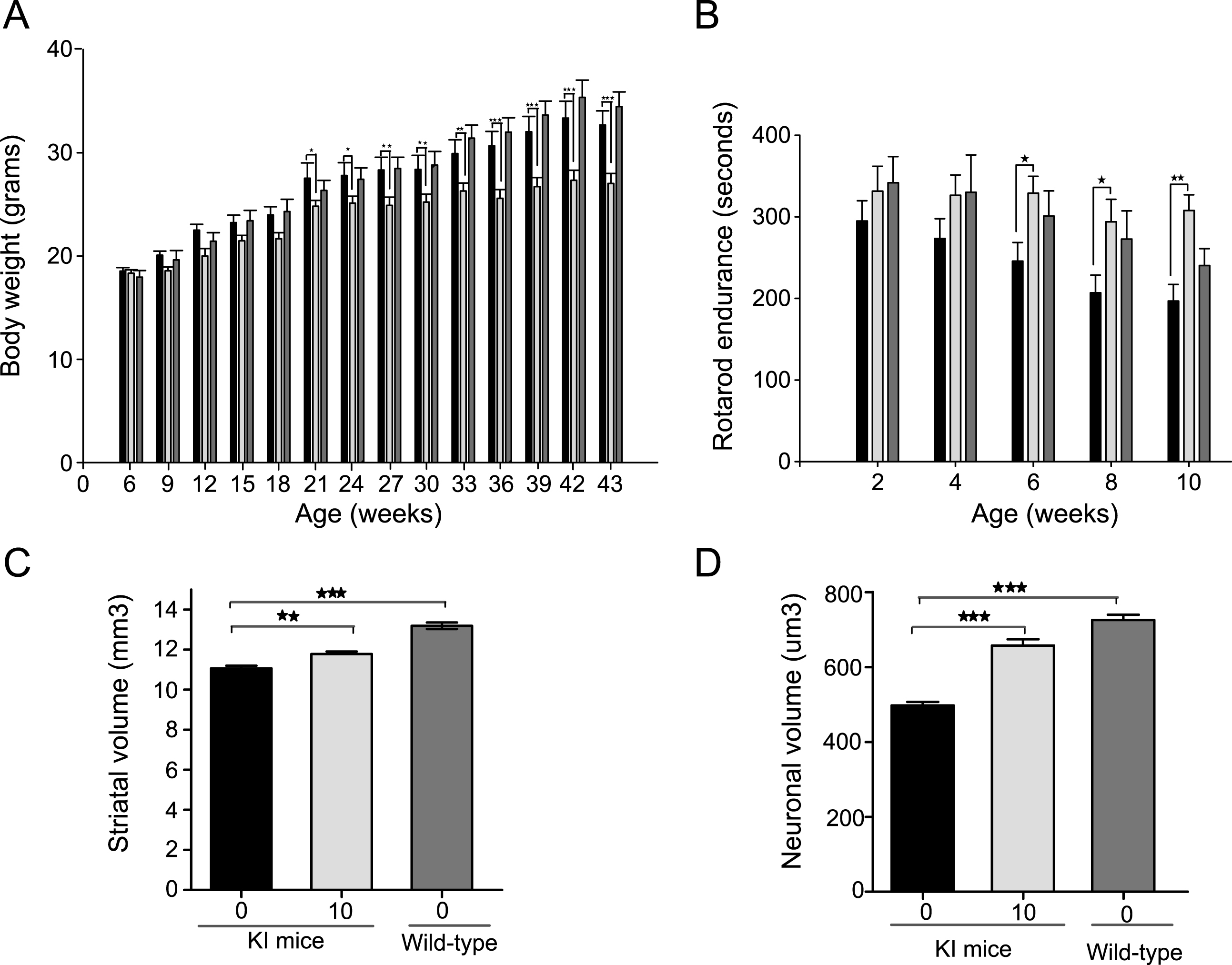
Based on the pilot tolerability studies, we selected 10 and 30 mg/kg, three times per week for studies intended to examine efficacy. Treatments were started from 28 days of age and continued until spontaneous death or euthanasia. Our higher dose of 30 mg/kg three times per week resulted in significant loss in body weight at 7 weeks of age (F2,82 = 10.46, p < 0.0001) and was subsequently reduced to 20 mg/kg three times per week; even then this dose was toxic as the mean survival of mice in this group was significantly lower than the vehicle-treated mice (Supplementary Figure 1). Some loss in body weight was also observed with the lower dose of 10 mg/kg at the later stage of disease (Fig. 3A). No loss in body weight was observed in wild-type mice dosed with 30 mg/kg three times a week up to 10 week of age (data not shown). There was no effect of LBH589 on the motor performance of R6/2 mice (F2,40 = 0.63, p = 0.53) (Fig. 3B). Unbiased stereological approaches were used to measure striatal and neuronal atrophy. Although striatal volume was unaltered, we found a significant reduction in neuronal cell body atrophy in R6/2 mice with LBH589-treatment (F2,25 = 9.330, p = 0.0009) (Fig. 3C-D). The average volume of striatal neuronal cell bodies in wild-type mice was 549.2±23.8μm3, as compared to 422.5±14.6μm3 in 13-week-old R6/2 mice. LBH589-treated (10 mg/kg) R6/2 mice had a mean striatal neuronal cell body volume of 525.6±25.4μm3, which was significantly greater (20%) than vehicle-treated R6/2 control mice. This neuroprotective effect at the highest tolerated dose in R6/2 mice raised the possibility that systemic toxicity in a fragile mouse model could have obscured finding more significant benefits. Since full-length mouse models of HD have a more protracted phenotype and are less sensitive to toxicity, we tested LBH589 further in the CAG140 knock-in model.
LBH589 ameliorates behavioral and neuropathological phenotype in CAG 140 knock-in mice
CAG 140 knock-in mice express the full-length human huntingtin gene, which causes a more gradual functional and neuropathological disease progression [28]. These mice have a normal lifespan and a phenotype analogous to presymptomatic and early symptomatic disease in humans. Mice were treated with 10 mg/kg LBH589 three times a week starting from 6 weeks of age. In the knock-in mice the drug was well tolerated up to 5 months without any significant loss of body weight; however, some loss in body weight was observed after 5 months of treatment (F1,26 = 10.01, p = 0.0039) (Fig. 4A). LBH589-treated mice performed significantly better on accelerating rotarod as compared to vehicle-treated knock-in mice (Fig. 4B). Rotarod testing revealed a significant benefit of treatment (F1,24 = 5.78, p = 0.0243). Latency to fall was improved by 25% at 6 months (246±23 s in vehicle-treated mice, 329±21 s in LBH589-treated mice, p = 0.018) by 30% at 8 months (207±22 s vehicle-treated mice, 294±28 s in LBH589-treated mice, p = 0.013) and by 36% at 10 months of age (197±20 s in vehicle-treated mice, 305±21 s in LBH589-treated mice, p = 0.004). Although it is feasible the reduction in body weight could have contributed to an improvement in rotarod performance of the mice, the magnitude of improvement in motor performance was quite large compared to the limited reduction in body weight, suggesting a more direct benefit. Next we examined whether LBH589 could reduce neuronal degeneration in knock-in mice (KI), in addition to R6/2 mice. All analyses were performed blind using unbiased stereological approaches. There were modest but significant reductions in striatal atrophy (F2,13 = 54.81, p < 0.0001) and striatal neuronal volume (F2,13 = 68.39, p < 0.0001), confirming the neuroprotective effects of LBH589-treatment (Fig. 4 C-D). The total striatal volume of vehicle-treated KI mice (11.1±0.1μm3) was considerably smaller than in wild type littermates (13.2±0.2μm3). However, LBH589-treatment significantly mitigated that striatal volume loss (11.8±0.1μm3). Unbiased measurement of neuronal cell body volumes were obtained from the neostriatum using the nucleator method (see methods). The average volume of striatal neuronal cell bodies in wild-type mice was 726±13.9μm3, as compared to 497±9.9μm3 in 10 month old CAG140 KI mice (p < 0.0001). LBH589-treatment (10 mg/kg) significantly reduced striatal neuronal atrophy by 25%, compared to vehicle-treated knock-in mice, for a mean volume of 657±16.6μm3.
DISCUSSION
Pan-HDAC inhibition has been validated as a therapeutic target for HD using a variety of compounds and pre-clinical models [29, 30]. Although significant effort has been made in the identification and development of class and isoform selective HDAC inhibitors with favorable CNS characteristics, many challenges remain. LBH589 is a novel hydroxamic acid-based deacetylase inhibitor that potently inhibits all Class I, II and IV HDAC enzymes at low nanomolar concentrations, that has been examined in humans in numerous phase II/III clinical trials for various oncology indications and was recently approved (Panobinostat) by the FDA for treating multiple myeloma [18, 24]. LBH589 is at least 10-fold more potent than SAHA, another hydroxamic acid HDAC inhibitor that has previously been shown to improve motor impairment in R6/2 mice [5, 18]. In the present study, we evaluated its potential for HD in two preclinical mouse models utilizing relevant neuropathological and behavioral outcome measures. Collectively, the data we report helps validate LBH589 preclinically as a potential disease modifying therapy for HD. LBH589 is an available medication for which there is existing clinical experience and safety database and its efficacy suggests that non-selective HDAC inhibition may be sufficient for clinical development for HD.
We found that LBH589 crosses the blood brain barrier, increases brain histone acetylation levels and prevents regional and neuronal atrophy of the striatum in R6/2 mice. Surprisingly, motor performance was not improved, despite the amelioration of neuropathology, We reasoned that even relatively low doses of LBH589 could have a deleterious impact on motor phenotype in the fragile R6/2 model given the observed limits of tolerability. Indeed, motor performance benefits related to LBH589-treatment, as well as neuropathologic benefits were observed in the more tolerant full-length CAG 140 knock-in HD mice, which have a more subtle and less rapidly progressing disease analogous to presymptomatic and early symptomatic disease in humans. Motor performance of LBH589-treated knock-in mice progressively diverged by 25%, 30% and 36% at 6, 8 and 10 months of age respectively compared to vehicle-treated controls. Stereologic measurement of total striatal volume and striatal neuronal cell body volumes were significantly higher in CAG140 mice treated thrice weekly with 10 mg/kg of LBH589.
Though demonstration of neuroprotective efficacy of LBH589 in two models (a fragment and a full-length model) is supportive of clinical development, especially if these findings are replicated independently, the mechanisms of neuroprotection by HDAC inhibition continue to be uncertain. Although we show that LBH589 increases histone acetylation, which could restore altered gene expression, which genes might be relevant to disease modification is unknown and other acetylase activities targeting non-histone proteins could be operative such as those involved in DNA repair (Ku70), cytoskeletal regulation (α- tubulin), and protein trafficking and turnover (Hsp90). Enhancement in mutant huntingtin clearance by altering its acetylation with an HDAC inhibitor has also been reported [31]. It is likely that more than one pathway contributes to the observed beneficial effect of LBH589, and perhaps its non-selectivity is advantageous.
Extensive literature is available on safety, pharmacology and toxicity of LBH589 in humans [32– 34]. It is currently being tested in phase II/ III clinical studies in Cutaneous T Cell lymphoma (CTCL), Hodgkin lymphoma, acute myeloid leukemia (AML), breast cancer, prostate cancer and AIDS. LBH589 has also shown beneficial effects for spinal muscular atrophy and for childhood brain tumor-DIPG in the preclinical studies, and is currently being developed as a phase I drug for children with DIPG through the Pediatric Brain Tumor Consortium. It has recently been approved by FDA as a combination therapy with bortezomib and dexamethasone in patients with recurrent multiple myelomas. Knowledge gained from the widening clinical use of LBH589, could enable its testing in HD patients. Although our data demonstrating disease-modifying benefits in two mouse models suggests that LBH589 is a potential therapeutic candidate for clinical assessment in HD patients, tolerability and long-term safety remain concerns. Significant side effects encountered in clinical studies of LBH589 include fatigue, nausea, vomiting, diarrhea, thrombocytopenia and prolonged QTc intervals [33– 34]. These could be mitigated by lower or more intermittent dosing for HD. Carefully planned early phase trials would be needed to optimize the dosing of LBH589 for HD patients and to assess the feasibility of further development. Examining the molecular pathways targeted by LBH589 in HD and their pharmacodynamics could help establish an optimum dose regimen and identify useful biomarkers.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Lorraine Vollor and Teal Connor for maintaining mouse colonies and help in behavioral and neuropathological studies. We also thank Dr. Jonathan Fox for his help in statistical analysis. This work was supported by the Beat HD Collaborative grant from the Novartis Institutes for BioMedical Research (NIBR).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JHD-160226.
REFERENCES
[1] | Humbert S , Saudou F . Huntington’s disease: Intracellular signaling pathways and neuronal death. J Soc Biol. (2005) ;199: (3):247–51. |
[2] | Zuccato C , Valenza M , Cattaneo E . Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. (2010) ;90: (3):905–81. |
[3] | Cha JH . Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. (2000) ;23: (9):387–92. |
[4] | Kazantsev AG , Hersch SM . Drug targeting of dysregulated transcription in Huntington’s disease. Prog Neurobiol. (2007) ;83: (4):249–59. |
[5] | Hockly E , Richon VM , Woodman B , Smith DL , Zhou X , Rosa E , et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. (2003) ;100: (4):2041–6. |
[6] | Ferrante RJ , Kubilus JK , Lee J , Ryu H , Beesen A , Zucker B , et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. (2003) ;23: (28):9418–27. |
[7] | Gardian G , Browne SE , Choi DK , Klivenyi P , Gregorio J , Kubilus JK , et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. (2005) ;280: (1):556–63. |
[8] | Zadori D , Geisz A , Vamos E , Vecsei L , Klivenyi P . Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol Biochem Behav. (2009) ;94: (1):148–53. |
[9] | Smith MR , Syed A , Lukacsovich T , Purcell J , Barbaro BA , Worthge SA , et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum Mol Genet. (2014) ;23: (11):2995–3007. |
[10] | Thomas EA , Coppola G , Desplats PA , Tang B , Soragni E , Burnett R , et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. (2008) ;105: (40):15564–9. |
[11] | Jia H , Pallos J , Jacques V , Lau A , Tang B , Cooper A , et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis. (2012) ;46: (2):351–61. |
[12] | Giralt A , Puigdellivol M , Carreton O , Paoletti P , Valero J , Parra-Damas A , et al. Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum Mol Genet. (2011) ;21: (6):1203–16. |
[13] | Moumne L , Campbell K , Howland D , Ouyang Y , Bates GP . Genetic knock-down of HDAC3 does not modify disease-related phenotypes in a mouse model of Huntington’s disease. PLoS One. (2012) ;7: (2):e31080. |
[14] | Bobrowska A , Paganetti P , Matthias P , Bates GP . Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PLoS One. (2011) ;6: (6):e20696. |
[15] | Benn CL , Butler R , Mariner L , Nixon J , Moffitt H , Mielcarek M , et al. Genetic knock-down of HDAC7 does not ameliorate disease pathogenesis in the R6/2 mouse model of Huntington’s disease. PLoS One. (2009) ;4: (6):e5747. |
[16] | Mielcarek M , Landles C , Weiss A , Bradaia A , Seredenina T , Inuabasi L , et al. HDAC4 reduction: A novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. (2013) ;11: (11):e1001717. |
[17] | Burli RW , Luckhurst CA , Aziz O , Matthews KL , Yates D , Lyons KA , et al. Design, synthesis, and biological evaluation of potent and selective class IIa histone deacetylase (HDAC) inhibitors as a potential therapy for Huntington’s disease. J Med Chem. (2013) ;56: (24):9934–54. |
[18] | Atadja P . Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. (2009) ;280: (2):233–41. |
[19] | Duvic M , Dummer R , Becker JC , Poulalhon N , Ortiz Romero P , Grazia Bernengo M , et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: Results of a phase II trial. Eur J Cancer. (2013) ;49: (2):386–94. |
[20] | Younes A , Sureda A , Ben-Yehuda D , Zinzani PL , Ong TC , Prince HM , et al. Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: Results of a phase II study. J Clin Oncol. (2012) ;30: (18):2197–203. |
[21] | Tan P , Wei A , Mithraprabhu S , Cummings N , Liu HB , Perugini M , et al. Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Cancer J. (2014) ;4: :e170. |
[22] | Rathkopf DE , Picus J , Hussain A , Ellard S , Chi KN , Nydam T , et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. (2013) ;72: (3):537–44. |
[23] | Hogh Kolbaek Kjaer AS , Brinkmann CR , Dinarello CA , Olesen R , Ostergaard L , Sogaard OS , et al. The histone deacetylase inhibitor panobinostat lowers biomarkers of cardiovascular risk and inflammation in HIV patients. AIDS. (2015) ;29: (10):1195–200. |
[24] | Garnock-Jones KP . Panobinostat: First global approval. Drugs. (2015) ;75: (6):695–704. |
[25] | Mangiarini L , Sathasivam K , Mahal A , Mott R , Seller M , Bates GP . Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. (1997) ;15: (2):197–200. |
[26] | Moller A , Strange P , Gundersen HJ . Efficient estimation of cell volume and number using the nucleator and the disector. J Microsc. (1990) ;159: (Pt 1):61–71. |
[27] | Mangiarini L , Sathasivam K , Seller M , Cozens B , Harper A , Hetherington C , et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) ;87: (3):493–506. |
[28] | Menalled LB , Sison JD , Dragatsis I , Zeitlin S , Chesselet MF . Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. (2003) ;465: (1):11–26. |
[29] | Sadri-Vakili G , Cha JH . Histone deacetylase inhibitors: A novel therapeutic approach to Huntington’s disease (complex mechanism of neuronal death). Curr Alzheimer Res. (2006) ;3: (4):403–8. |
[30] | Gray SG . Targeting histone deacetylases for the treatment of Huntington’s disease. CNS Neurosci Ther. (2010) ;16: (6):348–61. |
[31] | Jeong H , Then F , Melia TJ Jr , Mazzulli JR , Cui L , Savas JN , et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. (2009) ;137: (1):60–72. |
[32] | Khot A , Dickinson M , Prince HM . Panobinostat in lymphoid and myeloid malignancies. Expert Opin Investig Drugs. (2013) ;22: (9):1211–23. |
[33] | Richardson PG , Harvey RD , Laubach JP , Moreau P , Lonial S , San-Miguel JF . Panobinostat for the treatment of relapsed or relapsed/refractory multiple myeloma: Pharmacology and clinical outcomes. Expert Rev Clin Pharmacol. (2016) ;9: (1):35–48. |
[34] | Srinivas NR . Clinical pharmacokinetics of panobinostat, a novel histone deacetylase (HDAC) inhibitor: Review and perspectives. Xenobiotica. (2016) ;1–15. |
Figures and Tables
Fig.1
Pharmacokinetic profile of LBH589. A) Brain and plasma levels of LBH589 in C57/BL6 mice after single ip administration of 10 mg/kg. Levels were determined 0.08, 0.5, 1, 2, 4, 8, and 24 hrs post-dosing. n = 3, error bars = S.D. B) Brain and plasma levels of LBH589 in R6/2 HD mice after single i.p. administration of 10 mg/kg LBH589. Levels were determined after 2 hrs of dosing. n = 5, error bars = SE.
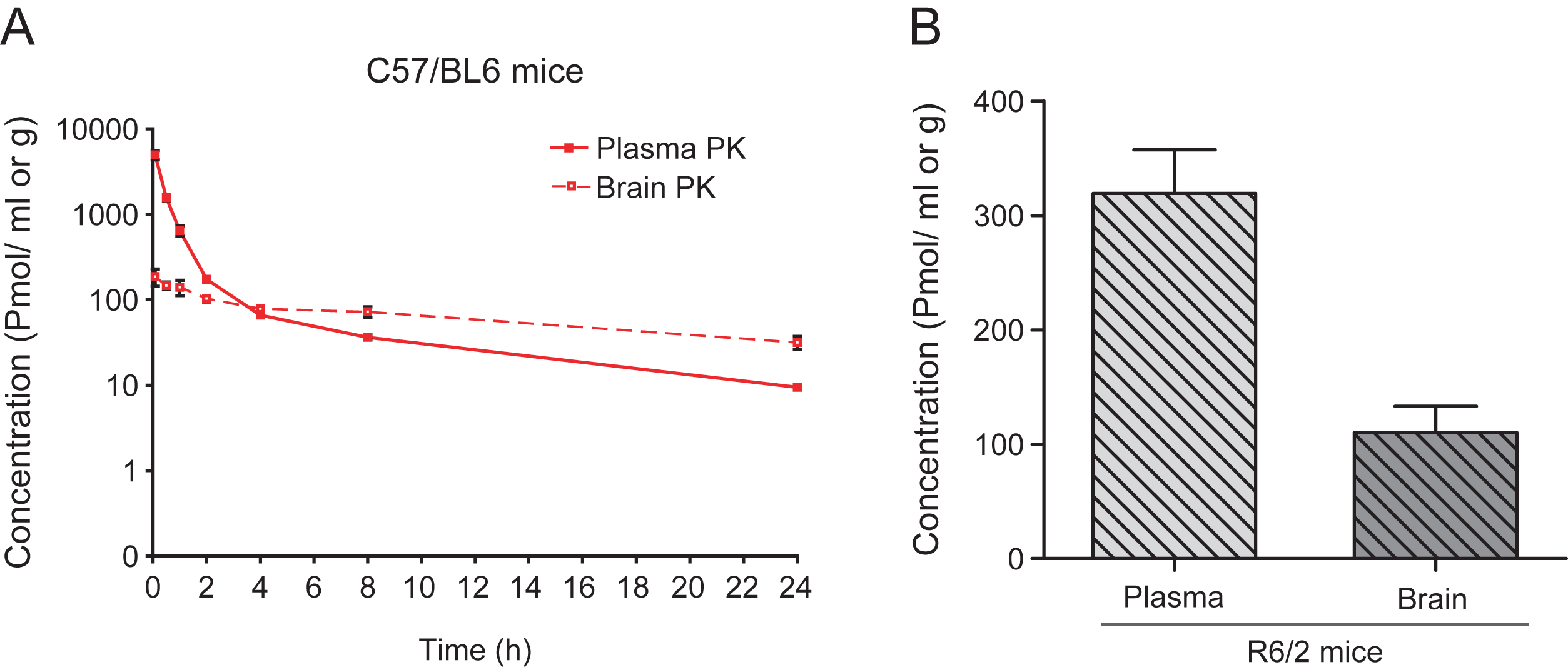
Fig.2
Pharmacodynamic response with LBH589 treatment. A-B) Western blots showing levels of histone H3 acetylation in cortical tissue (A) and muscle (B) of R6/2 mice treated with LBH589 or vehicle. Mice were treated with LBH589 at the dose rate of 20 mg/kg/three times per week for 3 weeks and acetylation levels were determined two hrs after the last injection. LBH589 treatment significantly increases total H3 acetylation in both brain and muscle tissue. GAPDH and Actin are used as loading controls and the level of AcH3 was normalized to total histone H3 level. n = 4-5. P value *p < 0.05; **p < 0.01. Error bars = S.E.
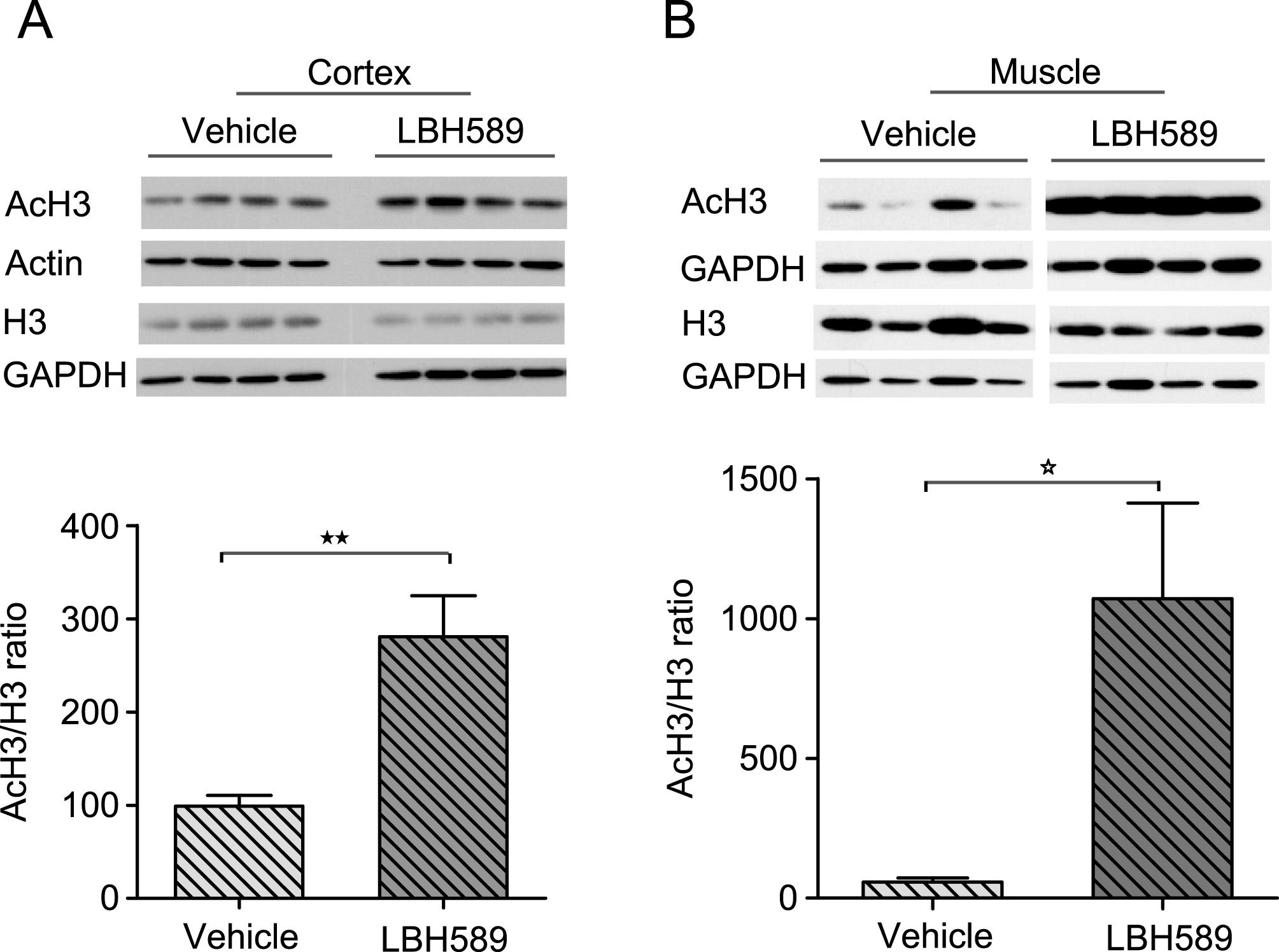
Fig.3
Efficacy of LBH589 in R6/2 HD mouse model. A-B. Bars: solid black = HD vehicle; light gray = low dose, dark gray = high dose, white = wild-type vehicle-treated. A) Higher dose (30 mg/kg) of LBH589 has a negative effect on body weight and a small negative effect of lower dose (10 mg/kg) was also observed at later stages of 11-12 weeks. B-C) LBH589-treatment does not improve the motor performance or striatal volume of R6/2 HD mice D) Stereological quantification of striatal neuronal cell body volume revealed small but significant reduction in neuronal cell body atrophy with the dose of 10 mg/kg. Error bars represent SEM, n = 9-10; **p < 0.01.
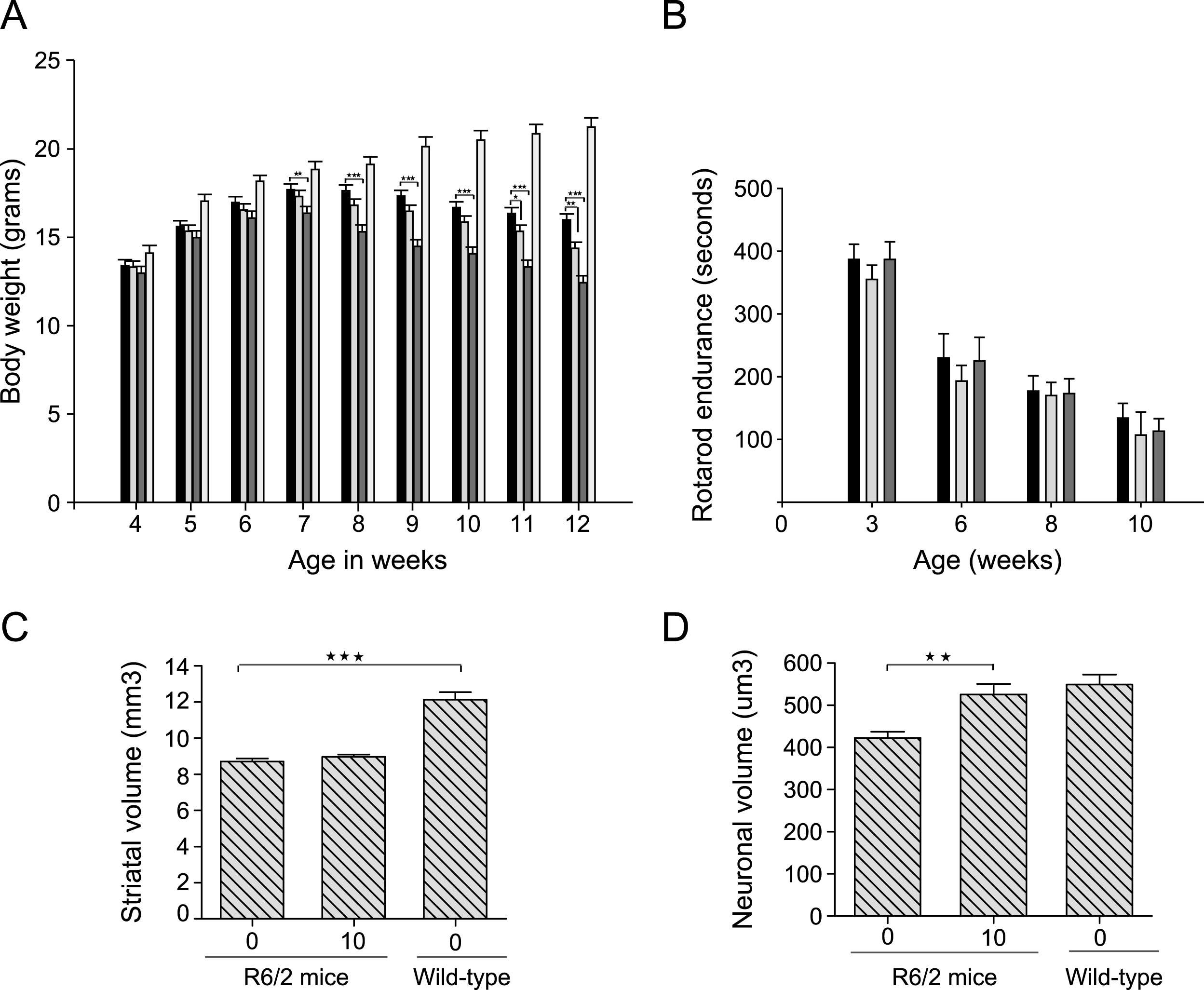
Fig.4
LBH589 rescues HD phenotype in CAG140 KI HD mice A-D. Bars: solid black = KI vehicle; light gray = LBH589 10 mg/kg and dark gray = wild-type vehicle-treated. A) Body weight data showed that LBH589 is well tolerated up to 18 weeks of age however a small negative effect of LBH589 on body weight was observed at later stages of treatment. B) LBH589 treatment ameliorates behavioral deficits in CAG 140 KI mice. Latency to fall was increased by 25, 30 and 36% at 6, 8, and 10 months of age. n = 10– 14 (C-D) Stereological quantification of total striatal volume and neuronal cell body volume. LBH589 treatment improves the total striatal volume of HD mice and rescues the shrinkage of striatal neuronal cell body volume. Data represent mean±SEM, n = 6 for KI mice and 4 for wild-type mice; *p < 0.05, **p≤0.01; ***p≤0.001.